1. Introduction
Drug discovery is the process of identifying new molecules with a certain therapeutic activity. This process is very expensive in terms of money and time. Translating basic research to the market (going through drug discovery, preclinical and clinical studies) takes tens of years and costs billions of dollars. The average cost to develop a new molecular entity is estimated to be
$1.8 billion and requires about 13.5 years [
1]. However, the usage of computational techniques at various stages of the drug discovery process could reduce that cost [
2]. Hence, computer-aided drug discovery/design (CADD) methods are becoming very popular and during the last three decades have played a major role in the development of therapeutically important molecules [
3,
4]. CADD techniques cover several aspects of the drug discovery pipeline, ranging from the selection of candidate molecules to the optimization of lead compounds. For instance, virtual profiling (VP) methods can predict the biological profile as well as mechanisms of action (MoA) of a certain molecule; molecular modelling techniques, such as docking and molecular dynamics (MD), can predict ligand–target interactions in terms of binding mode and/or binding strength, allowing discrimination between candidate compounds [
5,
6]; virtual screening (VS) methods are able to find analogues (similar molecules) for a given compound(s) and/or build compound libraries from an input molecule(s); hit to lead (H2L) optimization techniques are used to design new molecules, improving an existing compound; absorption, distribution, metabolism, excretion and toxicity (ADMET) prediction techniques are able to predict the physicochemical properties of a given compound, i.e., information that can be coupled to H2L techniques in order to design better and safer drugs before synthetizing them.
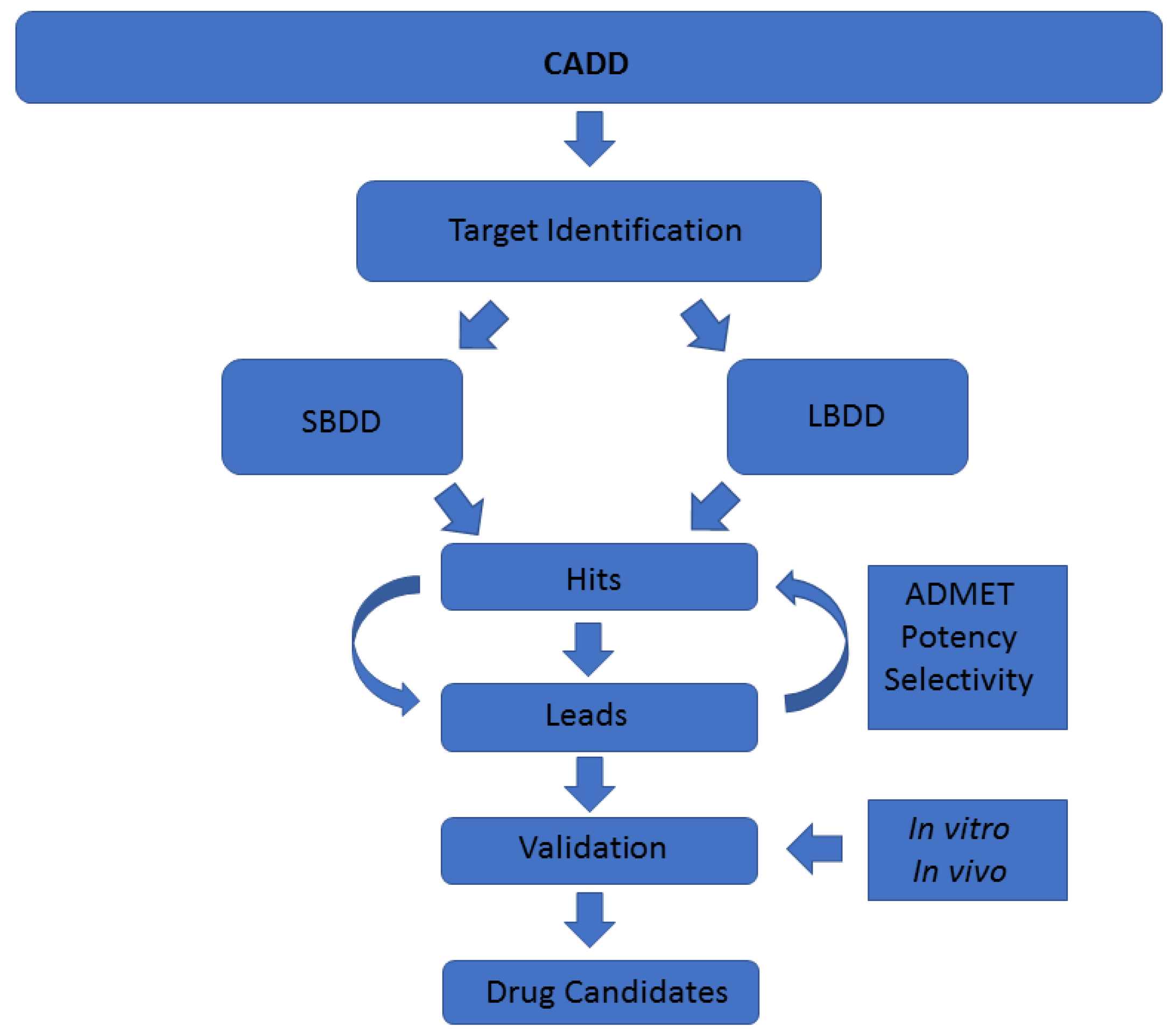
A common classification of these techniques is based on the nature of the input molecule. In this sense, there are two general types of CADD approaches: structure-based drug design (SBDD) and ligand-based drug design (LBDD). In SBDD, macromolecular three-dimensional (3D) target structures, usually proteins, are analysed with the aim of identifying compounds that could interact (block, inhibit or activate) with them. In LBDD, chemical compounds are analysed in order to, for instance, find chemical analogues, explore their biological and/or toxicological profile, or improve their physicochemical and pharmacological characteristics with the aim of developing drug-like compounds (
Figure 1) [
7,
8].
Historically, most new drugs have been designed from natural products (secondary metabolites) and/or from compounds derived from them [
9]. Natural products have thus been a rich source of compounds for drug discovery, and often, feature biologically relevant molecular scaffolds and pharmacophore patterns that have evolved as preferred ligand–protein binding motifs. The United States Food and Drug Administration (US FDA) revealed that between 1981 and 2010, 34% of those medicines approved were based on small molecules from natural products or direct derivates of them [
10,
11]. The identification of natural products that are capable of modulating protein functions in pathogenesis-related pathways is one of the most promising lines followed in drug discovery [
12]. Therefore, natural products constitute a huge source of inspiration in drug design [
13].
An example is Alzheimer’s disease (AD), a neurodegenerative pathology that constitutes the most common type of dementia (60–80% of the total cases), characterized by the presence of neurofibrillary tangles (NFT) primarily composed of abnormal phosphorylated tau and senile plaques (SP). Nowadays, despite its high incidence, there is still no specific treatment approved to cure this disease. Tau phosphorylation is regulated by a balance between tau kinase and phosphate activities. Splitting of this balance was considered to cause tau hyperphosphorylation and thereby its aggregation and NTF formation [
14,
15]. Due to that fact, inhibition of specific tau kinases or kinases involved in tau phosphorylation pathway, could be one of the key strategies to reverse tau phosphorylation and, ultimately, fight AD [
16].
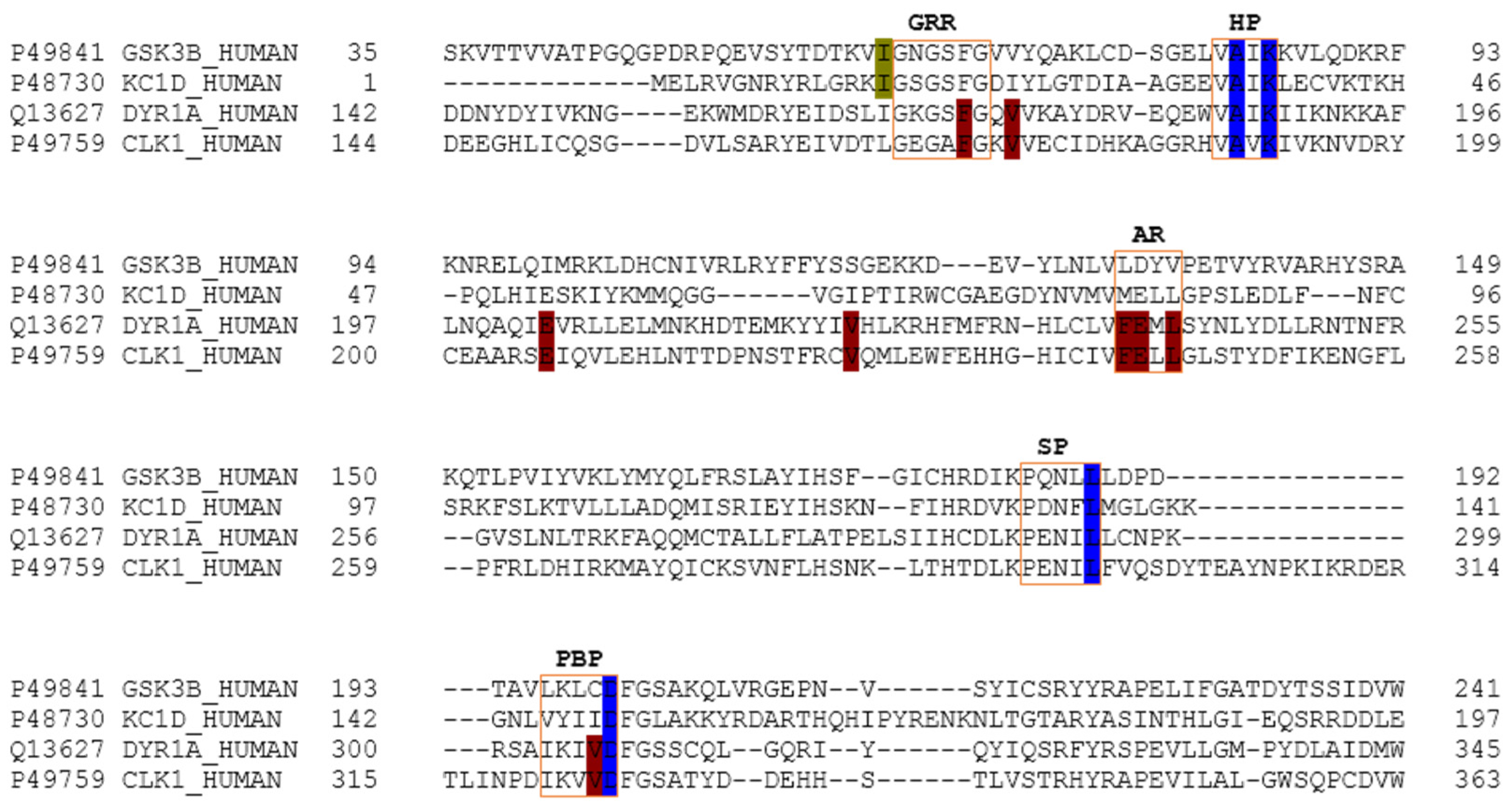
The main relevant protein kinases involved in tau phosphorylation have been grouped into two classes: tau protein kinases and dual specificity kinases. The first group contains proteins such as glycogen synthetase kinase-3 beta (GSK3β), that phosphorylates tau at different sites (specifically at 42 sites, 29 of them phosphorylated in AD brains) and casein kinase 1 delta (CK1δ), a non-proline-directed protein kinase (non-PDPK) that regulates the microtubule dynamics through tau phosphorylation at 46 sites (25 of them phosphorylated in AD brains). The second group contains proteins such as dual specificity tyrosine phosphorylation regulated kinase 1 (DYRK1) that self-catalyse their autophosphorylation and behave as serine/threonine kinase that phosphorylates tau and the transcription factor cyclic adenosine monophosphate-response element binding (cAMP-CREB), and an evolutionarily conserved group of dual specificity kinases cdc2-like kinases (CLKs), which play an important role in the regulation of ribonucleic acid RNA splicing and are involved in the pathology of AD by phosphorylating the serine residues in arginine-rich (SR) proteins [
14,
15,
17,
18,
19].
Among natural products, those of unexplored marine world origin are of great interest in the discovery of novel chemical structures, since they harbour most of the biodiversity of the world [
20,
21]. For instance, compounds from marine invertebrates may possess interesting pharmacological activities. Examples include Porifera, Cnidaria, Bryozoa, Mollusca and Tunicata [
22,
23]. However, although very interesting and useful from a pharmacological point of view, obtaining these compounds is difficult, both from technical and biological points of view; technically, because specimens have to be collected by hand using scuba diving or by trawling (both expensive, logistically difficult, and time consuming), and biologically, due to their marine habitats and due to the fact that they are usually unculturable [
23]. All these factors, together with the adequate implementation of the Nagoya Protocol and the bioavailability of marine natural products, result in CADD contributions being highly relevant, since no biological sample is needed to perform an in silico analysis [
24]. This also alleviates some of the marine drug discovery difficulties, such as the quantity of natural product necessary to be used in further clinical studies.
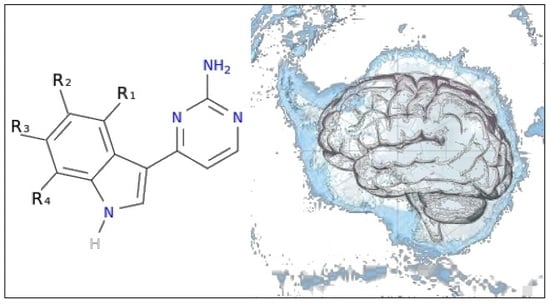
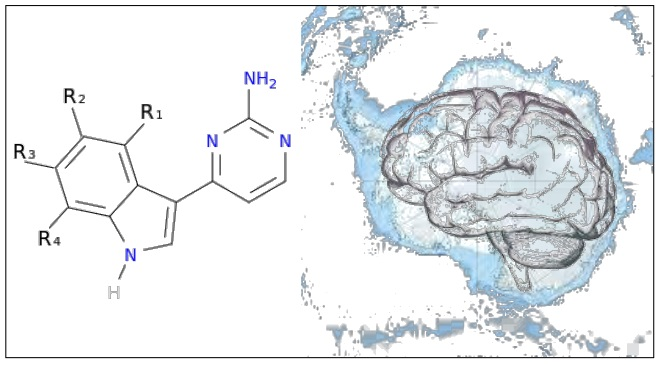
To exemplify and highlight the power of CADD techniques in marine drug discovery, as part of an ongoing study of bioactive marine molecules from benthic invertebrates, in this paper we evaluated and reported the inhibitory activity found in meridianins A–G (
Figure 2), a group of marine indole alkaloids consisting of an indole framework connected to an aminopyrimidine ring, isolated from specimens of the tunicate genus
Aplidium, against various protein kinases involved in AD.
3. Discussion
CADD techniques have an enormous potential in drug discovery, especially when they originate from marine natural products, as they do not waste natural resources. As mentioned, there are numerous different methodologies enclosed within the term CADD [
2,
4]. Usually the methodology is chosen based on its applicability, advantages/drawbacks, previous studies in the field, and also the expertise of the authors. In that sense, general methods such as docking, MD or ligand similarity searches have been developed, as well as more specific techniques such as disease or target models [
33,
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44]. Each technique requires a specific input and gives a specific output, aiming to solve one step of the drug discovery pipeline (
Figure 1). However, although individual CADD methods can provide insight and solve many questions, their power is their strength when combined, as we show here. With the techniques employed in this study, we have mostly covered the drug discovery process able to be coped computationally. The methodologies we show in this work, as well as the way and the order in which we have used them, are addressed to cover a plausible general pipeline, which in our opinion is of general interest regarding marine molecules discovery. In previous years, many resources have been invested in biodiscovery (for instance, European funded projects such as PharmaSea, MaCuMBA, SeaBiotech, BlueGenics or MicroB3) and some lead compounds have been designed, but a lot of information remains stored [
45,
46,
47,
48,
49]. Using CADD techniques, this information could be easily analysed and, potentially, employed to find drug candidates. In summary, we have shown how starting from a molecule, we were able to provide lead compounds (although in this case we provide insights to construct them instead of fully designed compounds) against a certain disease. In that sense, and as we have commented above, we exemplified the role of CADD tools applied to marine drug discovery in general, and in this particular case, analysing the role of meridianins in AD, even more specifically, against four protein kinases involved in its pathology.
The four protein kinases studied here were previously described by other authors as meridianin targets [
25,
32,
50,
51]. This constitutes an excellent validation of our computational, blind, approach to identify the biological profile of meridianins. However, although in the literature the possible anti-AD activity of meridianins was reported and several compounds have been designed from them [
25,
32,
50,
51], several aspects have not been taken into account and analysed, from a target-based (structural) perspective, as we have done here.
A common observed feature of protein kinases inhibitors is that most of them usually interact with the phosphate binding groove, in the innermost part of the pocket. This is a rich polar region, with groups such as arginine or aspartate, that consequently can create hydrogen bonds with small molecules acting as inhibitors [
52]. We observed that meridianins also show this trend, supporting their already mentioned general kinase inhibitory capacity. This, in addition to the fact that most of the meridianin binding residues are previously described as binders of known inhibitors, as well as the enzymatic assays that validated meridianin binding against the four studied kinases, also reinforce their tau protein and dual specificity kinase inhibitory capacity. As mentioned above, to exert this inhibitory capacity, meridianins show general binding trends against protein kinases in general and the studied targets in particular, but also specific features related to the nature of each of the targets. The understanding of these interactions (meridianin–target) and the identification of which of these characteristics are the most important to obtain good interactions is key in the design of meridianin-derived kinase inhibitors.
It was observed that for GSK3β, the best scored meridianins C, D, E, and F (
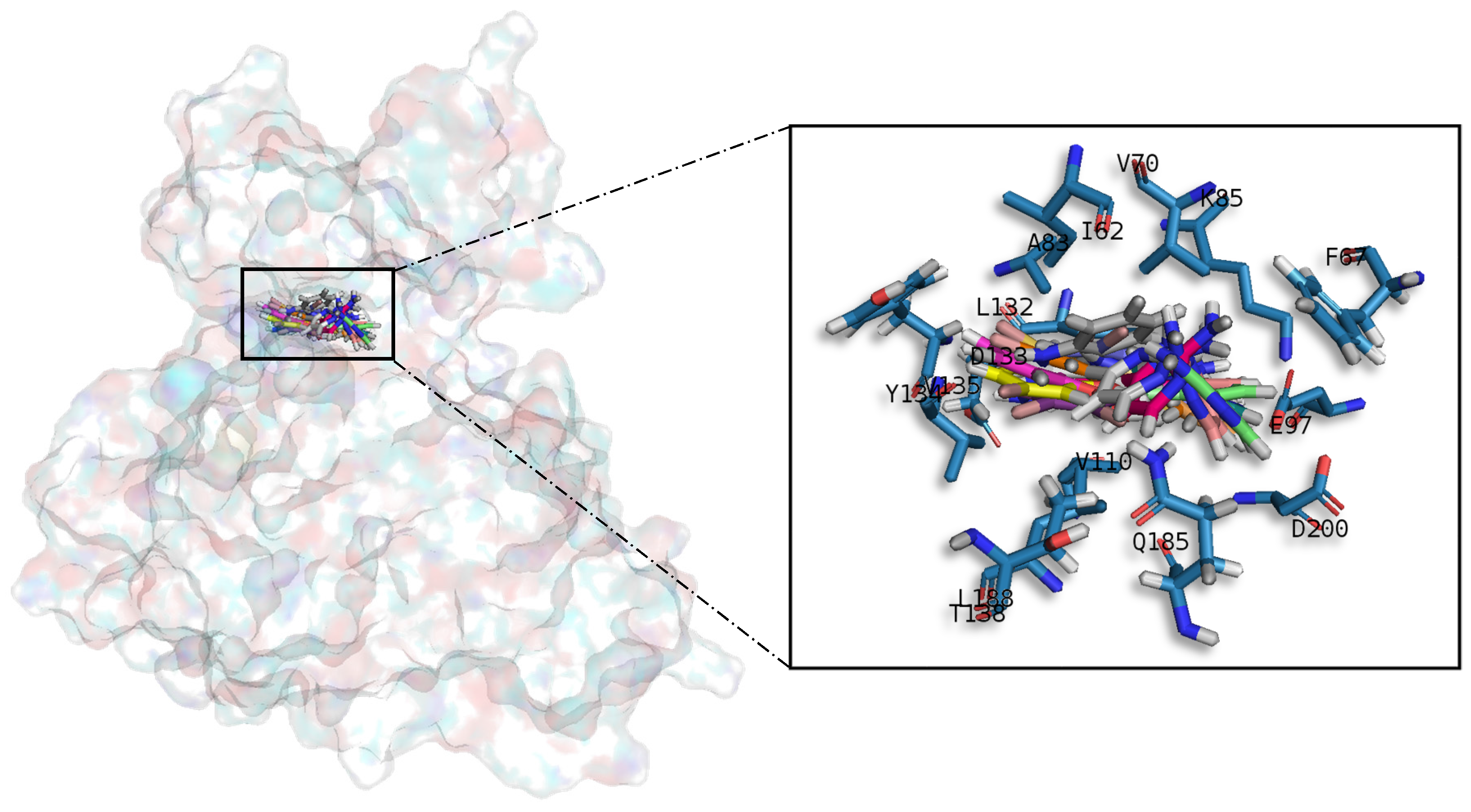
Table A1) establish hydrophobic contacts within the aminopyrimidine ring, revealing that this scaffold could be important in having optimal interactions. This highlights the fact that the most important interactions between GSK3β and meridianins were on the glycine rich loop and the hydrophobic and phosphate pockets. For CK1δ, analysing our in silico binding results, we observed that for the best scored meridianins C, D and F (
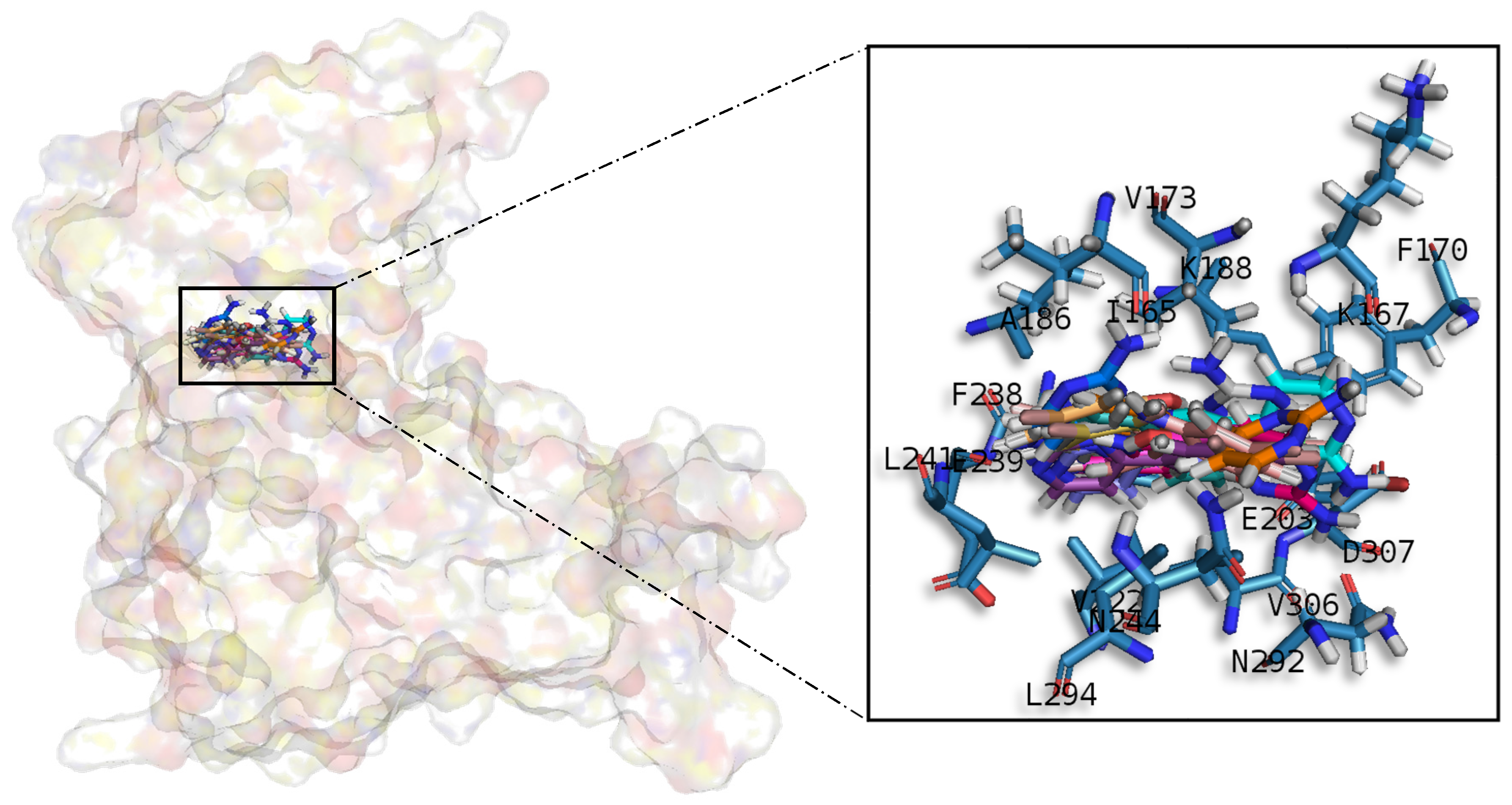
Table A1), it seems that to increase the affinity of the ligand on this receptor, the aminopyrimidine moiety should be oriented towards the top of the hydrophobic pocket at the N-terminal region. Also, key interactions were observed in the adenine and sugar-phosphate pockets. Regarding DYRK1A, meridianins mostly tend to be located over phosphate and sugar pockets as well as the adenine motif FEML rather than the glycine rich loop. Best scored meridianins B, C, E, and F (
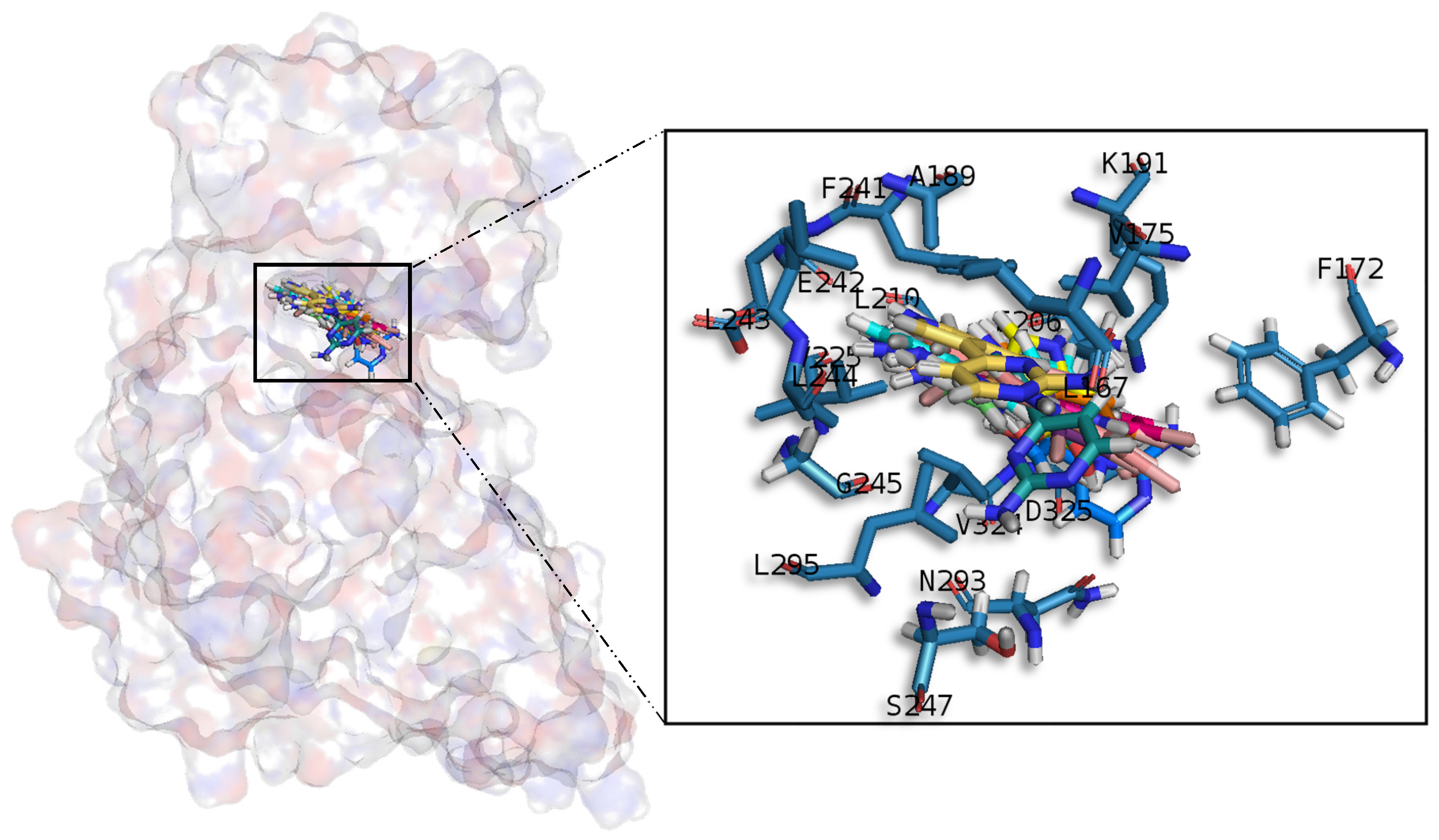
Table A1), share similar conformations but with different orientation with respect to the rest of the analysed meridianins, a fact that could be exploited for future developments together with meridianins preferential placement over the phosphate and sugar pocket. For CLK1, our molecular modelling studies have revealed that the best interacting meridianins B, C, D and F (
Table A1) tend to be located near the glycine rich loop and the sugar pocket.
In general, the orientation of meridianin indole scaffolds differs from one complex to another. Its preferential positioning is directed by hydrophobic interactions and steric effects, due to the aminopyrimidine ring position. In some models, it occupies hydrophobic region I, similar to many potent serine/threonine or tyrosine kinase inhibitors [
27]. It must also be mentioned that for GSK3β and CLK1, the preferred meridianin binding zones were located over the glycine rich loop (N-terminal). Nevertheless, over CK1δ and DYRK1A, meridianins tend to be located over the sugar and phosphate region (both over the C-terminal region), correlating this fact with the slightly highest interacting energy observed after in silico binding experiments (
Table A1). This could establish a new insight into future development of inhibitors.
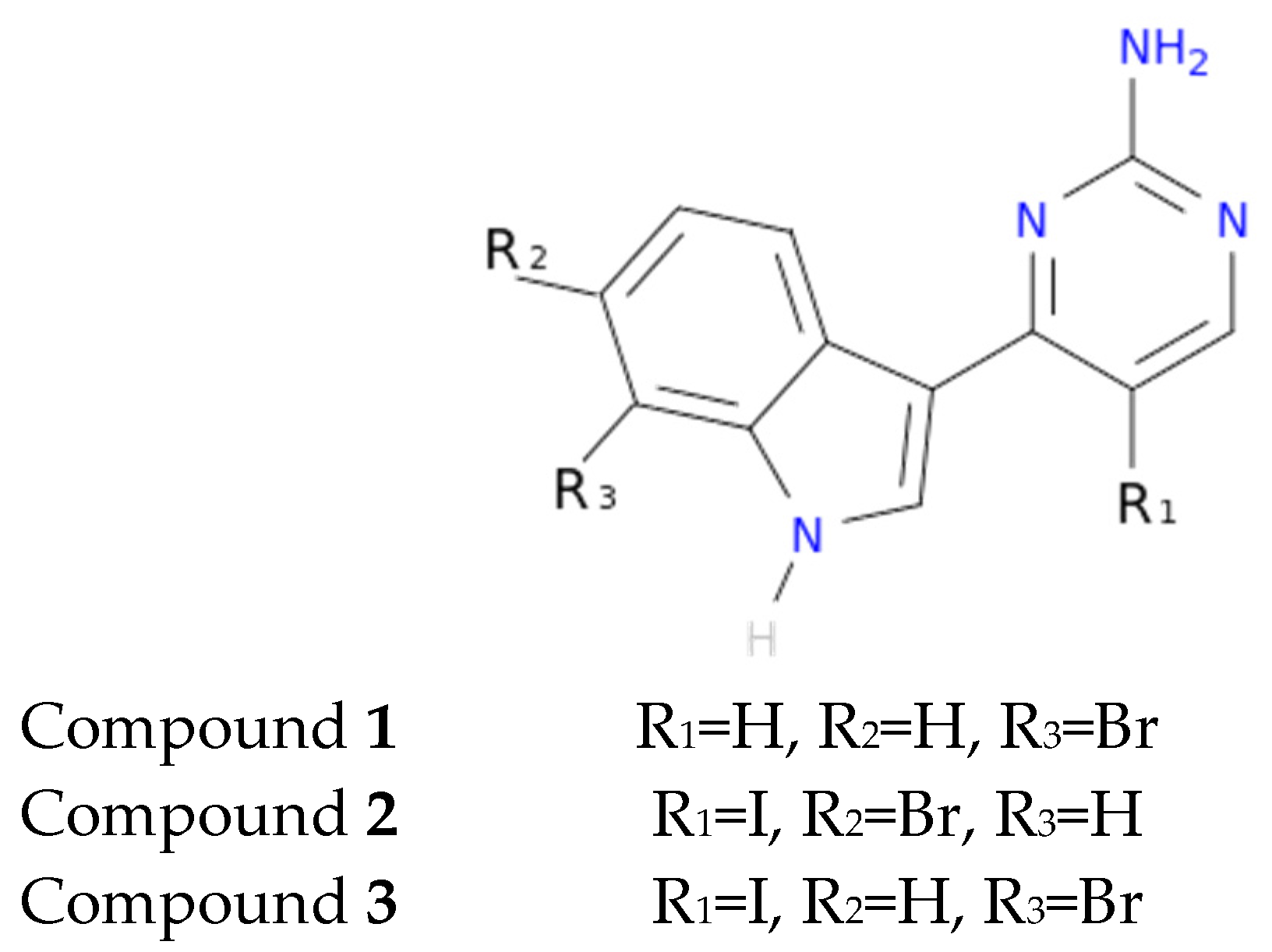
Another interesting feature observed with respect to the meridianin binding mode is the presence of bromine. When present, interaction energies seem to be higher. The perfect example is meridianin F, which has two Br at R
2 and R
3, and has the best interaction energies for each of the studied targets with respect to the rest of meridianins. Emphasizing this issue, a pattern was observed within the two classes of kinases. For CK1δ meridianins C (Br = R
2), D (Br = R
3) and F present the best interaction energies. In GSK3β, meridianins D and F are among the three best interacting compounds. On DYRK1A, meridianins B (Br = R
3), C and F are three of the four best interacting compounds and in CLK1, meridianins B, C, D and F are the ones that show the best energies. All these facts led us to hypothesize that Br on R
2 and R
3 on meridianins could be synonymous with potency and has to be taken into account for the design of new lead compounds against tau and dual-specificity kinases, in particular, and protein kinases in general. Interestingly, the most promising meridianin-derived compounds already designed (by Bharate and co-workers and Giraud and co-workers), are bromine-iodo derivates (compounds
2 and
3) and noniodinated bromine analogues (compound
1) (
Figure 9) [
25,
32]. This fact supports our hypothesis about the influence of Br in the potency of binding showed by meridianins. According to our binding results, the derived compounds do not interact with target kinases stronger than do the meridianins. Therefore, we hypothesized that to design more potent inhibitors, the presence of Br atoms is key, but it is not enough. Playing with the different orientations and binding residues implicated in the observed patterns in meridianins-kinase binding should be also taken into account.
As protein kinases are a wide family of proteins involved in many cellular events, being selective against the desired ones is key, probably even more important than having a potent inhibitor, to avoid undesired effects. In that sense, our results show that both meridianins and the compounds reported by Bharate and co-workers, as well as Giraud and co-workers, could bind to different protein kinases with a similar strength [
25,
32]. In addition to that, the reported selectivity of the derived compunds for DYRK1A and CLK1 respect to GSK3β and CK1δ is observed, but it is not extensible to all the tested kinases. Going deeply into the results (
Table A8), it could be observed that for IDH1 and PRKG1, the binding energies are slightly lower in comparison with the other targets. This fact is very relevant and could be explained because IDH1 is not a protein kinase. We put it in the pool of tested targets to see if out of the studied family, some selectivity could be observed. Regarding PRKG1, despite that it is a protein kinase member, the employed 3D structure contains an amino acid sequence that does not cover the kinase region. It was included to see what happened if despite being a protein kinase family member, the catalytic hinge region was not present. These findings allowed us to hypothesize that, despite meridianins do not show specific selectivity against any of the protein kinases tested, they do have a preferred binding to protein kinases. Moreover, this study validates the hypothesis that meridianins can act as protein kinase inhibitors. However, the low selectivity observed with respect to meridianins indicate that none of them is selective enough to properly act as AD therapeutic agent, even if able to inhibit the desired kinases. Although they could be a good starting point to design new drugs against AD, their selectivity should still be improved. To achieve that improvement, the presence of Br atoms is not enough. A rational design based on the structural differences and binding patterns observed along all meridianins should be carried out to obtain selective compounds that could have options to become an anti-AD drug. In that sense, the analysed derived compounds constitute an excellent example of how to improve meridianins to become therapeutic agents, but a new design is needed to overcome broader selectivity issues.
Potency and selectivity are important characteristics of a drug, but fulfilling certain ADMET requirements is also very important. The characterization of ADMET for the molecules being pursued as potential drug candidates is essential, as clinical failures of about 50% of the drugs under investigation are due to their inadequate ADMET attributes. In this regard, we have analysed the behaviour of all the studied meridianins and also the three compounds designed by Giraud and co-workers to evalute if the implemented modifications improve the properties of the meridianins (
Table A9,
Table A10 and
Table A11) [
25,
32].
Meridianins and the three derived compounds show a potentially high, oral and intestinal, absorbance as well as reasonable low skin permeability. Probably one of the most relevant findings is that any of the studied compounds is able to cross the BBB by itself, which is essential for a drug that should act in the brain. Good penetration was not shown in the CNS in general. In addition to CNS entrance, the Pgp that seems to play a role in amyloid beta (Aβ) transport across the BBB and its modulation (inhibition) has been designed as a mechanism to improve CNS pharmacotherapy [
53,
54,
55,
56]. Unfortunately, any of the studied compounds has been predicted as an inhibitor, but as a substrate, which reinforces their inability to cross the gate into the CNS. Also, in relation to distribution properties, high PPB probabilities were observed as well as a low VDss, which means these compounds will have a lot of difficulties in diffusing or traversing cell membranes.
These compounds are also able to interact with cytochrom P450, acting as inhibitors and even substrates of some isoforms, as described in the results. As it is well known that CYP450 drug metabolism can induce clinical effects, these properties should be carefully analysed in order to design lead compounds from the herein studied molecules [
57]. Moreover, toxicology predictors show that the studied molecules tend to have bad toxic effects, except meridianins A and E, for which no toxicity was predicted and the maximum tolerated dose increases with respect to the rest of the studied compounds.
Together, the obtained results suggest the necessity of performing a H2L optimization, in order to improve the absorption, distribution, metabolism and toxicity of the studied compounds, as well as their selectivity, with the aim of obtaining lead compounds able to become effective anti-AD drugs.
4. Materials and Methods
4.1. Virtual Profiling
VP techniques are computational tools aimed to elucidate the biological profile of a given molecule, for instance, therapeutic indications or targets of a chemical compound could be estimated. These techniques can be ligand- or target-based. Ligand-based approaches are able to automatically evaluate very large libraries or databases of compounds containing diverse information, for example, compound–target-bioactivities associations, using a chemical structure as a seed. As a result, similar molecules (restricted by a cut-off) are found and for instance, plausible targets to the input molecule selected. In this study, meridianin A was used as a seed. To run LBDD experiments, Cabrakan and Hurakan (Mind the Byte SL, Barcelona, Spain) software tools were employed [
58,
59]. Cabrakan is a two-dimensional (2D) ligand-based VP tool that compares molecules, through the use of 2D fingerprints, over a reference database and the assignment of biological activity. It allows the identification of similar chemical compounds (analogues) to the input molecule. Hurakan is a three-dimensional (3D) VP tool that compares a query molecule with the structures present in a reference database using Comparative Molecular Similarity Indices Analysis (CoMSIA) fields on a 3D grid. Hurakan can compare molecules according to their relationship with their environment, thus obtaining biomimetic compounds with different chemical structures. ChEMBL, which contains around 1,300,000 chemical compounds with detailed information including target data, was employed as the reference database [
60]. A target was counted once when it appeared as both 2D and 3D hit during ligand-based VP experiments.
Here, we have employed similarity search based techniques, as they are simple, fast and accurate. However, they have the limitation imposed by the reference database employed. If there are no similar molecules to the input compound in the database, no results will be returned. This limitation is shared with other LBDD techniques such as quantitative structure–activity relationship (QSAR) or quantitative structure–property relationship (QSPR). The choice of these software tools and not another ones is based basically on the deep knowledge we have about the algorithm, the database and their performance.
Target-based approaches are able to, through knowledge of the 3D structures, evaluate huge databases that contain cavity information of these structures and after a binding site identification, docking calculations can be performed. As a result, the binding energy of every possible interaction is returned, which allows the classification and prediction of the best targets. In this study, meridianin A was used as a seed. Ixchel (Mind the Byte SL, Barcelona, Spain) is a structure-based VP tool that performs docking calculations of a molecule (spatial data file (SDF) or simplified molecular input line entry specification (SMILE) file) against an in-house developed database comprising almost 9000 protein cavities (binding-sites) curated from Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB) according to UniProt Knowledgebase (UniProtKB) human entries [
61,
62,
63].
To run target (or virtual) profiling experiments related to SBDD, docking is the most used technique. MD simulations or related techniques could be also employed, but they are much too computationally expensive for these kinds of techniques, with docking the preferred option. There are several variants of the docking techniques, but as we have commented for LBDD, the main limitation is the reference database. In our case, we have selected a technique whose algorithm is well known and it also incorporates a curated database of which we have a deep understanding. A deep knowledge of the employed techniques is basic and based on that, we have selected Ixchel to run our experiments.
4.2. Structure Modelling
The meridianin structures were modelled from the 2D chemical structure published by Núñez-Pons, Avila and co-workers [
26]. The three meridianins derived compounds used for the selectivity test were modelled from Giraud and co-workers and Bharate et al. [
25,
32].
Prior to any calculation, all the structures of the selected targets, for the binding and the selectivity analysis, were modelled from their crystal structures available from the Protein Data Bank (RCSB PDB). All of them represent human targets. As obtaining good structures is crucial, the best 3D structures were selected; the structures and chains that cover the maximum amino acid region sequence, in general, and the binding region of each of the selected targets in particular.
GSK3β was modelled from the crystallographic 3D structure with a PDB ID 3PUP that contains the crystallographic ligand OS1. It is stored in the PDB database as a homodimer, but only chain B was considered for further studies since GSK3β biological assembly is in monomeric form [
31]. CK1δ was modelled from the 3D crystallographic structure corresponding to the entry 4KBK that contains the crystallographic ligand 1QG. Only chain B, since it is naturally a monomer, was considered for further studies [
64]. DYRK1A was modelled from the crystal 3D structure with a PDB ID 4AZE that contains the crystallographic ligand 3RA. In the PDB database, we found 3 chains (A, B and C), but only chain A was considered for further studies as DYRK1A biological assembly is in a monomeric form [
52]. CLK1 was modelled from the crystallographic 3D structure with a PDB code 2VAG with V25 as a crystallographic ligand. As this protein is naturally a monomer, there is only one chain in the PDB database, so further studies were performed against chain A [
52].
To test selectivity, for all the PDB crystallographic structures selected, chain A was used in all cases. Structures were modelled from their respective crystallographic 3D structure: Fibroblast growth factor receptor 1 (FGFR1); 1AGW containing SU2 as a ligand, cAMP-dependent protein kinase catalytic subunit alpha (PRKACA); 2GU8 containing 796 as a ligand, hexokinase-2 (HK2); 2NZT containing BG6 as a ligand, dual specificity mitogen-activated protein kinase 1 (MAP2K1); 3DY7 containing ATP as a ligand, phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform (PIK3CG); 3IBE containing L64 as a ligand, PRKG1; 3OCP containing CMP as a ligand, serine/threonine-protein kinase N1 (PKN1); 4OTI containing MI1 as a ligand and one non protein-kinase IDH1; 4I3K containing NDP as a ligand.
To test the binding of meridianins and their selectivity, molecular modelling experiments were performed using the 3D structural models of meridianins A–G, and the models generated from the crystallographic structures available in the PDB (PDB ID 3PUP, 4KBK, 4AZE and 2VAG, respectively) and the PDB ID structures 1AGW, 2GU8, 2NZT, 3DY7, 3IBE, 3OCP, 4OTI and 4I3K, respectively.
4.3. Docking Calculations
Docking calculations constitute a simulation method, which predicts the preferred orientation of one molecule (ligand) to a second (target). When only the movements of the first molecule are allowed, the docking is considered classical or rigid; when both molecules are allowed to move, docking is considered flexible. Generally, docking, without any other specification, refers to classical (rigid) docking [
7]. Docking, in the context of small-molecule drug discovery, concerns the study of binding process of small molecules (ligands) and targets (proteins), i.e., a candidate binding mode (pose) is predicted when ligand and receptor bind to each other. Scoring functions allow us to classify and rank, based on their calculated binding energies, the most favourable pose. In that sense, flexible docking has advantages over the rigid version of the technique. The dynamics is an intrinsic characteristic of proteins, necessary to carry out any of their functions. Flexibility incorporation within the binding mode prediction is key to obtain results capable of being correlated with experimental data. However, not all are advantages, as the predicted binding energies could worsen. The inclusion of additional degrees of freedom to simulate protein flexibility could increase the difficulty of accurately predicting the free energy of binding. This complication could arise because more contributions to the free energy must be considered, for instance, the interaction between flexible residues and the core of the protein, and typically, these additional contributions also introduce additional inaccuracies [
65].
Another option to add flexibility is the post-processing of docking results, which means, for instance, docking validation and/or refinement by MD simulations. Rigid docking can predict the optimal placement of a ligand within the binding site of a receptor, but not all the key interactions between the ligand and receptor are usually depicted accurately. Hence, MD simulations can optimize the predicted binding mode and also check the stability of the docked complex, as a bad docking pose will generate an unstable MD trajectory, during which the ligand could even leave the binding site [
34,
36]. In this study, we have employed a pipeline aimed to simulate a flexible docking protocol in a similar way to other studies reported in the literature, in that we post-processed the obtained docking poses [
66]. We selected this approximation as this two-step protocol constitutes a (probably the most) practical and convenient approach to address the docking problem [
67]. It is in general less computational expensive and provides the results that we need in an accurate way, comparable to “real” flexible docking methodologies (such as ensemble-based or flexible induced-fit docking). In general, using MD as a post-processing tool, a smaller fraction of the conformational space is usually covered, but without the several limitations that affect sampling and scoring algorithms for docking.
All docking calculations were performed using Itzamna and Kin software tools (Mind the Byte SL, Barcelona, Spain) [
68,
69] to perform classical and blind docking calculations, respectively. Itzamna is used to carry out docking calculations and needs the structure of the molecule to dock, as well as the cavity where it should be placed as an input. Kin is a software tool designed to perform blind docking calculations. It involves a cavity search and a (best) cavity selection prior to performing the binding calculation; a difference of Itzamna is that the docking cavity is given as an input to the calculation. When the employed crystal structures were co-crystallized with a ligand, the cavity defined by the ligand was employed. As mentioned above, the modelled structures of the meridianins and the selected targets were used. Two runs were carried out for each calculation to avoid false positives.
Results obtained from docking calculations were ranked based on their calculated binding affinities, and the best poses summarized in
Table A1 and
Table A8.
4.4. Molecular Dynamics Simulations
One of the principal tools in the computational studies of biomolecules are MD simulations, a theoretical method for studying the physical movements of atoms and molecules. MD calculates the time dependent behaviour of a molecular system, which means that atoms and molecules are allowed to interact for a fix period of time, giving a view of the dynamic evolution of the system.
Short (1 nanosecond (ns)) MD simulations were performed using NAMD program version 2.11 over the best-docked complexes, which were selected based on ΔG
bind [
70]
. The Amber ff99SB-ILDN and the General Amber Force Field (GAFF) set of parameters were employed for modelling receptors and ligands, respectively [
71,
72]. The election of these force-fields was based on the fact that both have been extensively tested, being two of the most used for protein and protein-ligand simulations [
71,
72,
73,
74]. It has been shown that ff99SB-ILDN correlates consistently well with experimental data, and the GAFF force-field can conveniently and quickly produce reasonable ligand (especially organic molecules) parameters. Moreover, as amber force-fields, both are compatible, giving combined satisfactory results in several studies. Ligand GAFF parameters were obtained using Antechamber, whereas the receptor structures were modelled using the leap module of Amber Tools [
75,
76]. Simulations were carried out in explicit solvent using the TIP3P water model with the imposition of periodic boundary conditions via a cubic box [
77]. Electrostatic interactions were calculated by the particle-mesh Ewald method using constant pressure and temperature conditions. Each complex was solvated with a minimum distance of 10 Å from the surface of the complex to the edge of the box. Temperature was kept at 300 Kelvin (K) using a Langevin Piston barostat. The time step employed was 2 femtoseconds (fs). Bond lengths to hydrogens were constrained with the SHAKE algorithm [
78]. Before production runs, the system was energy minimized. Next, the solvent surrounding the protein was equilibrated at the target temperature using harmonic position restraints on the heavy atoms. Finally, the system was submitted to a slow heating-up phase, from 0 to 300 K. For the production run, all position restraints were removed.
4.5. Molecular Mechanics/Generalized Born Surface Area (MM/GBSA)
The so-called reweighting techniques are computational approaches to estimate the alchemical free energy of interaction (ΔG
bind) between small ligands and biological macromolecules. In the literature, MM/GBSA is usually employed to estimate ligand-binding affinities based on docking or MD simulations to get a more realistic view of the interaction of docked complexes. The obtained energies are more realistic than the docking interaction values, allowing a better ranking of the analysed compounds, although they cannot be biologically comparable. In our case and following similar approaches, we applied reweighting techniques, specifically MM/GBSA, over the generated MD trajectories for post-processing docking results [
34,
66,
79].
MM/GBSA rescoring was performed using the MMPBSA python algorithm contained within the Amber Tools suite [
80]. The snapshots generated in the 1 ns MD simulation were imputed into the post-simulation MM/GBSA calculation of binding free energy. MM/GBSA was chosen over other techniques such as molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA), linear interaction energy (LIE), thermodynamics integration (TI) or free energy perturbation (FEP) because of its good balance between accuracy and computational cost.
Rigorous thermodynamic pathway approaches, such as TI or FEP, provide more accurate predicting binding free energies, whereas LIE, MM/GBSA and MM/PBSA constitute the so-called end-point methods that in general are less accurate. Each of these methods has its own strengths and limitations, and their computational requirements and speed are inversely correlated with their accuracy. TI and FEP, which outperform end-point approaches, are very useful, especially for ranking molecules inside a chemical series. Consequently, and regardless of their computational cost but given the computational advances, these techniques are gradually being more frequently used in the drug discovery pipeline, especially in guiding lead optimisation. However, in this study, our aim is not to provide a detailed library of lead compounds, and thus we have employed a less rigorous, but very popular approach in SBDD, alternative as the MMGBSA approach. The main problem of these techniques could be that the efficacy of the method is usually system dependent. However, it is generally accepted that they outperform docking results, so a better ranking of the analysed compounds will be always obtained, although, as commented above, the obtained binding energies could be far from being experimentally comparable.
4.6. Interaction Analysis
To analyse the key residues of the active site involved in the inhibitor binding, we examined the obtained binding modes after molecular modelling studies with already known binders of each of the targets. These binders (residues that have been revealed as necessary for the binding of known substrates/inhibitors) were identified through an evidence-based interaction analysis. It was carried out through a bibliographical search plus a database analysis. The bibliographical search was conducted using several studies in which inhibitors against the selected kinases were identified describing each compound binding mode [
25,
31,
32,
50,
51,
52,
64,
81,
82]. The database search was done using an in-house, recently constructed database. It was built by crossing ChEMBL and the RCSB PDB [
62], and it contains all PDB structures per UniProtKB ID with active compounds (by now there are only PDBS with compounds not competing against cofactors). Moreover, the database also contains the residues to which each active compound (per PDB) is bound. Thus, it allows the user, after docking or an MD calculation, to easily check whether the analysed molecules behave as a binder.
4.7. Sequence Analysis
The four targets were aligned using the UniProtKB clustal omega interface from the amino acid sequence associated with each UniProtKB entry.
4.8. Selectivity Analysis
Docking calculations of meridianins, as well as the three selected compounds (derived from them and described in the literature), against twelve protein kinases were performed. These meridianins derived molecules were obtained from the papers of Bharate et al. and Giraud et al. [
25,
32], and have shown interesting inhibitory concentration (IC
50) values in the micro and sub-micromolar range, and a good selectivity for DYRK1A and CLK1. We selected them to see how the selectivity was taken into account in the design of these compounds as they strongly resemble the original meridianins scaffolds that we suspect are not selective enough.
To test the selectivity, we choose seven protein kinases, specifically, FGFR1, PRKACA, HK2, MAP2K1, PIK3CG, PRKG1 (for which the selected crystal structure do not contain the catalytic hinge), PKN1 and one non-protein kinase, IDH1. Thus, we tested if the selected compounds are selective between different protein kinases, belonging to different subfamilies, and between protein and non-protein kinases. Moreover, we explored if without the catalytic hinge, binding could be produced.
4.9. ADMET Properties Prediction
For the meridianins and the three derived compounds, ADMET properties prediction was carried out using proprietary machine-learning (ML) models and the pkCSM webserver [
83,
84]. The proprietary ML models covered logS (molecular aqueous coefficient), logP (octanol/water partition coefficient), Caco2 permeability, BBB penetration and PPB. The first two models were generated by super vector regression (SVR) techniques and the last three employed supper vector machines (SVM). For training and testing the models, Chembl (logS, logP, Caco2) and Huuskonen (logS) datasets were employed, and for BBB and PPB, the datasets described by Muehlbacher et al. and Zhu and coworkers [
85,
86,
87]. The pkCSM webserver allows the prediction of PK properties based on (I) compound general properties (including molecular properties, toxicophores and pharmacophore) and (II) distance-based graph signatures. Given an input molecule, both sources of information are used to train and test machine learning-based predictors. The webserver is composed of 28 (not all employed in this work) regression and classification ML models that have been generated and trained against 30 datasets (described at Pires et al.) [
84].
The use of proprietary models, some of which are also covered by pkCSM, is because these methods, similar to other such as VS or VP, strongly rely on the employed reference dataset. As we have a deeper knowledge of our methods, we prefer to use them when possible. Only for Caco2 did we employ both models, ours and the pkCSM model, because for two compounds, our model is not good enough to make a reliable prediction (they are out of the applicability domain as they are too different with respect to the molecular fragments contained in the dataset employed to generate and train the model. If less than 90% of the molecular fragments in that the input molecule can be discomposed are not in the database, the prediction is not done). pkCSM predicted properties for all the compounds; however, it does not indicate if a prediction is out of the applicability domain.
In summary, we have analysed 21 ADMET properties, 5 of which were studied with our proprietary ML models and 17 with pkCSM. One of these properties, Caco2, was analysed twice using both our proprietary model and the pkCSM model.
4.9.1. Absorption Properties
Caco2 permeability, LogS, intestinal absorption (human), P-glycoprotein substrate, P-glycoprotein I/II inhibitor and skin permeability. Caco-2 permeability is used to predict the absorption of orally administered drugs. A high permeability is assessed when the predicted valued is >0.90 for the pkCSM model, or high (H), in the proprietary model. LogS reflects the solubility of the molecule in water at 25 °C and also reflects the bioavailability of a given compound; it is represented by the logarithm of the molar concentration (log mol/L). Intestinal absorption indicates the portion of compounds absorbed through the human intestine; a molecule with an absorbance (intestinal absorption) of less than 30% is considered to be poorly absorbed. Pgp acts as a biological barrier by extruding toxins and xenobiotics out of cells, although it could have other, transport mediated, functions in certain tissues and organs. The predictor assesses whether a given compound is likely to be a substrate of Pgp. Pgp I and II inhibitors have significant PK implications for Pgp substrate, and the predictor will determine the inhibitory effect of a given compound against Pgp I/II, which could have advantages that can be exploited therapeutically, or result in contraindications. Skin permeability predicts if a given compound is likely to be skin permeable (logKp > −2.5).
4.9.2. Distribution
LogP, VDss, PPB, BBB and CNS permeability. LogP allows us to estimate the distribution of a drug within the body (lipophilicity). VDss, which is the theoretical volume that the total dose of a drug would need to be uniformly distributed to give the same concentration as in blood and plasma, is considered low if log VDss <−0.15 and high if >0.45 (the higher the VD, the greater the drug distribution in tissue rather than plasma). PPB estimates the probability (>90% is considered high) that a given molecule binds to a plasma protein, the less bound a drug is, the more efficiently it can traverse cell membranes or diffuse. BBB permeability describes the ability of a drug to cross into the brain. The predictor describes whether a compound is able to cross the BBB. CNS permeability measures blood brain permeability surface-area (logPS), and it is similar to BBB but more direct, as it lacks the systemic distribution effects that may distort brain penetration. Compounds with a logPS >−2 are considered to penetrate CNS, while those with logPS <−3 are considered unable to penetrate.
4.9.3. Metabolism
CYP450. Cytochrom P450 isoforms are important detoxification enzymes in the body and are essential for the metabolism of many medications. Drugs can be inhibitors of CYP450, blocking its metabolic activity, or can be metabolised (substrate) by them. CYP metabolism predictor assess whether a given molecule is likely to be metabolised or not and act as inhibitor of specific isoforms of CYP450; a specific inhibitor of CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 and/or substrate of CYPD26 and CYP3A4.
4.9.4. Excretion
Renal OCT2 substrate and Total Clearance. OCT2 is a renal uptake transporter that plays an important role in disposition and renal clearance of drugs and endogenous compounds. The OCT2 substrate predictor indicates if a given molecule is likely to be an OCT2 substrate, which provides not only clearance-related information but potential contraindications. Total clearance is related to bioavailability and is also important for determining dosing rates to achieve steady-state concentrations, and the predictor measures their value in log(mL/min/kg).
4.9.5. Toxicology
MRTD, AMES toxicity, hepatotoxicity, skin sensitization, hERG I/II inhibitors. MRTD provides an estimated of the toxic dose threshold of chemicals in humans, and results less than or equal to 0.477 log(mg/kg/day) are considered low, and high when greater than 0.477 log(mg/kg/day). AMES toxicity indicates if a compound could be mutagenic and therefore may act as a carcinogen. hERG I and II inhibitor predictors determine if a given compound is likely to be a hERG I/II inhibitor as the inhibition of potassium channels encoded by hERG could result in fatal pathologies (for instance it is the principal cause of the development of acquiring long QT syndrome, fatal arrhythmia) and the withdrawal of many substances from the pharmaceutical market. Hepatotoxicity predicts if a given molecule is likely to be associated with disrupted normal function of the liver. Skin permeability predicts if a given compound is likely to be associated with skin sensitisation.
4.10. Graphical Representations
Graphical representations of protein-ligand complexes were prepared using PyMOL version 1.7 [
88] and PLIP version 1.3.0 [
89].
5. Conclusions
Meridianins can be classified as kinase inhibitors and can be used as a starting point to design and develop novel anti-AD drugs. It has been demonstrated, in silico and in vitro, that they are able to bind specific tau (GSK3β and CK1δ) and dual-specificity (DYRK1A and CLK1) protein kinases. However, they are not selective enough to constitute a therapeutic treatment against AD by themselves. In fact, as they are demonstrated to be protein kinase inhibitors, they could probably inhibit several kinases involved in different diseases [
90]. In any case, they could serve as a starting scaffold to design new anti-AD drugs. To achieve that, a rational design taking advantage of the differences found in the binding patterns against different protein-kinases subfamilies, has to be carried out. In that sense, the presence of Br on R
2 and the R
3 position over the meridianin indole scaffold could be synonymous with potency. Besides, it seems that exploiting the C-terminal region (sugar and phosphate pocket) rather than the N-terminal side, could increase the strength of the interactions exerted by meridianins, and probably the potency shown by the designed compounds. However, although potency is important, and maintaining the presence of Br seems to be fairly accomplished [
25,
32], the selectivity between protein-kinase subfamilies is a crucial point to design proper anti-AD drugs, and even anti-cancer drugs. Meridianinsare not selective enough and should be improved to gain functionality and applicability. In addition, their measured ADMET properties indicate that they should be optimized in order to become a drug or at least a drug-lead compound. Therefore, the above-mentioned rational design in order to improve the potency and selectivity of meridianins should include H2L optimization cycles. The showed toxicity should be removed, and compounds interaction with Cytochrom P450 carefully analysed and, given the case, eliminated or modulated. Moreover, their distribution properties should be improved, lowering the PPB and VDss, to be able to diffuse and penetrate into cells easily. Besides, a mechanism to cross the BBB should be found and in that sense, modifying each compound to be Pgp inhibitors could be a possible strategy, although there are other mechanisms to overcome the BBB, including other protein binding and nanodelivery, that could be also exploited [
91,
92,
93].
Regarding meridianins specifically and CADD methods in general, we can conclude that these techniques, despite their drawbacks, are very helpful in drug discovery, constituting a powerful tool that could save time and money in experiments. Our study with meridianins is an example of this, since we have been able to find plausible targets, that in the case of AD and cancer we have already validated through the literature. The key role that these techniques could have in drug discovery is even higher for the discovery and development of marine drugs, since no sample is needed to run these virtual experiments. Moreover, since these methods could point out the best direction to follow and in which targets expand the low sample amount that usually is available, these are crucial technologies to maximize the success of marine prospection, as well as to protect biodiversity.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}