
The Role of Spongia sp. in the Discovery of Marine Lead Compounds



Abstract
:
1. Introduction
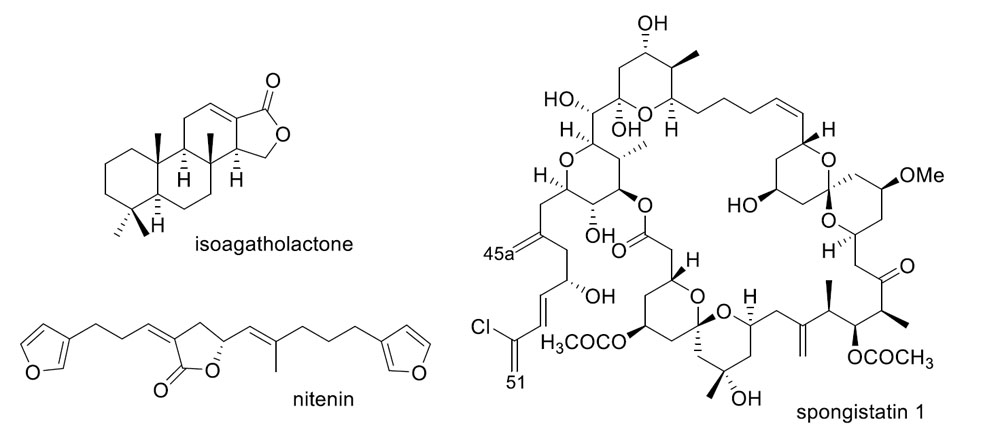
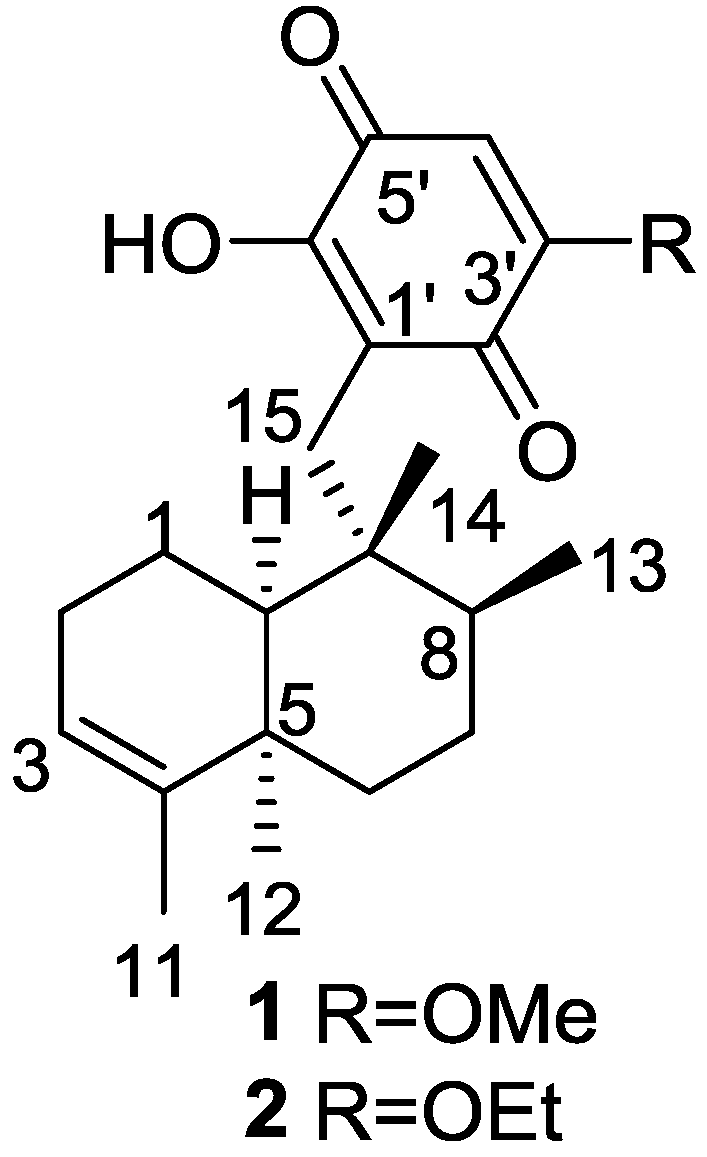
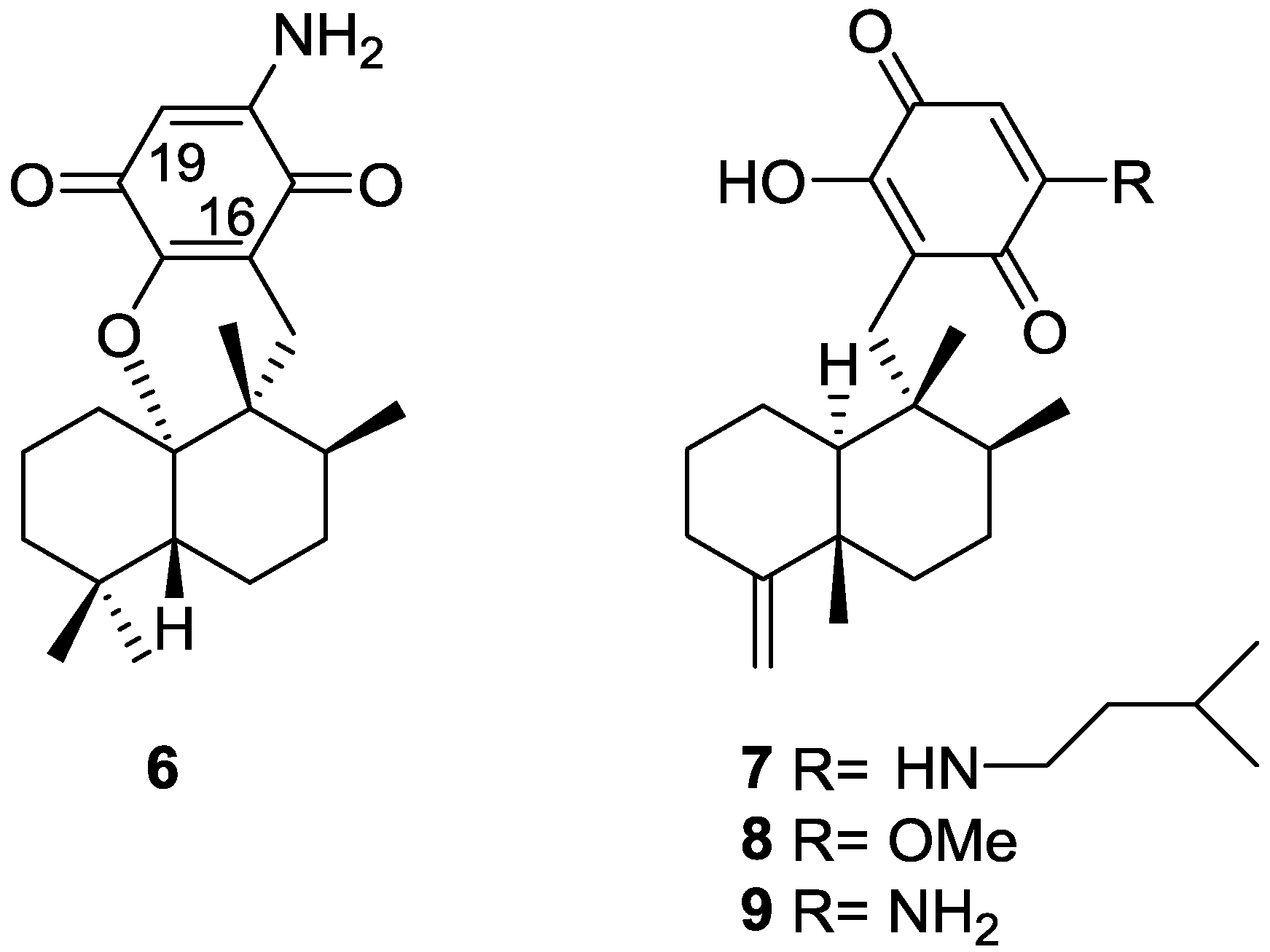
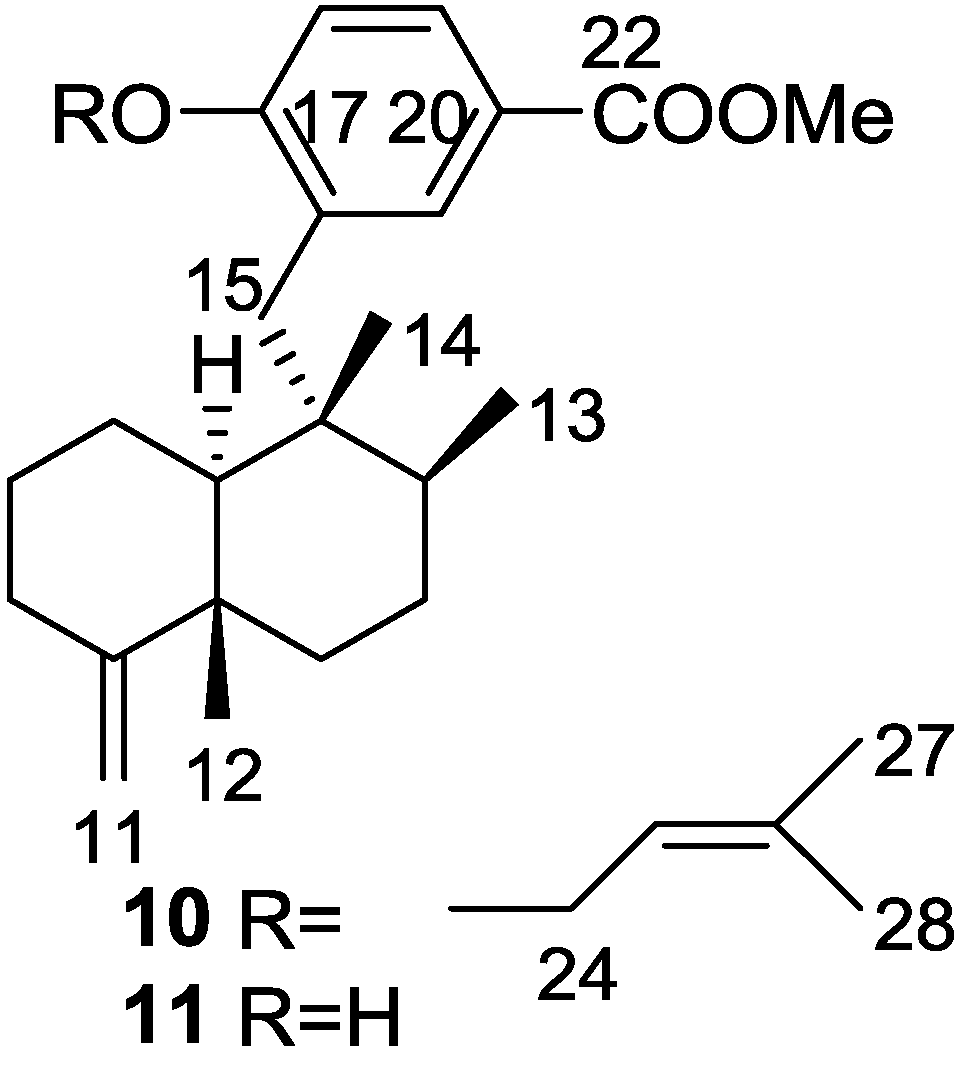
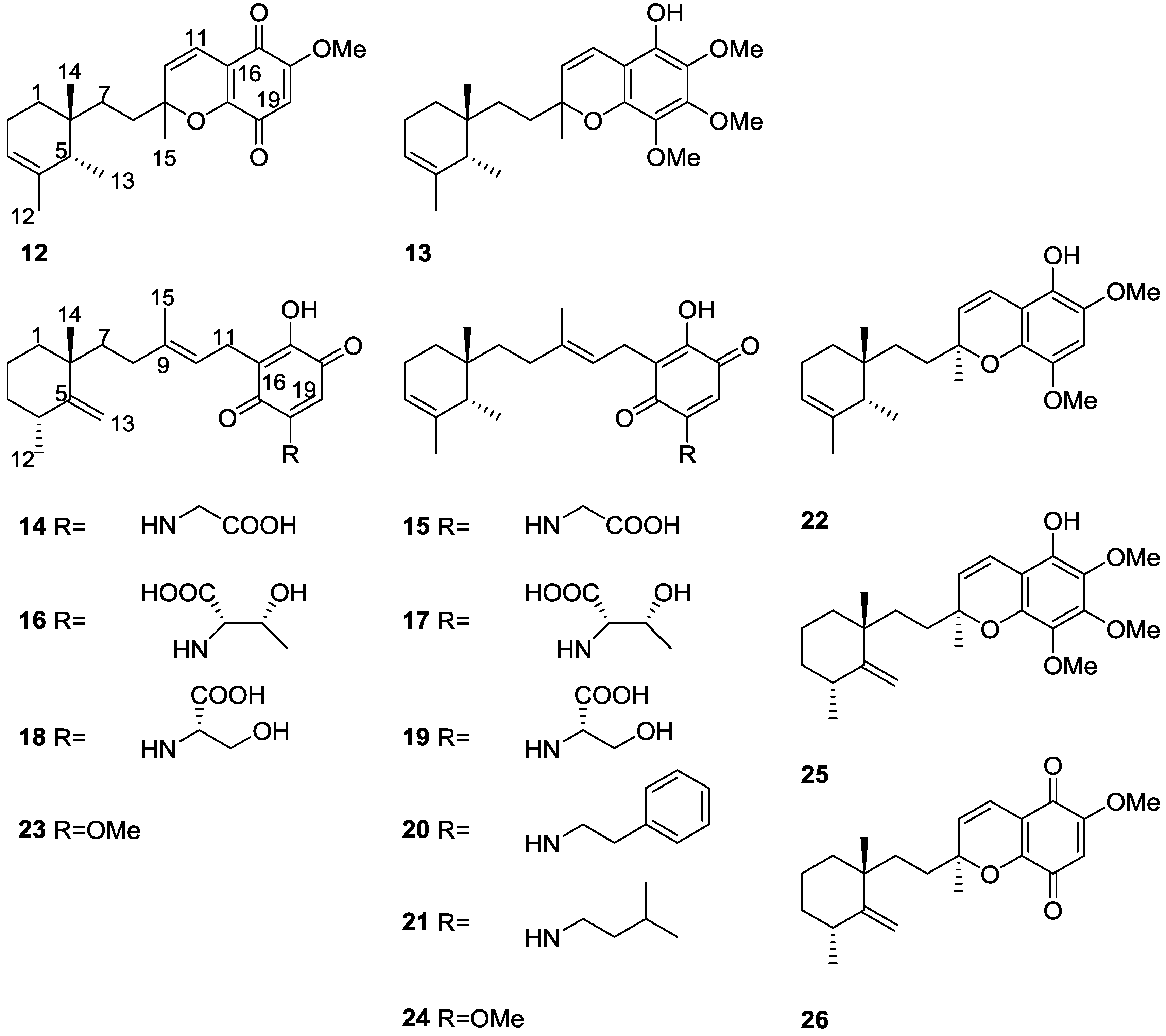
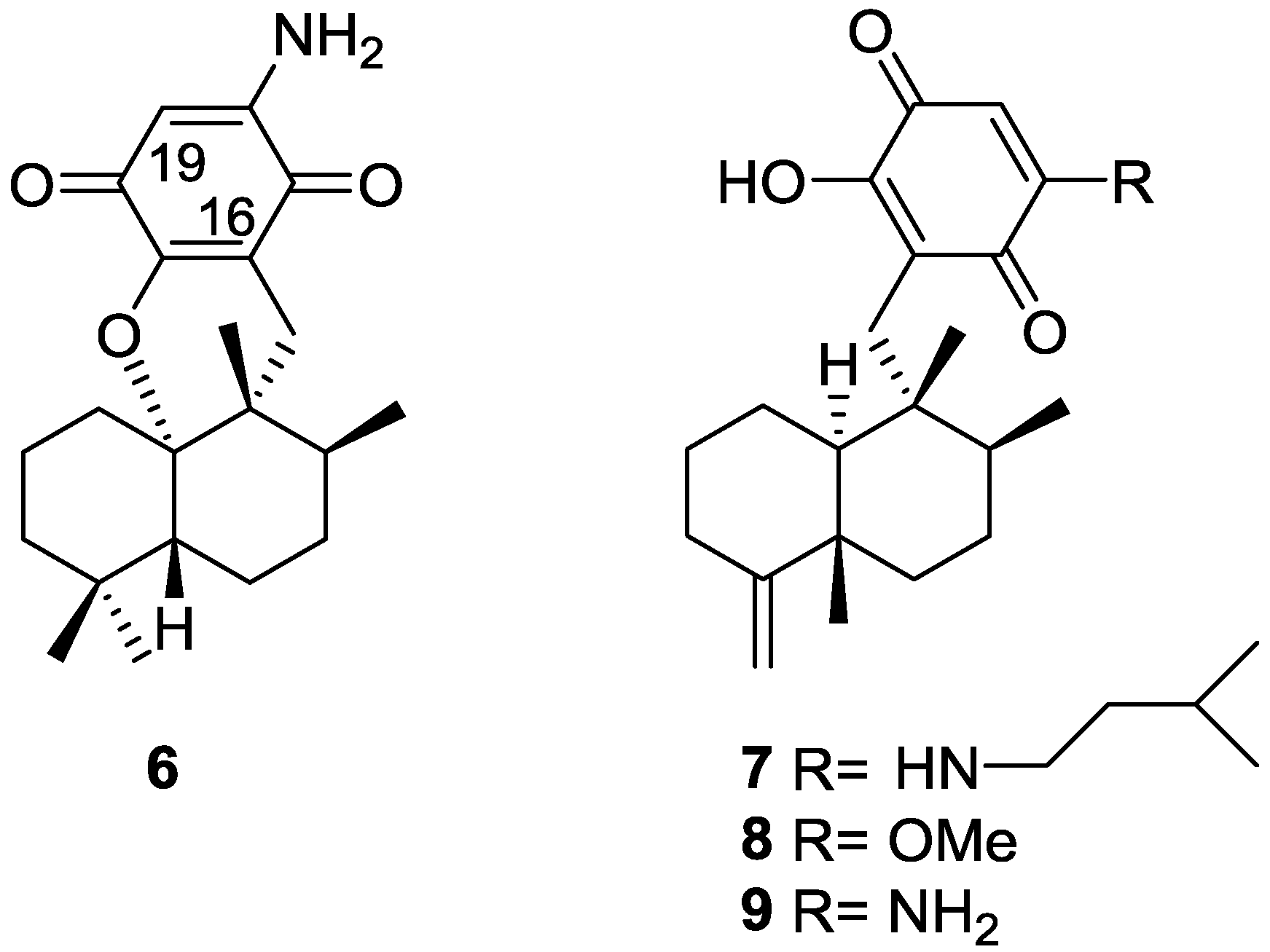
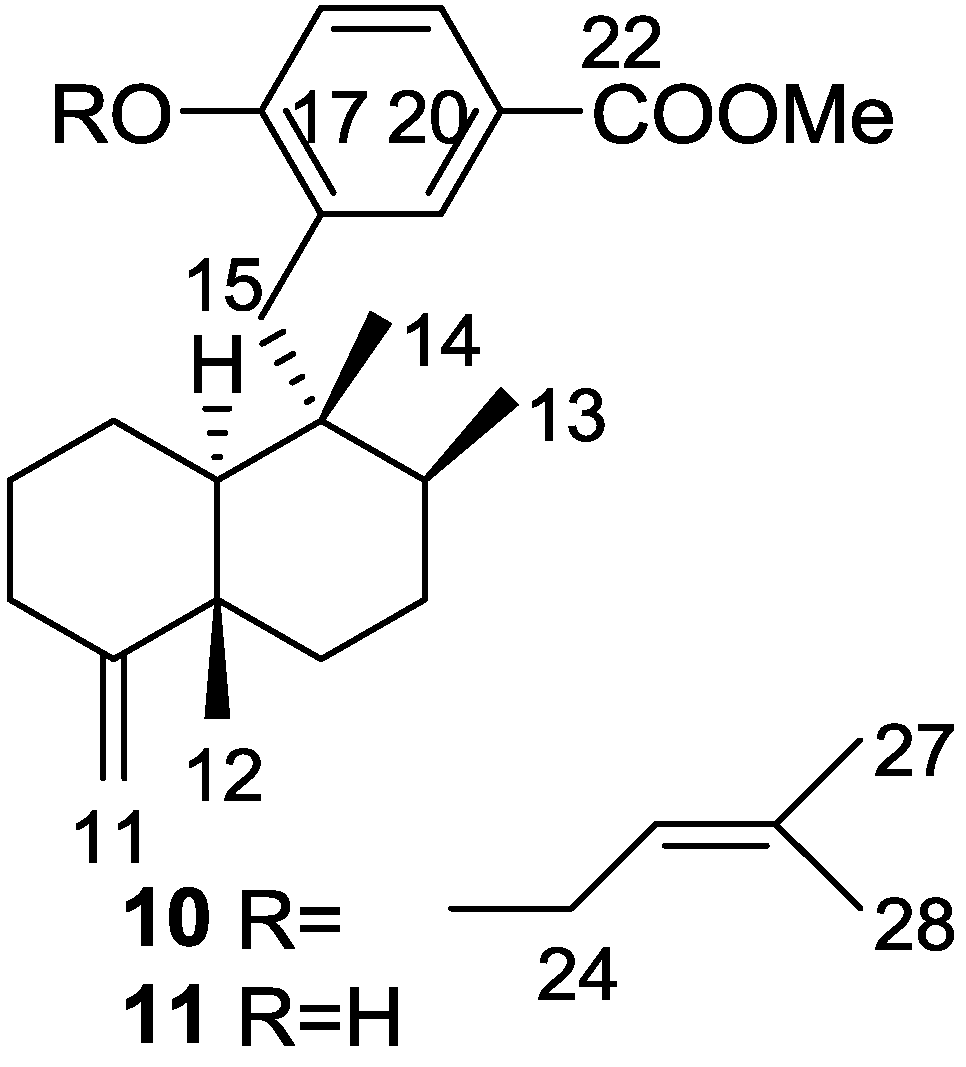
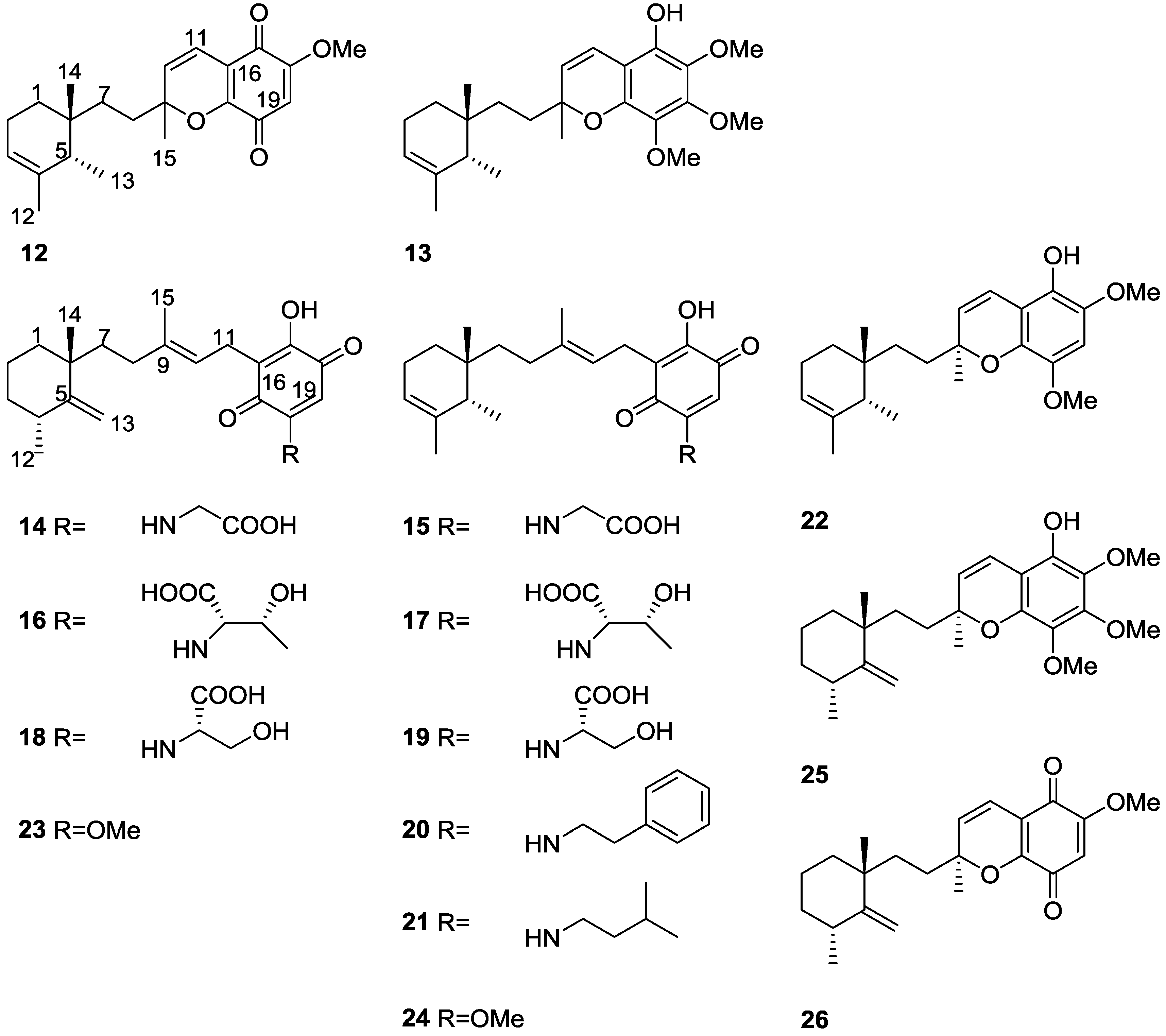
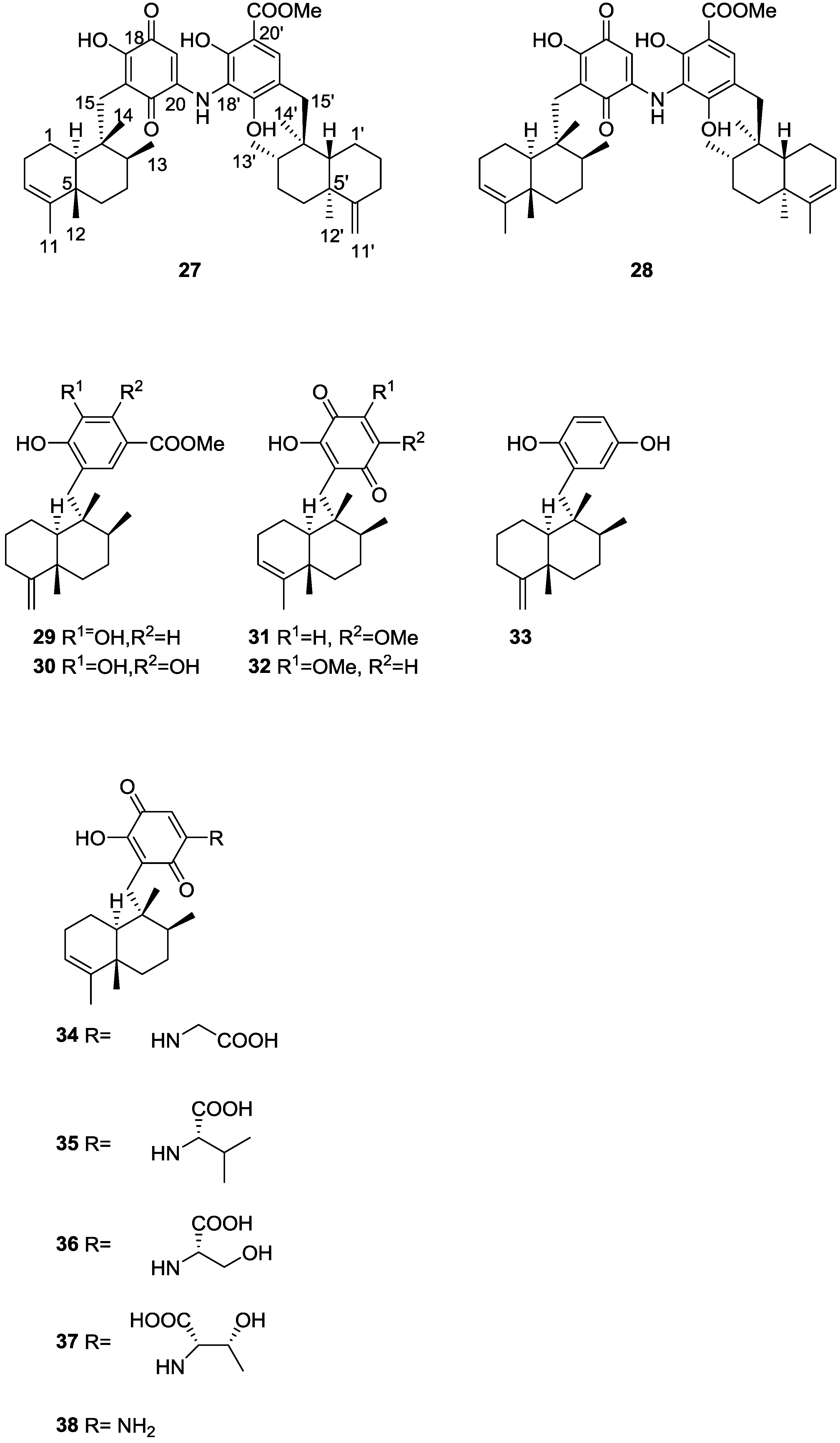
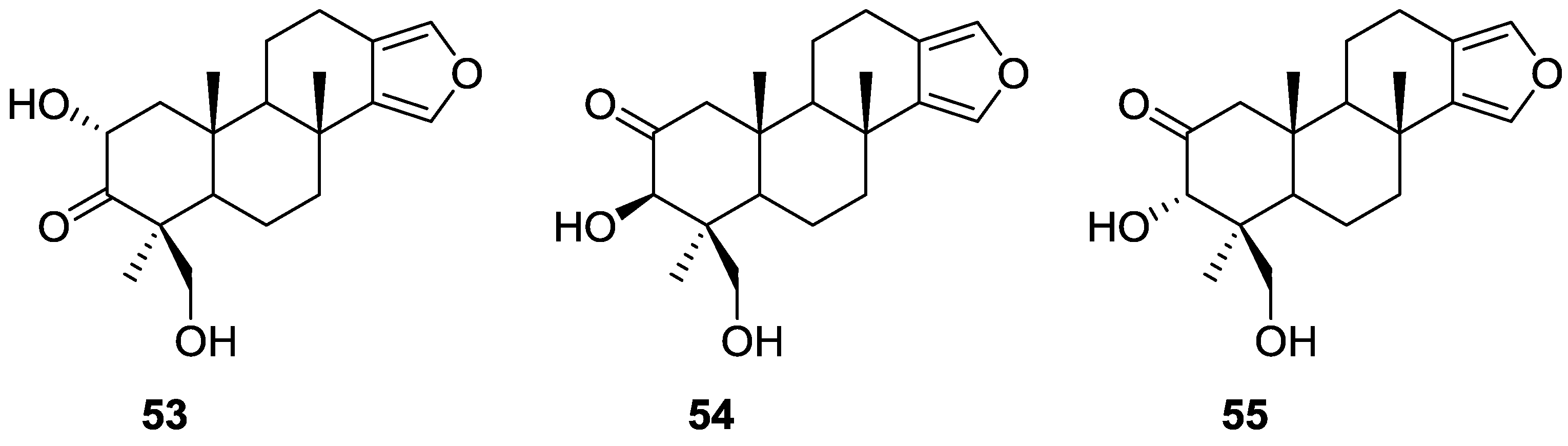
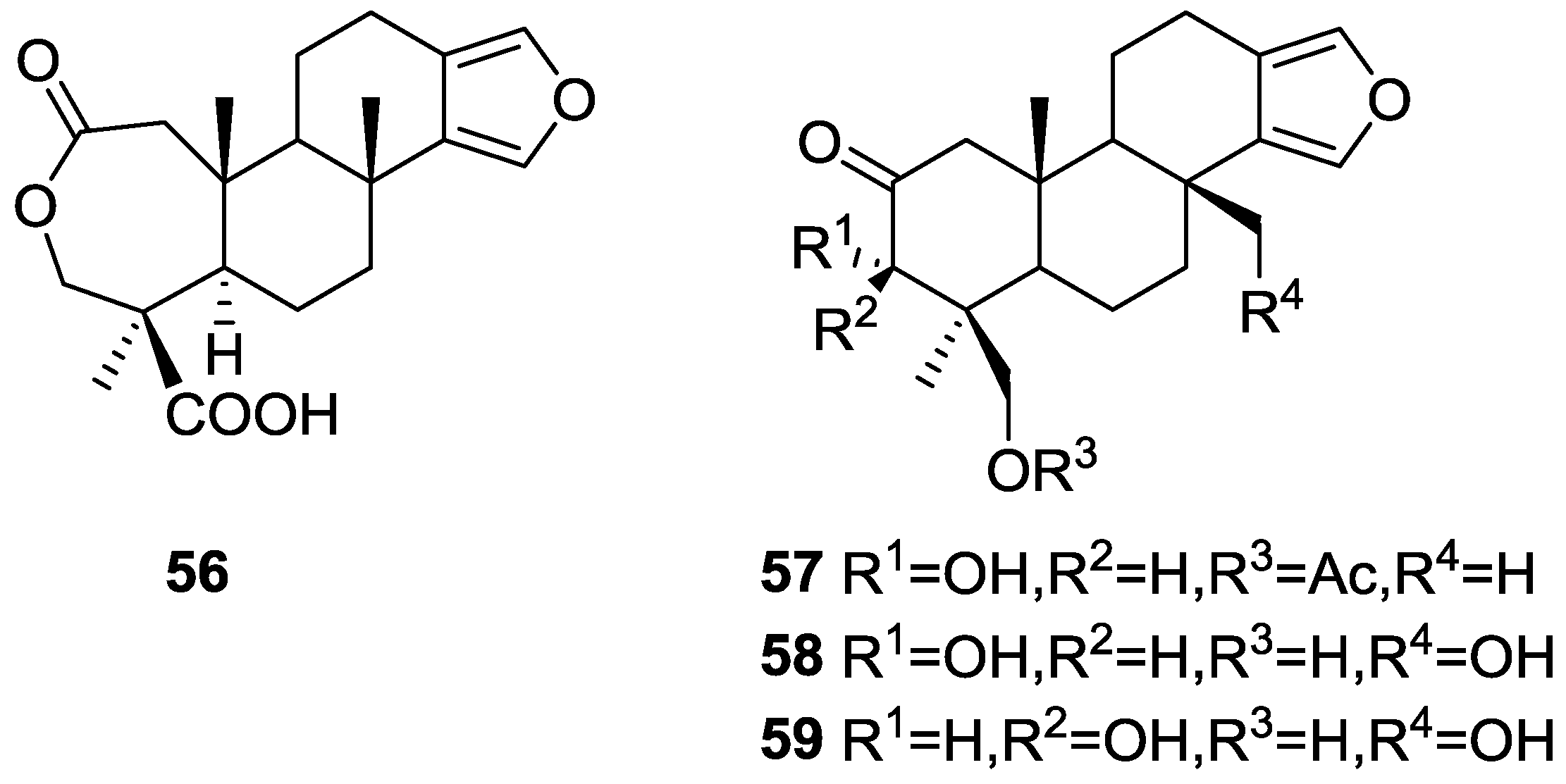
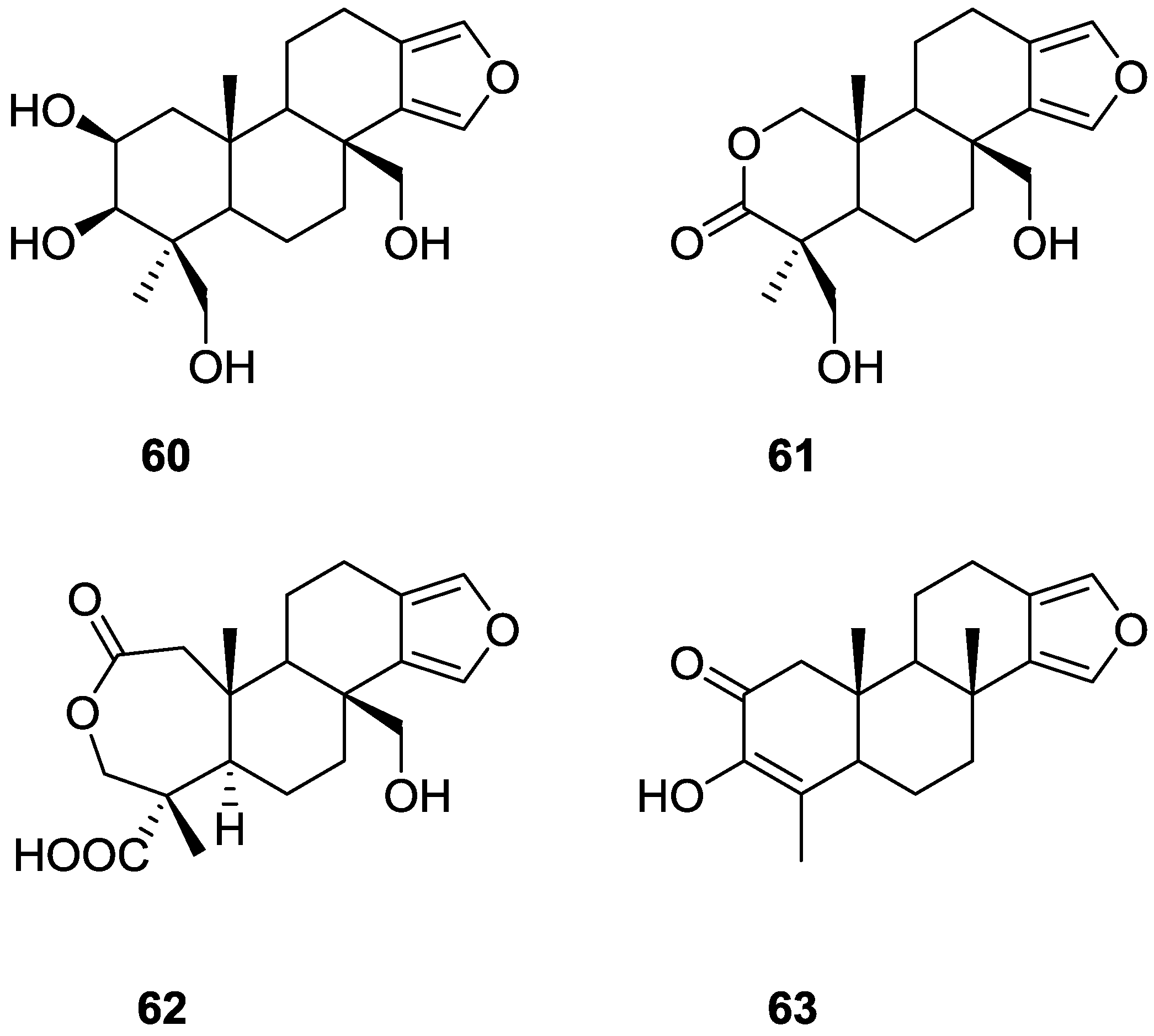
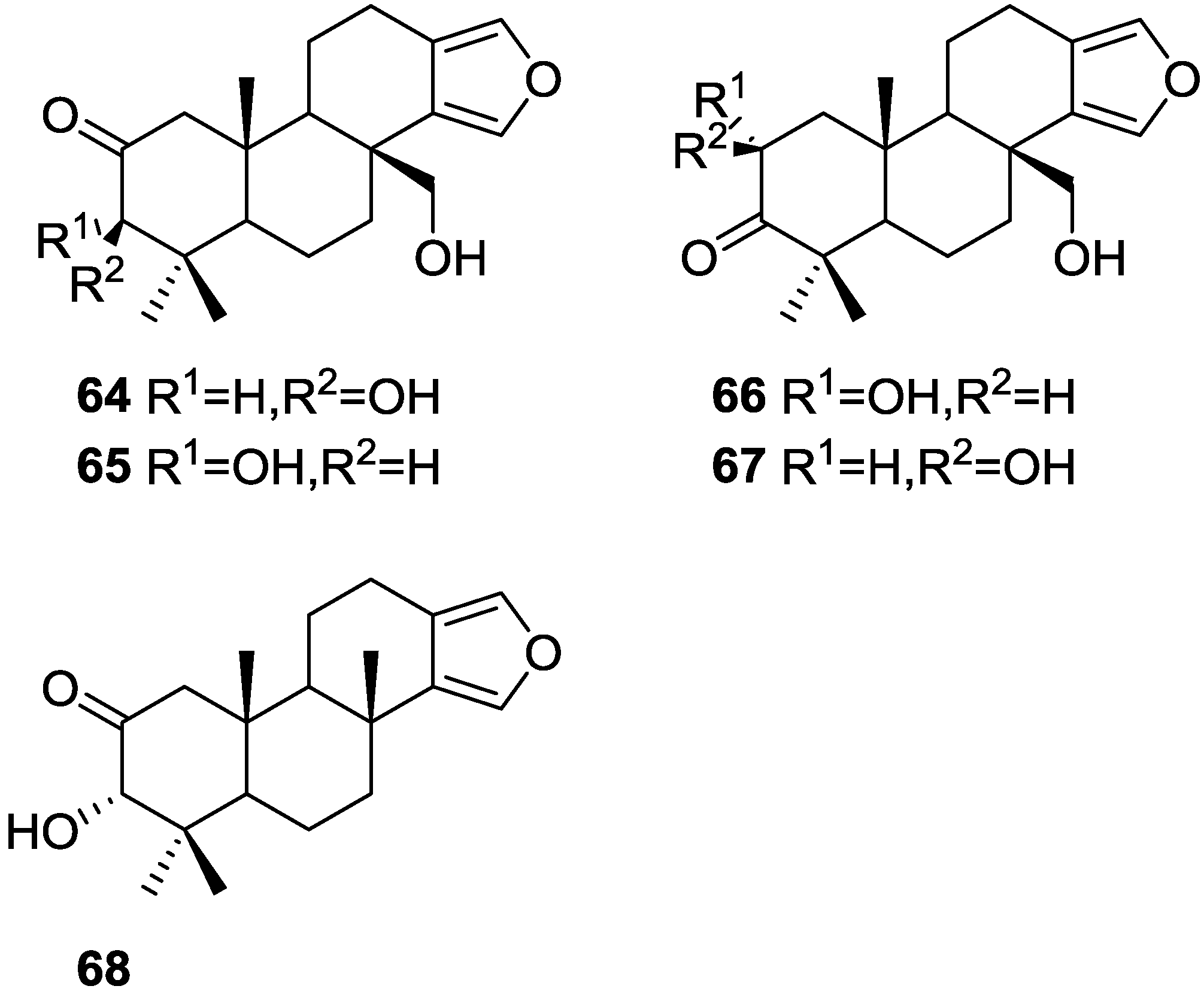
2. Sesquiterpene Quinones
Other Studies
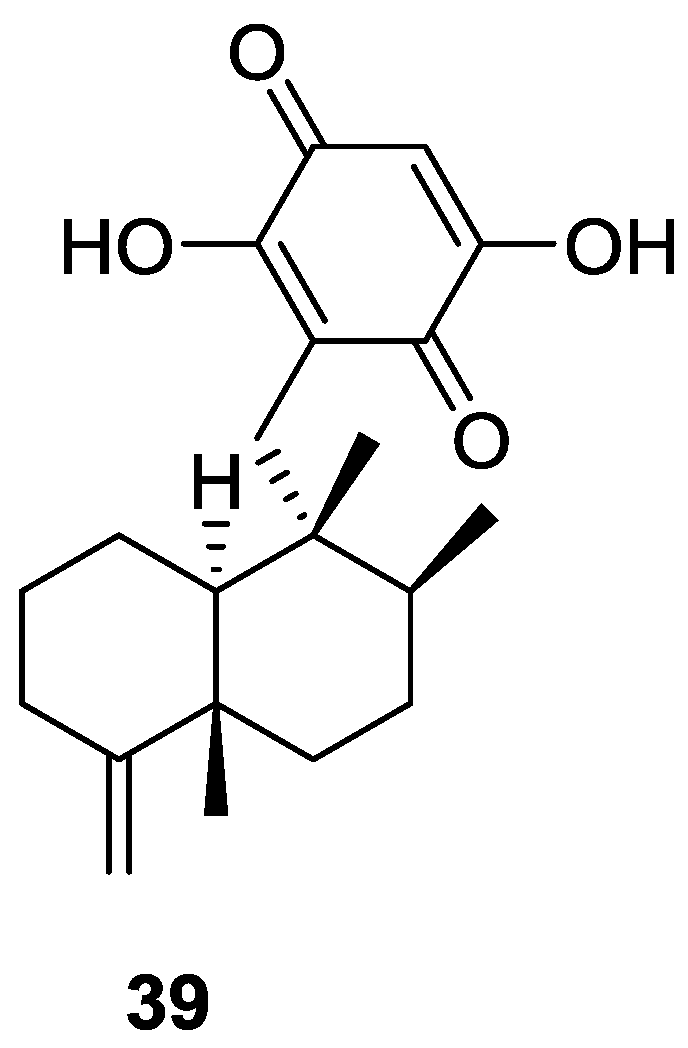
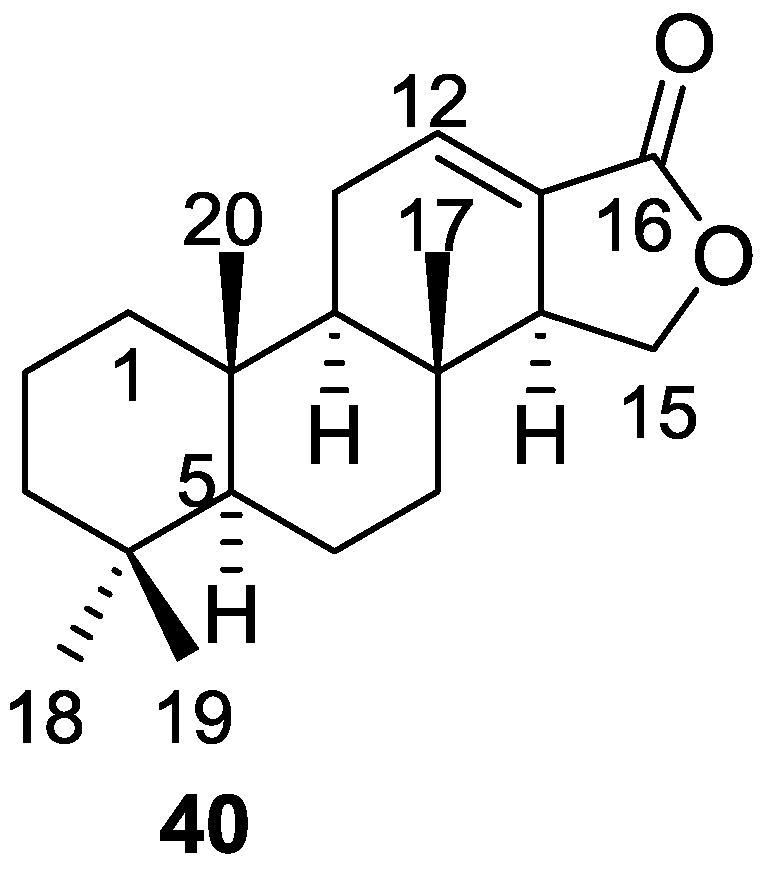
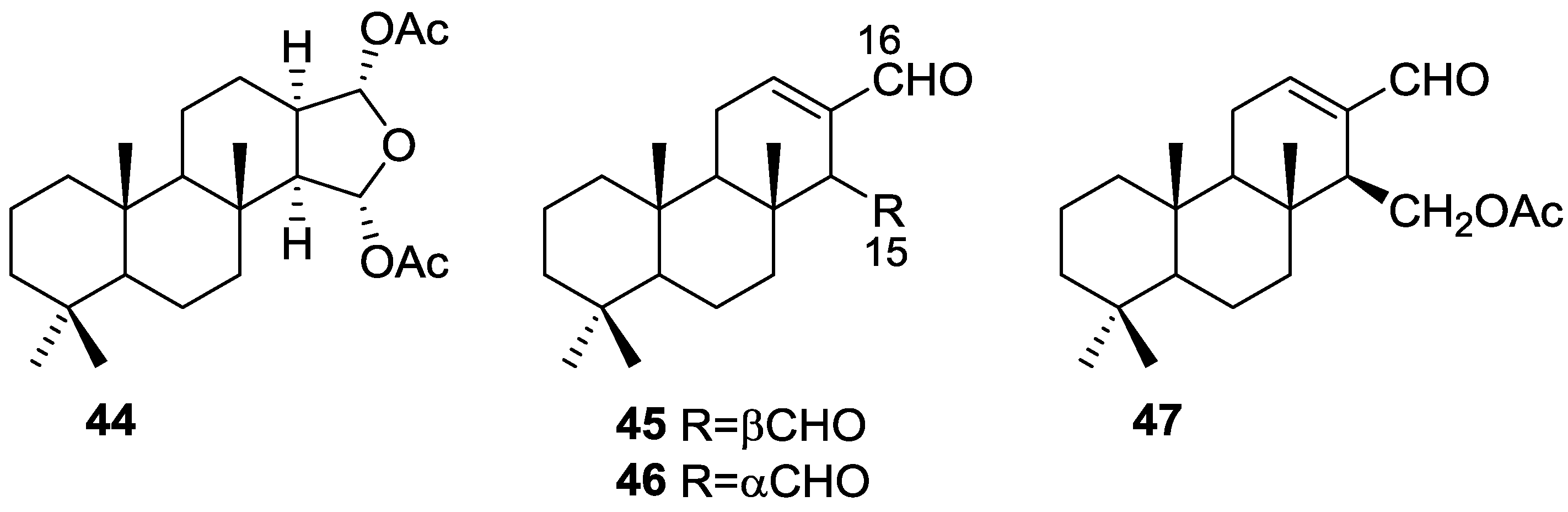
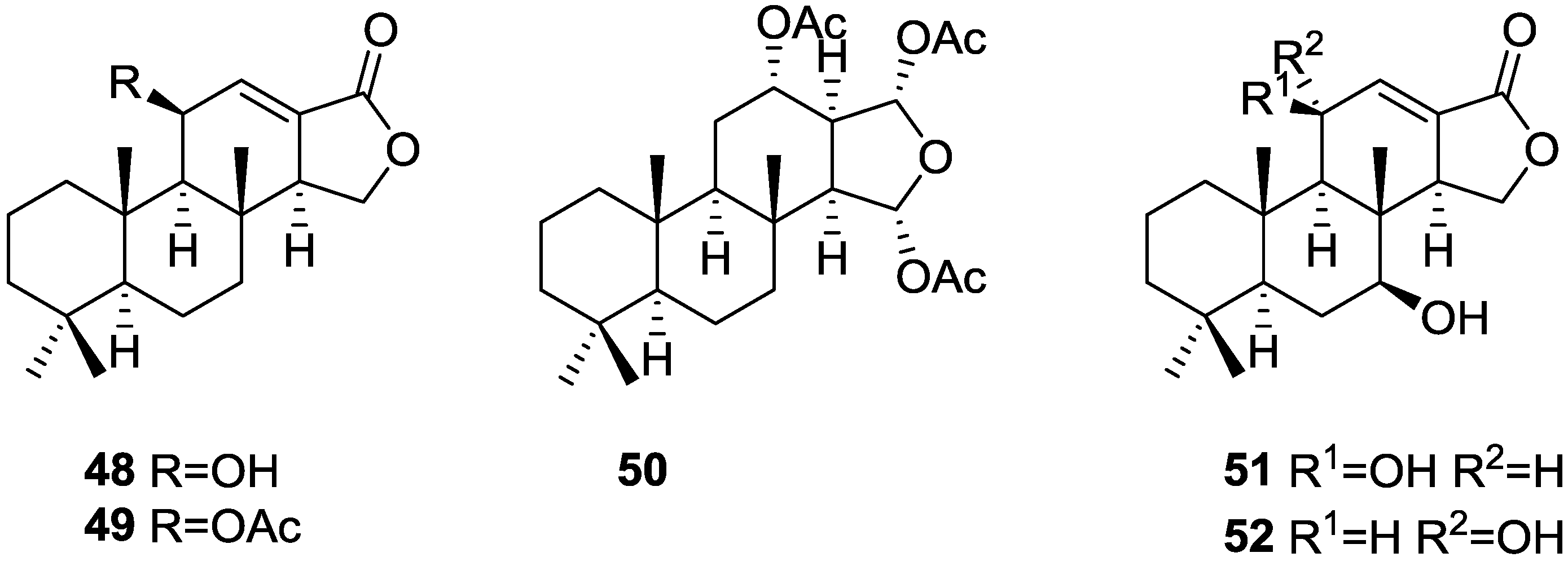
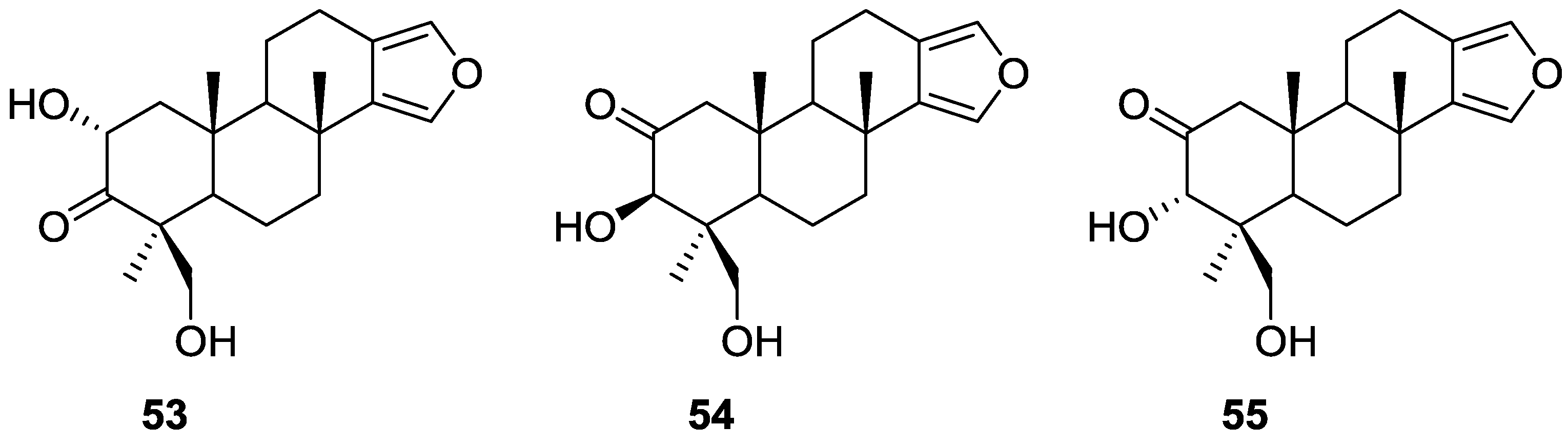
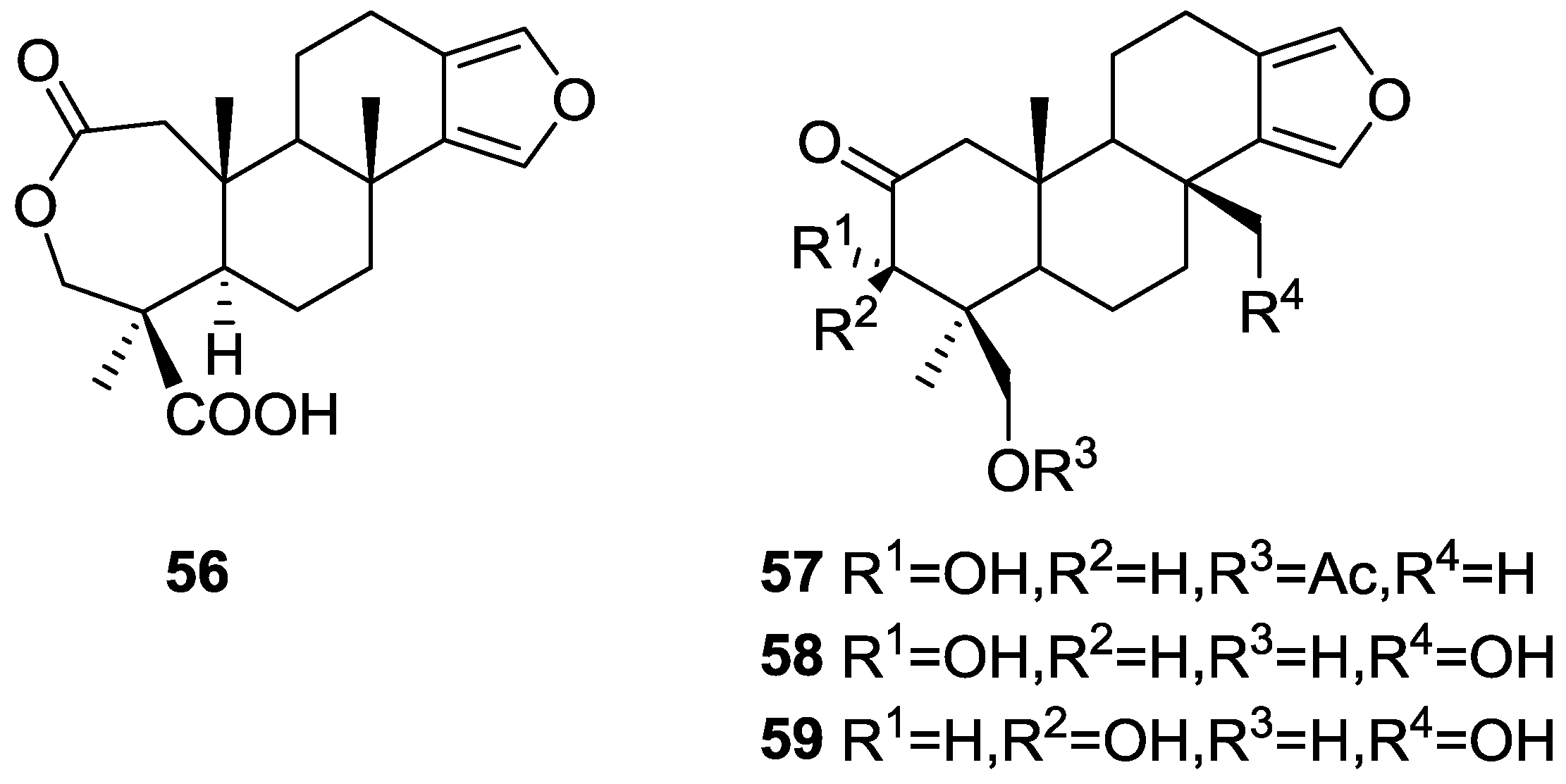
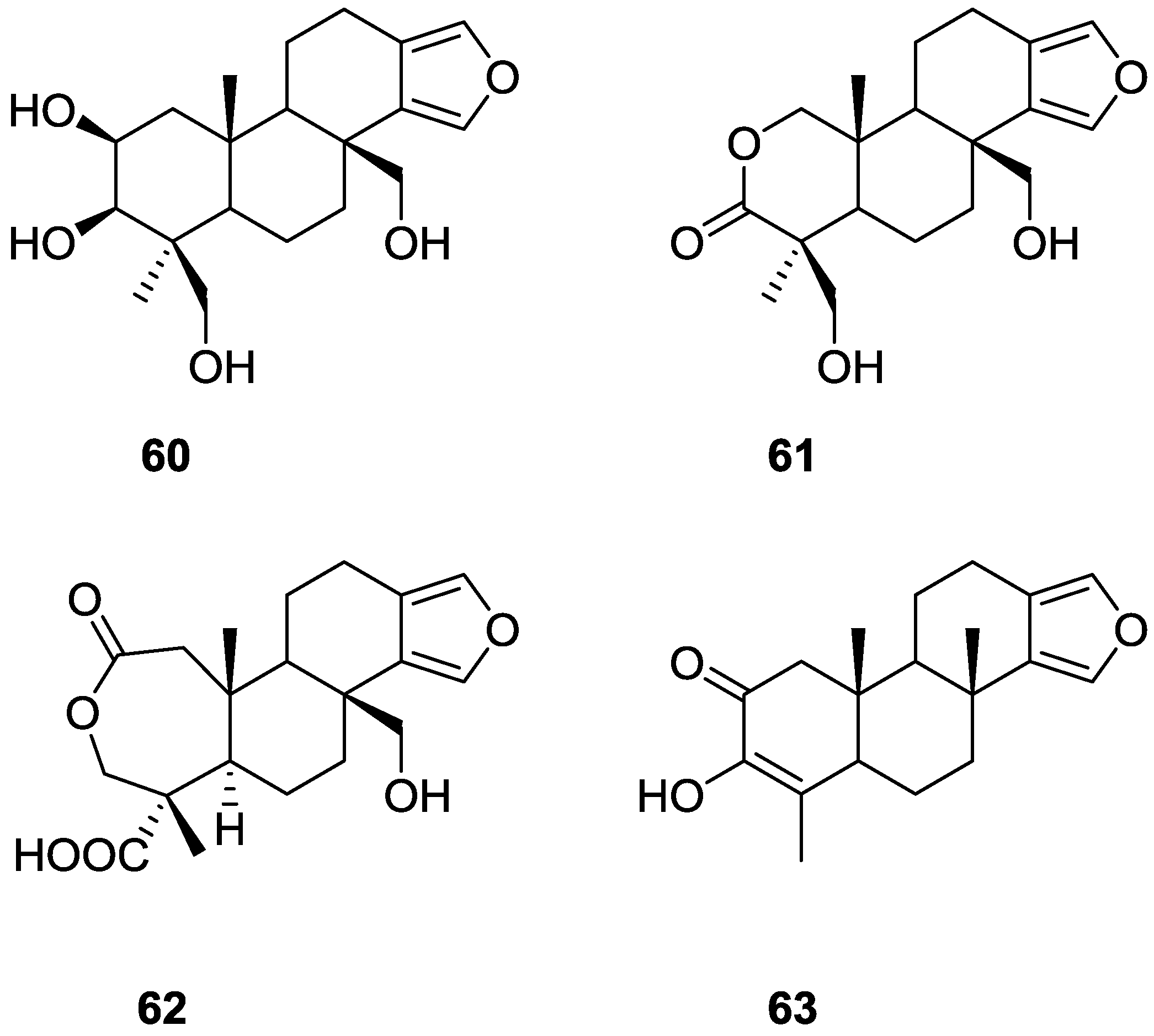
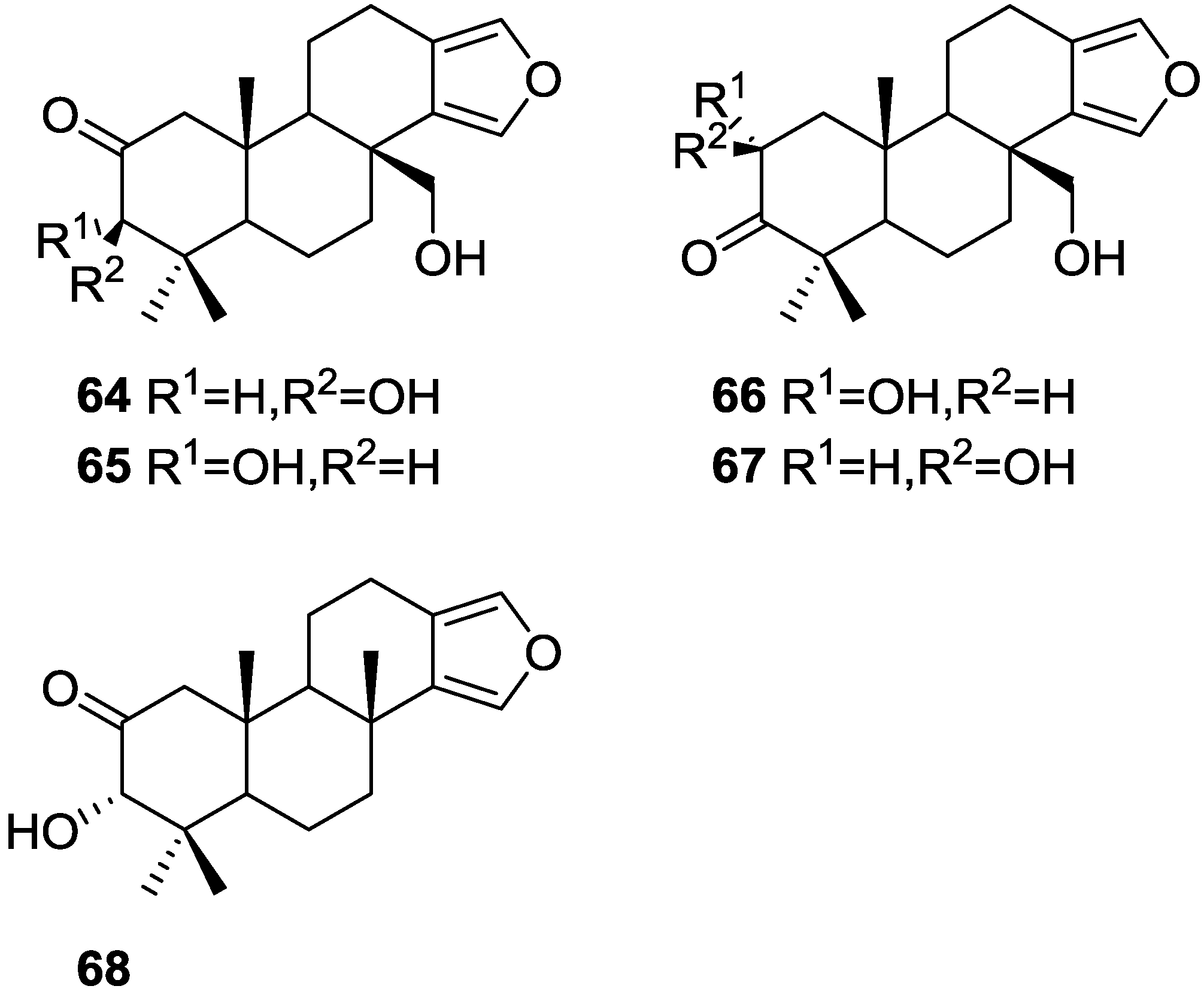
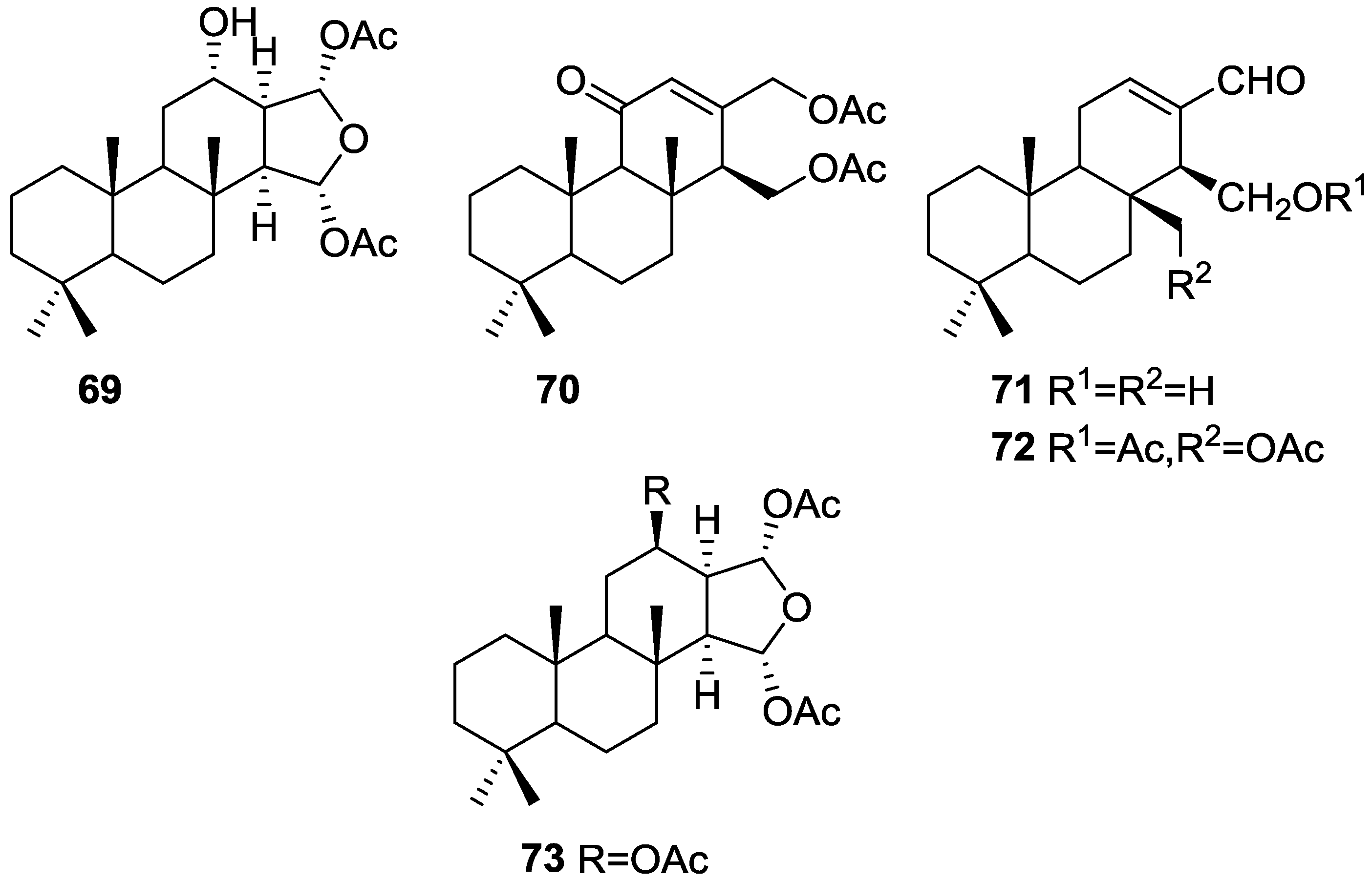
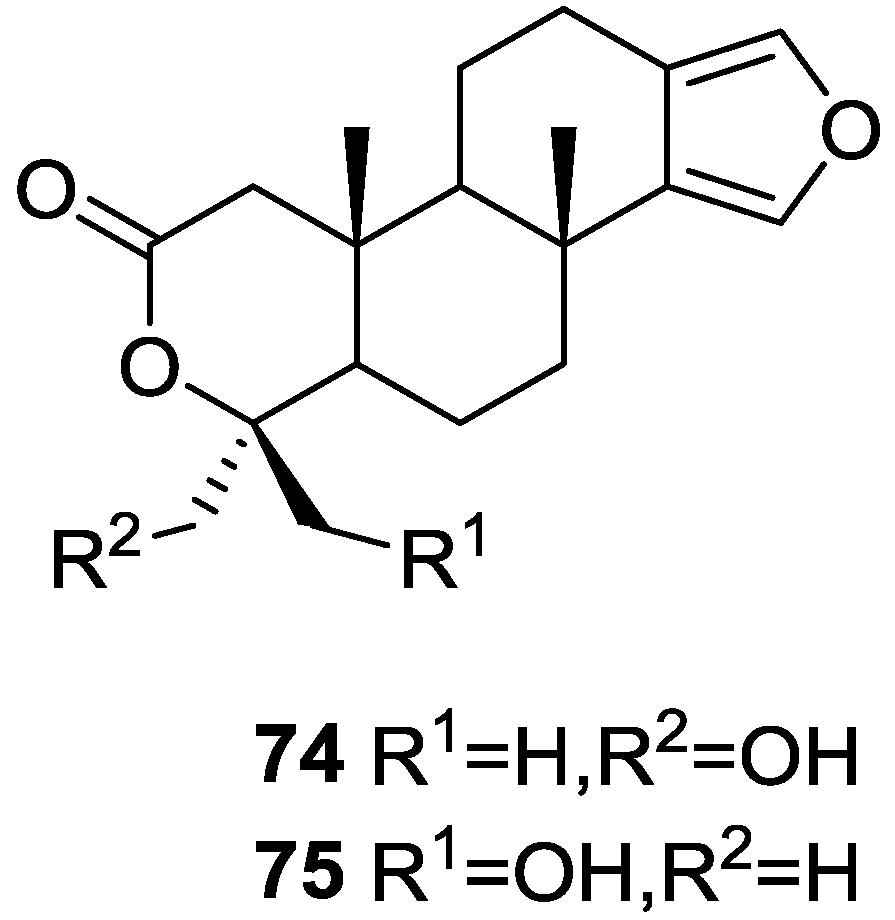
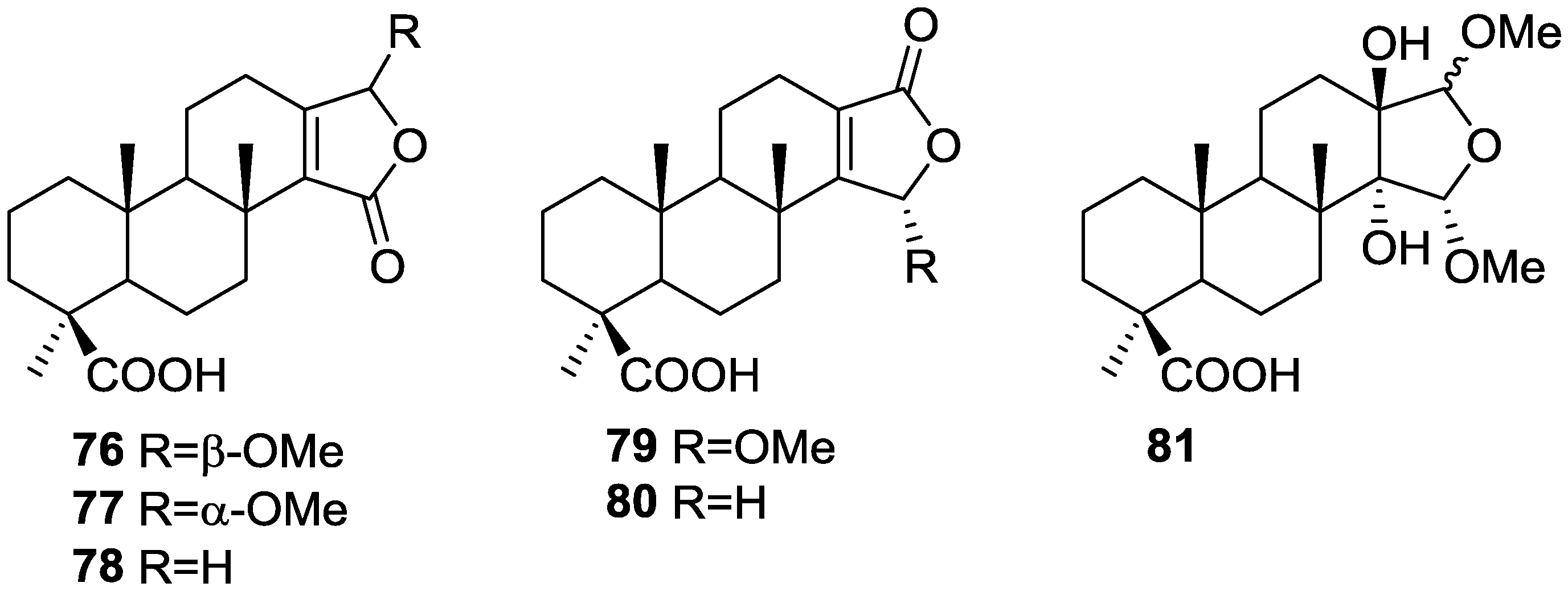
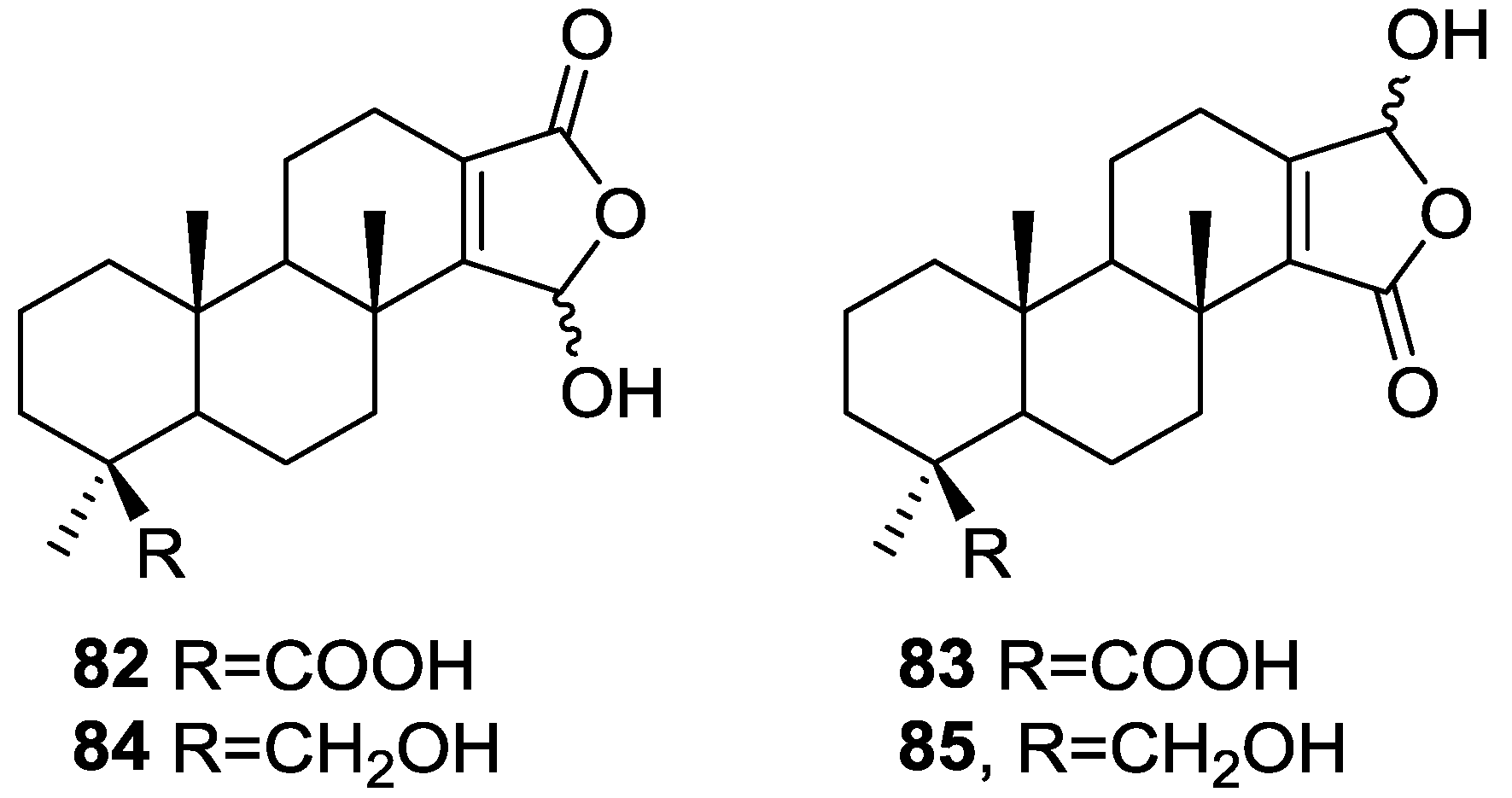
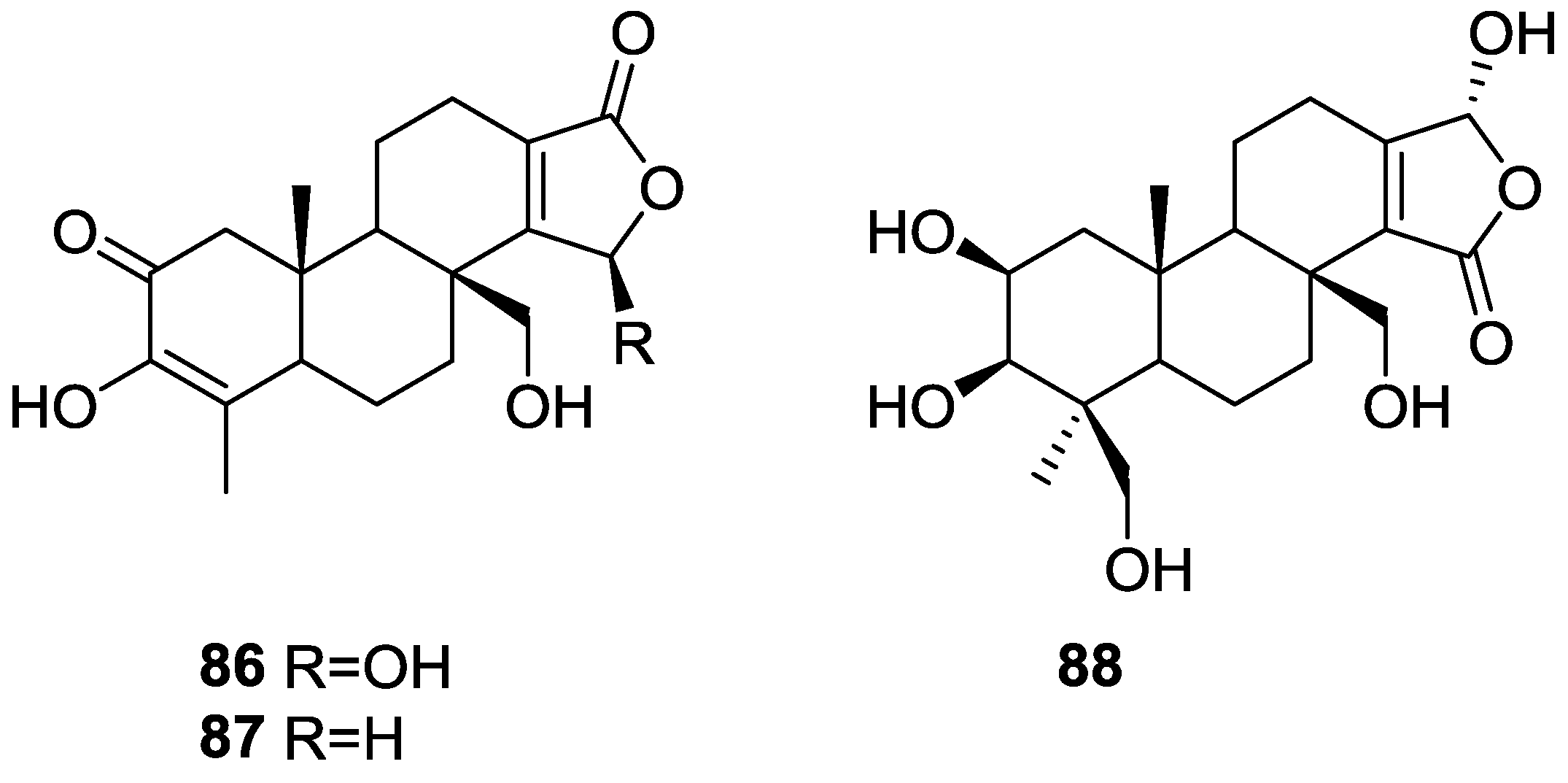
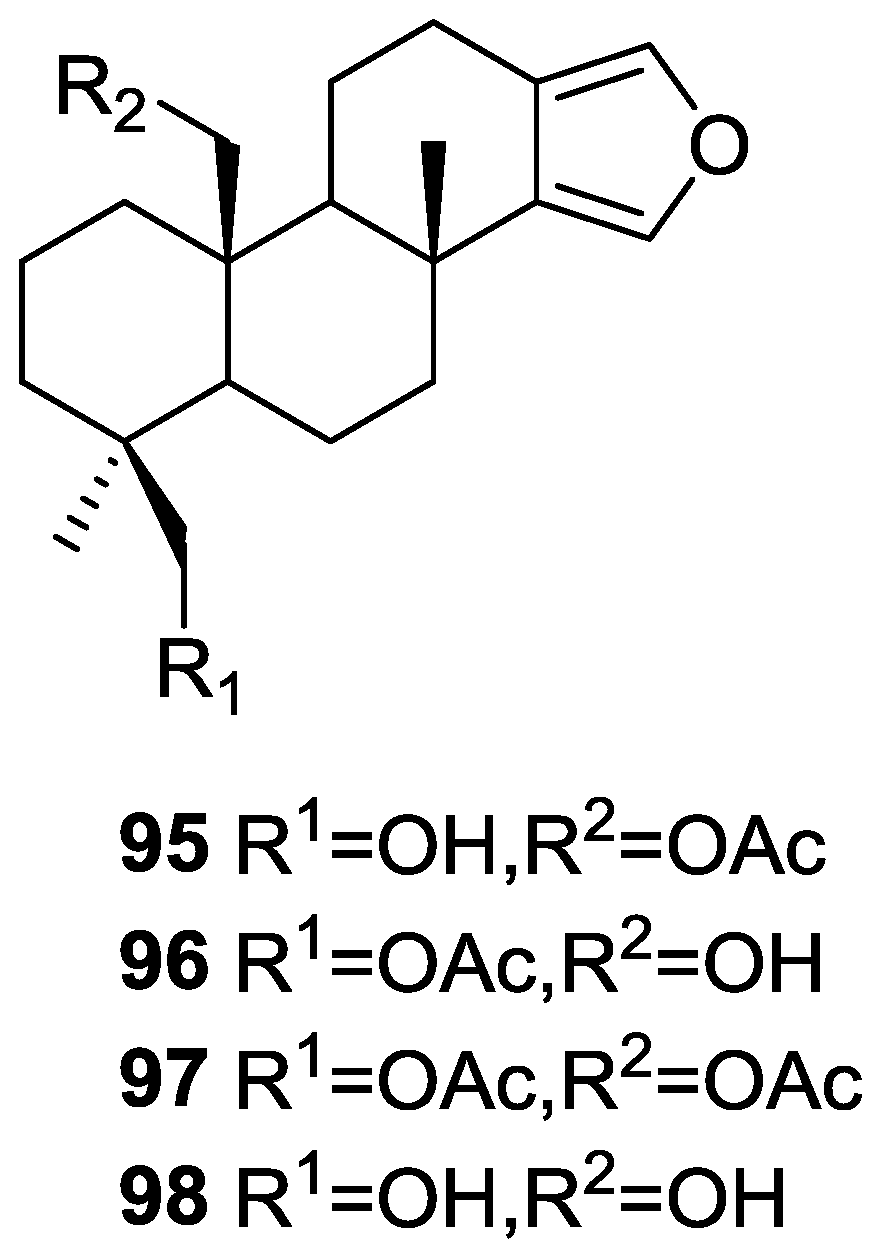
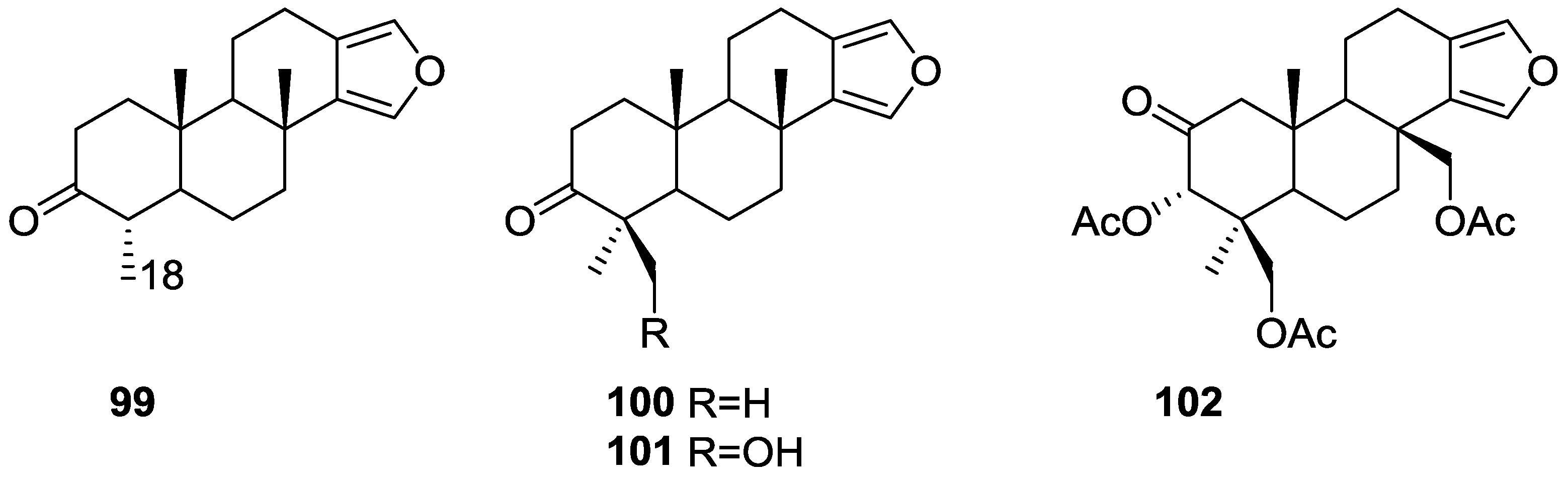
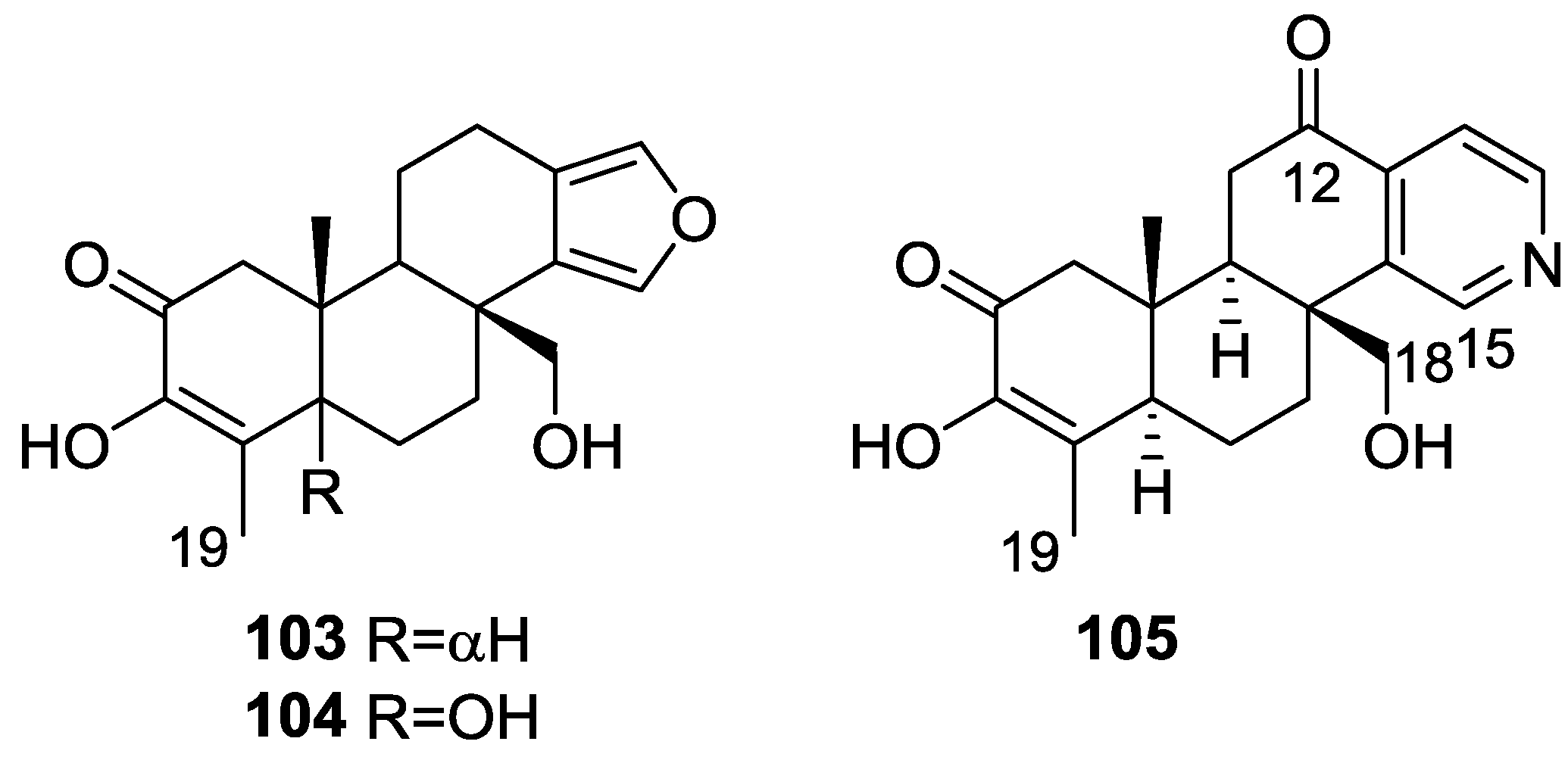
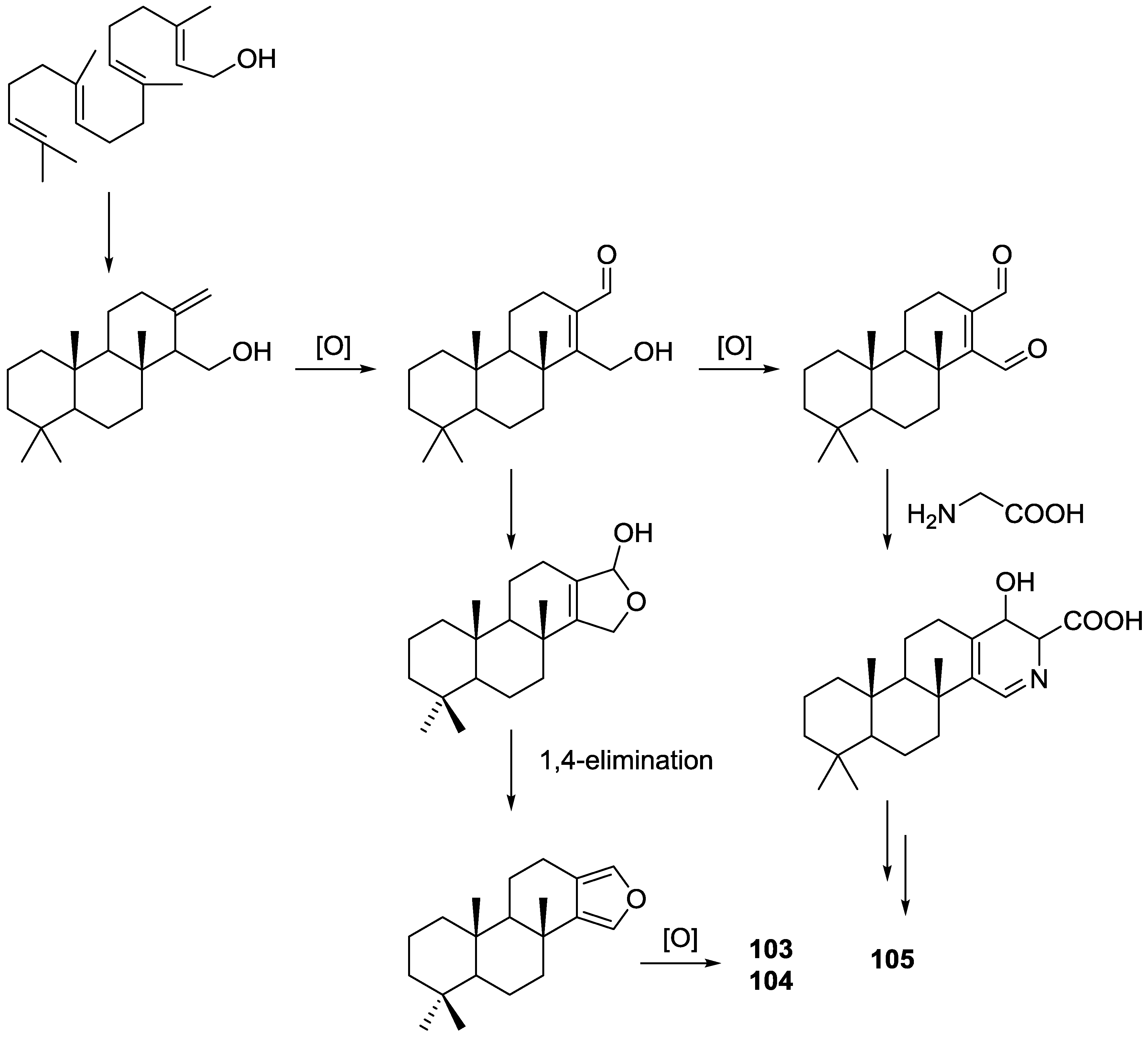
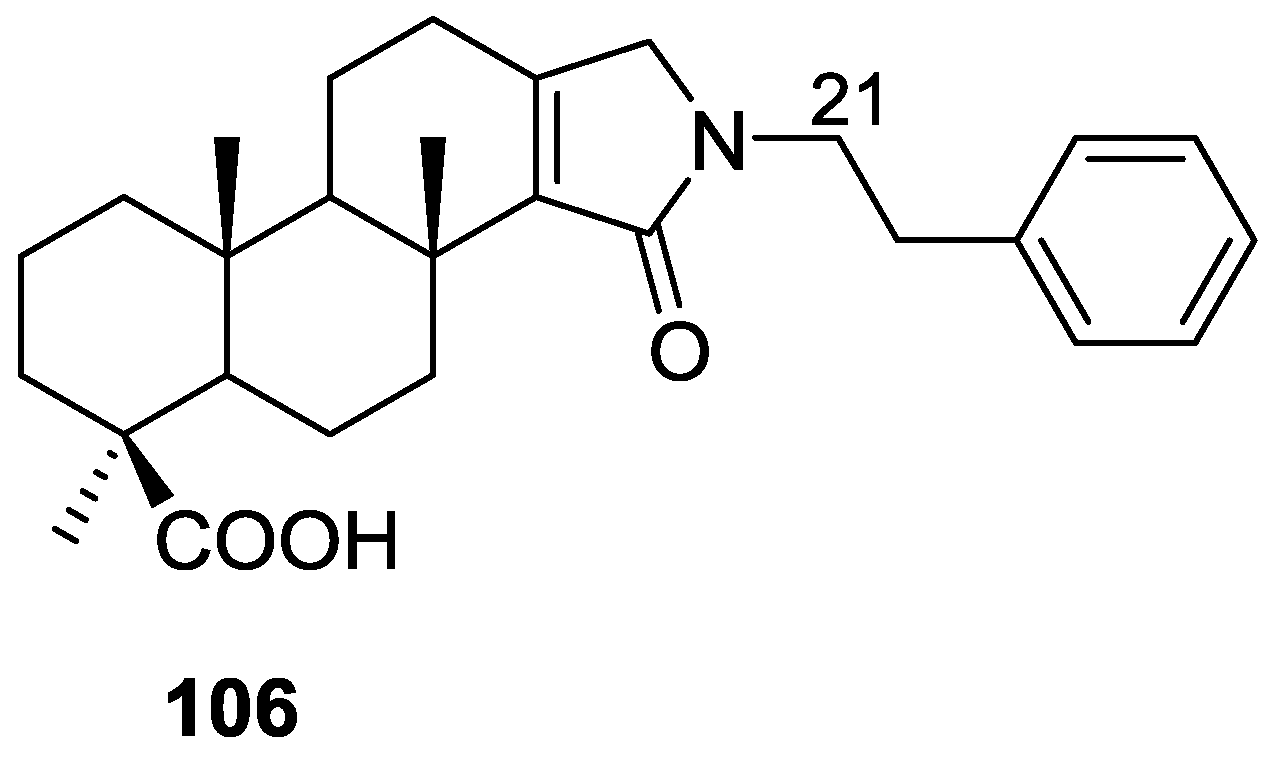
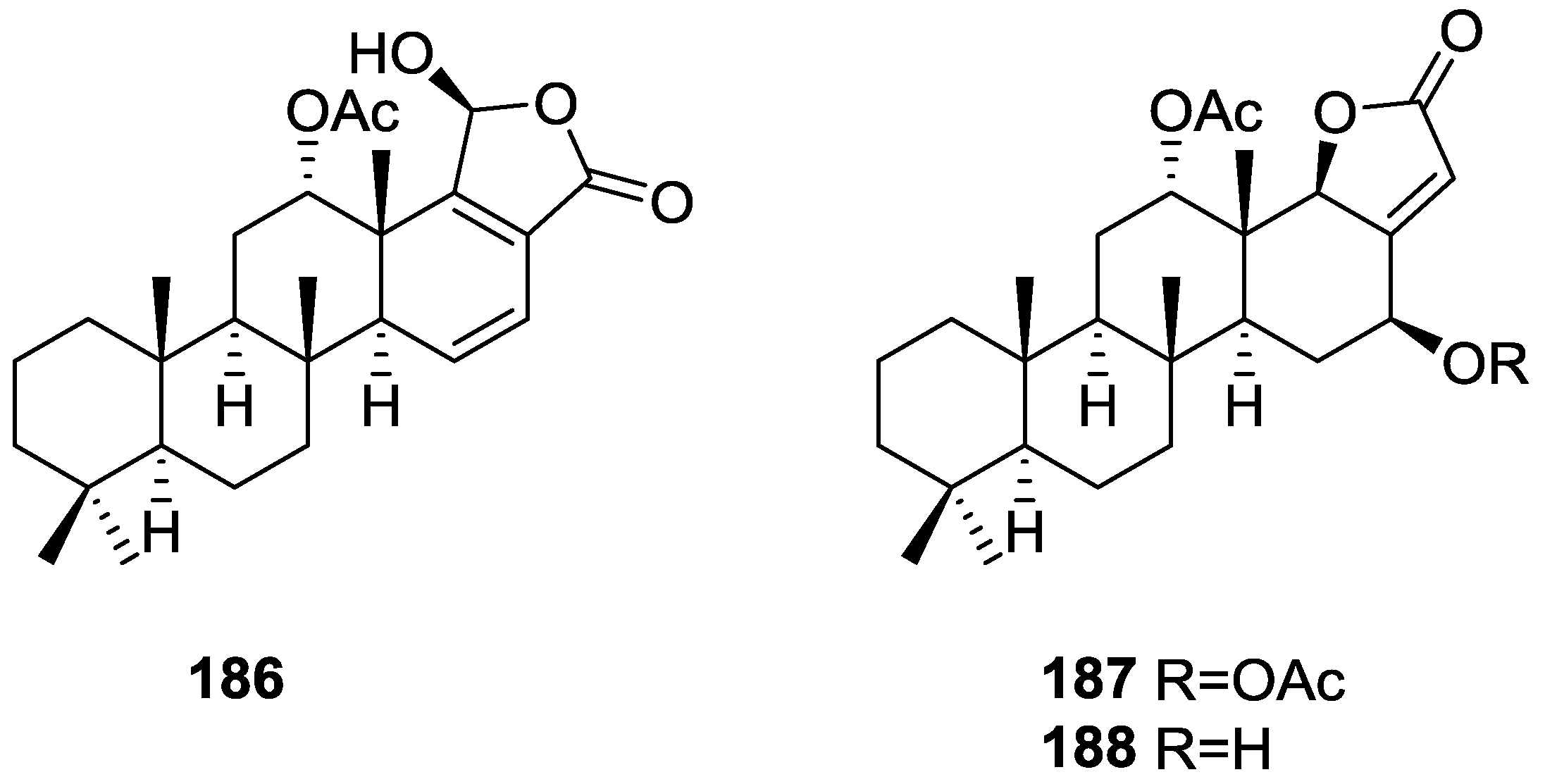
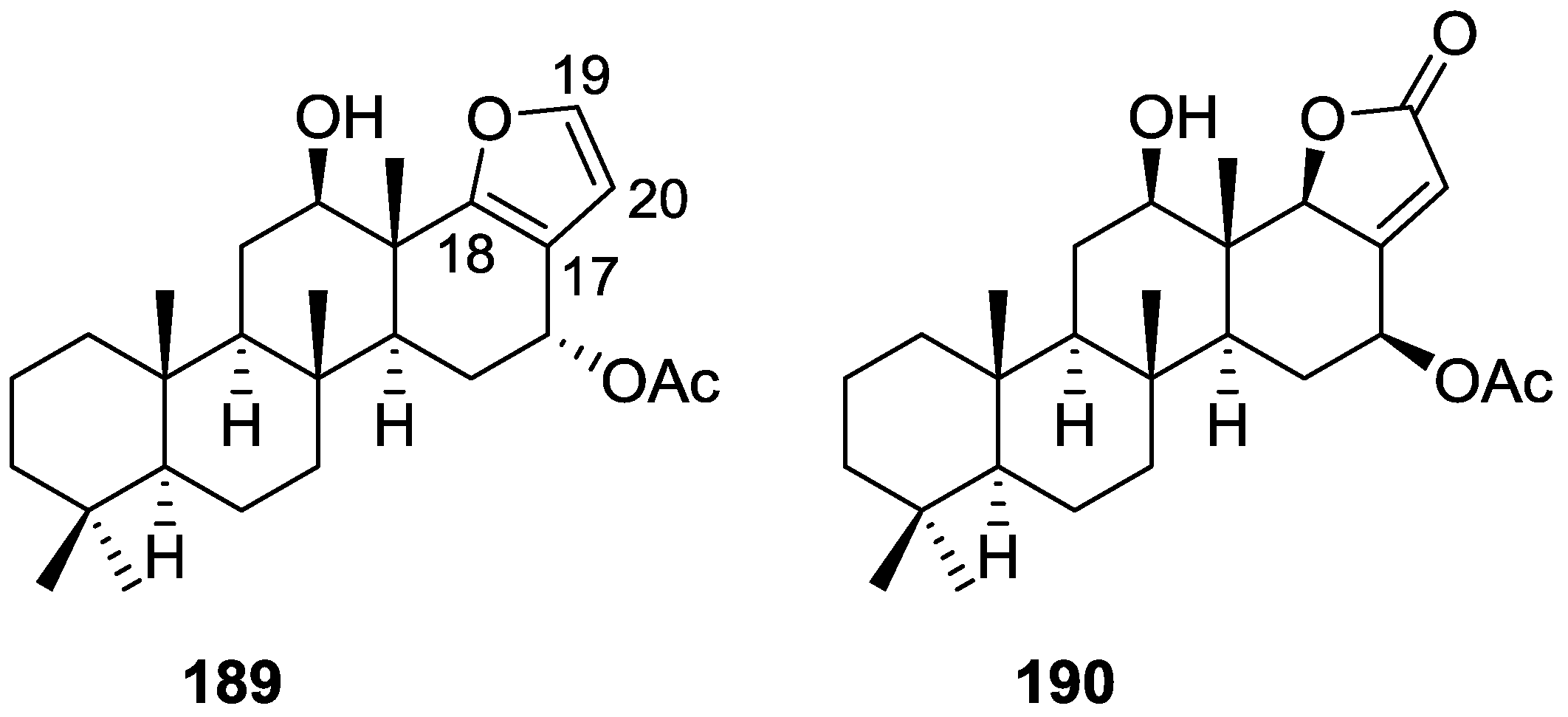
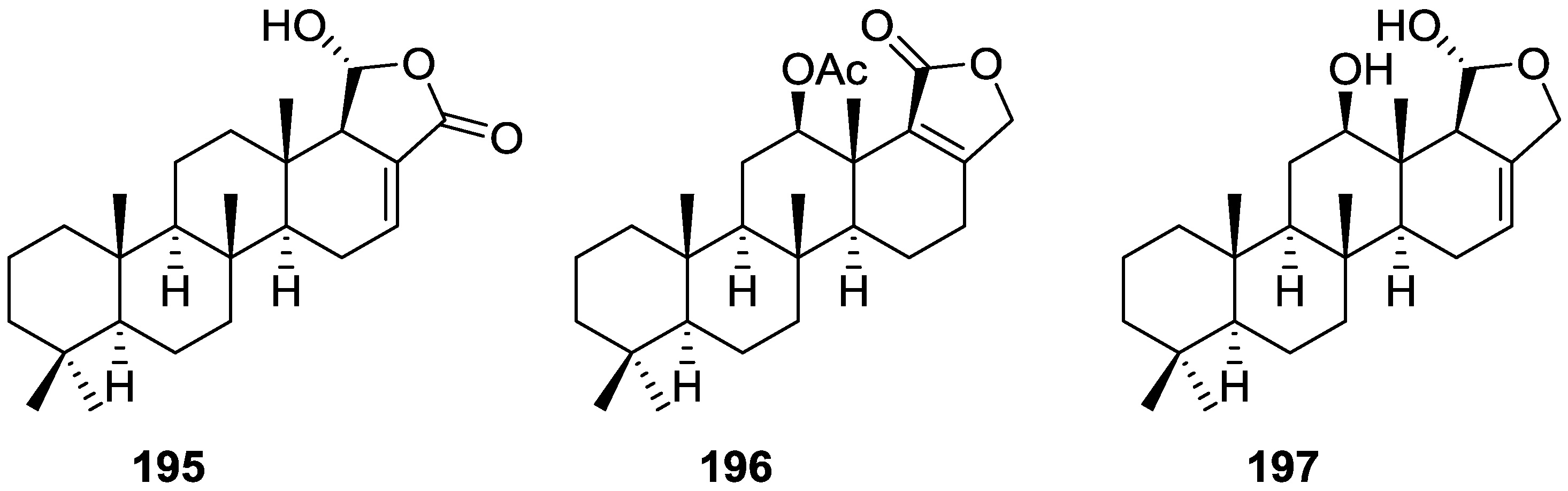
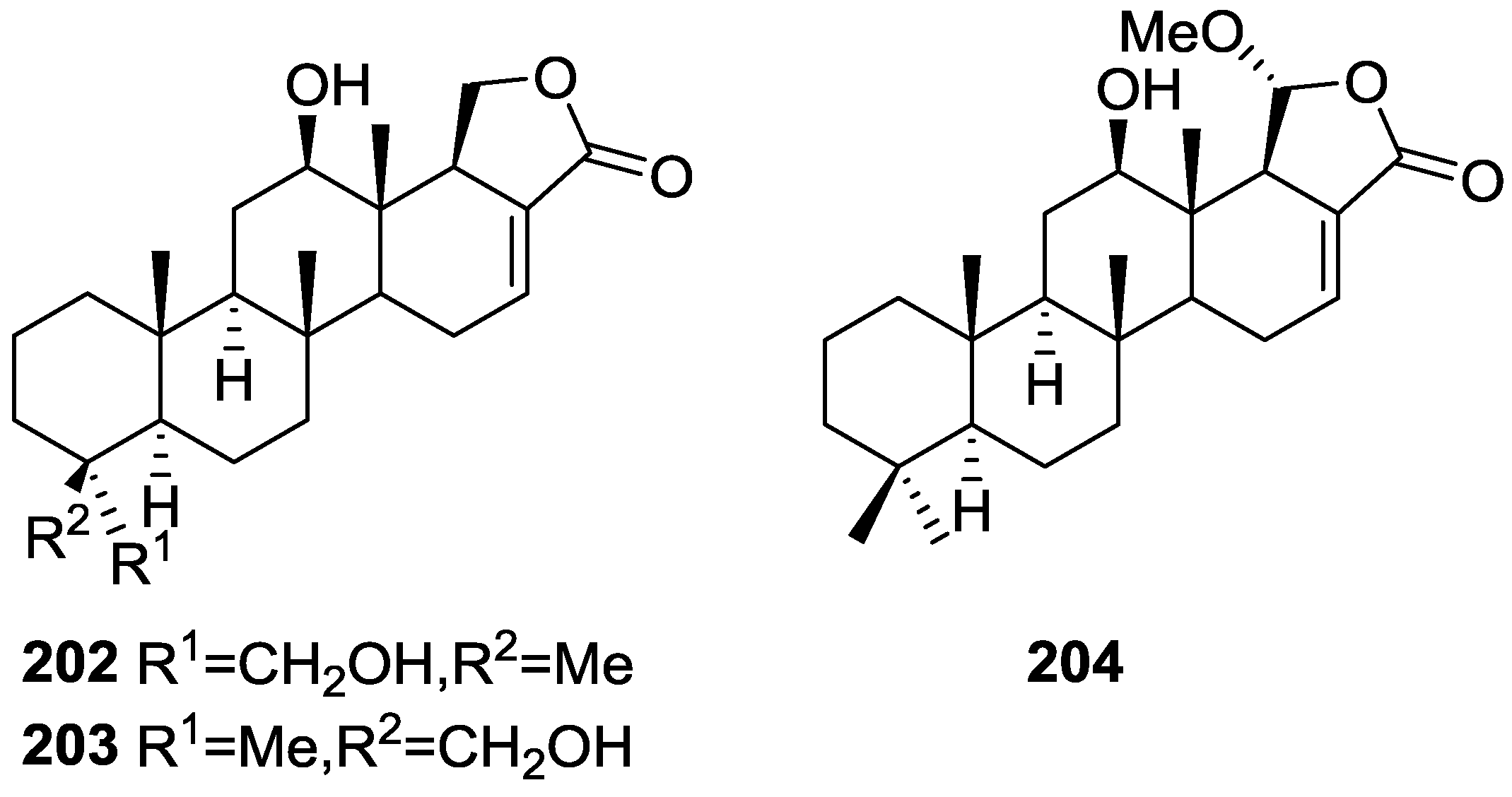
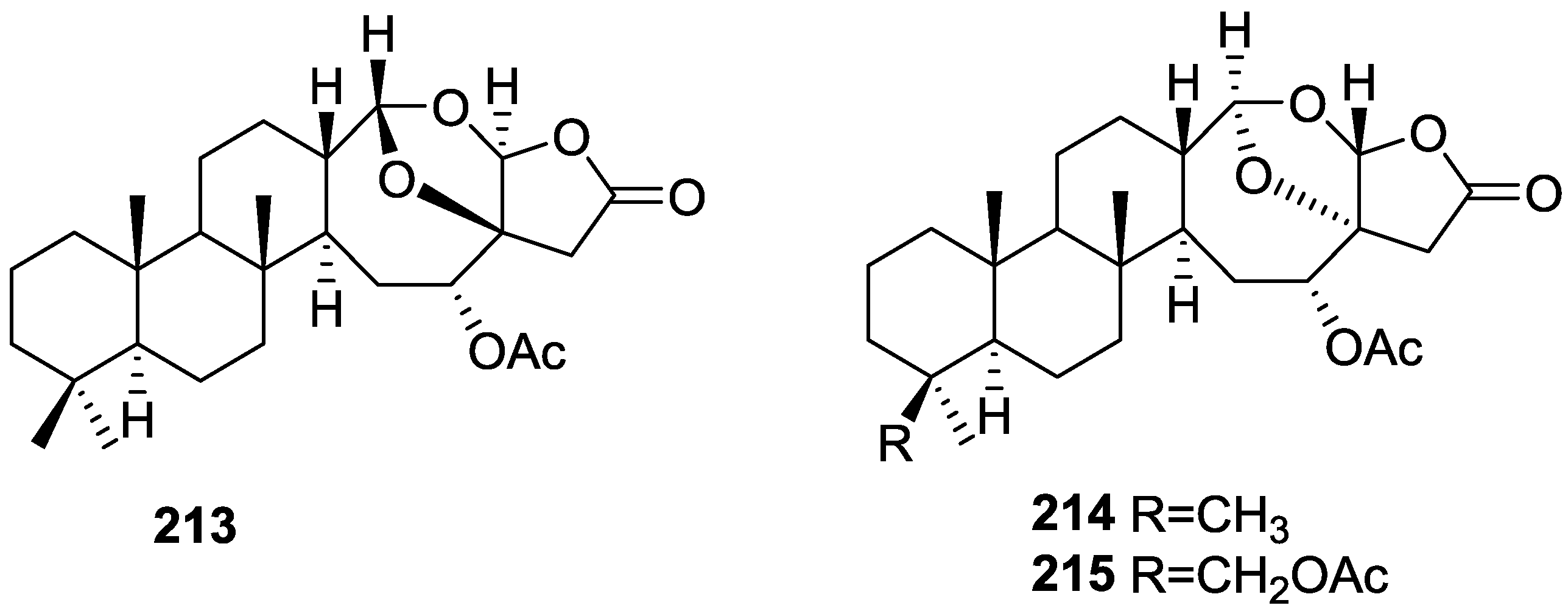
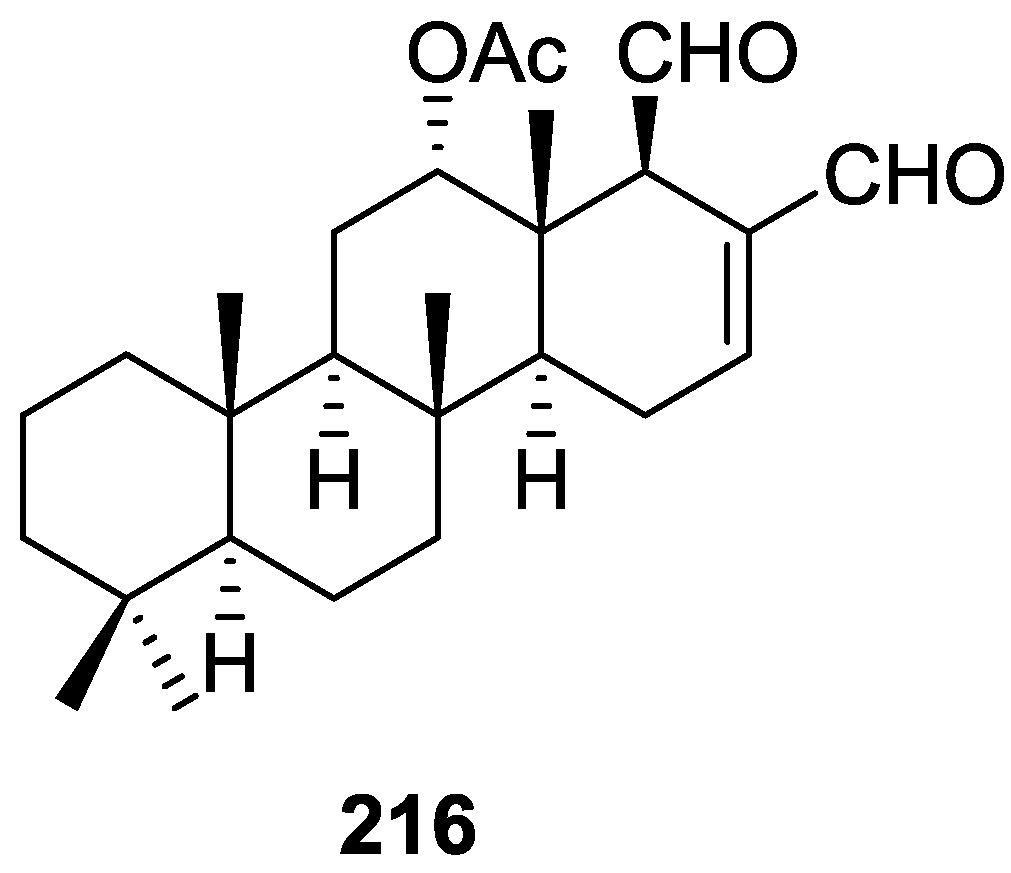
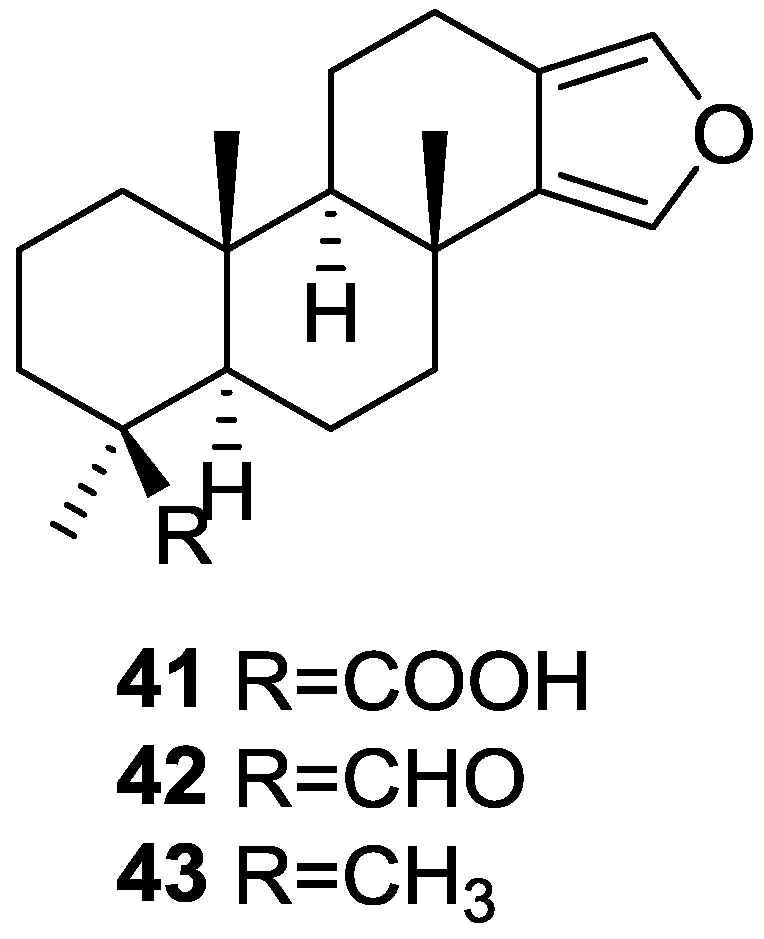
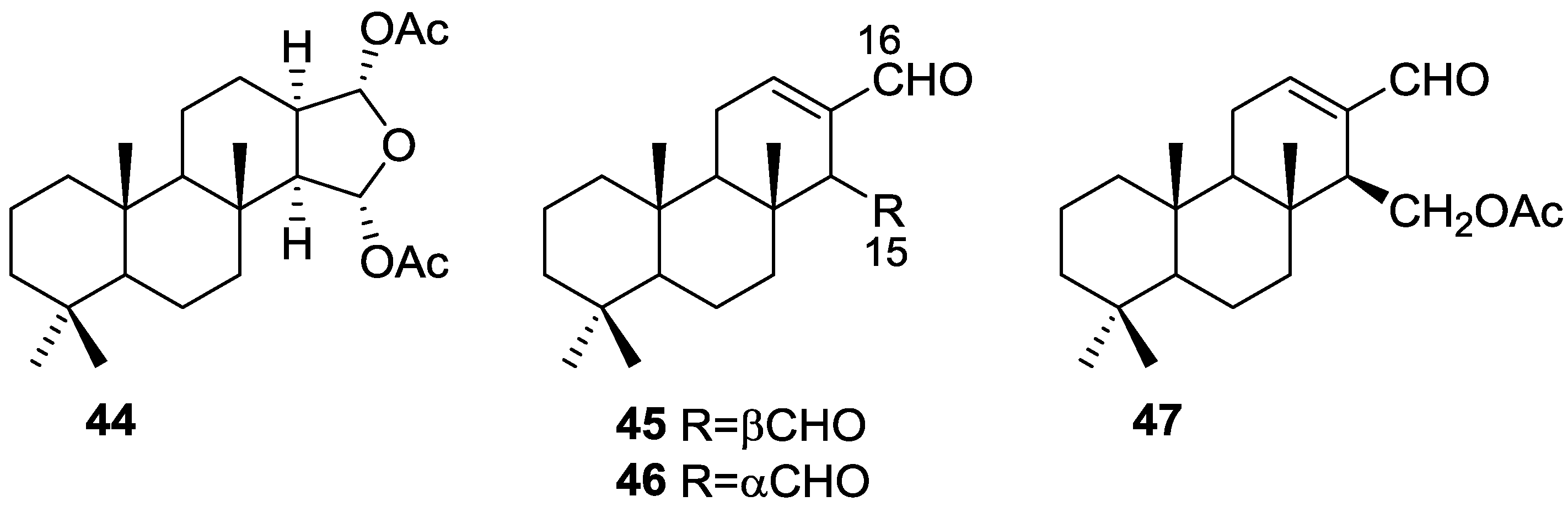
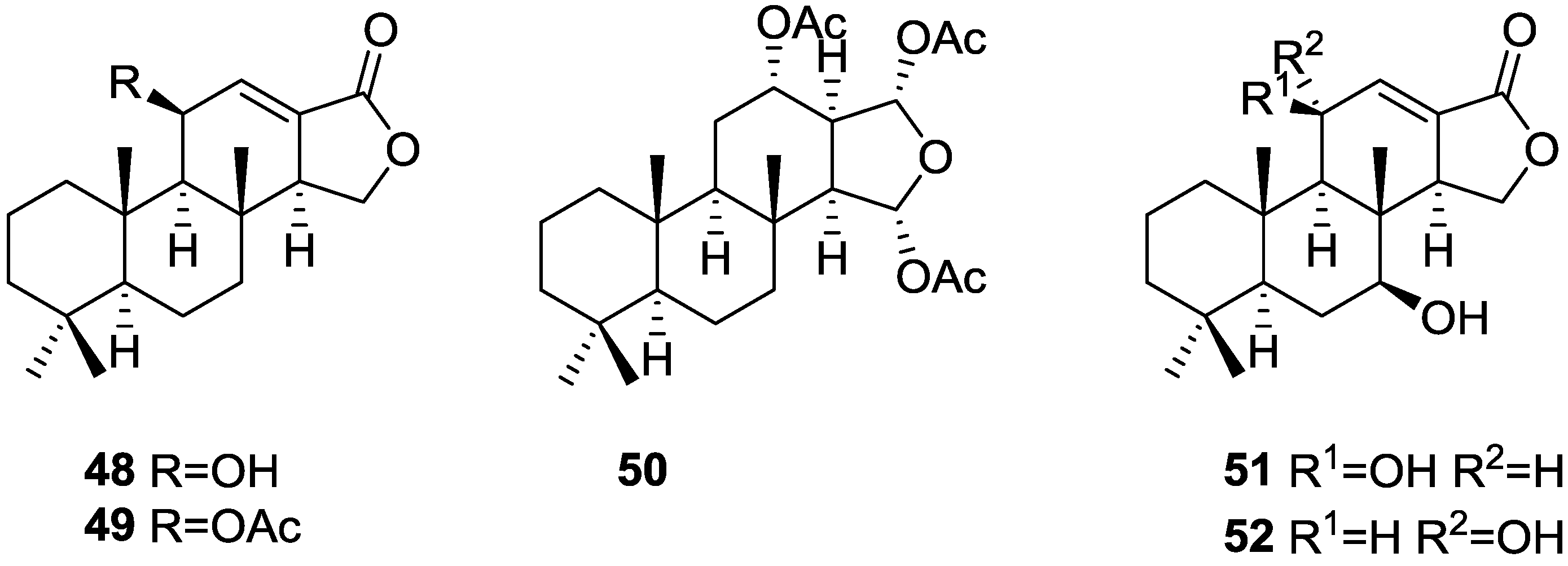
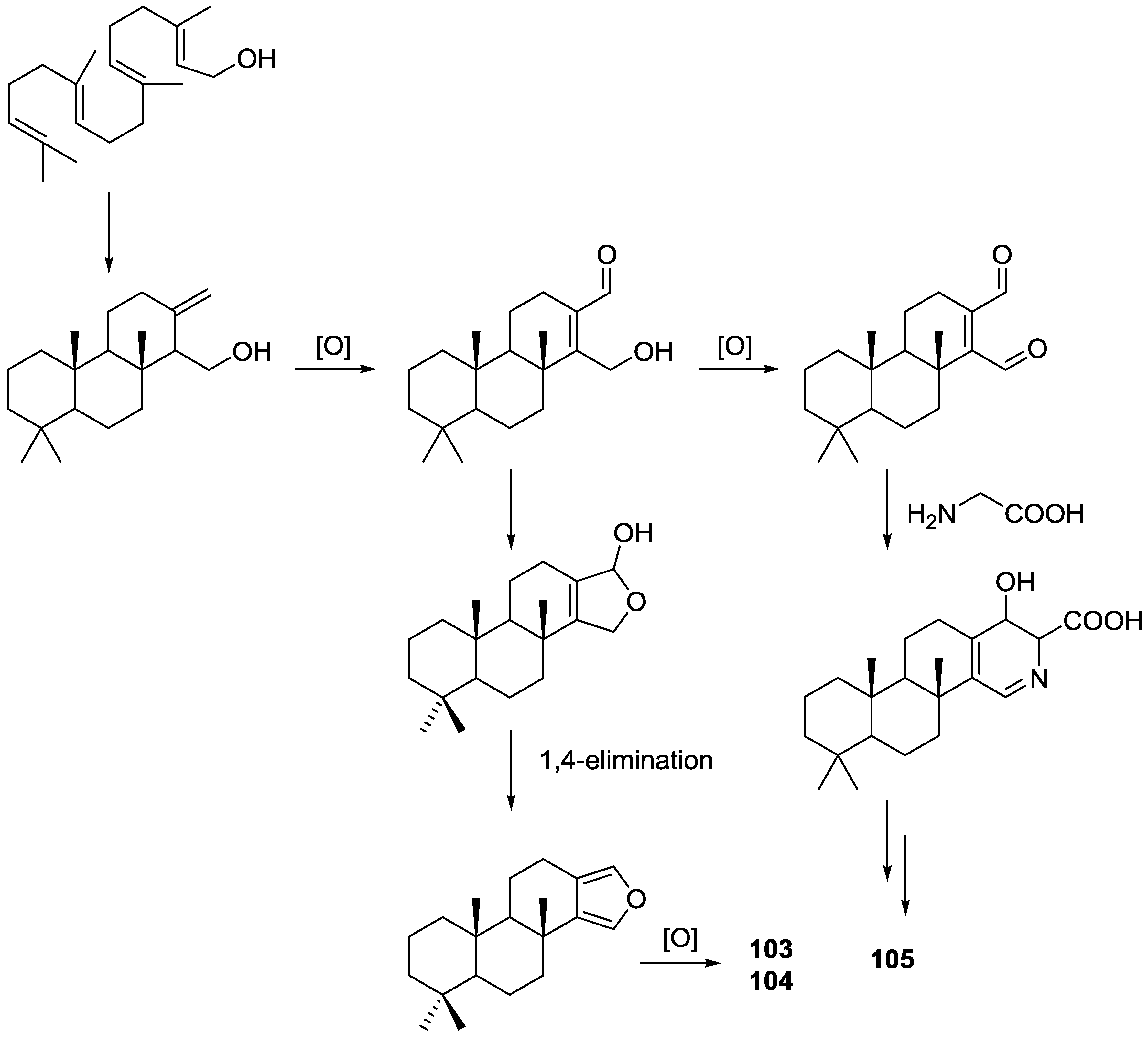
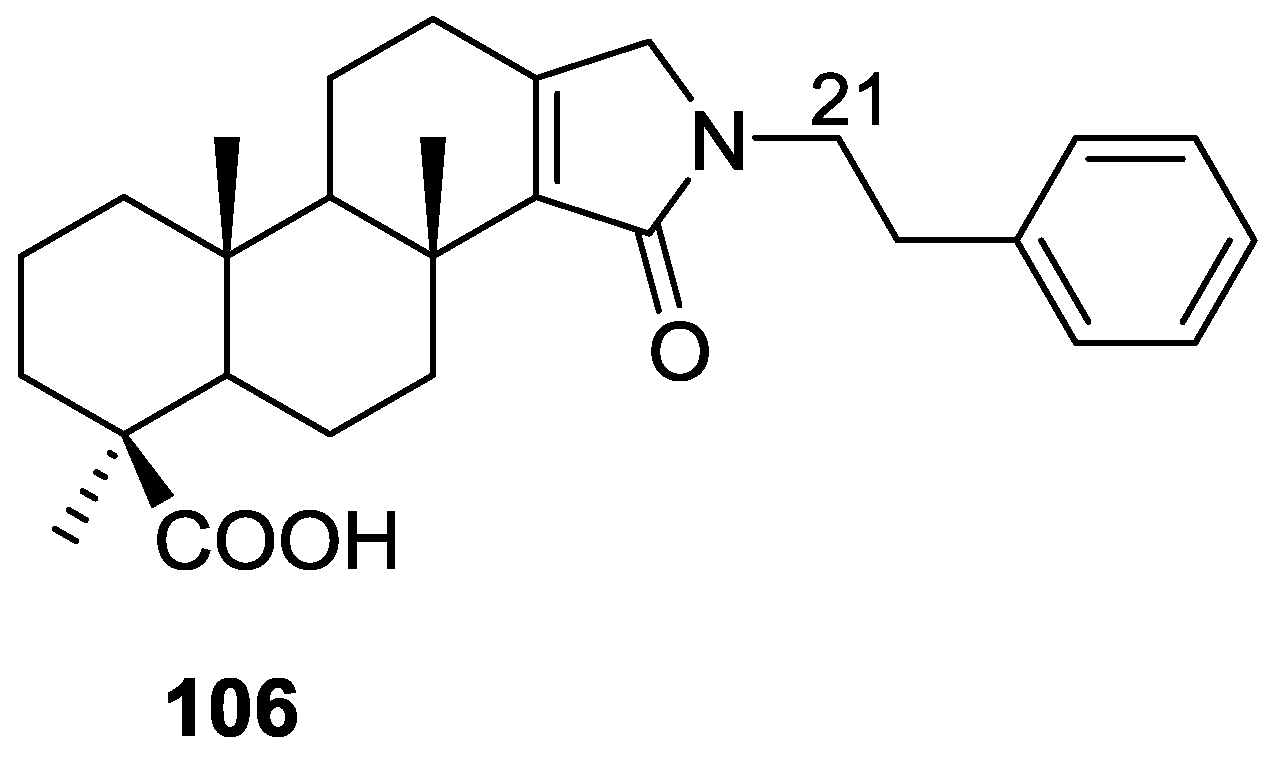
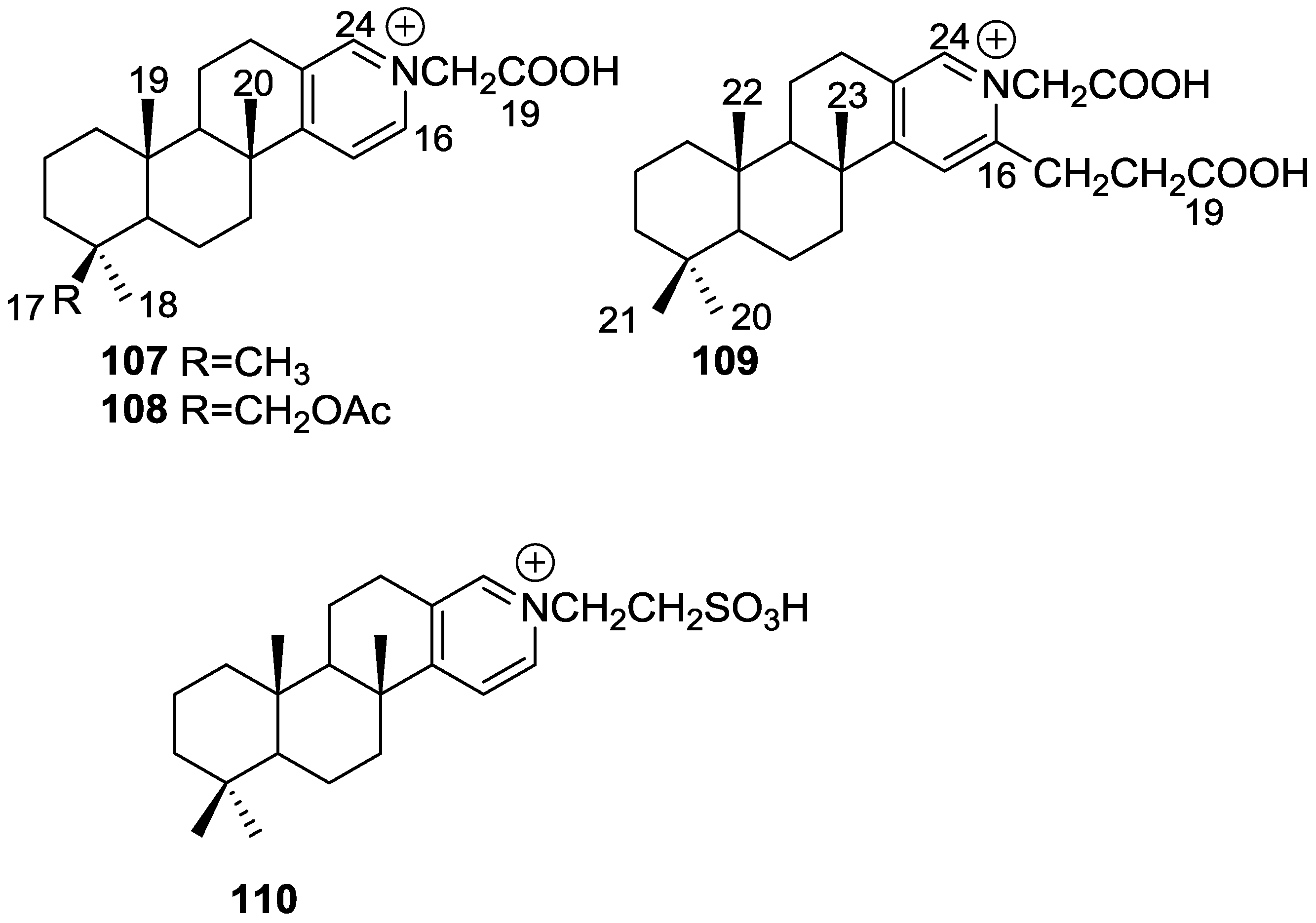
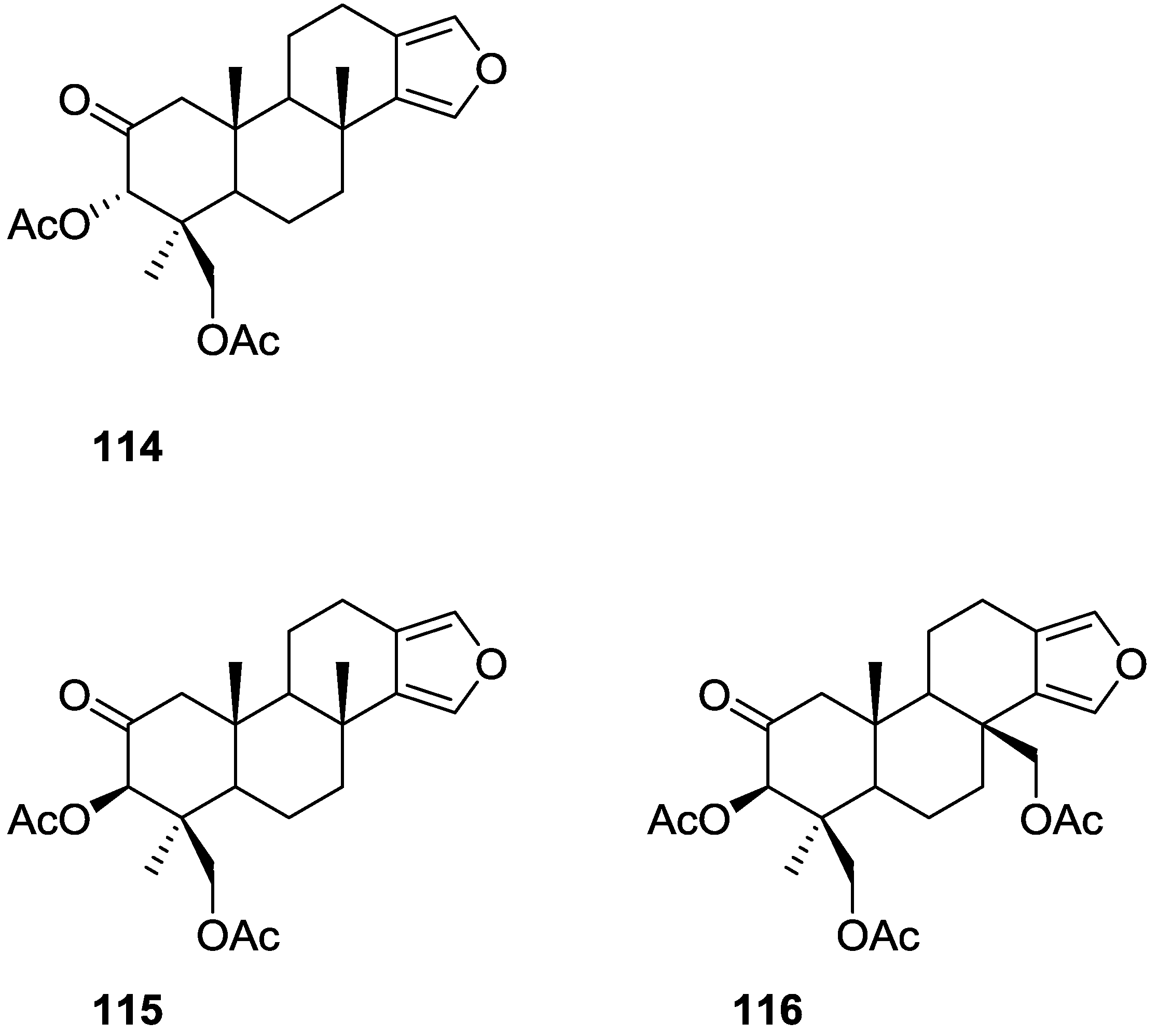
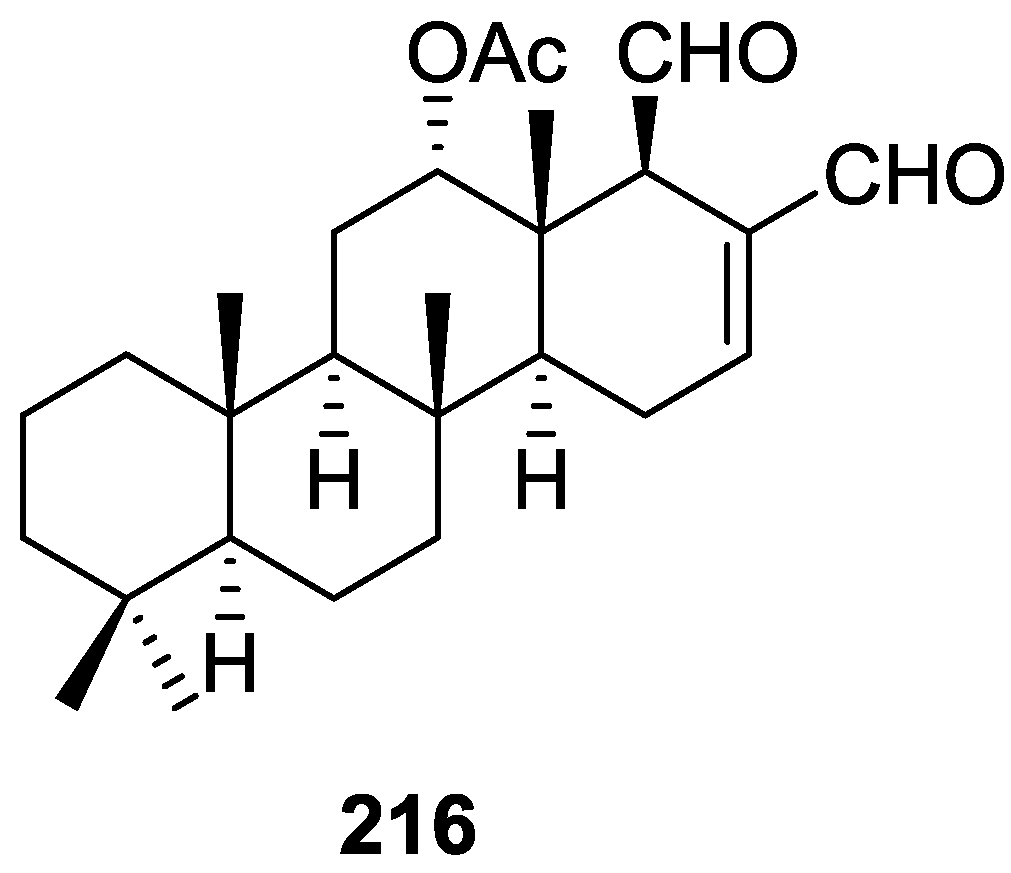
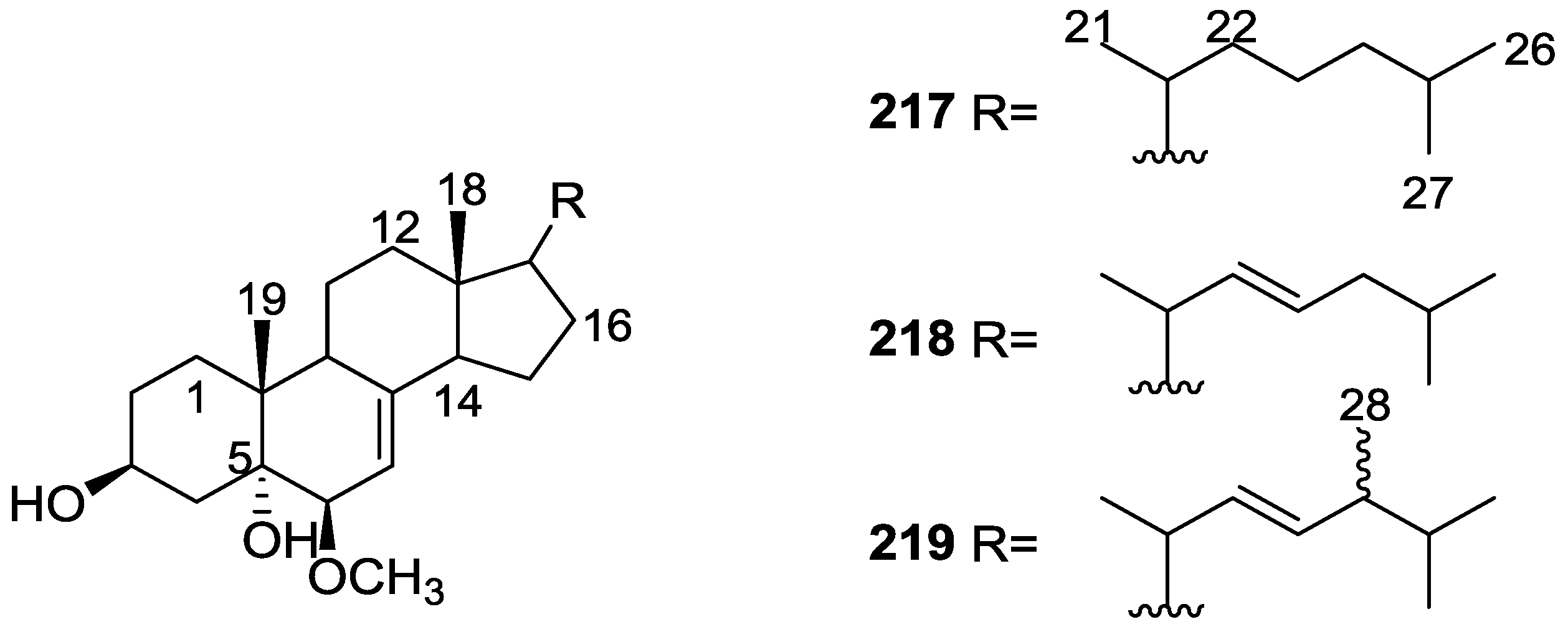
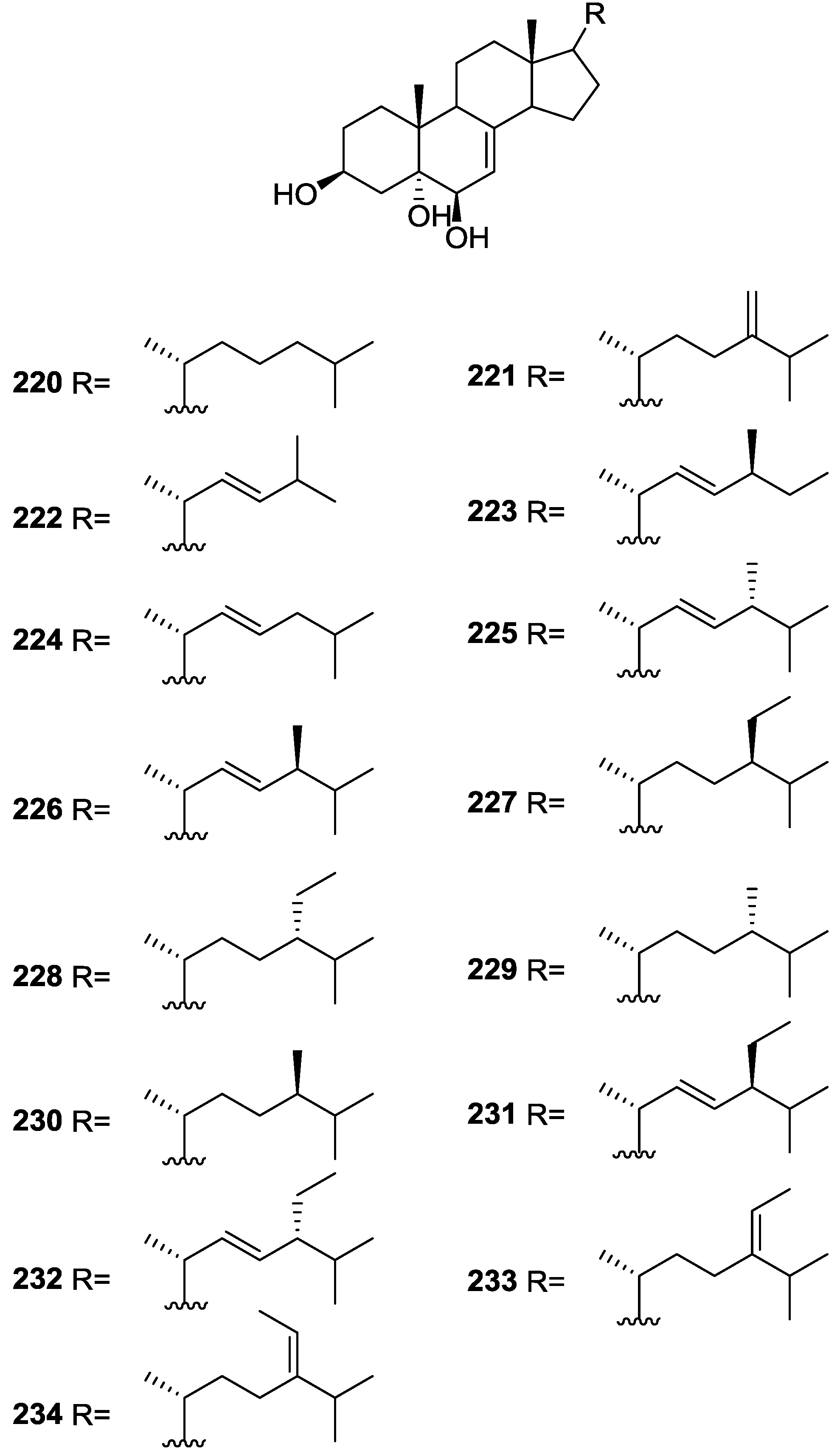
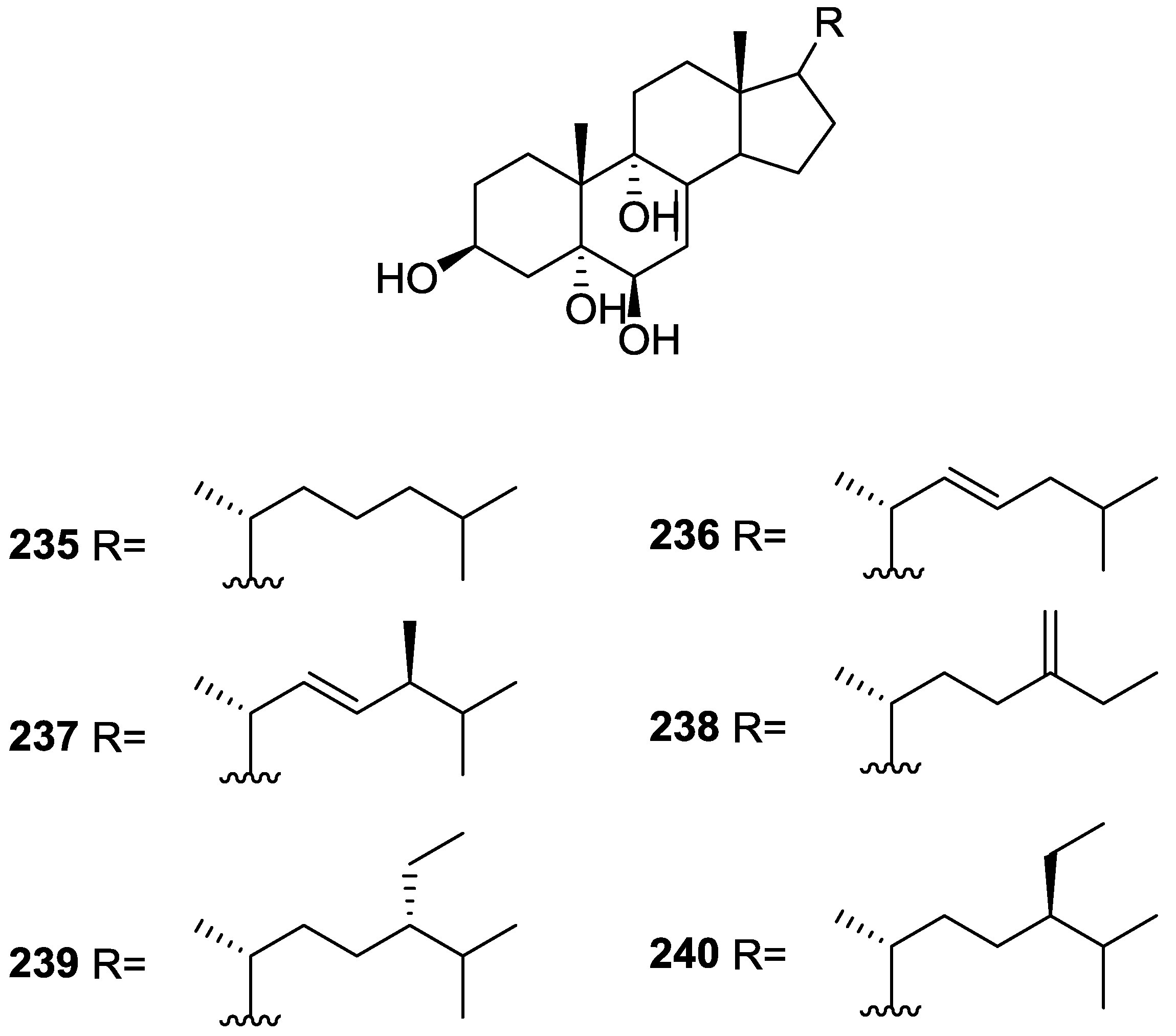
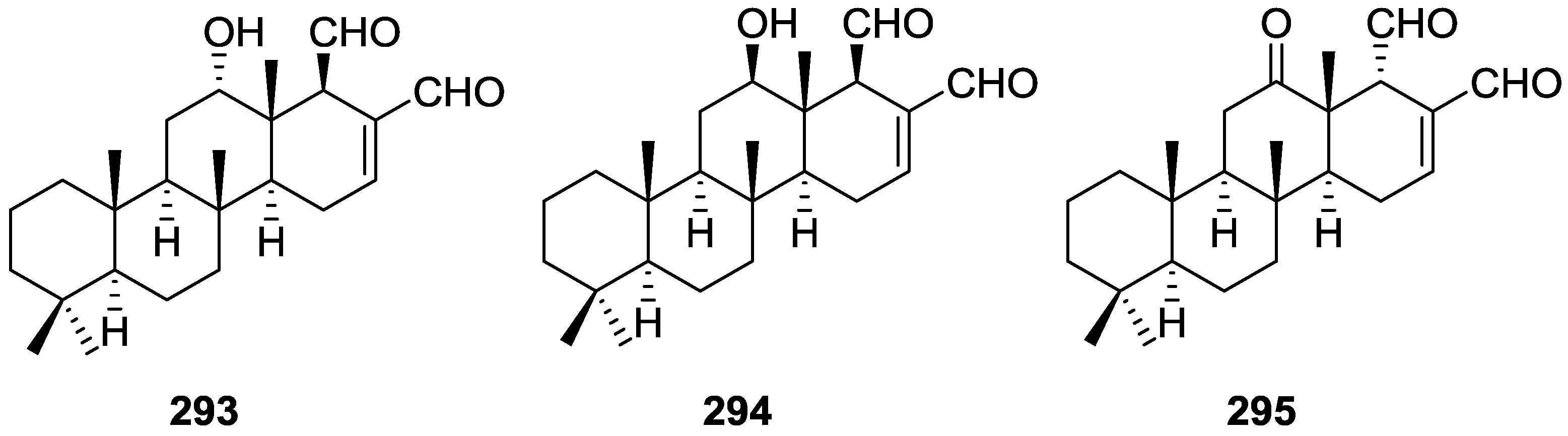
3. Diterpenes
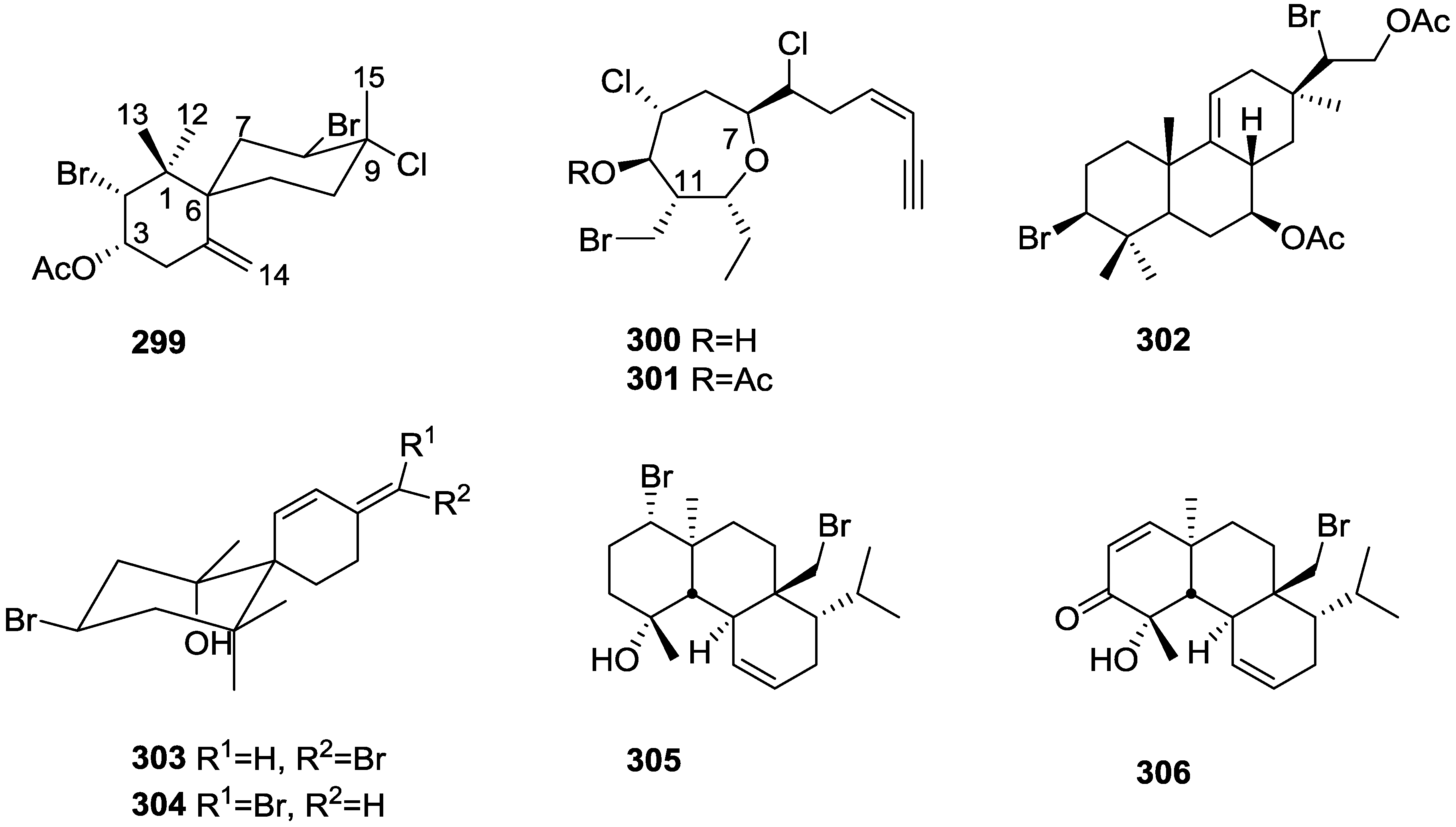
Other Studies
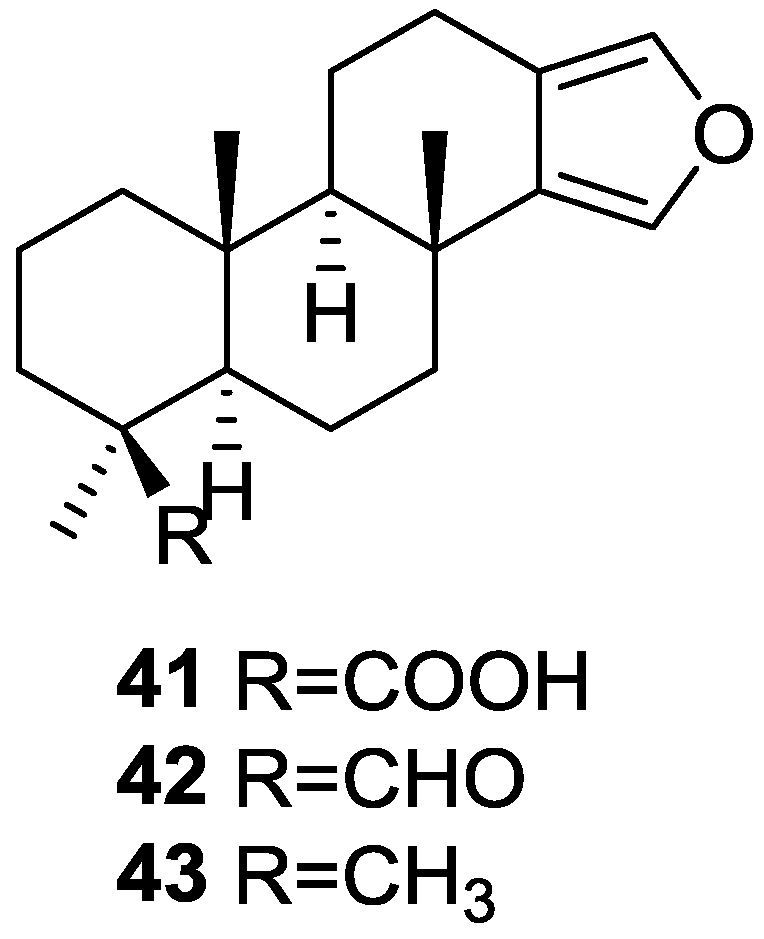
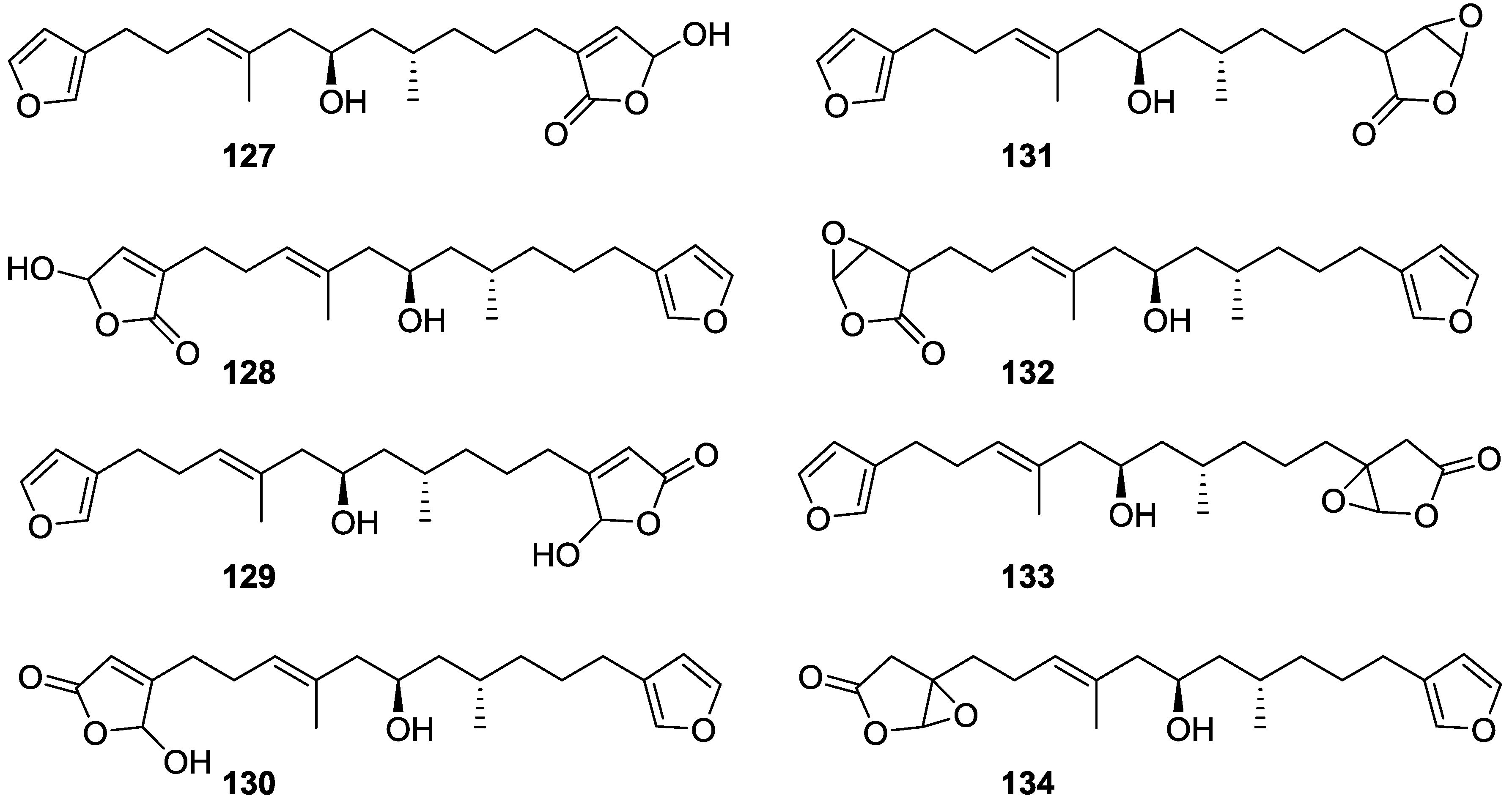
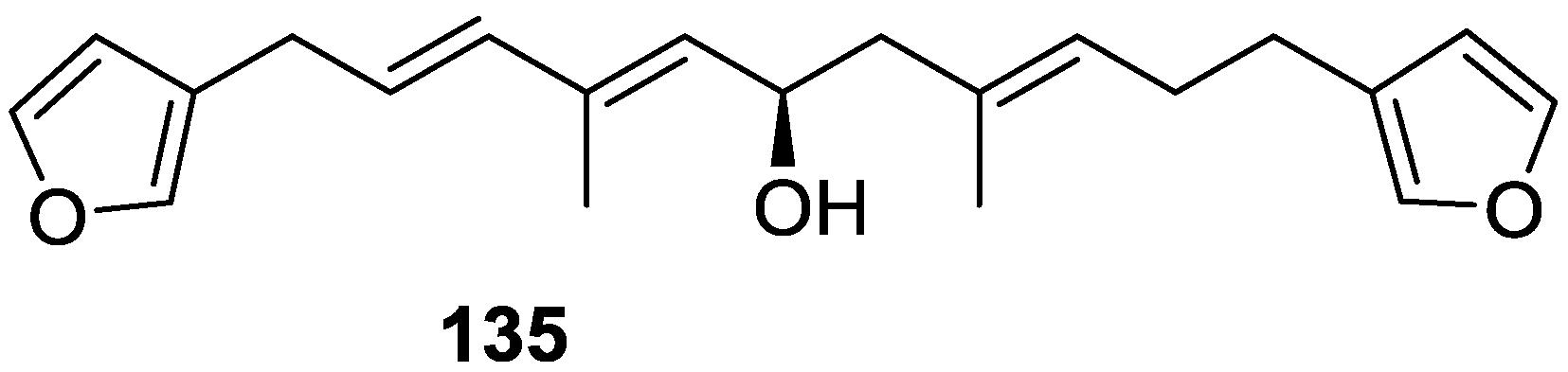
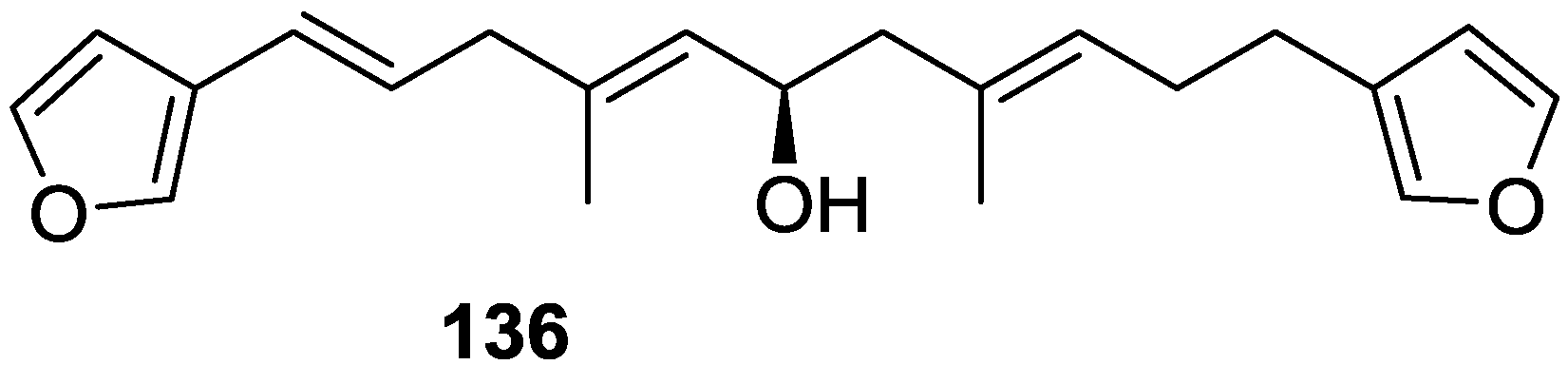
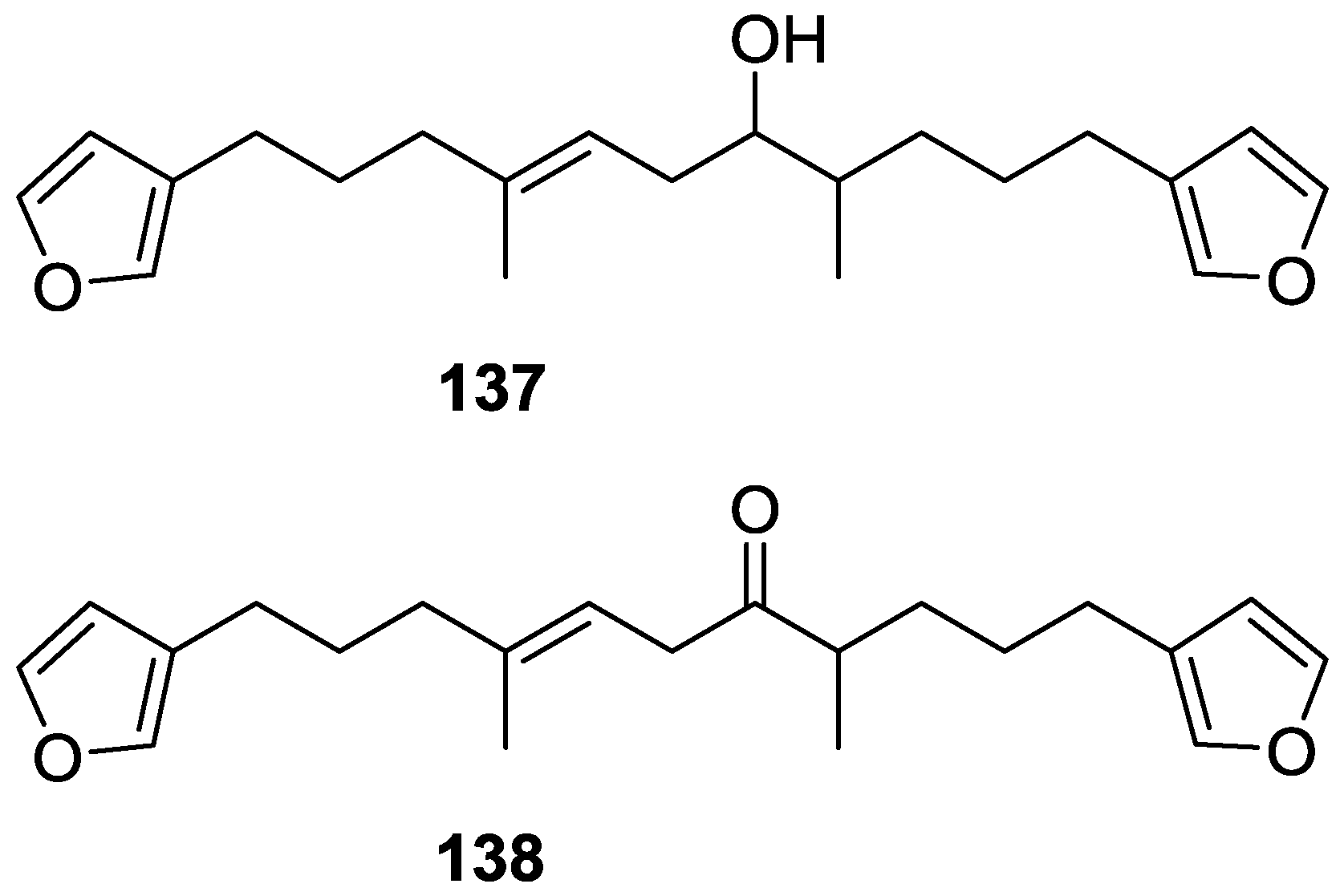
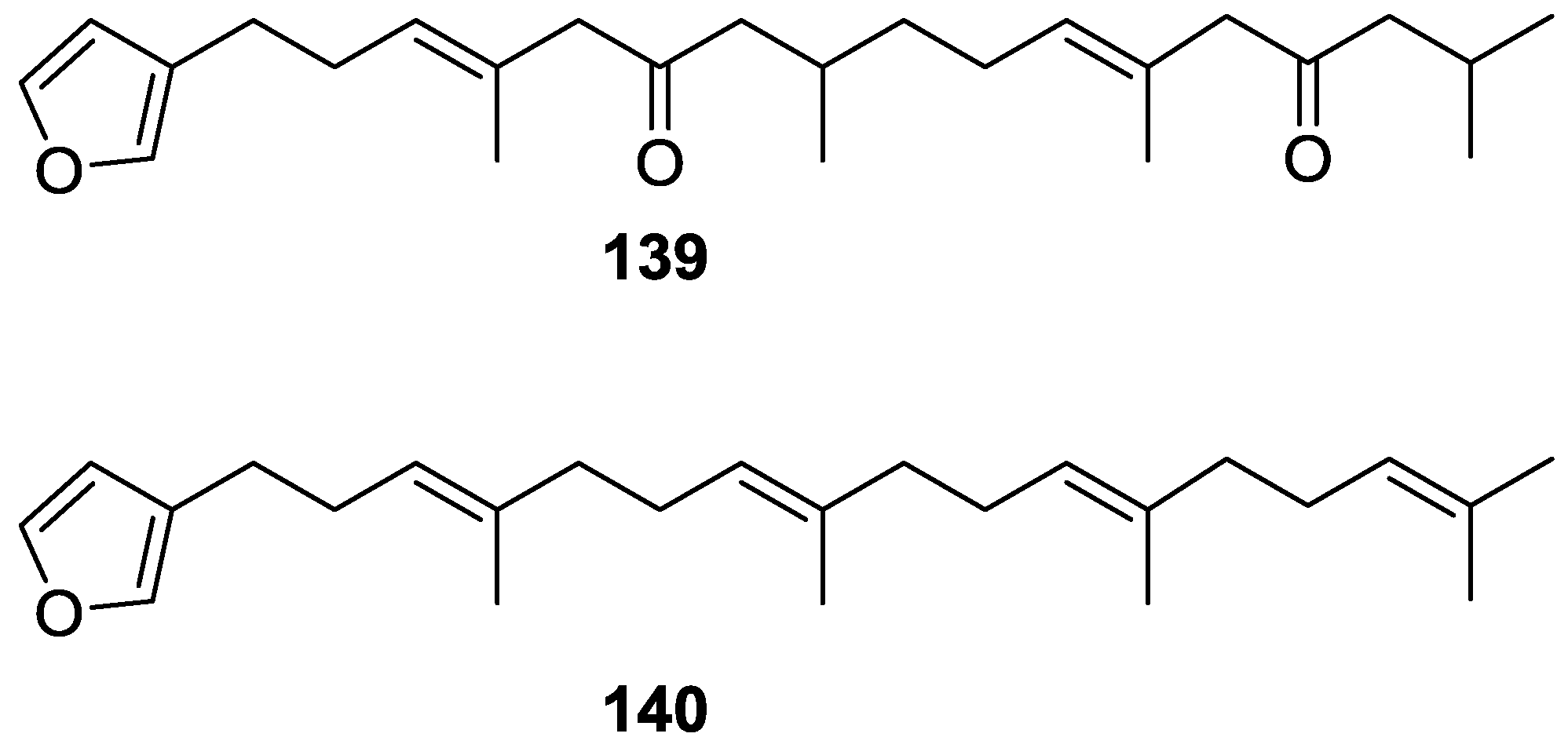
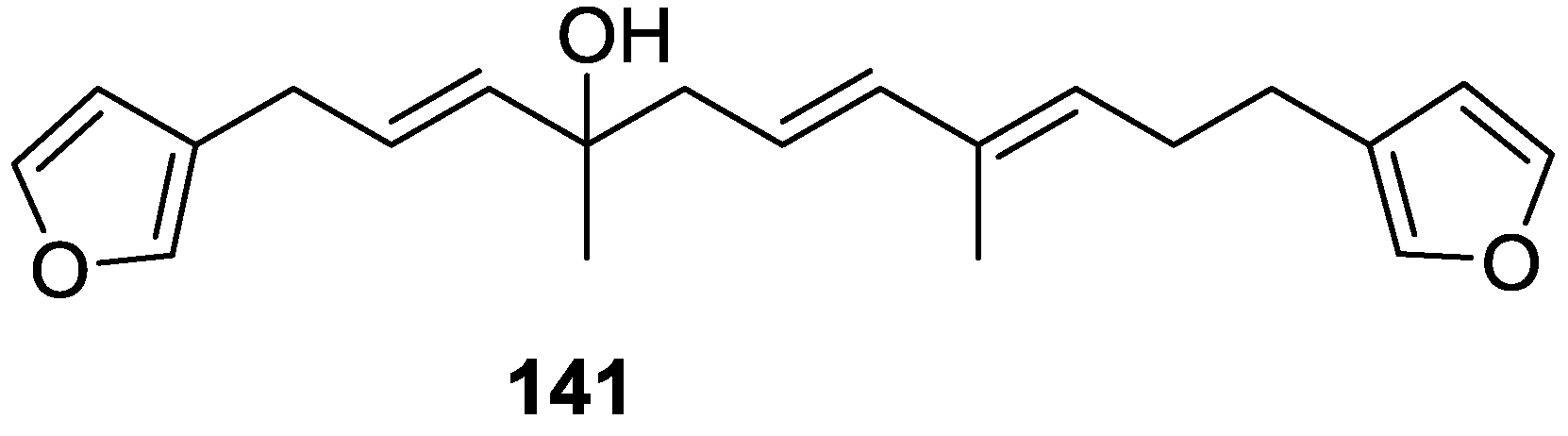
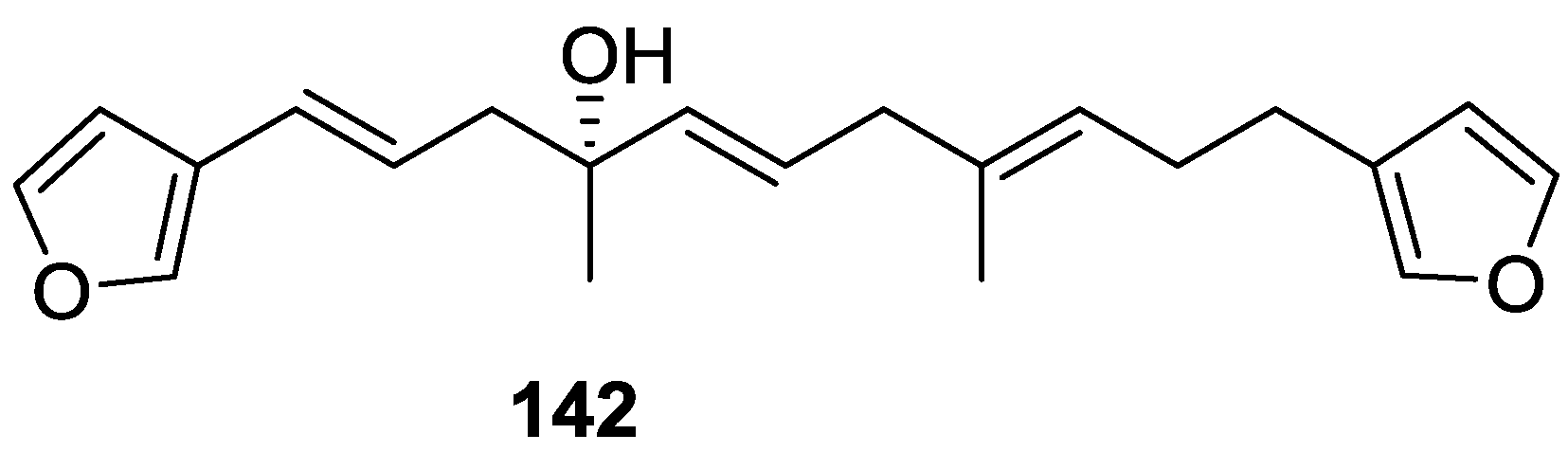
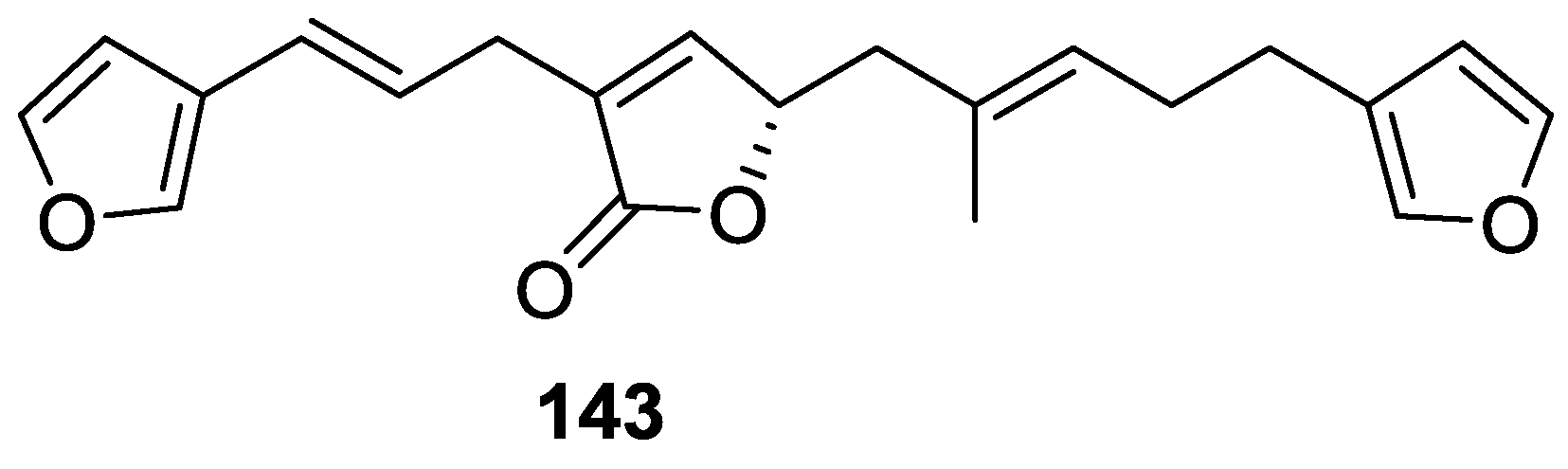
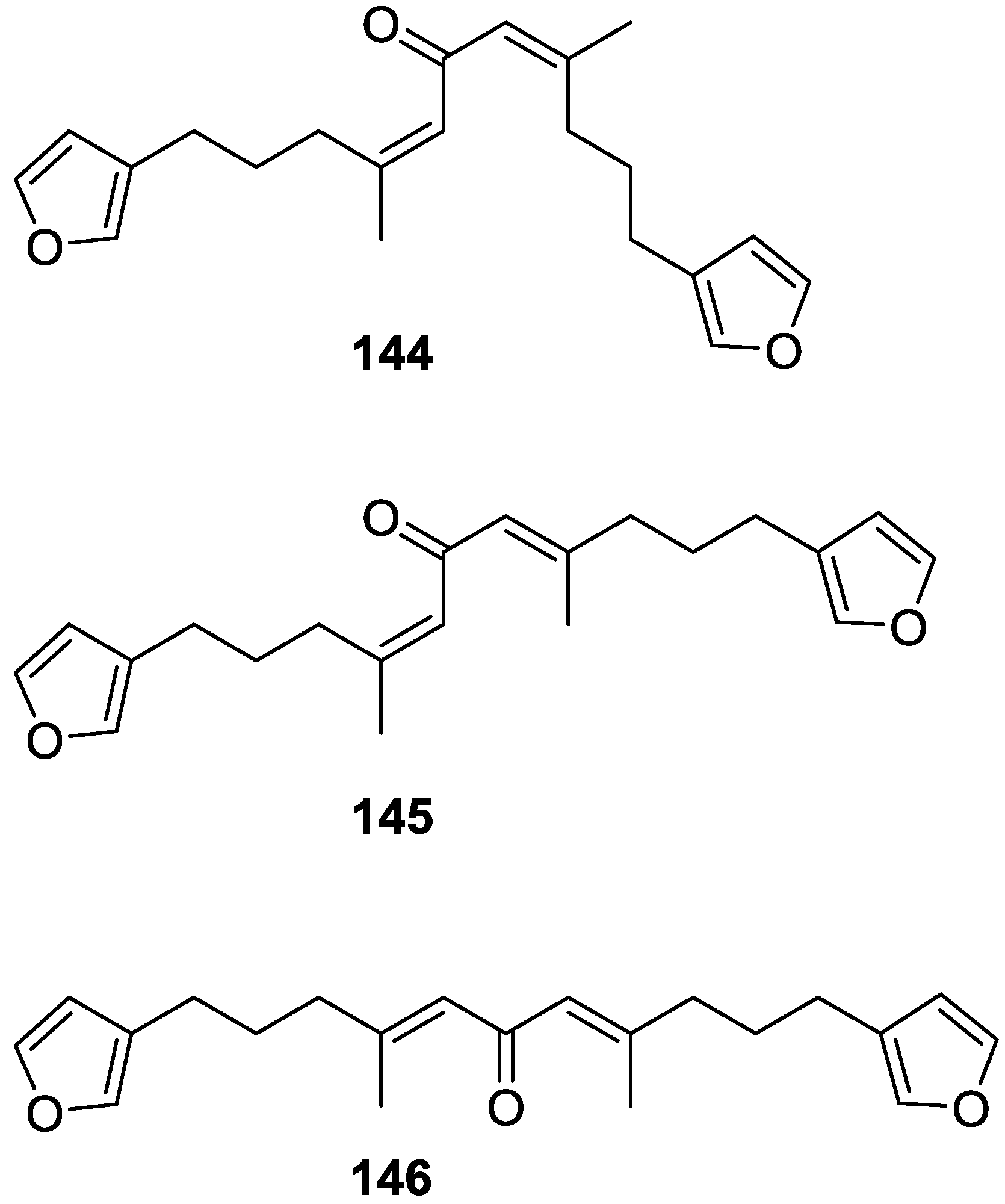
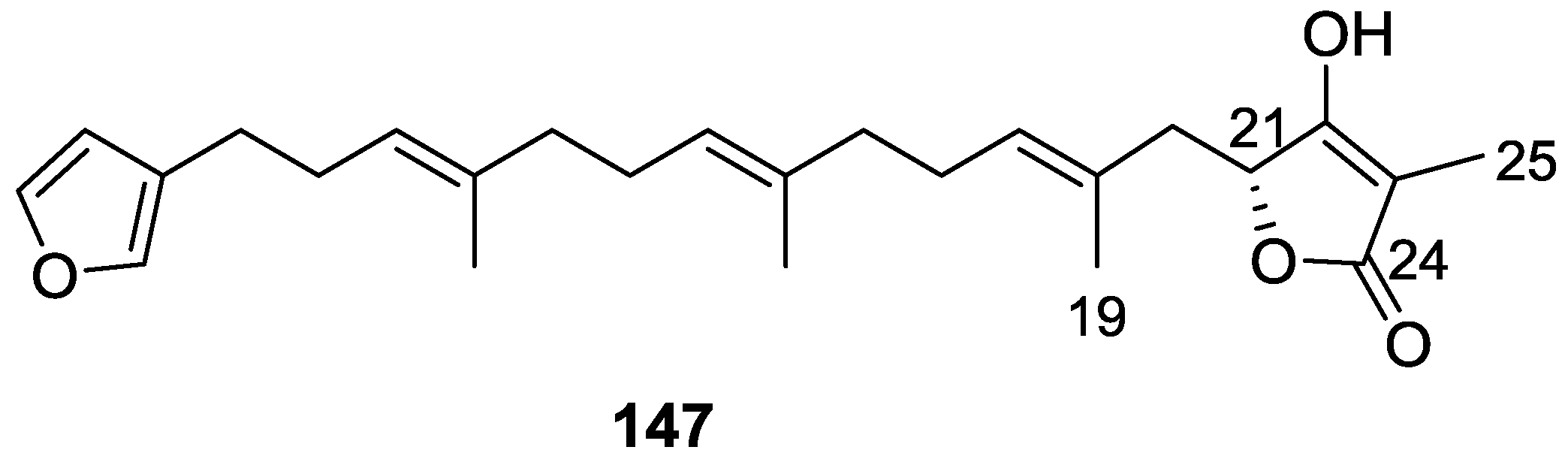
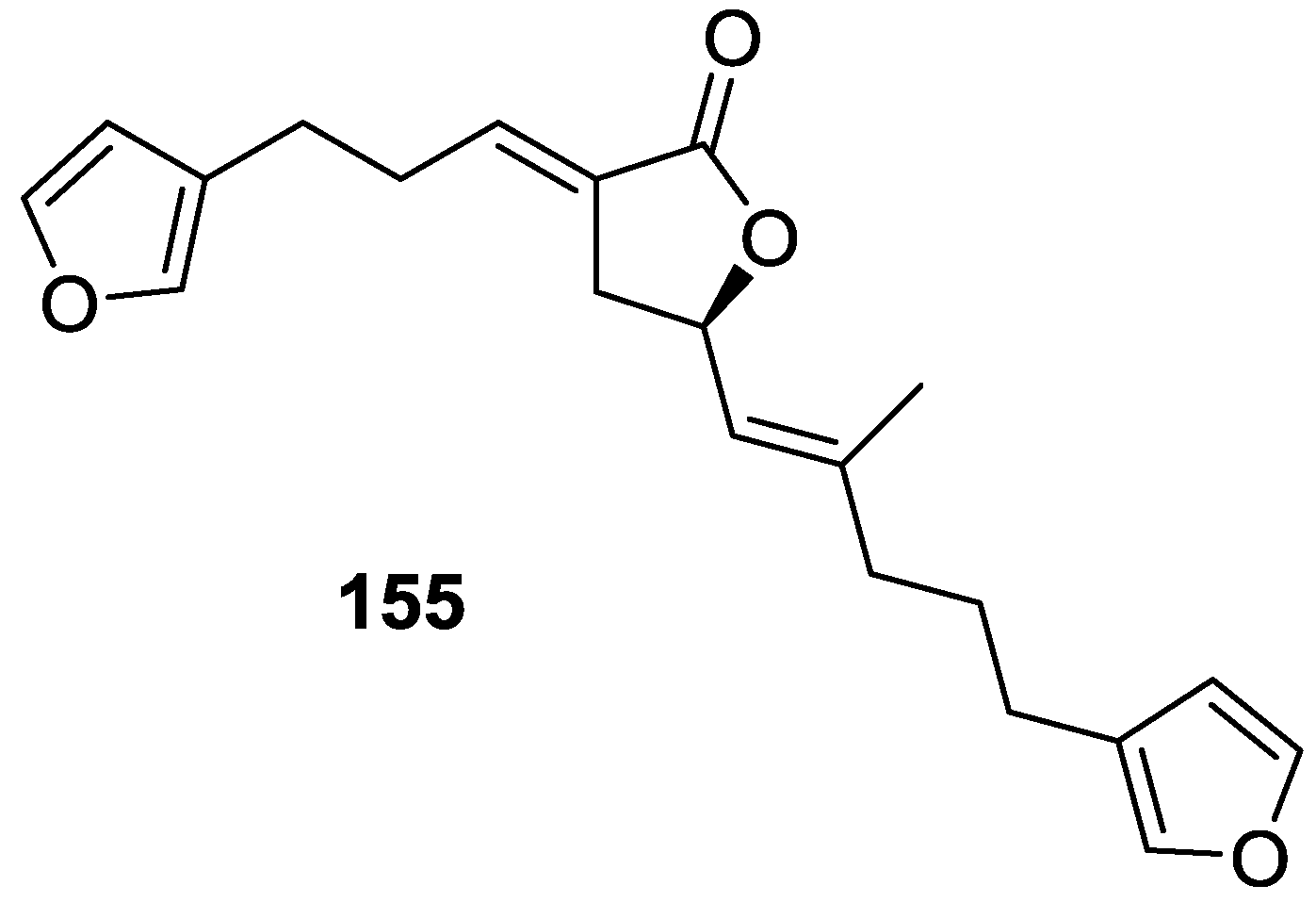
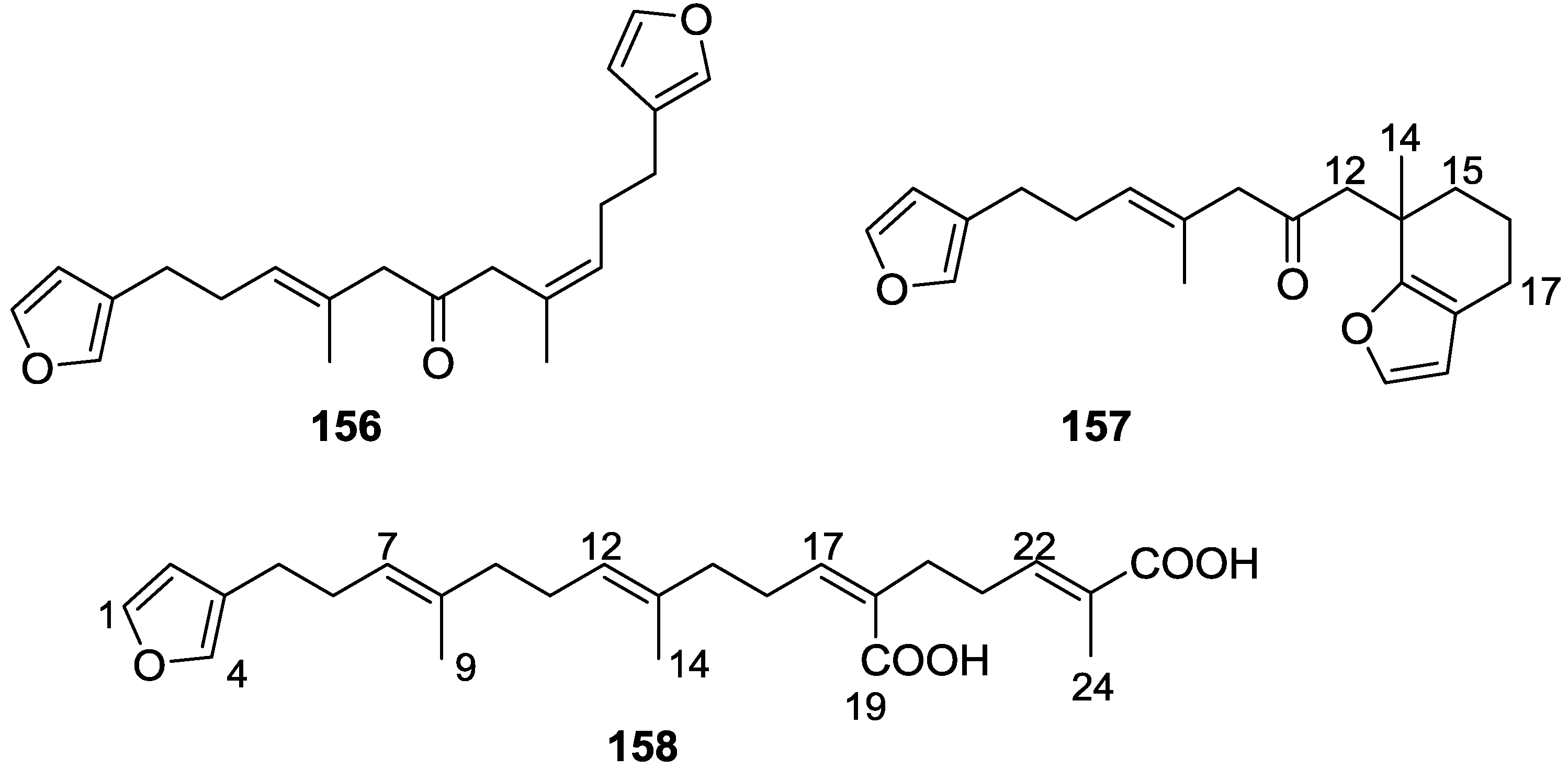
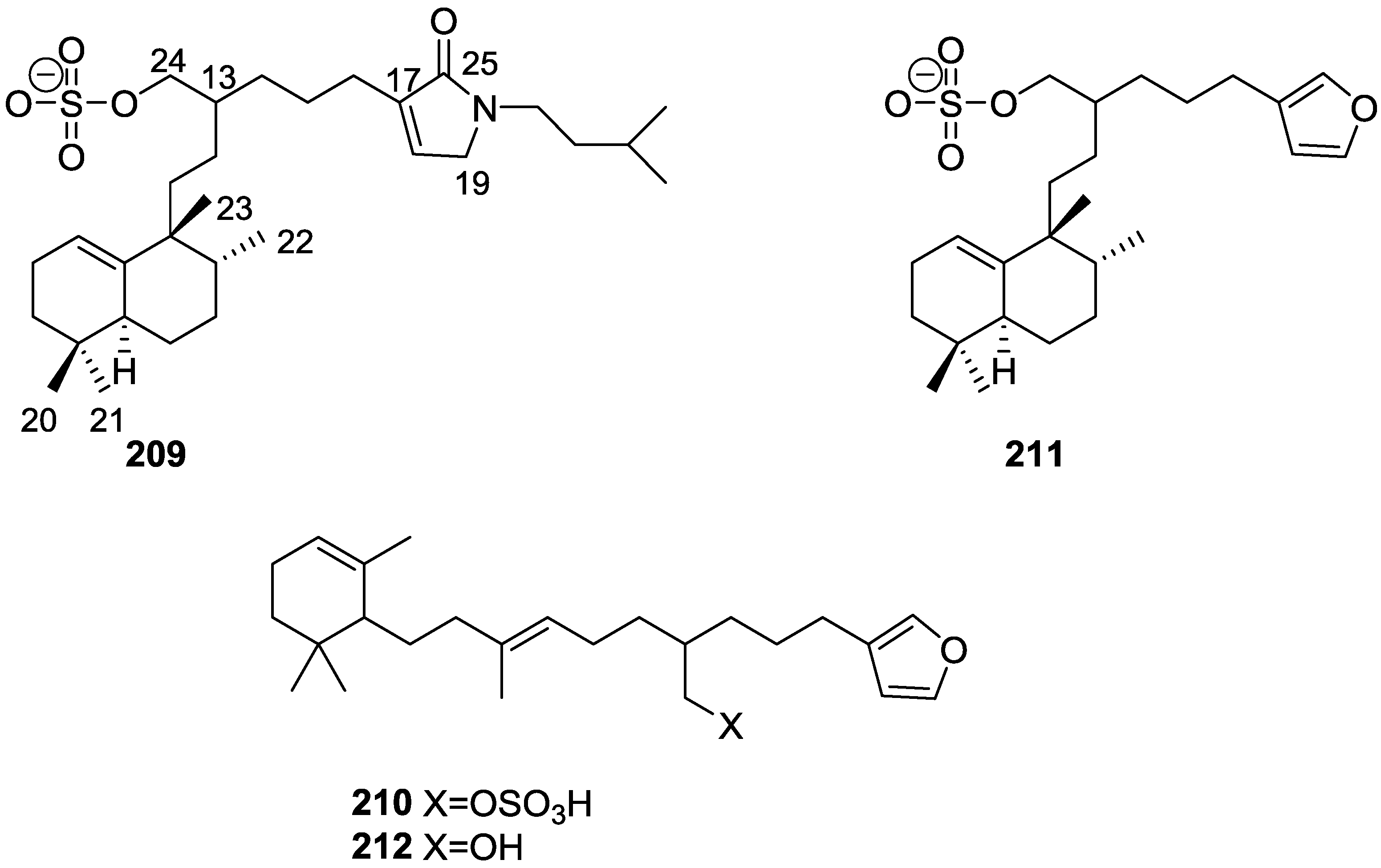
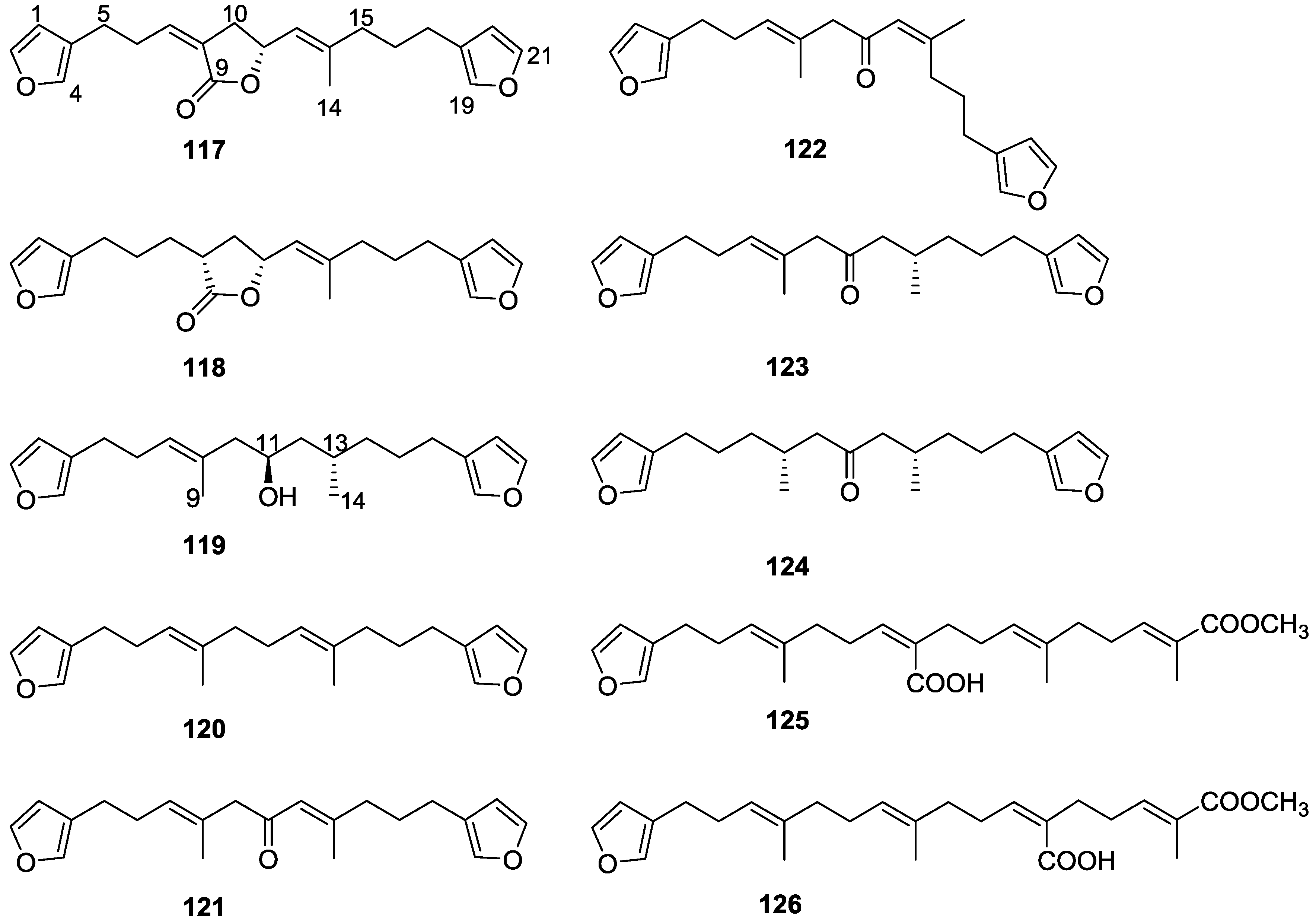
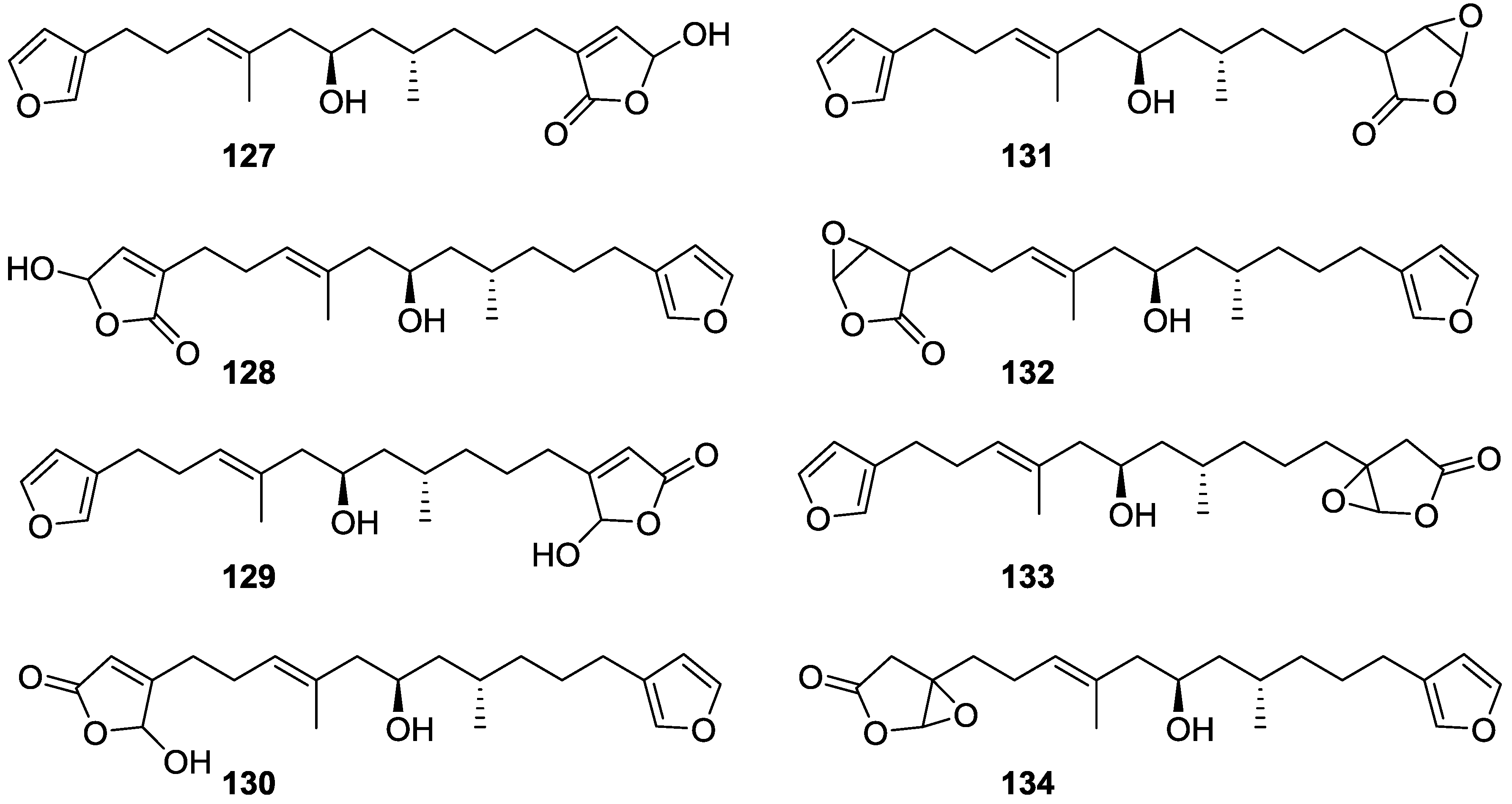
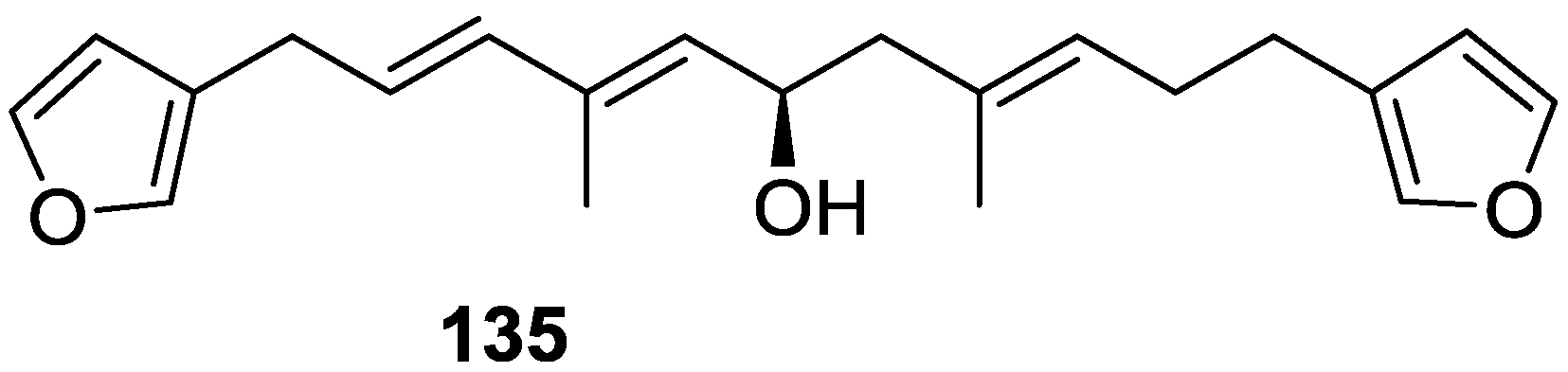
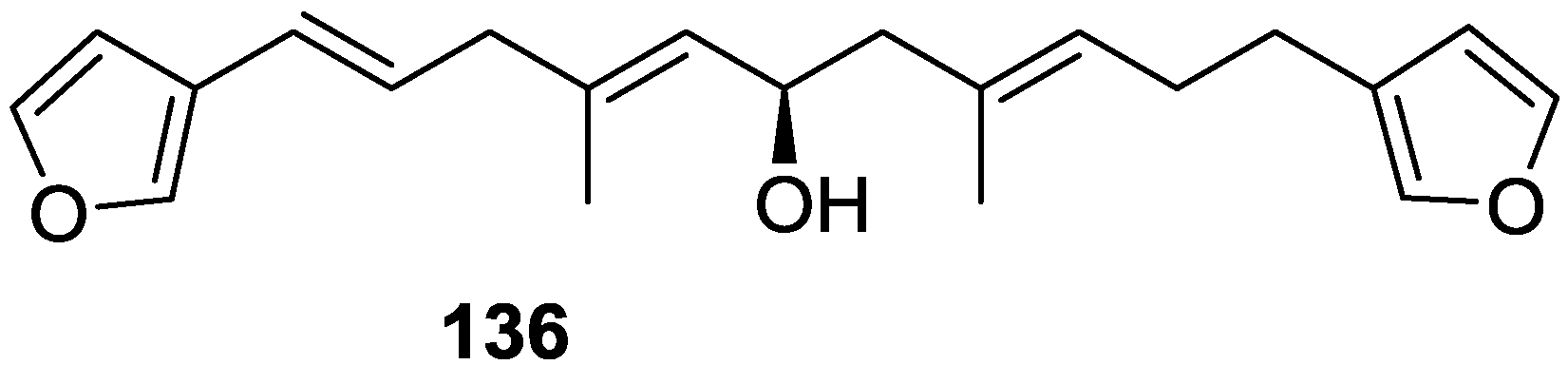
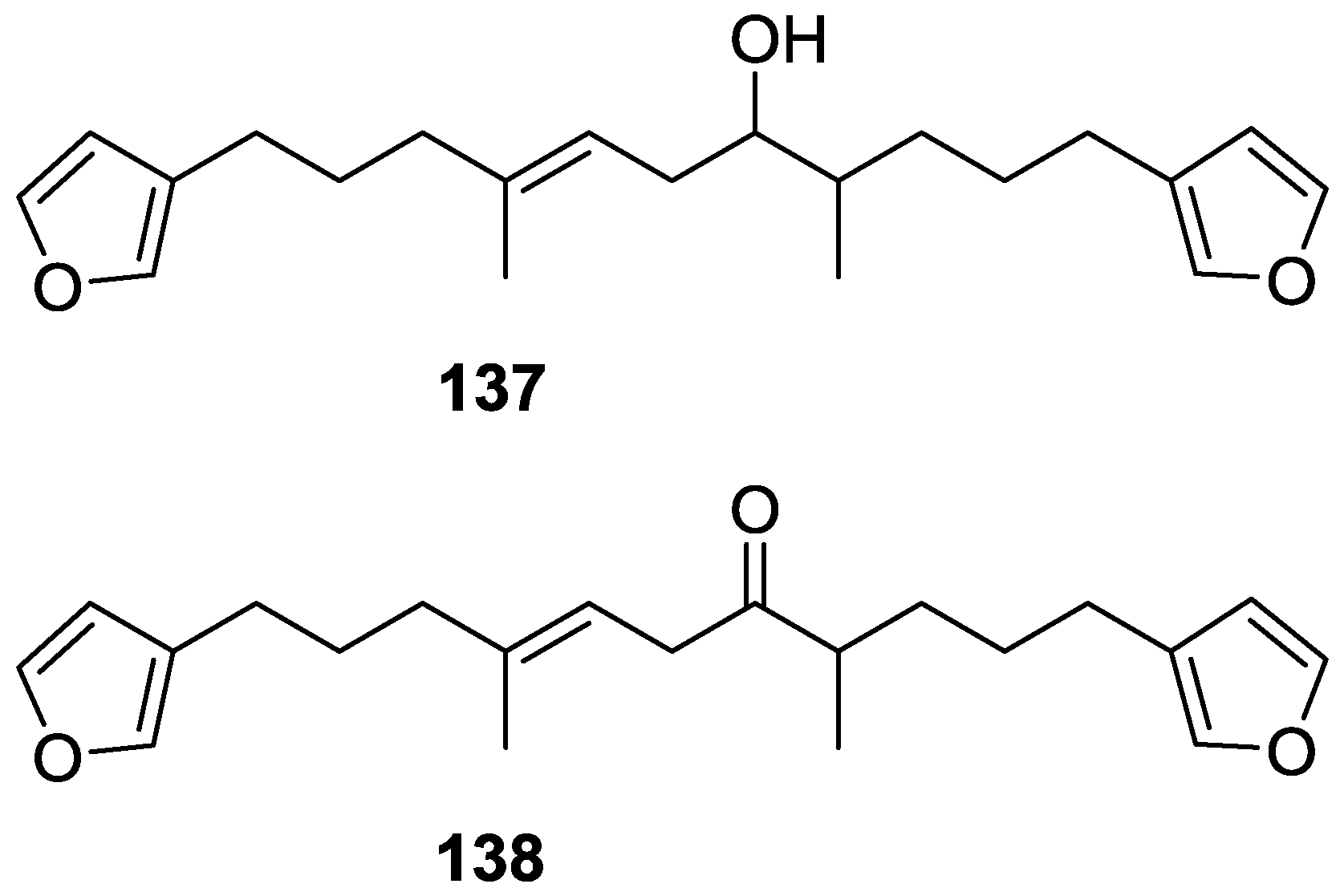
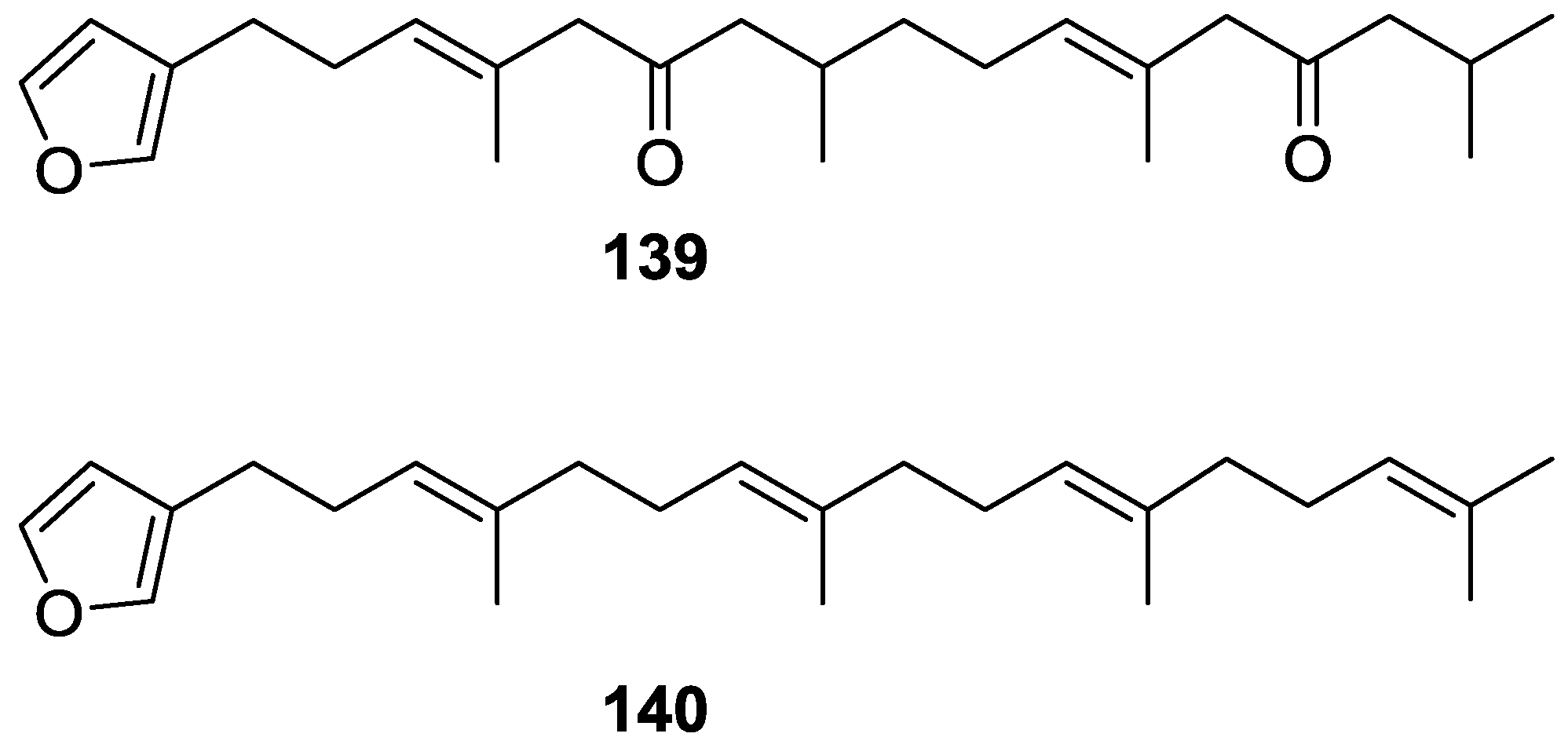
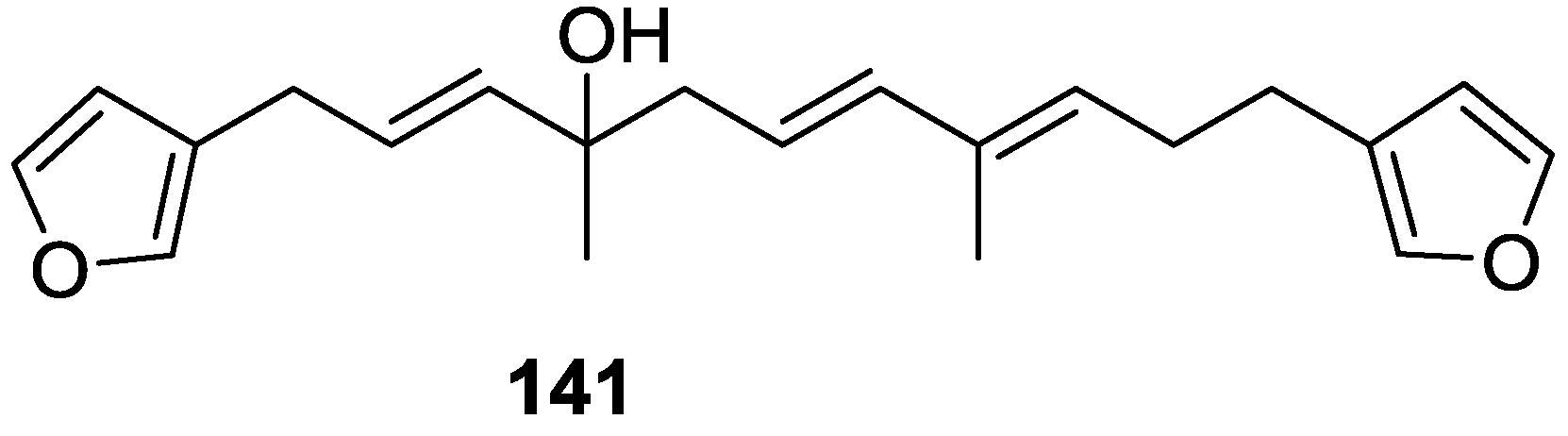
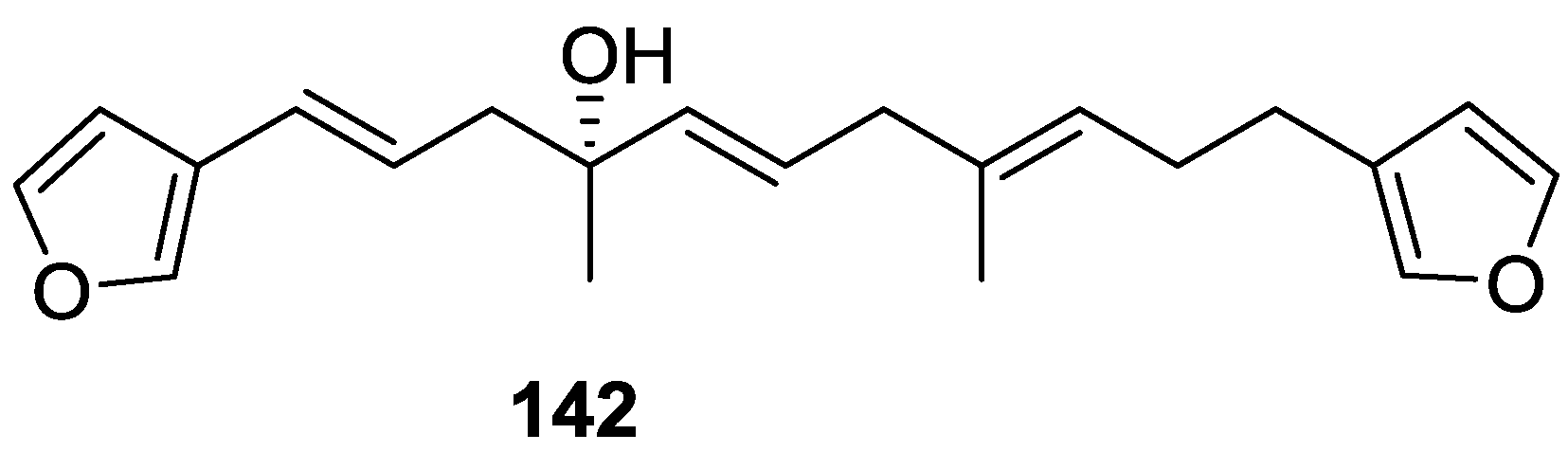
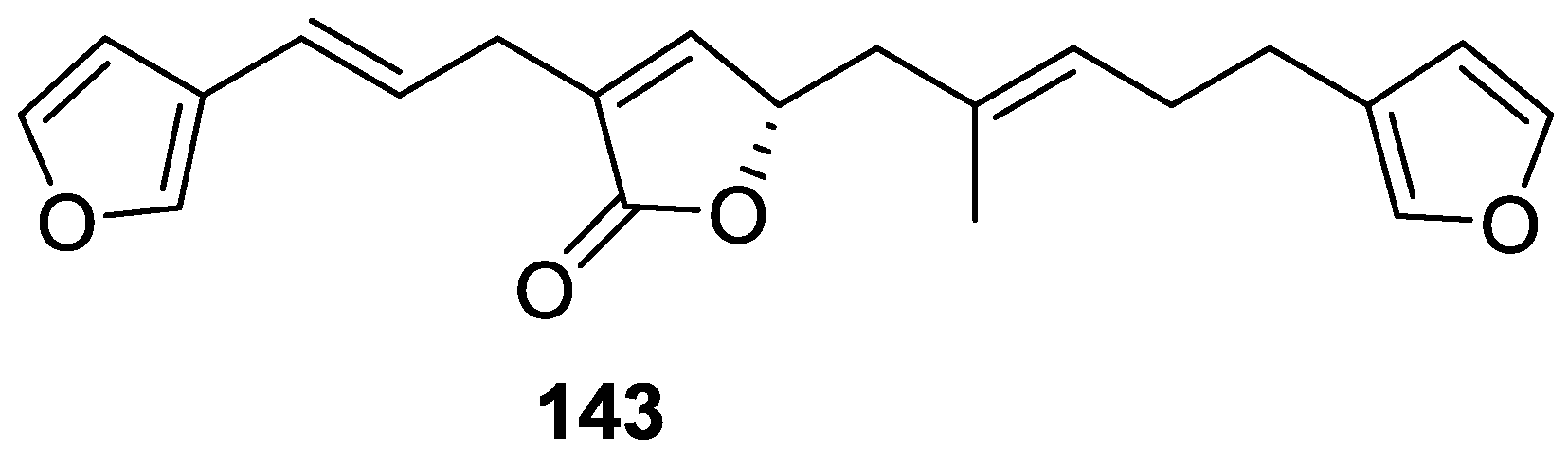
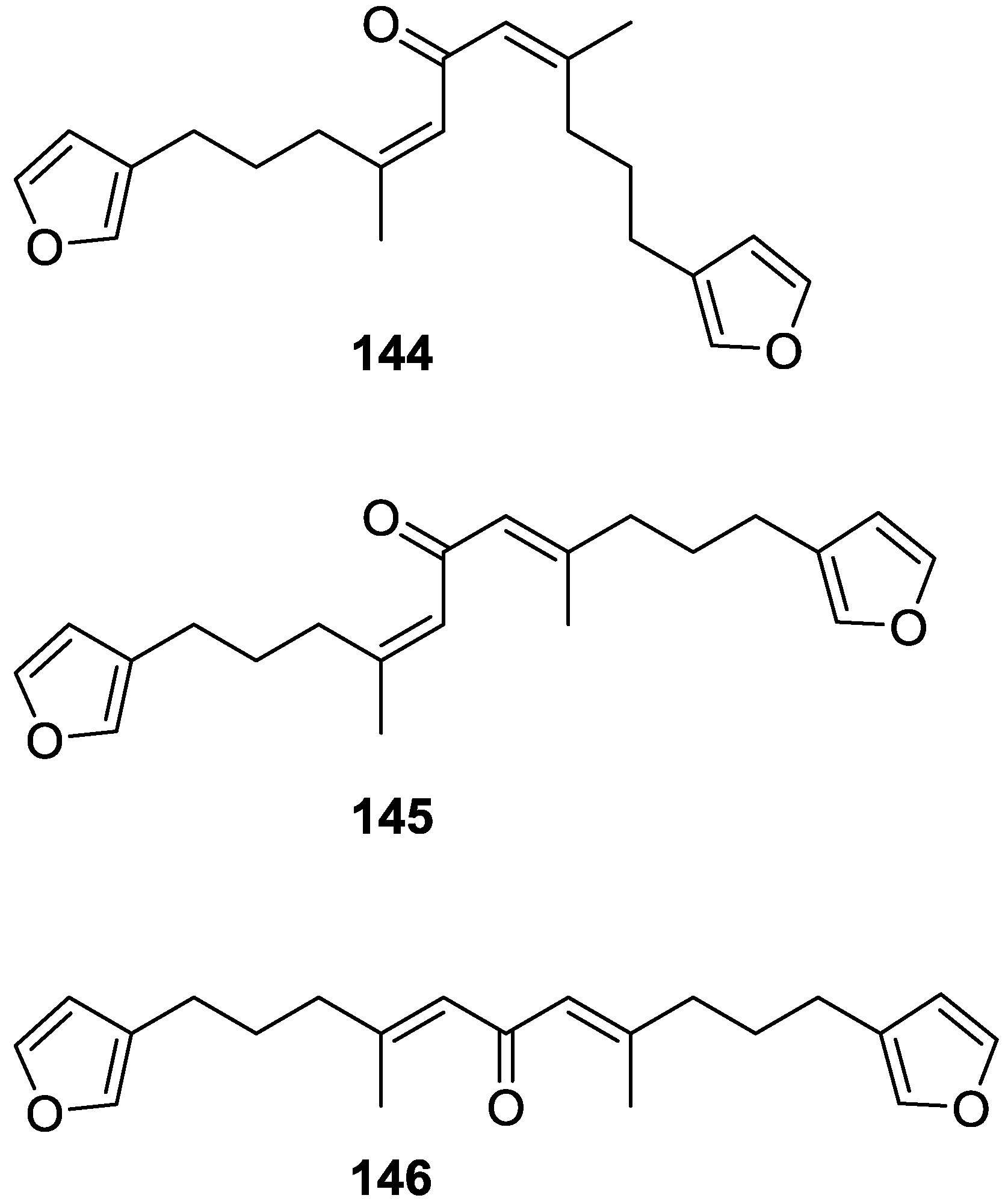
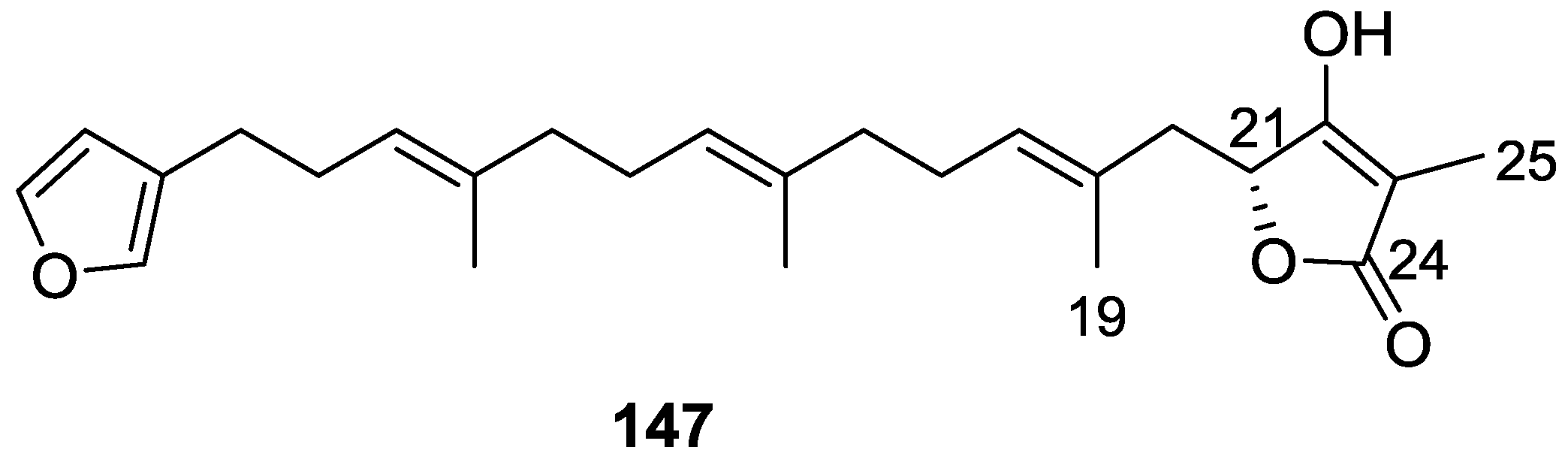
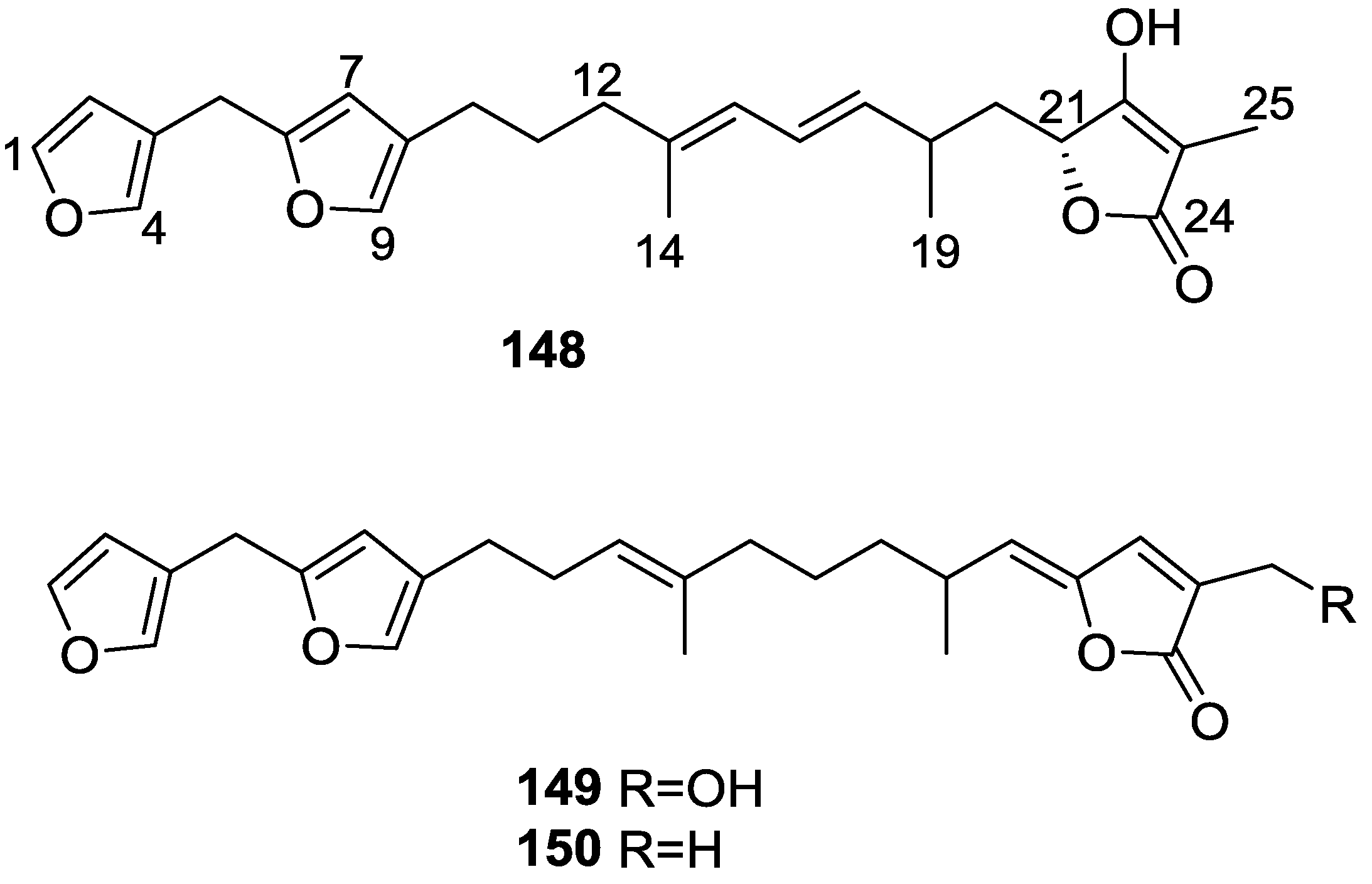
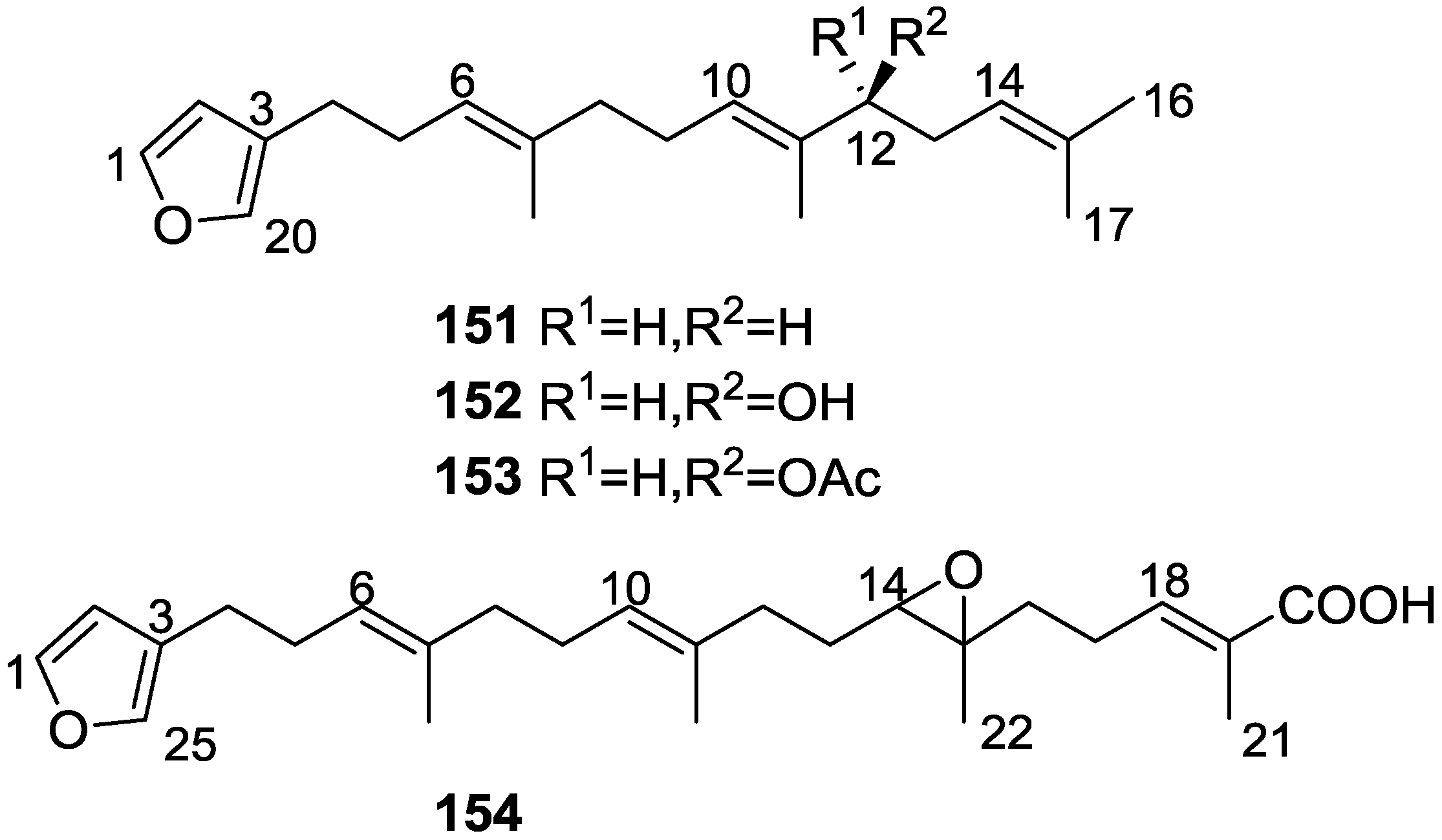
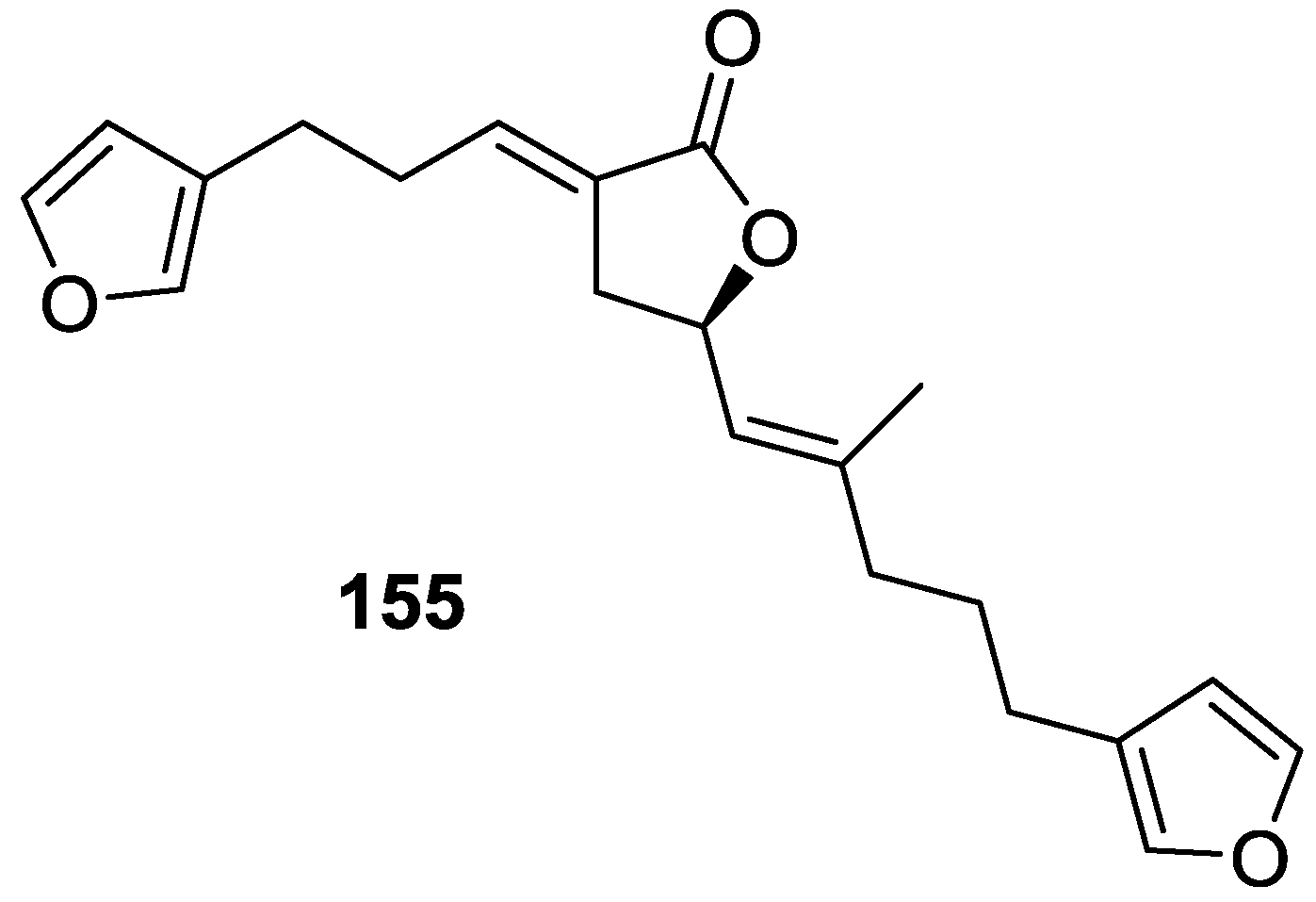
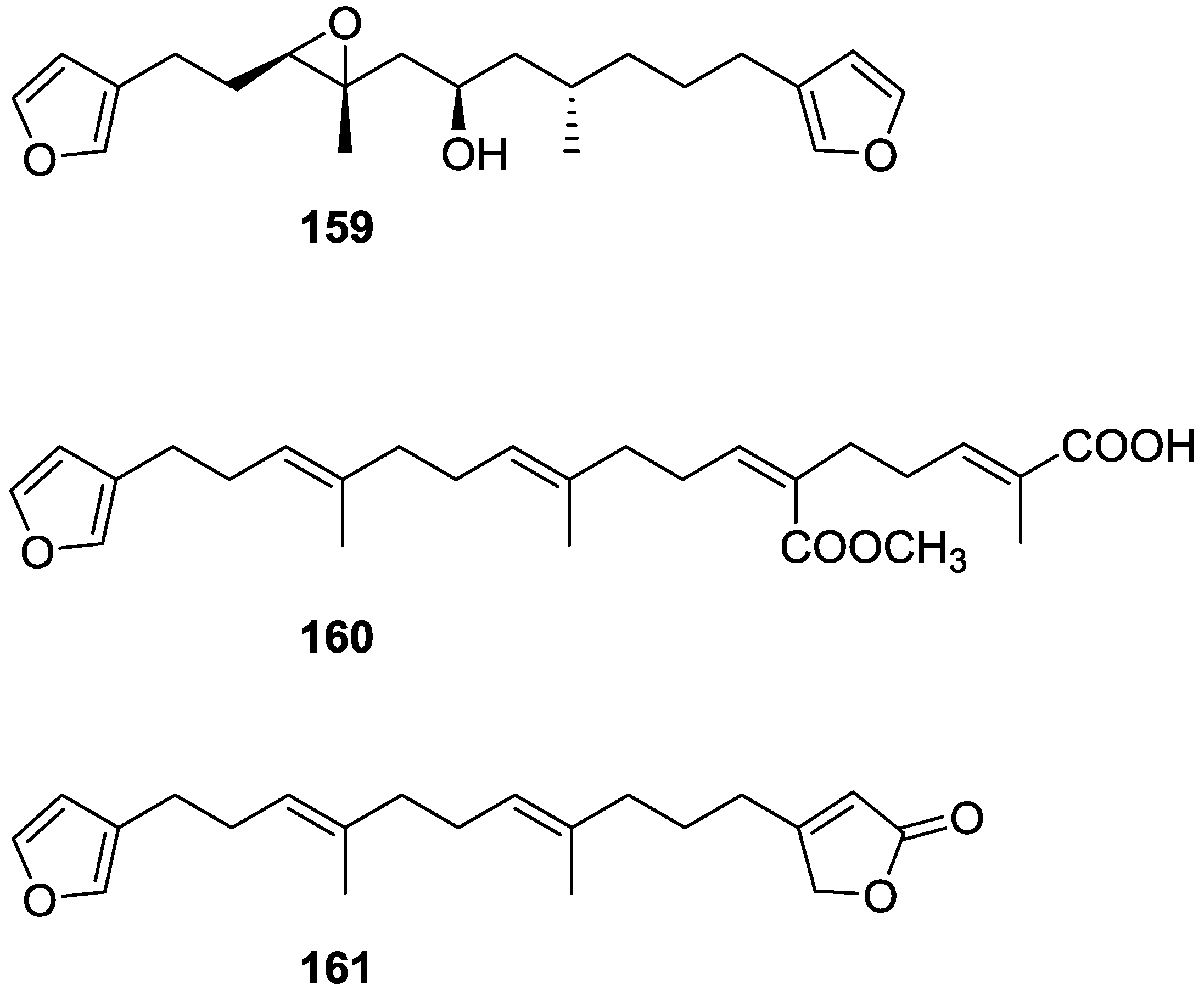
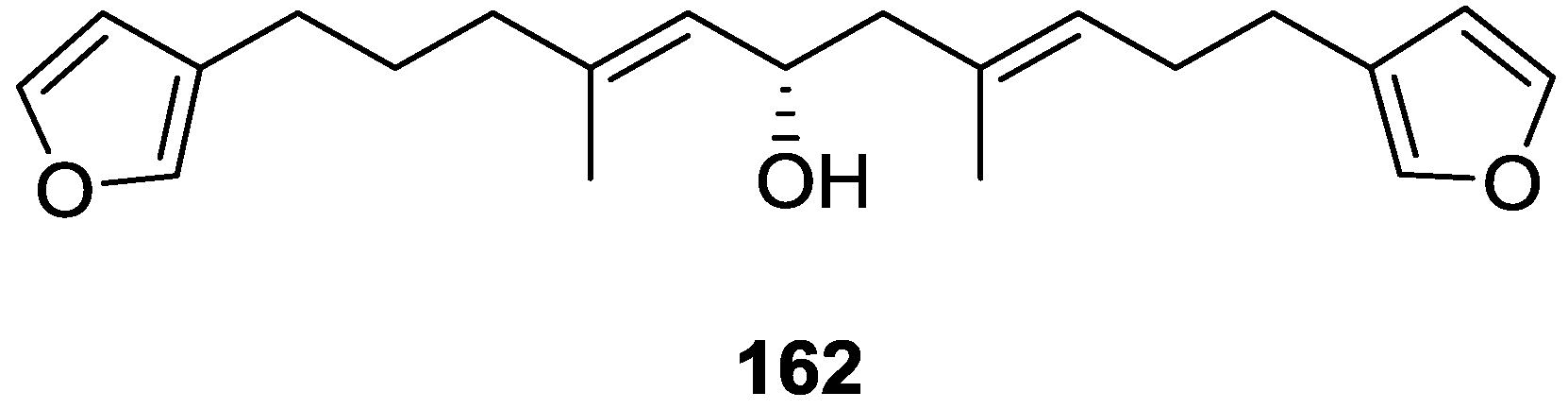
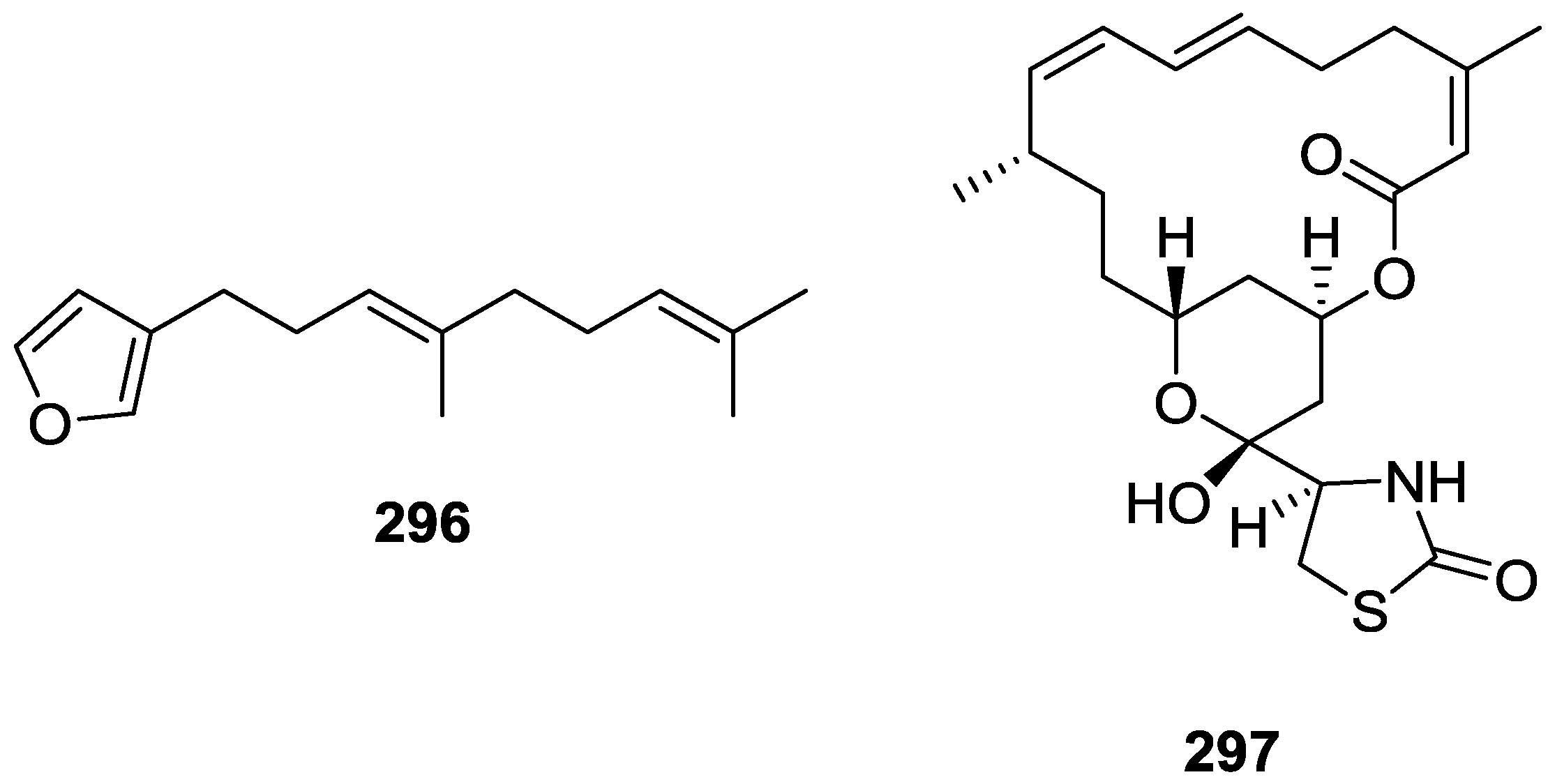
4. C21 and Other Linear Furanoterpenes
Other Studies
5. Sesterterpenes
Other Studies
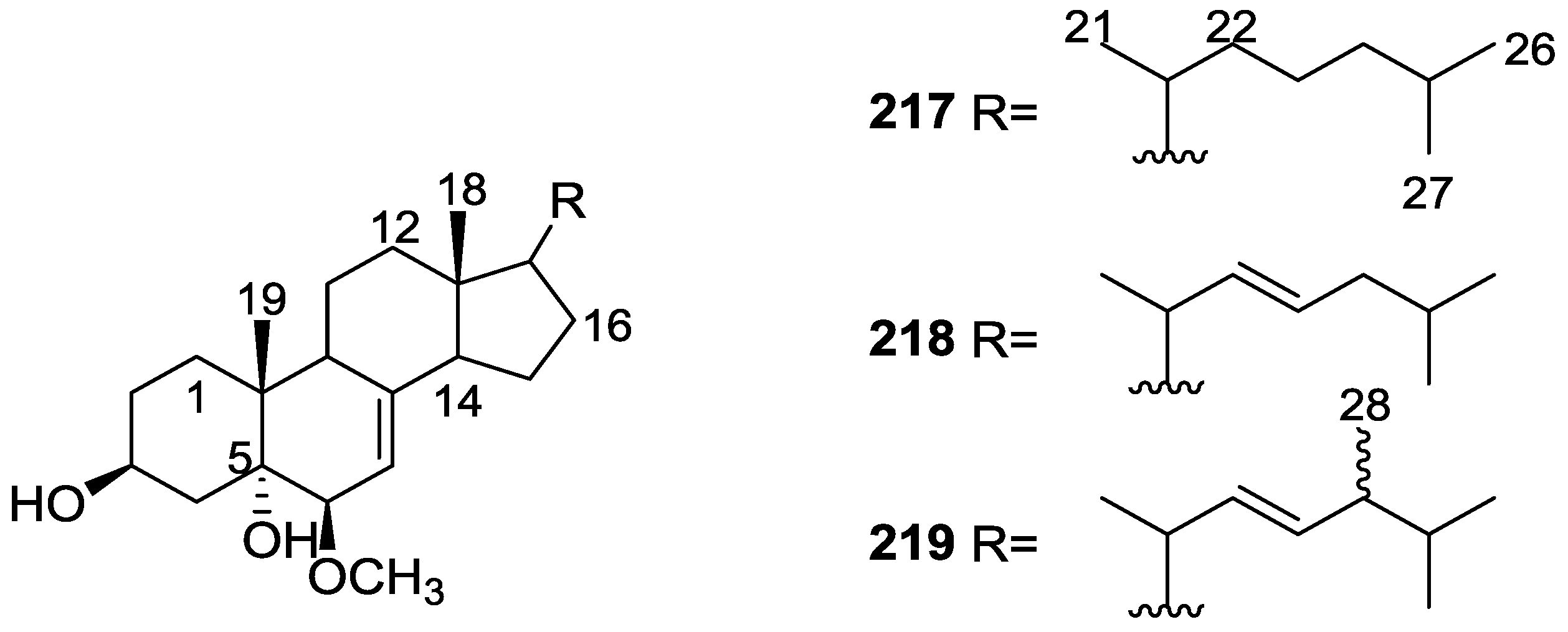
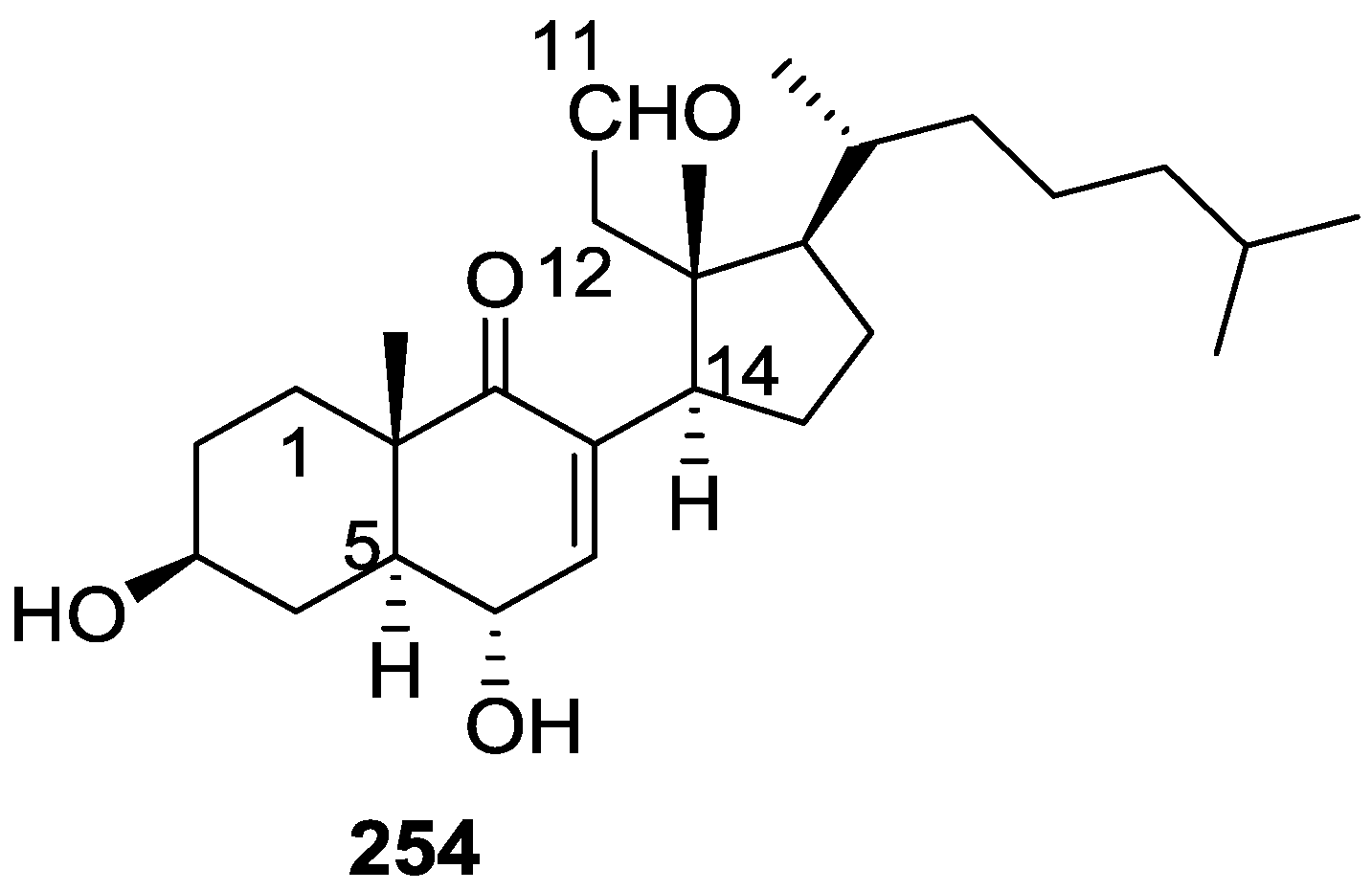
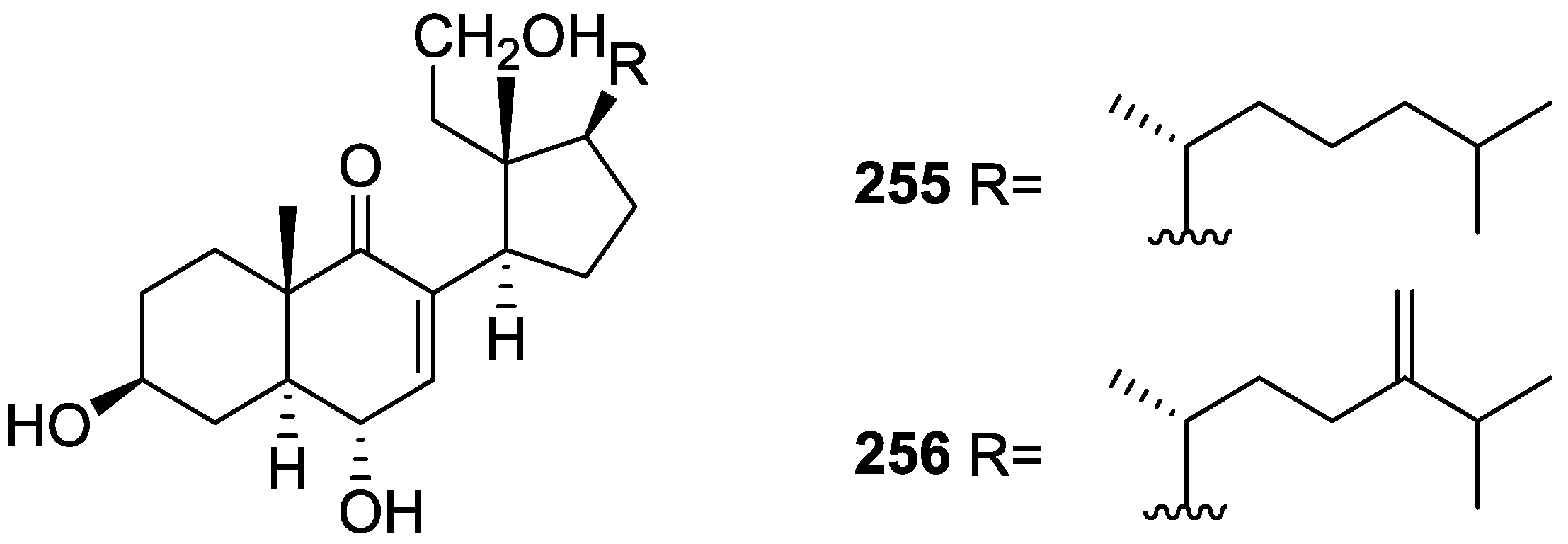
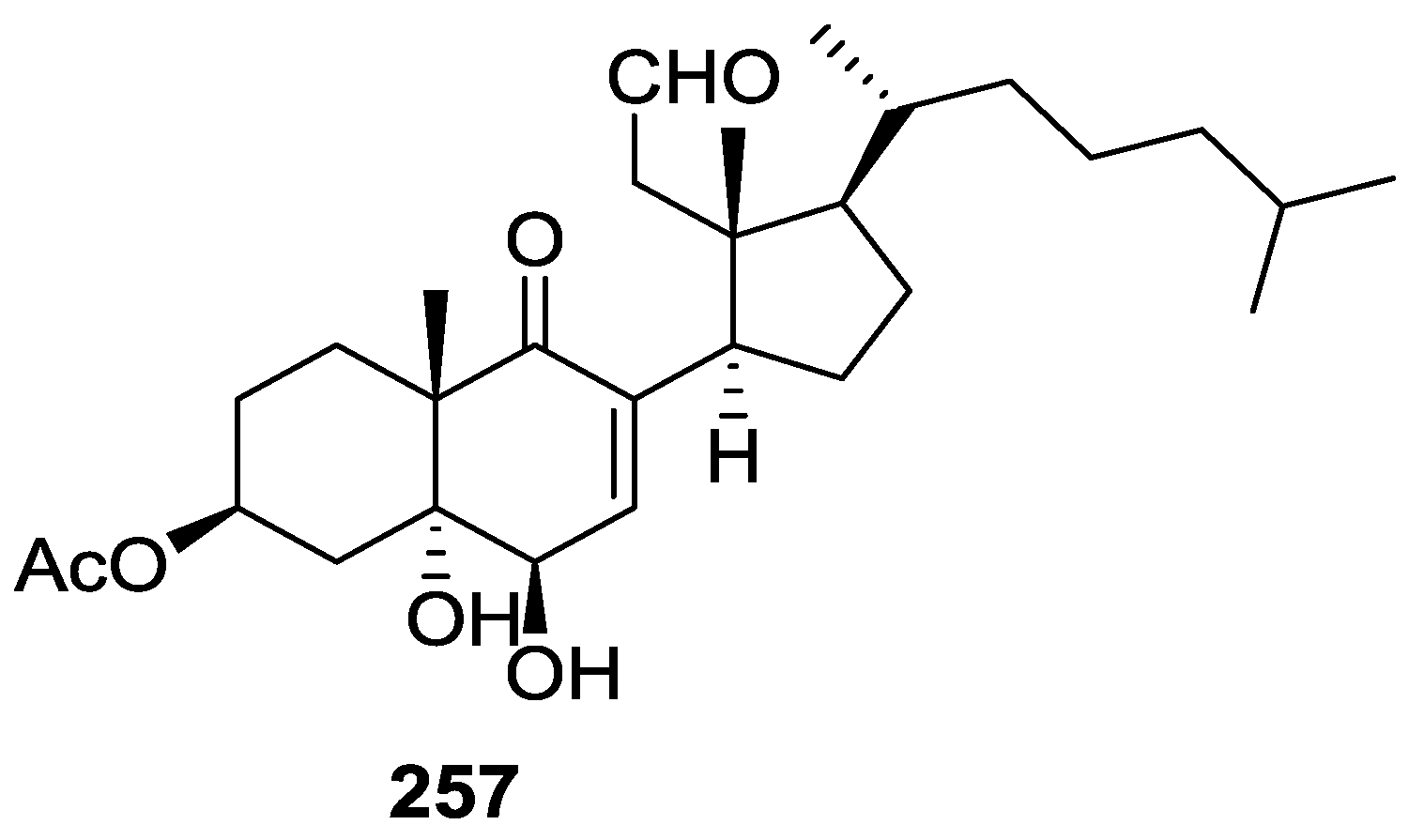
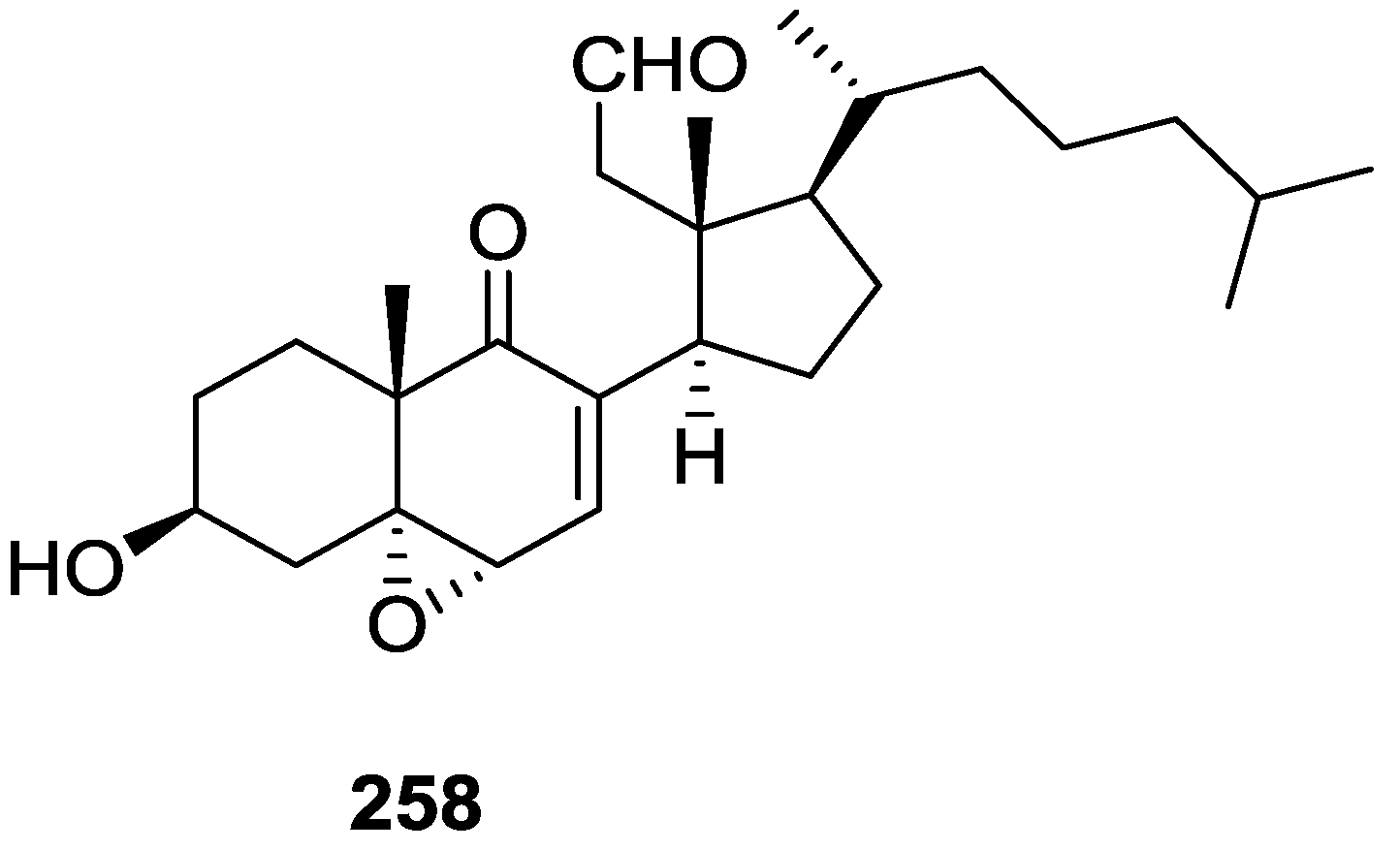
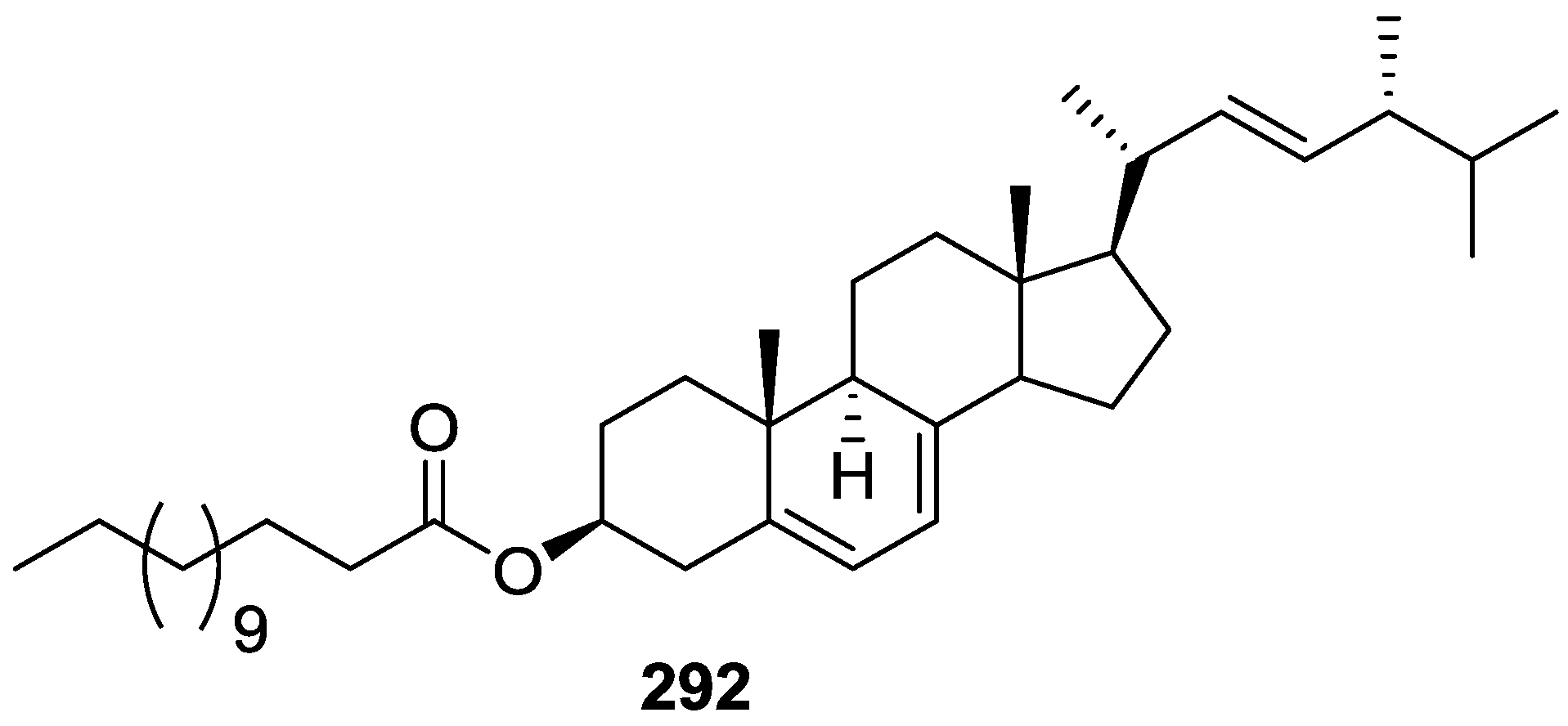
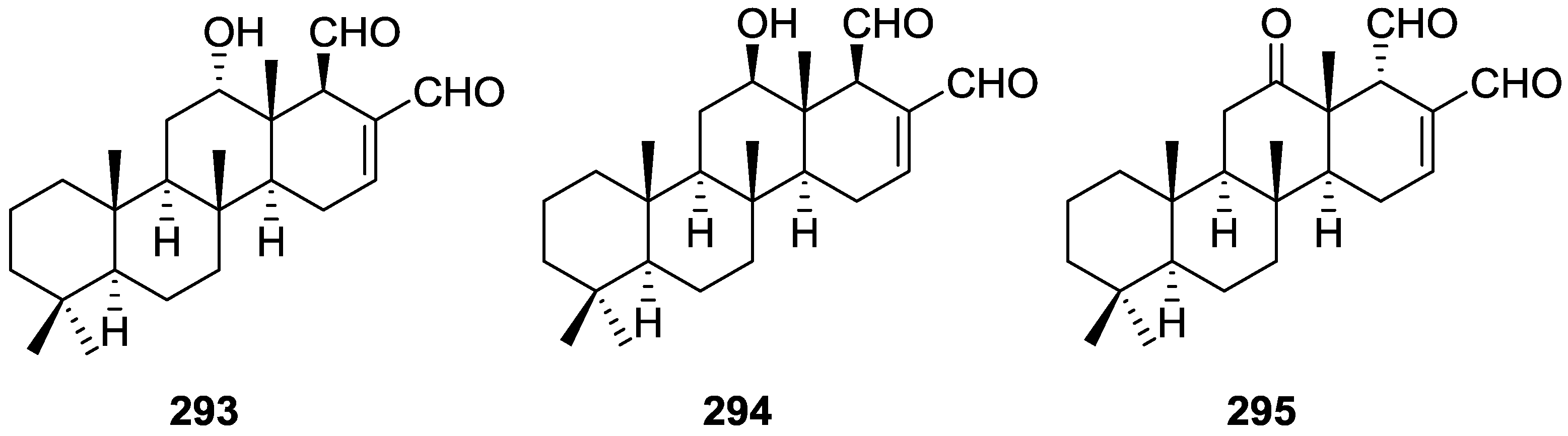
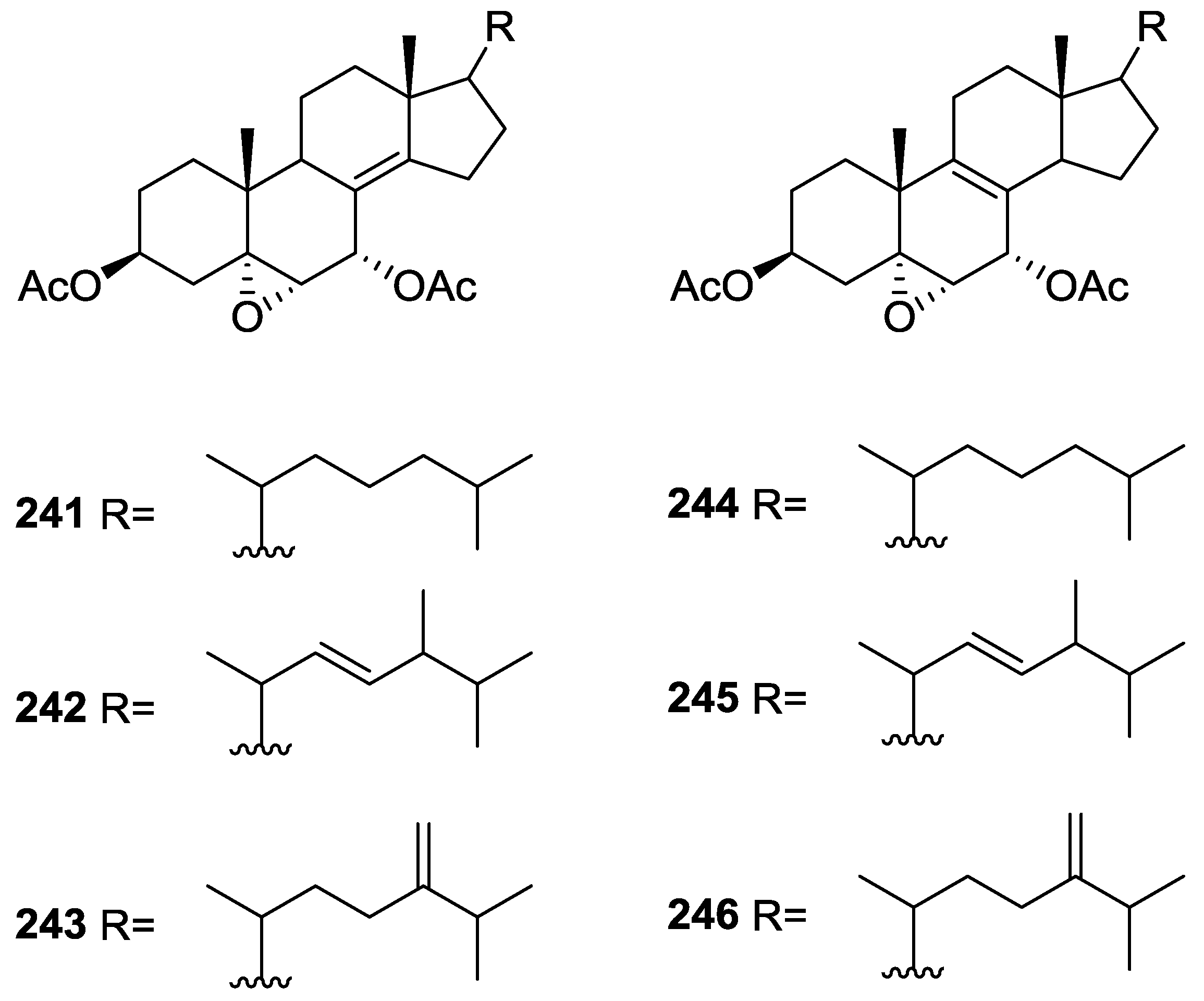
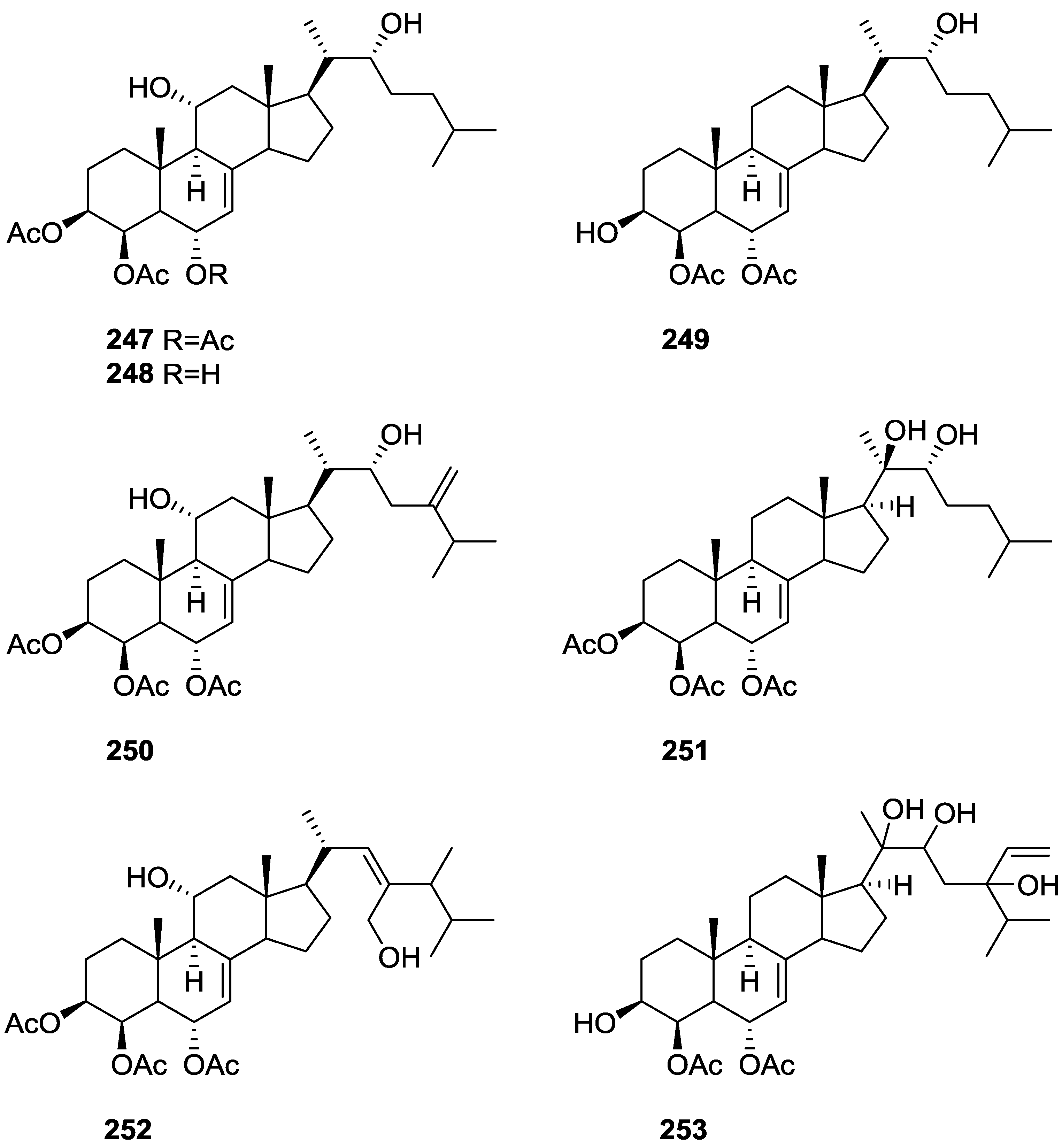
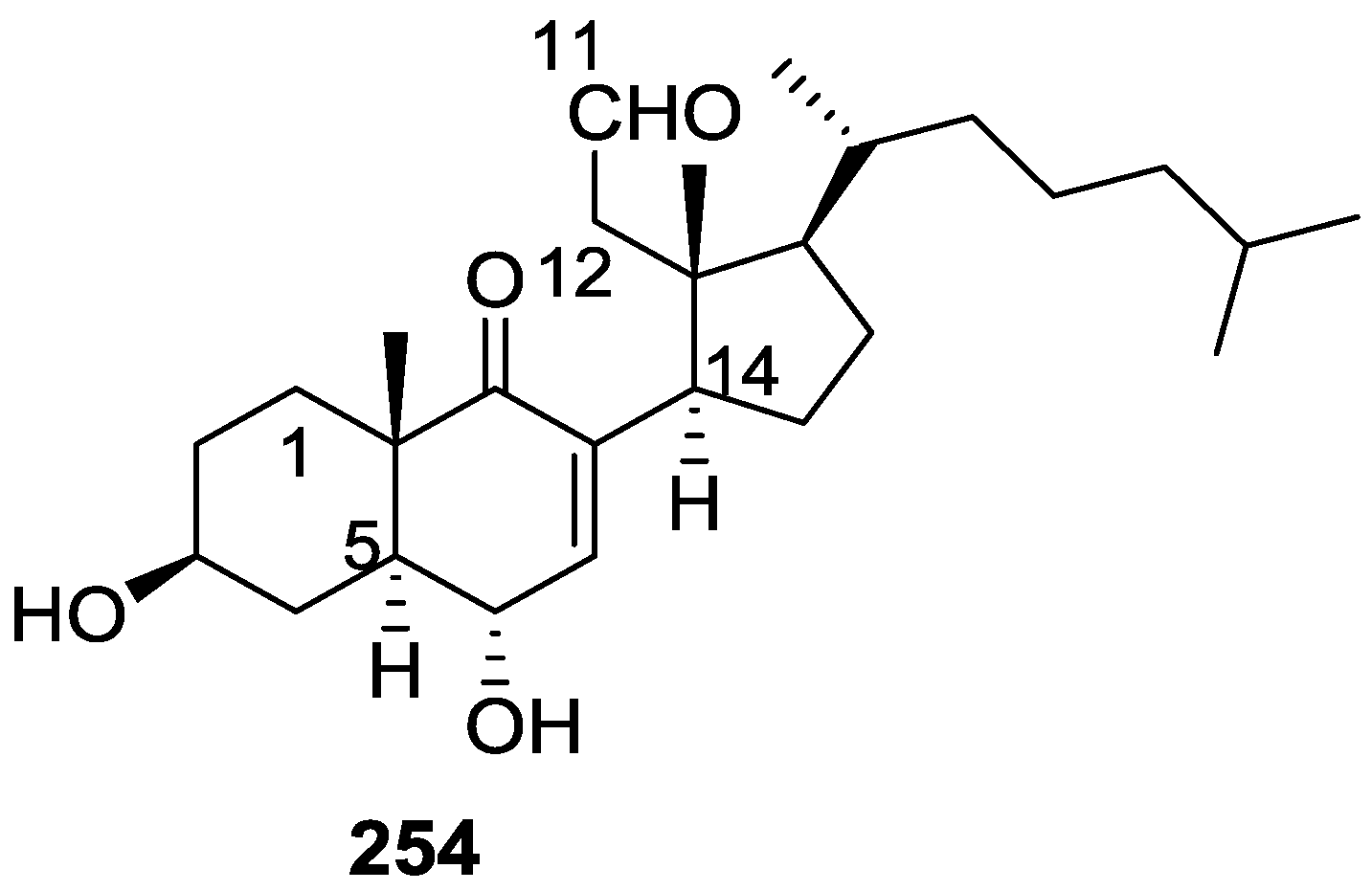
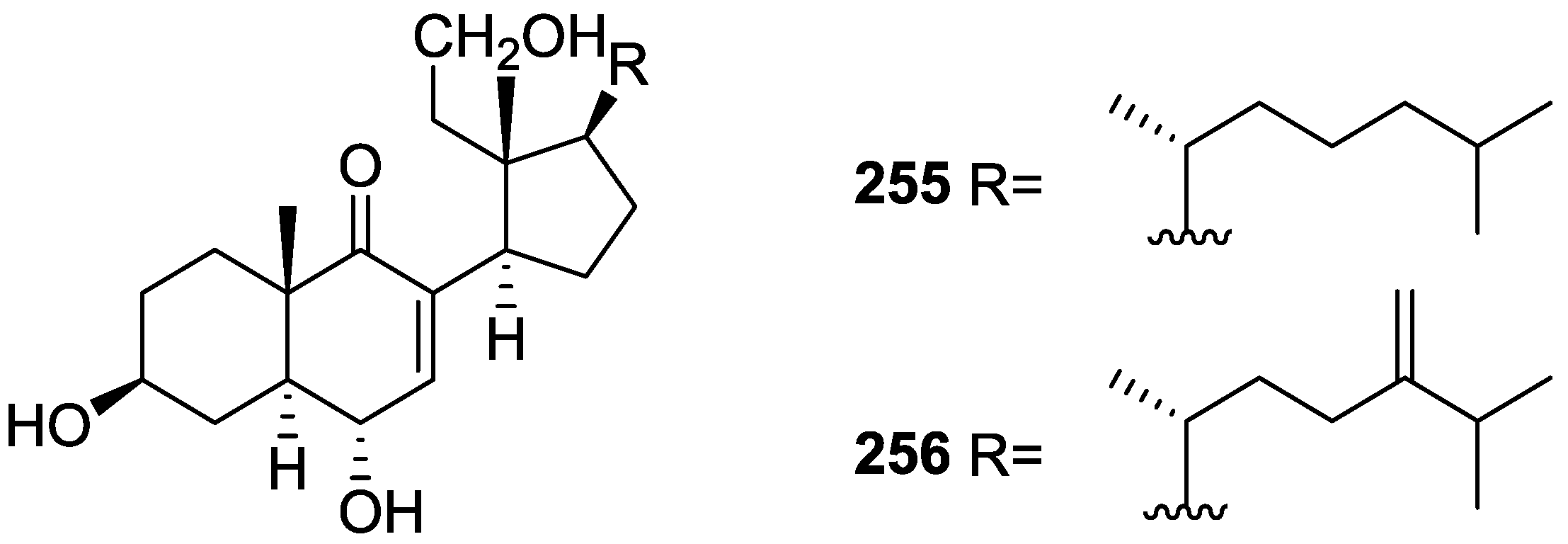
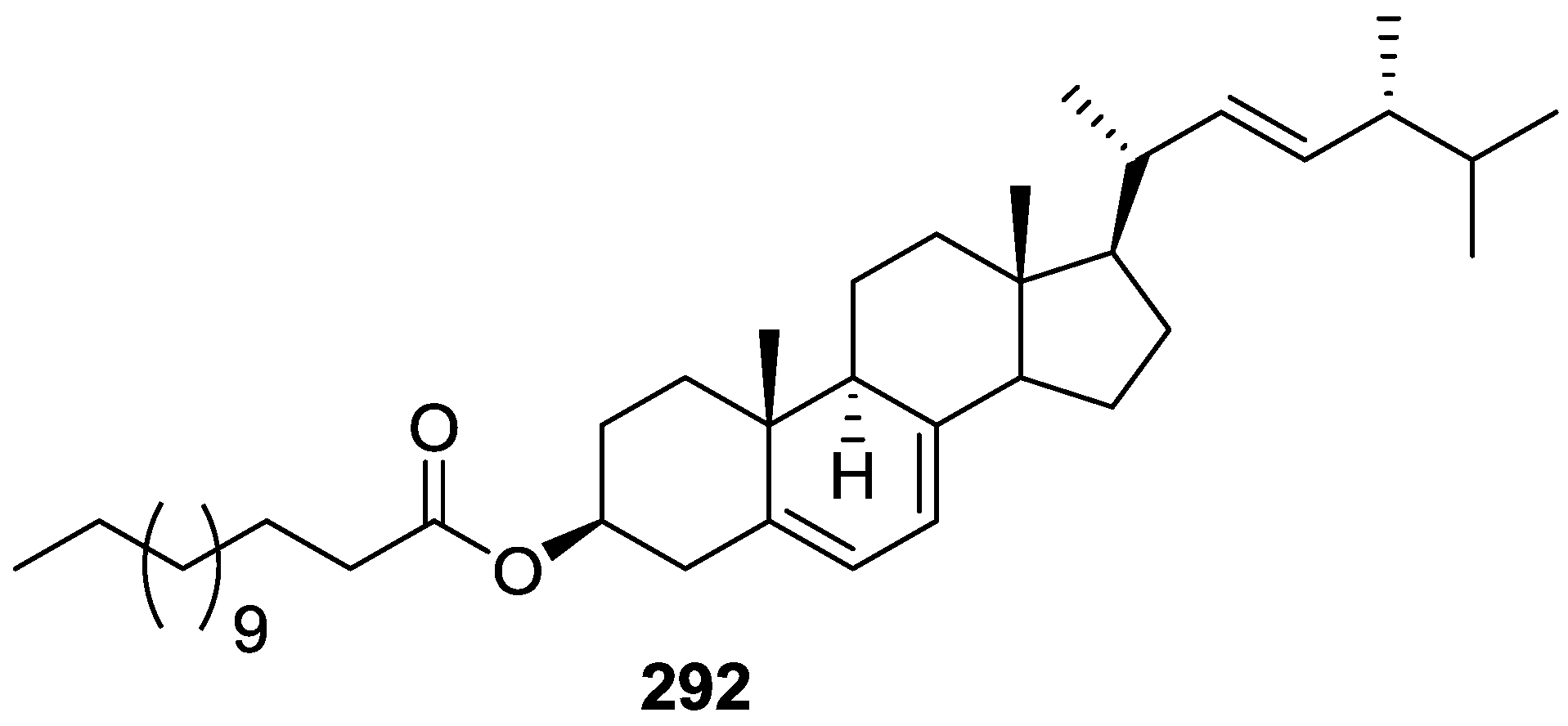
6. Sterols
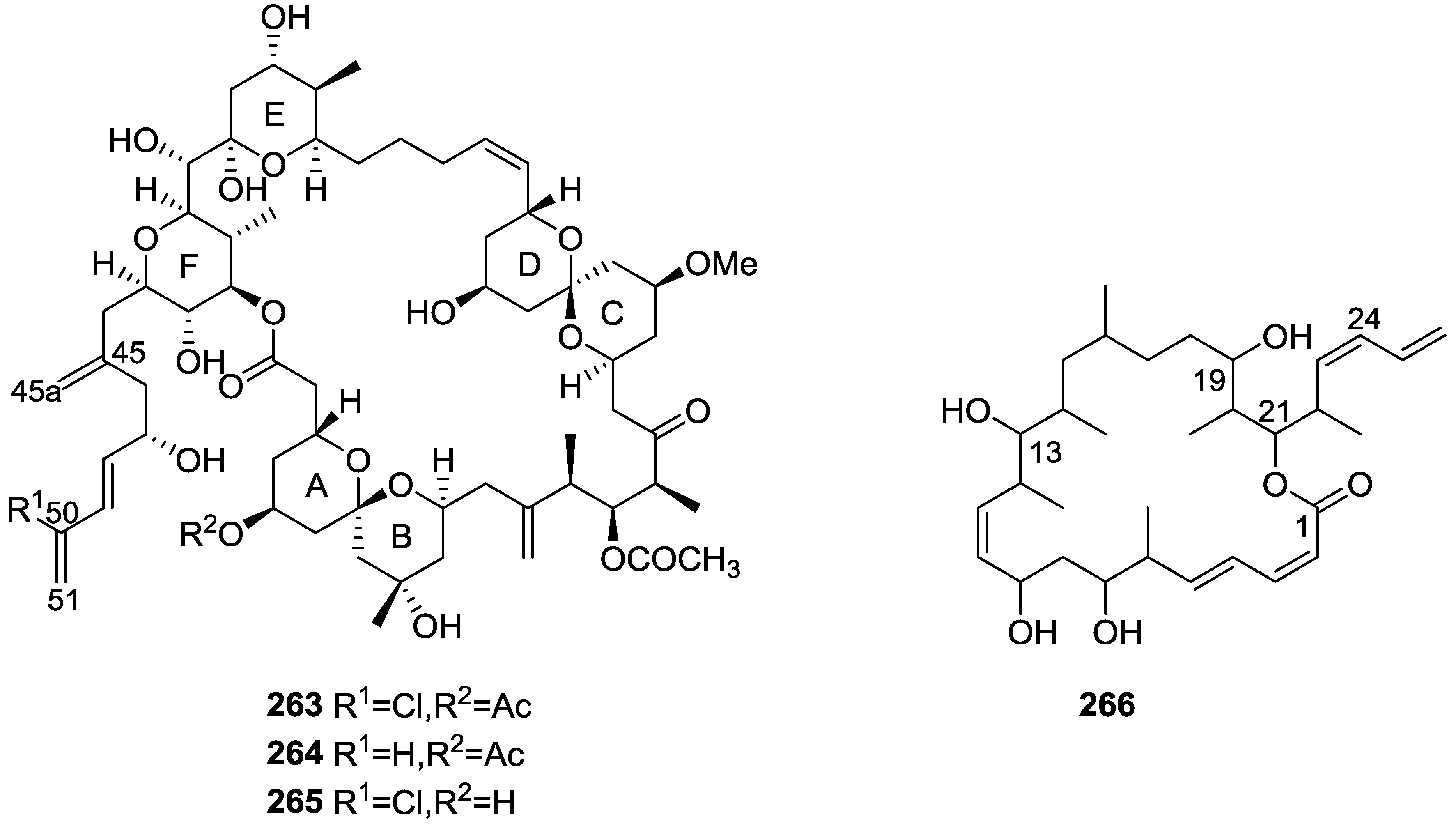
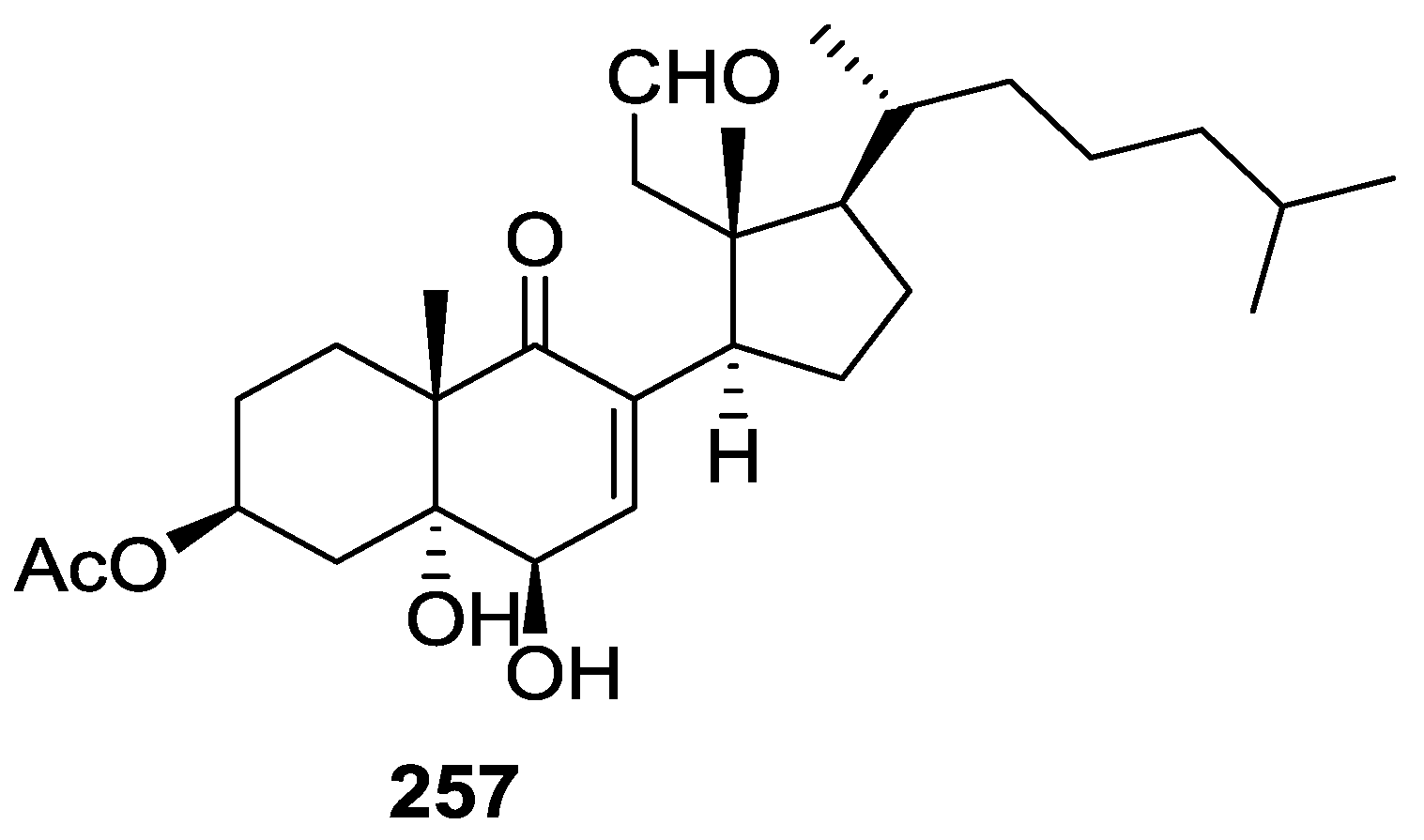
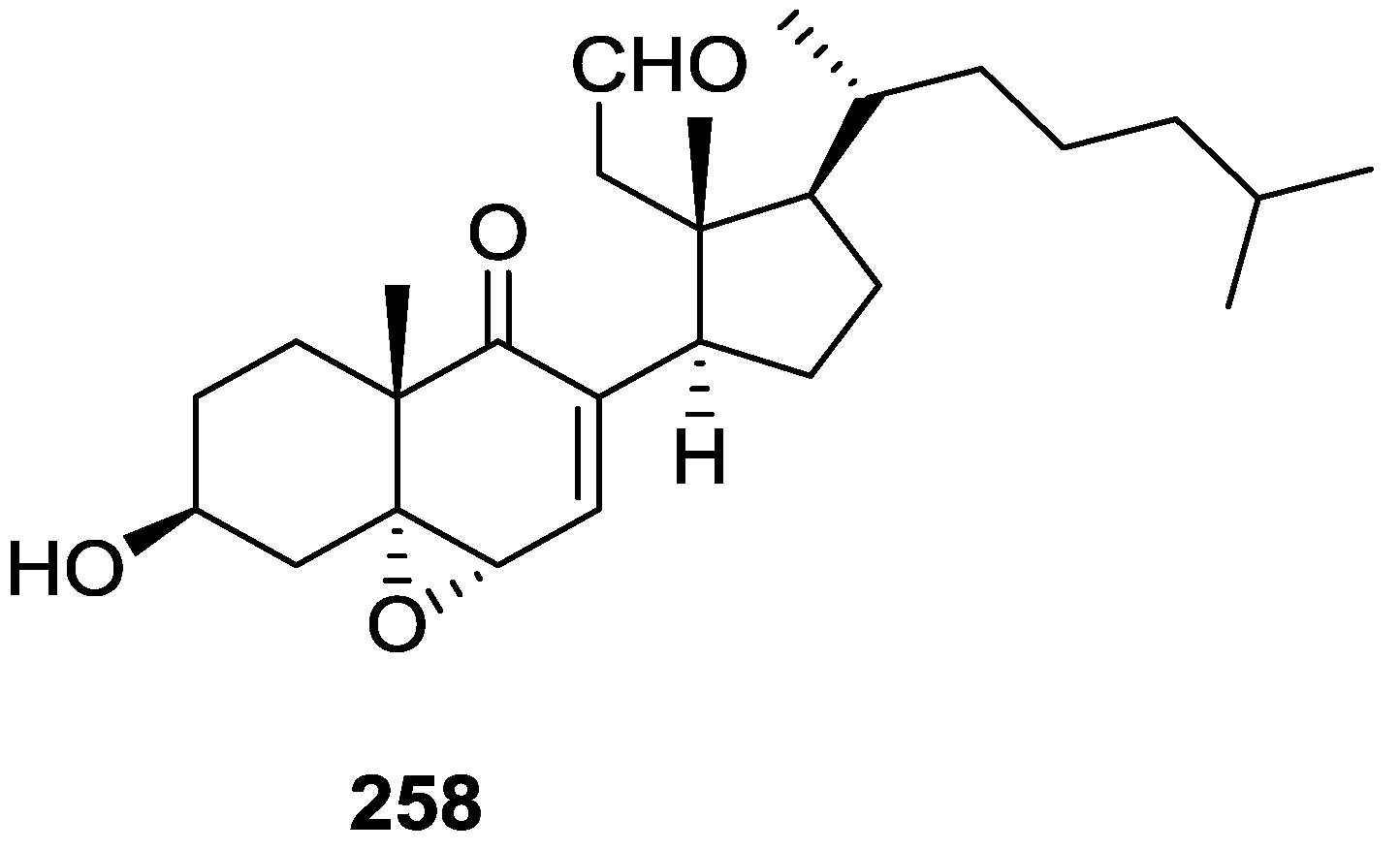
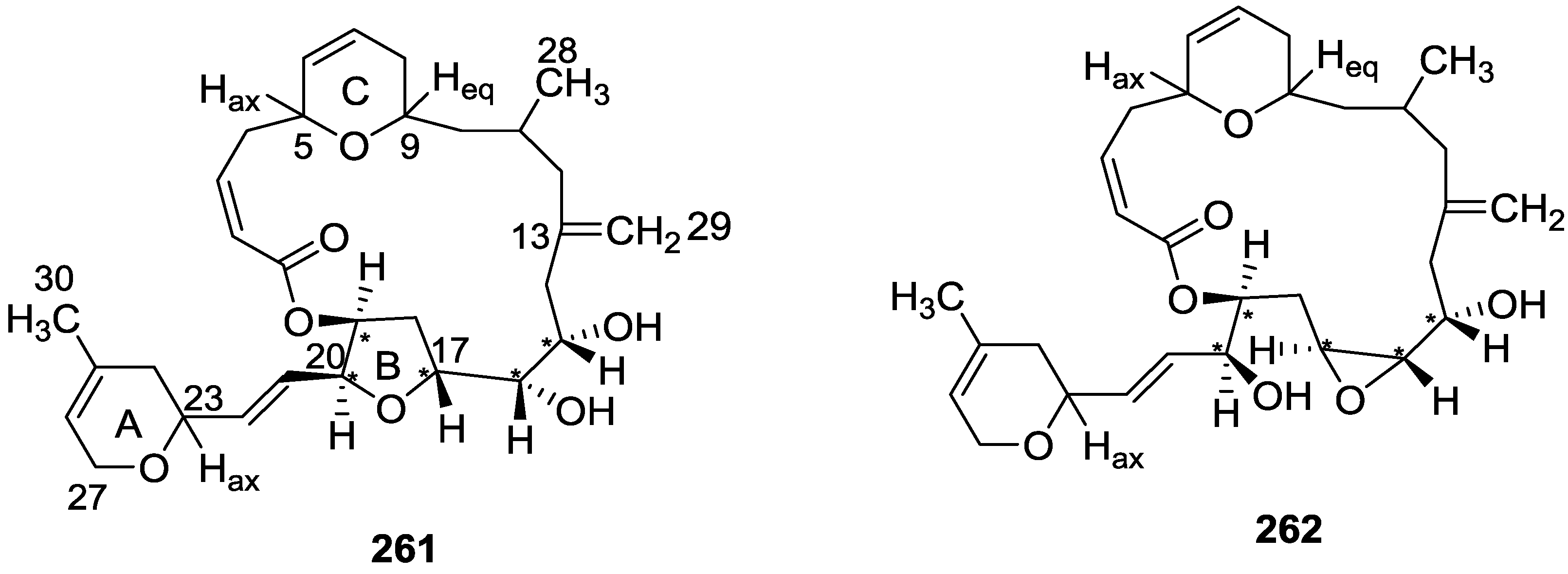
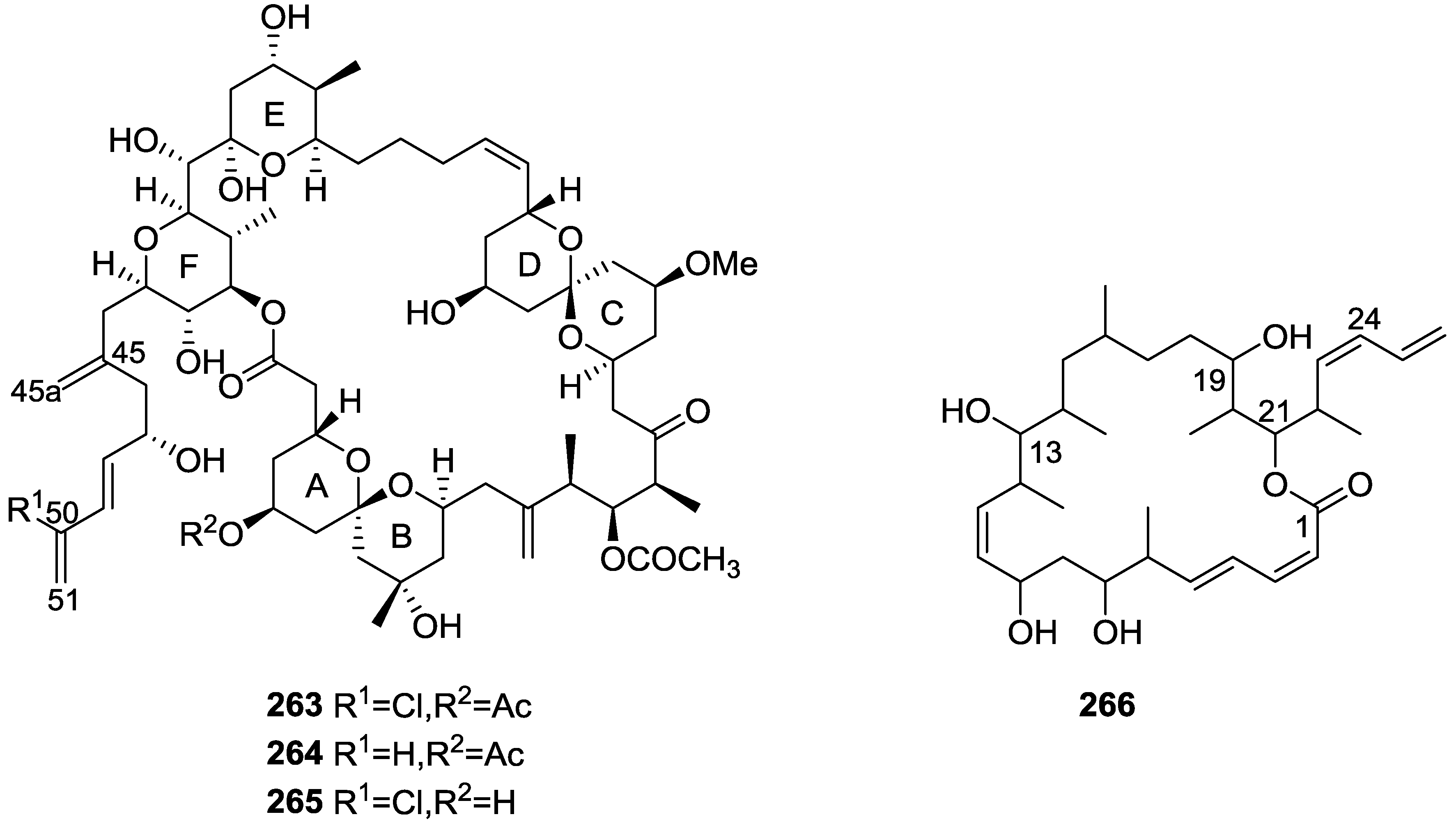
7. Macrolides
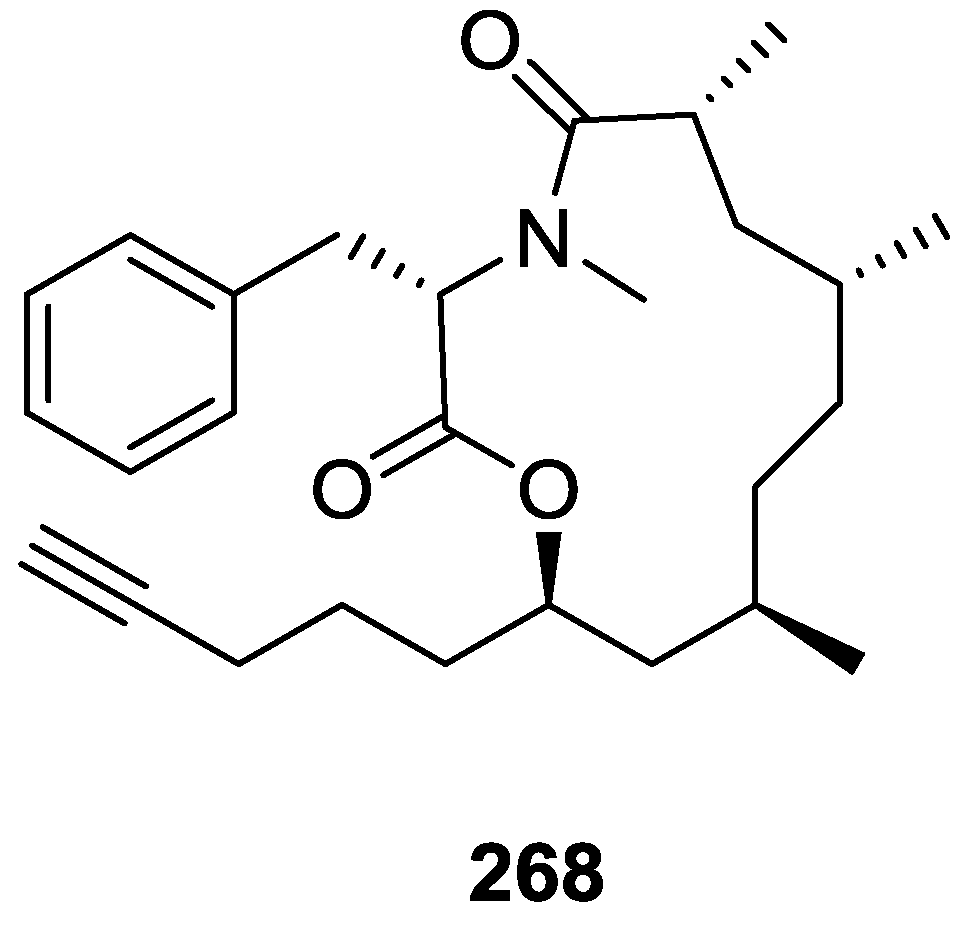
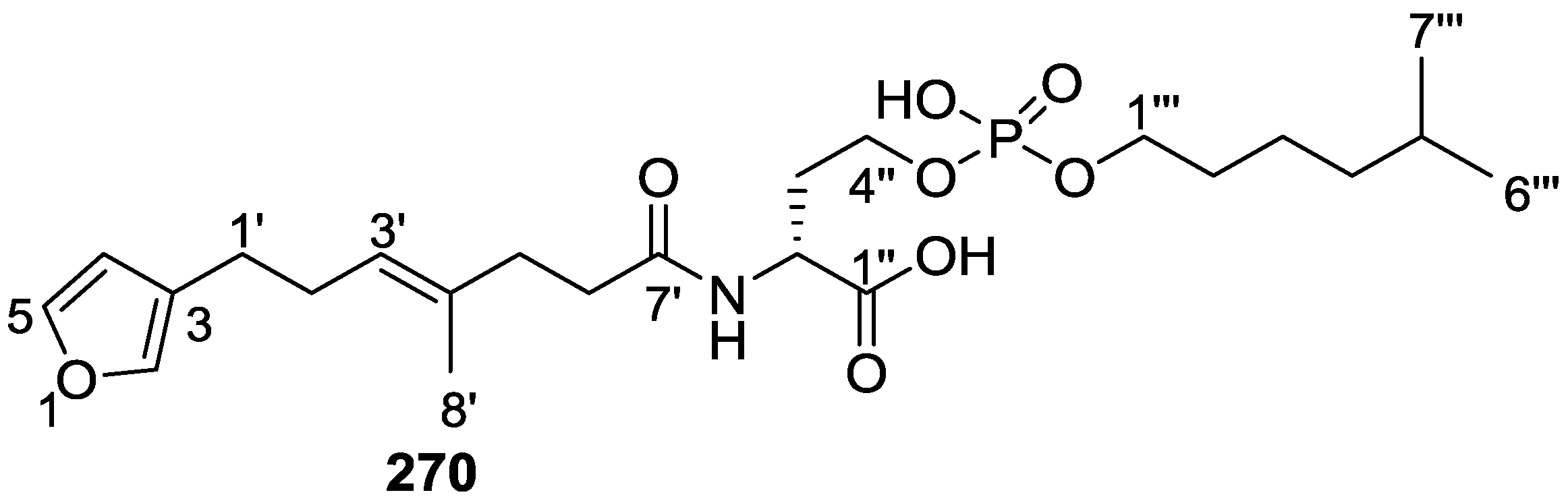
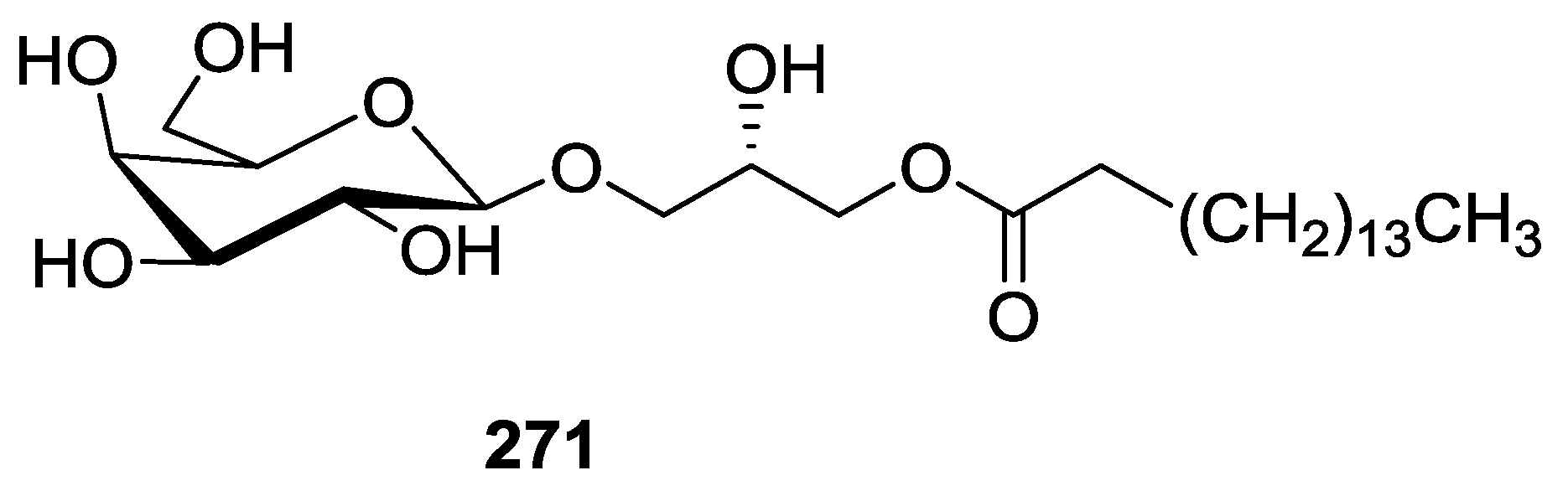
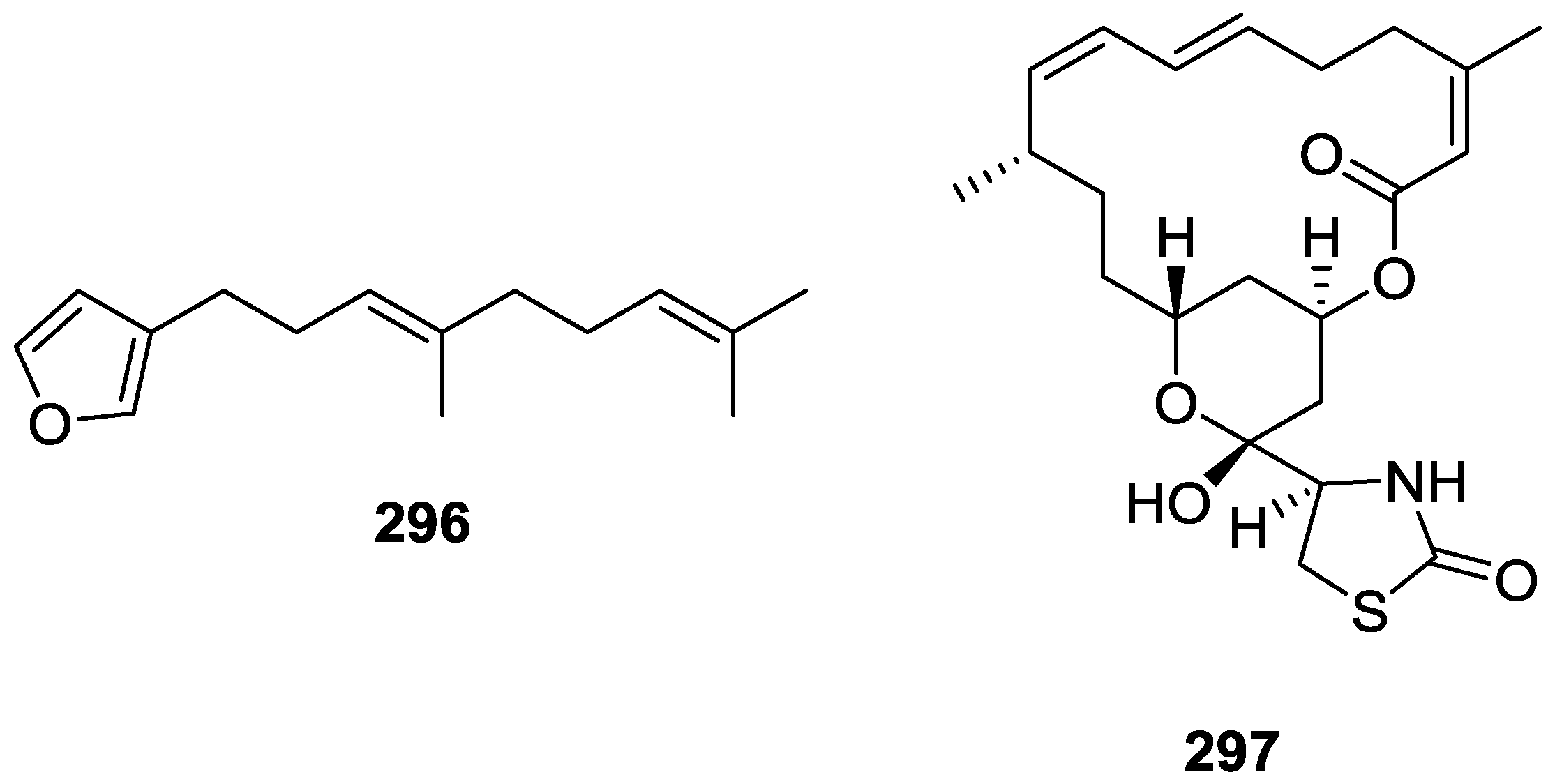
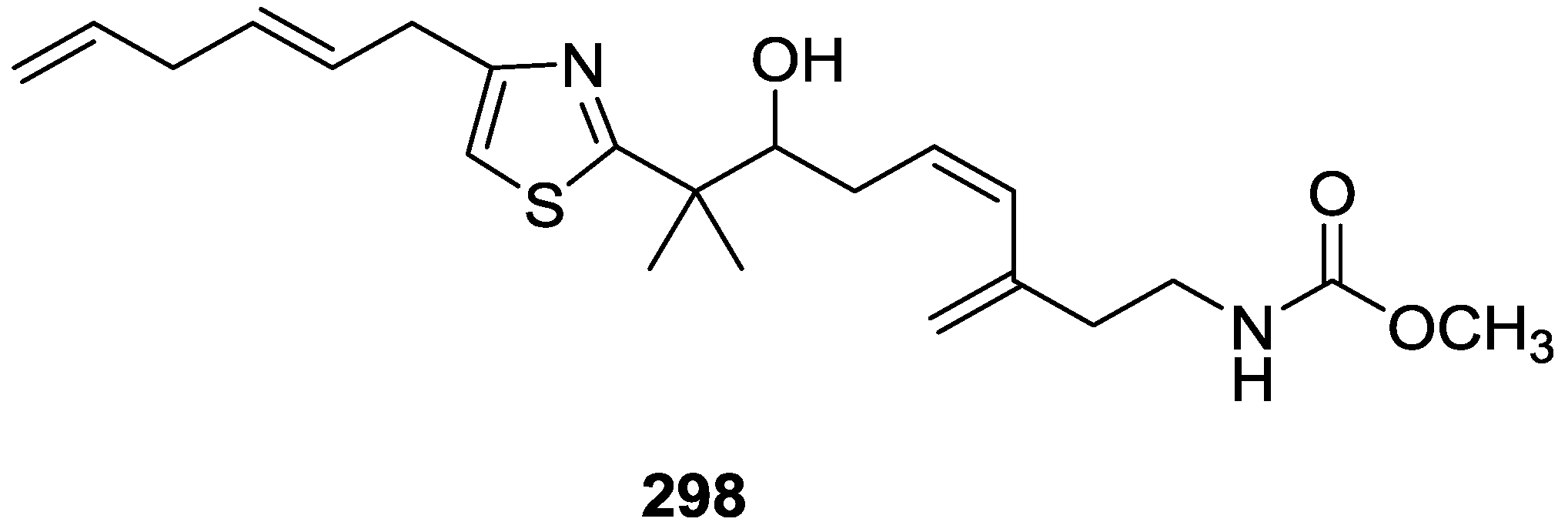
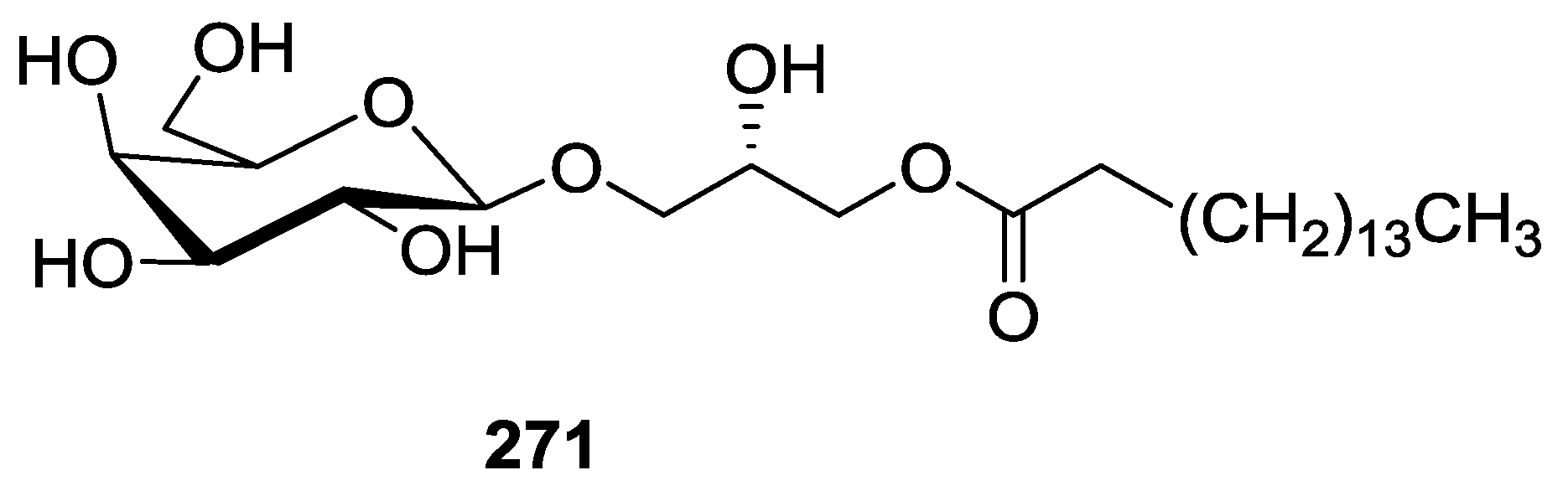
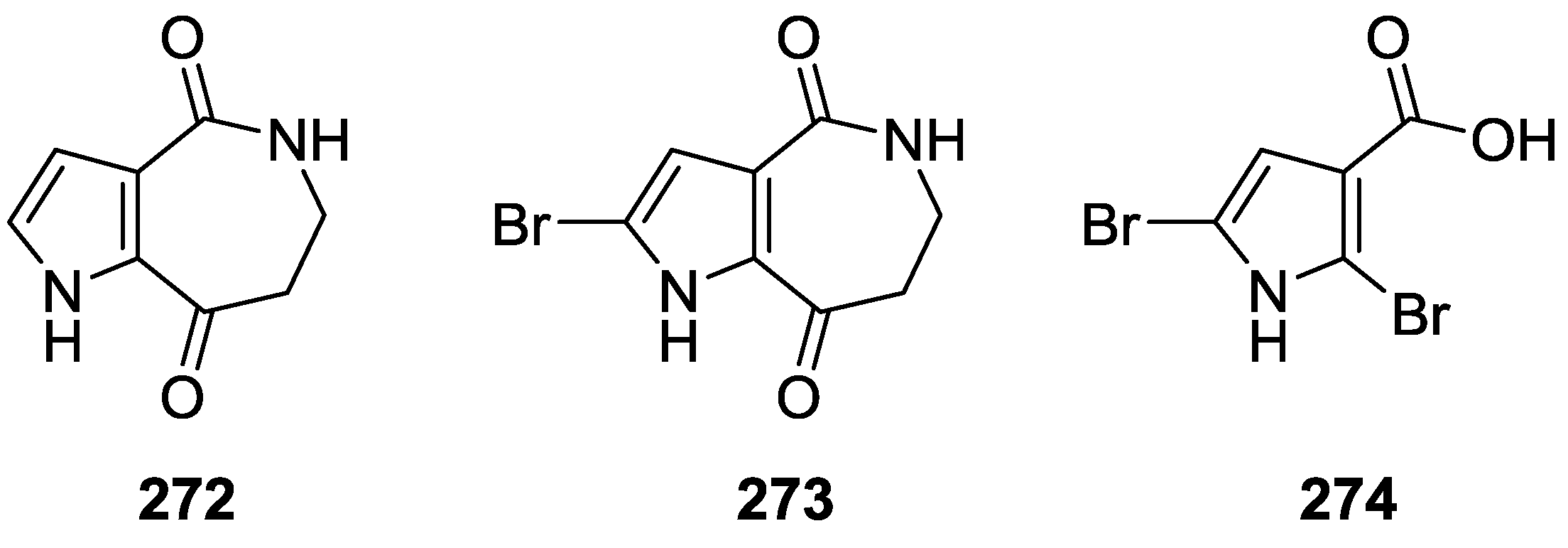
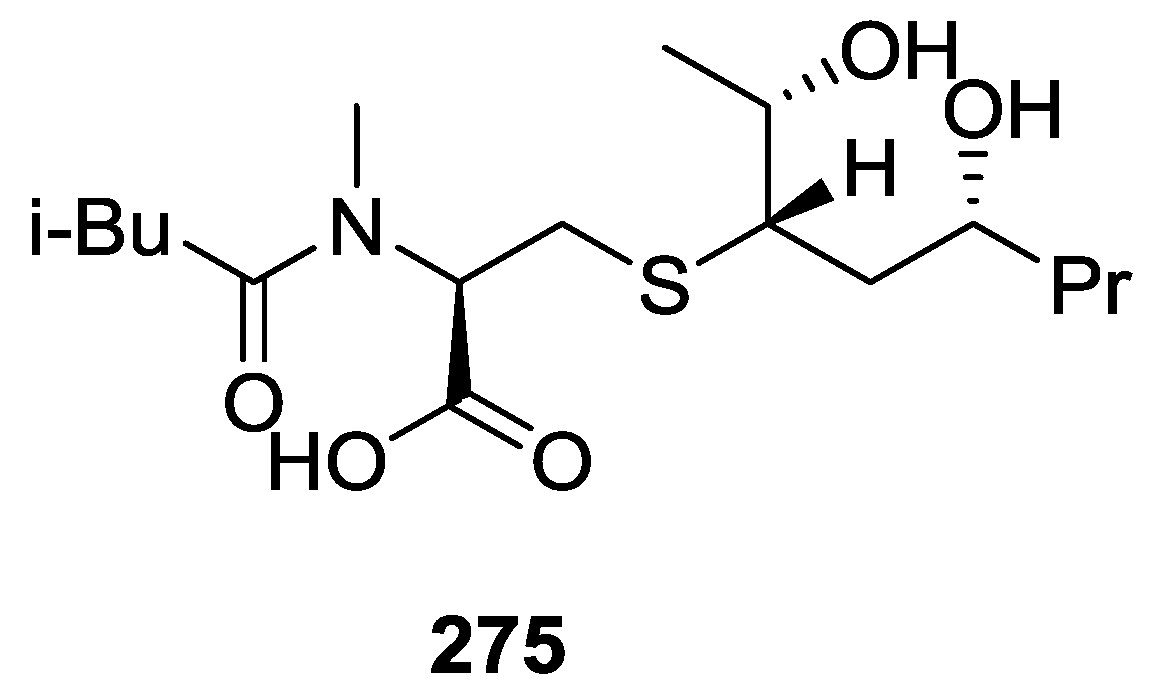
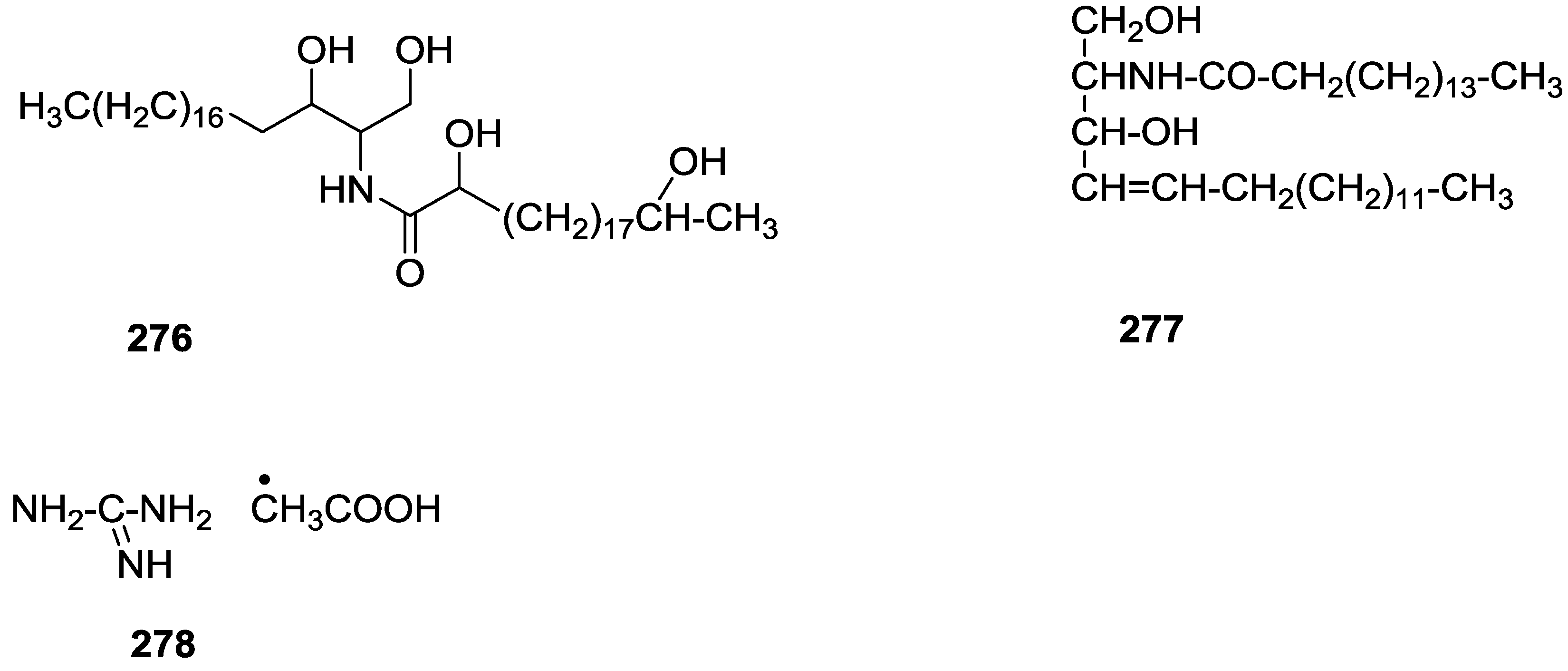
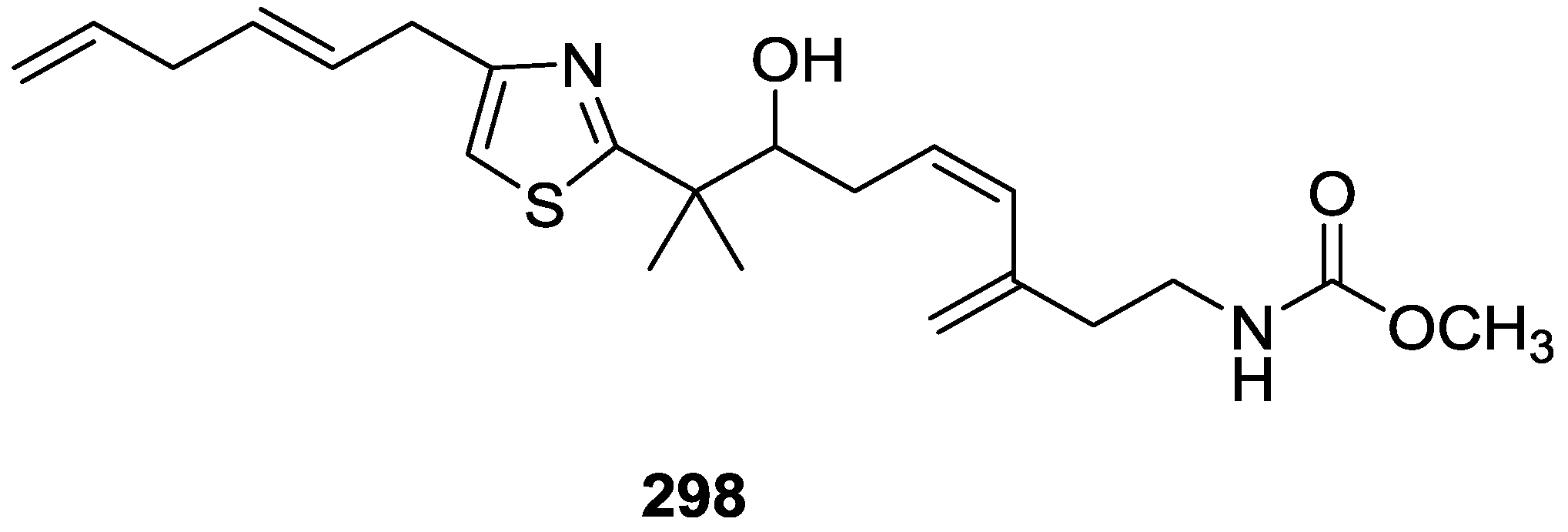
8. Miscellaneous Compounds
9. Other Reports
10. Biological Activity
- -
- the screening against clinical isolates of bacteria including multi-drug resistant (MDR) strains and fungi, where activity against MDR strains of Streptococcus pyogenes and Acinetobacter sp. was observed [129];
- -
- the anticonvulsant (using pentylenetetrazole seizure model) and analgesic (using writhing test in mice) activities of the extract (and its fractions) of the defensive secretion on the sponge, where analgesic activity in a dose dependent manner was observed for the extract [130];
- -
- the evaluation of antiproliferative (A549—lung cell carcinoma, HCT15—colon cell carcinoma and MCF7—breast adenocarcinoma) and anti-inflammatory (carrageenan-induced rat paw edema) activities of the extract (and its semi-purified fractions) of the defensive secretion of the sponge [131] and of the sponge methanol extract (and its semi-purified fractions) [132], where significant antiproliferative activities and anti-inflammatory activity were observed, and
- -
- the antiamoebic activity of extracts of S. officinalis var. ceylonensis against Entamoeba histolytica, where the alkaloids xestospongins and araguspongins where identified (by LCMS) as the major components of the active fraction [133].
11. Conclusions
Acknowledgments
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munroa, M.H.G.; Prinsepd, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed]
- Perdicaris, S.; Vlachogianni, T.; Valavanidis, A. Bioactive Natural Substances from Marine Sponges: New Developments and Prospects for Future Pharmaceuticals. Nat. Prod. Chem. Res. 2013, 1. [Google Scholar] [CrossRef]
- Pronzato, R.; Manconi, R. Mediterranean Commercial Sponges: Over 5000 Years of Natural History and Cultural Heritage. Mar. Ecol. 2008, 29, 146–166. [Google Scholar] [CrossRef]
- Voultsiadou, E.; Dailianis, T.; Antoniadou, C.; Vafidis, D.; Dounas, C.; Chintiroglou1, C.C. Aegean Bath Sponges: Historical Data and Current Status. Rev. Fish. Sci. 2011, 19, 34–51. [Google Scholar] [CrossRef]
- Noyer, C.; Agell, G.; Pascual, M.; Becerro, M.A. Isolation and Characterization of Microsatellite Loci from the Endangered Mediterranean Sponge Spongia agaricina (Demospongiae: Dictyoceratida). Conserv. Genet. 2009, 10, 1895–1898. [Google Scholar] [CrossRef]
- Noyer, C.; Becerro, M.A. Relationship between Genetic, Chemical, and Bacterial Diversity in the Atlanto-Mediterranean Bath Sponge Spongia lamella. Hydrobiology 2012, 687, 85–99. [Google Scholar] [CrossRef]
- Cunningham, E.; Dunne, N.; Walker, G.; Maggs, C.; Wilcox, R.; Buchanan, F. Hydroxyapatite Bone Substitutes Developed via Replication of Natural Marine Sponges. J. Mater. Sci. Mater. Med. 2010, 21, 2255–2261. [Google Scholar] [CrossRef] [PubMed]
- Fattorusso, E.; Minale, L.; Sodano, G.; Trivellone, E. Isolation and Structure of Nitenin and Dihydronitenin, New Furanoterpenes from Spongia nitens. Tetrahedron 1971, 27, 3909–3917. [Google Scholar] [CrossRef]
- Urban, S.; Capon, R.J. 5-epi-isospongiaquinone, A New Sesquiterpene/Quinone Antibiotic from an Australian Marine Sponge, Spongia Hispida. J. Nat. Prod. 1992, 55, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Capon, R.J.; Groves, D.R.; Urban, S.; Watson, R.G. Spongiaquinone Revisited-Structural and Stereochemical Studies on Marine Sesquiterpene Quinones from a Southern Australian Marine Sponge, Spongia sp. Aust. J. Chem. 1993, 46, 1245–1253. [Google Scholar] [CrossRef]
- Utkina, N.K.; Denisenko, V.A.; Scholokova, O.V.; Virovaya, M.V.; Prokof’eva, N.G. Cyclosmenospongine, a New Sesquiterpenoid Aminoquinone from an Australian Marine Sponge Spongia sp. Tetrahedron Lett. 2003, 44, 101–102. [Google Scholar] [CrossRef]
- Capon, R.J.; MacLeod, J.K. A Revision of the Absolute Stereochemistry of Ilimaquinone. J. Org. Chem. 1987, 52, 5059–5060. [Google Scholar] [CrossRef]
- Kondraki, M.-L.; Guyot, M. Smenospongine: A Cytotoxic and Antimicrobial Aminoquinone Isolated from Smenospongia sp. Tetrahedron Lett. 1987, 28, 5815–5818. [Google Scholar] [CrossRef]
- Utkina, N.K.; Denisenko, V.A.; Scholokova, O.V.; Makarchenko, A.E. Determination of the Absolute Stereochemistry of Cyclosmenospongine. J. Nat. Prod. 2003, 66, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Gao, Z.; Thomas, S.J.; Hecht, S.M.; Lazo, J.S.; Kingston, D.G.I. Marine Sesquiterpenoids that Inhibit the Lyase Activity of DNA Polymerase β. J. Nat. Prod. 2004, 67, 1716–1718. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Tsuda, M.; Fromont, J.; Kobayashi, J. Metachromins J and K, New Sesquiterpenoids from Marine Sponge Spongia Species. Heterocycles 2006, 67, 791–796. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kubota, T.; Fromontb, J.; Kobayashi, J. Metachromins L–Q, New Sesquiterpenoid Quinones with an Amino Acid Residue from Sponge Spongia sp. Tetrahedron 2007, 63, 8770–8773. [Google Scholar] [CrossRef]
- Takahashi, Y.; Yamada, M.; Kubota, T.; Fromont, J.; Kobayashi, J. Metachromins R–T, New Sesquiterpenoids from Marine Sponge Spongia sp. Chem. Pharm. Bull. 2007, 55, 1731–1733. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kubota, T.; Yamamoto, S.; Kobayashi, J. Inhibitory Effects of Metachromins L–Q and Its Related Analogs Against Receptor Tyrosine Kinases EGFR and HER2. Bioorg. Med. Chem. Lett. 2013, 23, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kubota, T.; Kobayashi, J. Nakijiquinones E and F, New Dimeric Sesquiterpenoid Quinones from Marine Sponge. Bioorg. Med. Chem. 2009, 17, 2185–2188. [Google Scholar] [CrossRef] [PubMed]
- Utkina, N.K.; Denisenko, V.A. Sesquiterpene Quinones from a Viet Nam Sea Sponge Spongia sp. Chem. Nat. Compd. 2011, 47, 135–137. [Google Scholar] [CrossRef]
- Kittiwisut, S.; Yuenyongsawad, S.; Mooberry, S.L.; Plubrukarn, A. DNA Damage Initiated by Merosesquiterpenes from the Sponge Spongia sp. Planta Med. 2012, 78, 1147. [Google Scholar] [CrossRef]
- Cimino, G.; Derosa, D.; Destefan, S.; Minale, L. Isoagatholactone, a Diterpene of a New Structural Type from Sponge Spongia officinalis. Tetrahedron 1974, 30, 645–649. [Google Scholar] [CrossRef]
- Capelle, N.; Braekman, J.C.; Daloze, D.; Tursch, B. Chemical Studies of Marine-Invertebrates .44. 3 New Spongian Diterpenes from Spongia officinalis. Bull. Des Soc. Chim. Belges 1980, 89, 399–404. [Google Scholar] [CrossRef]
- Cimino, G.; Morrone, R.; Sodano, G. New Diterpenes from Spongia officinalis. Tetrahedron Lett. 1982, 23, 4139–4142. [Google Scholar] [CrossRef]
- Gonzalez, A.G.; Estrada, D.M.; Martin, J.D.; Martin, V.S.; Perez, C.; Perez, R. New Antimicrobial Diterpenes from a Sponge Spongia officinalis. Tetrahedron 1984, 40, 4109–4113. [Google Scholar] [CrossRef]
- Kohmoto, S.; McConnell, O.J.; Wright, A.; Cross, S. Isospongiadiol, A Cytotoxic And Antiviral Diterpene From A Caribbean Deep-Water Marine Sponge, Spongia sp. Chem. Lett. 1987, 16, 1687–1690. [Google Scholar] [CrossRef]
- Hirsch, S.; Ashman, Y.K. Spongialactone A, a New Spongian Diterpene from Spongia arabica. J. Nat. Prod. 1988, 51, 1243–1245. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Schmitz, F.J. New Spongian Diterpenoids from a Great Barrier Reef Sponge, Spongia sp. J. Org. Chem. 1991, 56, 1250–1253. [Google Scholar] [CrossRef]
- Searle, P.A.; Molinski, T.F. Scalemic 12-Hydroxyambliofuran and 12-Acetoxyambliofuran, Five Furanoditerpenes and a Furanosesterterpene from Spongia sp. Tetrahedron 1994, 50, 9893–9908. [Google Scholar] [CrossRef]
- Zubia, E.; Gavagnin, M.; Scognamiglio, G.; Cimino, G. Spongiane and Ent.isocopalane Diterpenoids from the Mediterranean Sponge Spongia zimocca. J. Nat. Prod. 1994, 57, 725–731. [Google Scholar] [CrossRef]
- Li, C.-J.; Schmitz, F.J.; Kelly-Borges, M. New Diterpene Lactones from the Sponge Spongia matamata. J. Nat. Prod. 1998, 61, 546–547. [Google Scholar] [CrossRef]
- Li, C.-J.; Schmitz, F.J.; Kelly-Borges, M. Six New Spongian Diterpenes from the Sponge Spongia matamata. J. Nat. Prod. 1999, 62, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.S.; Harper, M.K.; Faulkner, D.J. Spongiabutenolides A–D: Minor γ-Hydroxybutenolide Diterpenoids from a Philippines Spongia sp. Tetrahedron 1999, 55, 10887–10892. [Google Scholar] [CrossRef]
- Zeng, L.M.; Guan, Z.; Su, J.Y.; Feng, X.L.; Cai, J.W. Two New Spongian Diterpene Lactones. Acta Chim. Sin. 2001, 59, 1675–1679. [Google Scholar]
- Su, J.Y.; Lin, C.W.; Zeng, L.M.; Yan, S.J.; Feng, X.L.; Cai, J.W. Separation and Structure Determination of a New 19-nor-spongian Diterpenoid. Chem. J. Chin. Univ. 2003, 24, 817–819. [Google Scholar]
- Ponomarenko, L.P.; Kalinovsky, A.I.; Afiyatullov, S.S.; Pushilin, M.A.; Gerasimenko, A.V.; Krasokhin, V.B.; Stonik, V.A. Spongian Diterpenoids from the Sponge Spongia (Heterofibria) sp. J. Nat. Prod. 2007, 70, 1110–1113. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Lamb, J.; Moni, R.; Hooper, J.N.A.; Quinn, R.J. Spongian Diterpenes with Thyrotropin Releasing Hormone Receptor 2 Binding Affinity from Spongia sp. J. Nat. Prod. 2008, 71, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, L.P.; Terent’eva, N.A.; Krasokhin, V.B.; Kalinovsky, A.I.; Rasskazov, V.A. Terpenoid Metabolites from Spongia spp. and Their Effects on Nucleic Acid Biosynthesis in Sea Urchin Eggs. Nat. Prod. Commun. 2011, 6, 773–776. [Google Scholar] [PubMed]
- Parrish, S.M.; Yoshida, W.Y.; Kondratyuk, T.P.; Park, E.-J.; Pezzuto, J.M.; Kelly, M.; Williams, P.G. Spongiapyridine and Related Spongians Isolated from an Indonesian Spongia sp. J. Nat. Prod. 2014, 77, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.T.; Carney, J.R.; Yoshida, W.Y.; Scheuer, P.J. Haumanamide, a Nitrogenous Spongian Derivative from Spongia sp. Tetrahedron Lett. 1992, 33, 1147–1148. [Google Scholar] [CrossRef]
- Marino, S.D.; Iorizzi, M.; Zollo, F.; Debitus, C.; Menou, J.-L.; Ospina, L.F.; Alcaraz, M.J.; Payá, M. New Pyridinium Alkaloids from a Marine Sponge of the Genus Spongia with a Human Phospholipase A2 Inhibitor Profile. J. Nat. Prod. 2000, 63, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Mori, D.; Kimura, Y.; Kitamura, S.; Sakagami, Y.; Yoshioka, Y.; Shintani, T.; Okamoto, T.; Ojika, M. Spongolactams, Farnesyl Transferase Inhibitors from a Marine Sponge: Isolation through an LC/MS-Guided Assay, Structures, and Semisyntheses. J. Org. Chem. 2007, 72, 7190–7198. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, R.; Murphy, P.T.; Wells, R.J.; Noack, K.; Oberhansli, W.E.; Schonholzer, P. New Series of Diterpenes from Australian Spongia Species. Aust. J. Chem. 1979, 32, 867–880. [Google Scholar] [CrossRef]
- Thompson, J.E.; Murphy, P.T.; Bergquist, P.R.; Evans, E.A. Environmentally Induced Variation in Diterpene Composition of the Marine Sponge Rhopaloeides Odorabile. Biochem. Syst. Ecol. 1987, 15, 595–606. [Google Scholar] [CrossRef]
- Puliti, R.; Mattia, C.A. Ent-Isocopal-12-ene-15,16-dialdehyde from Spongia officinalis. Acta Crystallogr. 1999, 55, 2160–2163. [Google Scholar]
- Yong, K.W.L.; Garson, M.J.; Bernhardt, P.V. Absolute Structures and Conformations of the Spongian Diterpenes Spongia-13(16),14-dien-3-one, Epispongiadiol and Spongiadiol. Acta Crystallogr. Sect. C 2009, 65, O167–O170. [Google Scholar] [CrossRef] [PubMed]
- Betancur-Galvis, L.; Zuluaga, C.; Arnó, M.; González, M.A.; Zaragozá, R.J. Cytotoxic Effect (on Tumor Cells) and in Vitro Antiviral Activity against Herpes Simplex Virus of Synthetic Spongiane Diterpenes. J. Nat. Prod. 2002, 65, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Cimino, G.; Stefano, S.D.; Minale, L. Furospongin-1, a New C-21 Furanoterpene from the Sponges Spongia officinalis and Hippospongia communis. Tetrahedron 1971, 27, 4673–4679. [Google Scholar] [CrossRef]
- Cimino, G.; Stefano, S.D.; Minale, L. Minor C-21 Furanoterpenes from the Sponges Spongia officinalis and Hippospongia communis. Tetrahedron 1972, 28, 267–273. [Google Scholar] [CrossRef]
- Cimino, G.; Stefano, S.D.; Minale, L. Further Linear Furanoterpenes from Marine Sponges. Tetrahedron 1972, 28, 5983–5991. [Google Scholar] [CrossRef]
- Fontana, A.; Albarella, L.; Scognamiglio, G.; Uriz, M.; Cimino, G. Structural and Stereochemical Studies of C-21 Terpenoids from Mediterranean Spongiidae Sponges. J. Nat. Prod. 1996, 59, 869–872. [Google Scholar] [CrossRef]
- Kobayashi, M.; Chavakula, R.; Murata, O.; Sarma, N.S. Marine Terpenes and Terpenoids. Part 16. Revised Structure of Marine Furanoterpene (+)-furospongin-1. J. Chem. Res. 1992, 366–367. [Google Scholar] [CrossRef]
- Cimino, G.; Stefano, S.D.; Minale, L. Oxidized Furanoterpenes from the Sponge Spongia officinalis. Experientia 1974, 30, 18–20. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Quinn, R.J.; Wells, R.J. Tetradehydrofurospongin-1, a New C21 Furanoterpene from a Sponge. Tetrahedron Lett. 1976, 16, 1331–1332. [Google Scholar] [CrossRef]
- Capon, R.J.; Ghisalberti, E.L.; Jefferies, P.R. A New Furanoterpene from a Spongia sp. Experientia 1982, 38, 1444–1445. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Quinn, R.J.; Wells, R.J. Two New Unsymetrically Oxygenated C21 furanoterpenes from a Sponge. Tetrahedron Lett. 1976, 16, 1333–1334. [Google Scholar] [CrossRef]
- Walker, R.P.; Thompson, J.E.; Faulkner, D.J. Sesterterpenes from Spongia idia. J. Org. Chem. 1980, 45, 4976–4979. [Google Scholar] [CrossRef]
- Capon, R.J.; Jenkins, A.; Rooney, F.; Ghisalberti, E.L. Structure Revision and Assignment of Absolute Stereochemistry of a Marine C21 Bisfuranoterpene. J. Nat. Prod. 2001, 64, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Higa, T. The Absolute Configuration of Kurospongin, a New Furanoterpene from a Marine Sponge, Spongia sp. Tetrahedron 1988, 44, 2805–2810. [Google Scholar] [CrossRef]
- Giulio, A.D.; Rosa, S.D.; Vincenzo, G.D. Terpenoids from the North Adriatic Sponge Spongia officinalis. J. Nat. Prod. 1989, 52, 1258–1262. [Google Scholar] [CrossRef]
- Lumsdon, D.; Capon, R.J.; Thomas, S.G.; Beveridge, A.A. A New Sesterterpene Tetronic Acid and a Pentaprenylated Para-Quinol from an Australian Marine Sponge, Spongia sp. Aust. J. Chem. 1992, 45, 1321–1325. [Google Scholar] [CrossRef]
- Urban, S.; Capon, R.J. Cometins (A–C), New Furanosesterterpenes from an Australian Marine Sponge, Spongia sp. Aust. J. Chem. 1992, 45, 1255–1263. [Google Scholar] [CrossRef]
- Lenis, L.A.; Nunez, L.; Jimenez, C.; Riguera, R. Isonitenin and Acetylhomoagmatine New Metabolites from the Sponges Spongia officinalis and Cliona Celata Collected at the Galician Coast (NW Spain). Nat. Prod. Lett. 1996, 8, 15–23. [Google Scholar] [CrossRef]
- Garrido, L.; Zubía, E.; Ortega, M.J.; Salvá, J. New Furanoterpenoids from the Sponge Spongia officinalis. J. Nat. Prod. 1997, 60, 794–797. [Google Scholar] [CrossRef]
- Manzo, E.; Ciavatta, M.L.; Villani, G.; Varcamonti, M.; Sayem, S.M.A.; Soest, R.; Gavagnin, M. Bioactive Terpenes from Spongia officinalis. J. Nat. Prod. 2011, 74, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Rueda, A.; Zubía, E.; Ortega, M.J.; Carballo, J.L.; Salvá, J. New Metabolites from the Sponge Spongia agaricina. J. Nat. Prod. 1998, 61, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Cimino, G.; Stefano, S.D.; Minale, L. Deoxoscalarin, a Further Sesterterpene with the Unusual Tetracyclic Carbon Skeleton of Scalarin, from Spongia officinalis. Experientia 1973, 29, 934–936. [Google Scholar] [CrossRef]
- Cimino, G.; Stefano, S.D.; Minale, L.; Trivellone, E. 12-epi-Scalarin and 12-epi-Deoxoscalarin, Sesterterpenes from the Sponge Spongia nitens. J. Chem. Soc. Perkin Trans. 1977, 13, 1587–1593. [Google Scholar] [CrossRef]
- Cimino, G.; Stefano, S.D.; Luccia, A.D. Further Sesterterpenes from the Sponge Spongia nitens: 12-epi-scalaradial and 12,18-diepi-scalaradial. Experientia 1979, 35, 1277–1278. [Google Scholar] [CrossRef]
- Cimino, G.; Rosa, S.D.; Stefano, S.D. Scalarolbutenolide, a New Sesterterpenoid from the Marine Sponge Spongia nitens. Experientia 1981, 37, 214–216. [Google Scholar] [CrossRef]
- Davis, R.; Capon, R.J. Two New Scalarane Sesterterpenes-Isoscalarafuran-A and Isoscalarafuran-B, Epimeric Alcohols from a Southern Australian Marine Sponge, Spongia hispida. Aust. J. Chem. 1993, 46, 1295–1299. [Google Scholar] [CrossRef]
- He, H.; Kulanthaivel, P.; Baker, B.J. New Cytotoxic Sesterterpenes from the Marine Sponge Spongia sp. Tetrahedron Lett. 1994, 35, 7189–7192. [Google Scholar] [CrossRef]
- Conte, M.R.; Fatorrusso, E.; Lanzotti, V.; Magno, S.; Mayol, L. Lintenolides, new pentacyclic bioactive sesterterpenes from the caribbean sponge Cacospongia cf. linteiformis. Tetrahedron 1994, 50, 849–856. [Google Scholar] [CrossRef]
- Lu, Q.; Faulkner, D.J. Two New Sesterterpenoids and a New 9,11-Secosterol from Spongia matamata. J. Nat. Prod. 1997, 60, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Miyamot, T.; Amano, H.; Higuchi, R.; Komori, T.; Sasaki, T. 34th Tennen Yuki Kagobutsu Toronkai Koen Yoshishu; Organizing Committee of the 34th Symposium on the Chemistry of Natural Products: Tokyo, Japan, 1992; Volume 34, Chem. Abstr. 173183; pp. 455–462. [Google Scholar]
- Tsukamoto, S.; Miura, S.; Soest, R.W.M.V.; Ohta, T. Three New Cytotoxic Sesterterpenes from a Marine Sponge Spongia sp. J. Nat. Prod. 2003, 66, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Tokue, T.; Miura, S.; Kato, H.; Hirota, H.; Ohta, T.; Tsukamoto, S. Neurotrophic Sesterterpenes Isolated from a Marine Sponge, Spongia sp. Heterocycles 2006, 69, 521–526. [Google Scholar] [CrossRef]
- Nam, S.-J.; Ko, H.; Shin, M.; Ham, J.; Chin, J.; Kim, Y.; Kim, H.; Shin, K.; Choi, H.; Kang, H. Farnesoid X-activated Receptor Antagonists from a Marine Sponge Spongia sp. Bioorg. Med. Chem. Lett. 2006, 16, 5398–5402. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.-J.; Ko, H.; Ju, M.K.; Hwang, H.; Chin, J.; Ham, J.; Lee, B.; Lee, J.; Won, D.H.; Choi, H.; et al. Scalarane Sesterterpenes from a Marine Sponge of the Genus Spongia and Their FXR Antagonistic Activity. J. Nat. Prod. 2007, 70, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Carr, G.; Raszek, M.; Soest, R.V.; Matainaho, T.; Shopik, M.; Holmes, C.F.B.; Andersen, R.J. Protein Phosphatase Inhibitors Isolated from Spongia irregularis Collected in Papua New Guinea. J. Nat. Prod. 2007, 70, 1812–1815. [Google Scholar] [CrossRef] [PubMed]
- Grassia, A.; Bruno, I.; Debitus, C.; Marzocco, S.; Pinto, A.; Gomez-Paloma, L.; Riccio, R. Spongidepsin, a New Cytotoxic Macrolide from Spongia sp. Tetrahedron 2001, 57, 6257–6260. [Google Scholar] [CrossRef]
- Aiello, A.; Ciminiello, P.; Fattorusso, E.; Magno, S. 3,5-Dihydroxy-6-methoxycholest-7-enes from the Marine Sponge Spongia agaricina. J. Nat. Prod. 1988, 51, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Madaio, A.; Piccialli, V.; Sica, D. New Polyhydroxysterols from the Dictyoceratid Sponges Hippospongia communis, Spongia officinalis, Ircinia variabilis, and Spongionella gracilis. J. Nat. Prod. 1989, 52, 952–961. [Google Scholar] [CrossRef]
- Migliuolo, A.; Notaro, G.; Piccialli, V.; Sica, D. New Tetrahydroxylated Sterols from the Marine Sponge Spongia officinalis. J. Nat. Prod. 1990, 53, 1414–1424. [Google Scholar] [CrossRef]
- Migliuolo, A.; Piccialli, V.; Sica, D.; Giordano, F. New Delta-8-5-alpha,6-alpha-epoxysterols and Delta-8(14)-5-alpha,6-alpha-epoxysterols from the Marine Sponge Spongia officinalis. Steroids 1993, 58, 134–140. [Google Scholar] [CrossRef]
- Aoki, S.; Yoshioka, Y.; Miyamoto, Y.; Higuchi, K.; Setiawan, A.; Murakami, N.; Chen, Z.-S.; Sumizawa, T.; Akiyama, S.; Kobayashi, M. Agosterol A, a Novel Polyhydroxylated Sterol Acetate Reversing Multidrug Resistance from a Marine Sponge of Spongia sp. Tetrahedron Lett. 1998, 39, 6303–6306. [Google Scholar] [CrossRef]
- Aoki, S.; Setiawan, A.; Yoshioka, Y.; Higuchi, K.; Fudetani, R.; Chen, Z.-S.; Sumizawa, T.; Akiyama, S.; Kobayashi, M. Reversal of Muitidrug Resistance in Human Carcinoma Cell Line by Agosterols, Marine Spongean Sterols. Tetrahedron 1999, 55, 13965–13972. [Google Scholar] [CrossRef]
- Chen, Z.S.; Aoki, S.; Komatsu, M.; Ueda, K.; Sumizawa, T.; Furukawa, T.; Okumura, H.; Ren, X.Q.; Belinsky, M.G.; Lee, K.; et al. Reversal of Drug Resistance Mediated by Multidrug Resistance Protein (MRP) 1 by Dual Effects of Agosterol A on MRP1 Function. Int. J. Cancer 2001, 93, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Migliuolo, A.; Piccialli, V.; Sica, D. Structure Elucidation and Synthesis of 3β,6α-dihydroxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al, a novel 9,11-secosterol from the Sponge Spongia officinalis. Tetrahedron 1991, 47, 7937–7950. [Google Scholar] [CrossRef]
- Migliuolo, A.; Piccialli, V.; Sica, D. 2 New 9,11-Secosterols from the Marine Sponge Spongia officinalis. Synthesis of 9,11-seco-3-beta,6-alpha,11-Trihydroxy-5-alpha-cholest-7-en-9-one. Steroids 1992, 57, 344–347. [Google Scholar] [CrossRef]
- Adinolfi, R.; Migliuolo, A.; Piccialli, V.; Sica, D. Isolation and Synthesis of a New 9,11-Secosterol from the Sponge Spongia officinallis. J. Nat. Prod. 1994, 57, 1220–1226. [Google Scholar] [CrossRef]
- Quinoa, E.; Kakou, Y.; Crews, P. Fijianolides, Polyketide Heterocycles from a Marine Sponge. J. Org. Chem. 1988, 53, 3642–3644. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gal, F.; Herald, C.L.; Boyd, M.R.; Schmidt, J.M.; Hooperlc, J.N.A. Isolation and Structure of Spongistatin 1. J. Org. Chem. 1993, 58, 1302–1304. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Heralds, C.L.; Boyd, M.R. Isolation and Structure of the Remarkable Human Cancer Cell Growth Inhibitors Spongistatins 2 and 3 from an Eastern Indian Ocean Spongia sp. J. Chem. Soc. Chem. Commun. 1993, 24, 1166–1168. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Boyd, M.R.; Schmidt, J.M. Isolation and Structure of the Cancer Cell Growth Inhibitor Dictyostatin 1. J. Chem. Soc. Chem. Commun. 1994, 9, 1111–1112. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Sakai, H.; Kawazoe, K.; Kihara, N.; Sasaki, T.; Kitagawa, I. Altohyrtin A, a Potent Anti-tumor Macrolide from the Okinawan Marine Sponge Hyrfios alturn. Tetrahedron Lett. 1993, 34, 2795–2798. [Google Scholar] [CrossRef]
- Fusetani, N.; Shinoda, K.; Matsunaga, S. Cinachyrolide A: A Potent Cytotoxic Macrolide Possessing Two Spiro Ketals from Marine Sponge Cinachyra sp. J. Am. Chem. Soc. 1993, 115, 3977–3981. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Cichacz, Z.A.; Gao, F.; Boyd, M.R.; Christie, N.D.; Schmidt, J.M. Antineoplastic Agents 293. the Exceptional Human Cancer Cell Growth Inhibitors Spongistatins 6 and 7. Nat. Prod. Lett. 1993, 3, 239–244. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Gato, K.; Kitagawa, I. Marine Natural Products. XXXVIII. Absolute stereostructures of Altohyrtins A, B, and C and 5-desacetylaltohyrtin A, a potent Cytotoxic Macrolides, from the Okinawan Marine Sponge Hyrtios altum. Chem. Pharm. Bull. 1996, 44, 2142–2149. [Google Scholar] [CrossRef]
- Guo, J.; Duffy, K.J.; Stevens, K.L.; Dalko, P.I.; Roth, R.M.; Hayward, M.M.; Kishi, Y. Total Synthesis of Altohyrtin A (Spongistatin 1): Part 1. Angew. Chem. Int. Ed. 1998, 37, 187–192. [Google Scholar] [CrossRef]
- Hayward, M.M.; Roth, R.M.; Duffy, K.J.; Dalko, P.I.; Stevens, K.L.; Guo, J.; Kishi, Y. Total Synthesis of Altohyrtin A (Spongistatin 1): Part 2. Angew. Chem. Int. Ed. 1998, 37, 192–196. [Google Scholar] [CrossRef]
- Bai, R.L.; Cichacz, Z.A.; Herald, C.L.; Pettit, G.R.; Hamel, E. Spongistatin-1, A Highly Cytotoxic, Sponge-Derived, Marine Natural Product that Inhibits Mitosis, Microtubule Assembly, and the Binding of Vinblastine to Tubulin. Mol. Pharmacol. 1993, 44, 757–766. [Google Scholar] [PubMed]
- Luduena, R.F.; Roach, M.C.; Prasad, V.; Pettit, G.R.; Cichacz, Z.A.; Herald, C.L. Interaction of 3 Sponge-Derived Macrocyclic Lactone Polyethers (Spongistatin-3, Halistatin-1 and Halistatin-2) With Tubulin. Drug Dev. Res. 1995, 35, 40–48. [Google Scholar] [CrossRef]
- Chen, J.; Forsyth, C.J. Total Synthesis and Structural Assignment of Spongidepsin through a Stereodivergent Ring-Closing-Metathesis Strategy. Angew. Chem. Int. Ed. 2004, 43, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Xu, X. Assignment of Absolute Stereochemistry and Total Synthesis of (−)-Spongidepsin. Org. Lett. 2004, 6, 2055–2058. [Google Scholar] [CrossRef] [PubMed]
- Kalidindi, R.S.; Yoshida, W.Y.; Palermo, J.A.; Scheuer, P.J. Pokepola ester: A Phosphat Diester from a Maui Sponge. Tetrahedron Lett. 1994, 35, 5579–5582. [Google Scholar] [CrossRef]
- Pettit, G.R.; Bond, T.J.; Herald, D.L.; Penny, M.; Doubek, D.L.; Williams, M.D.; Pettit, R.K.; Hooper, J.N.A. Isolation and Structure of Spongilipid from the Republic of Singapore Marine Porifera Spongia cf. hispida. Can. J. Chem. 1997, 75, 920–925. [Google Scholar] [CrossRef]
- Xu, S.H.; Cen, Y.Z.; Zeng, L.M.; Su, J.Y. Isolation and Structural Determination of Heterocyclic Alkaloidal Compounds. Chin. J. Org. Chem. 2000, 20, 248–250. [Google Scholar]
- Kobayashi, K.; Shimogawa, H.; Sakakura, A.; Teruya, T.; Suenaga, K.; Kigoshi, H. Spongiacysteine, a Novel Cysteine Derivative from Marine Sponge Spongia sp. Chem. Lett. 2004, 33, 1262–1263. [Google Scholar] [CrossRef]
- Lin, C.W.; Su, J.Y.; Zeng, L.M.; Wei, W.X.; Wei, T.Y. Compounds Containing Nitrogen from Spongia zimocca Aubspecles Irregularia (Lendenfeld). Chin. J. Org. Chem. 2005, 25, 225–228. [Google Scholar]
- Xu, S.H.; Yang, K. Three New Ceramides from the Sponge Spongia suriganensis. Chin. J. Org. Chem. 2006, 26, 56–59. [Google Scholar]
- Guan, Z.; Zeng, L. A New Ceramide from a New Species of Spongia Sponge. Chem. Nat. Compd. 2010, 46, 287–288. [Google Scholar] [CrossRef]
- Guan, Z.; Zeng, L. Chemical constituents of a new species of Spongia sponge. Zhongguo Zhong Yao Za Zhi 2010, 35, 1004–1008. [Google Scholar] [PubMed]
- Salim, A.A.; Rae, J.; Fontaine, F.; Conte, M.M.; Khalil, Z.; Martin, S.; Partonb, R.G.; Capon, R.J. Heterofibrins: Inhibitors of Lipid Droplet Formation from a Deep-water Southern Australian Marine Sponge, Spongia (Heterofibria) sp. Org. Biomol. Chem. 2010, 8, 3188–3194. [Google Scholar] [CrossRef] [PubMed]
- Rae, J.; Fontaine, F.; Salim, A.A.; Lo, H.P.; Capon, R.J.; Parton, R.G.; Martin, S. High-Throughput Screening of Australian Marine Organism Extracts for Bioactive Molecules Affecting the Cellular Storage of Neutral Lipids. PLoS ONE 2011, 6, e22868. [Google Scholar] [CrossRef] [PubMed]
- Carballeira, N.M.; Emiliano, A.; Morales, R. Positional Distribution of Octadecadienoic Acids in Sponge Phosphatidylethanolamines. Lipids 1994, 29, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Junqua, S.; Lemonnier, M.; Robert, L. Glycoconjugates from Spongia officinalis (Phylum Porifera). Isolation, Fractionation by Affinity-Chromatography on Lectins and Partial Characterization. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1981, 69, 445–453. [Google Scholar] [CrossRef]
- Noyer, C.; Thomas, O.P.; Becerro, M.A. Patterns of Chemical Diversity in the Mediterranean Sponge Spongia lamella. PLoS ONE 2011, 6, e20844. [Google Scholar] [CrossRef] [PubMed]
- Terem, B.; Scheuer, P.J. Scalaradial Derivatives from the Nudibranch Chromodoris youngbleuthi and the Sponge Spongia oceania. Tetrahedron 1986, 42, 4409–4412. [Google Scholar] [CrossRef]
- Kakou, Y.; Crew, P. Dendrolasin and Latrunculin A from the Fijan Sponge Spongia mycofijiensis and an Associated Nudibranch Chromodoris lochi. J. Nat. Prod. 1987, 50, 482–484. [Google Scholar] [CrossRef]
- Crews, P.; Kakou, Y.; Quinoa, E. Mycothiazole, a Polyketide Heterocycle from a Marine Sponge. J. Am. Chem. Soc. 1988, 110, 4365–4368. [Google Scholar] [CrossRef]
- Guella, G.; Mancini, I.; Chiasera, G.; Pietra, F. Rogiolol Acetate: A Novel β-chamigrene-type Sesquiterpene Isolated from a Marine Sponge. Helv. Chim. Acta 1990, 73, 1612–1620. [Google Scholar] [CrossRef]
- Guella, G.; Pietra, F. Rogiolenyne-A, Rogiolenyne-B, and Rogiolenyne-C—The first Branched Marine C-15 Acetogenins. Isolation from the Red Seaweed Laurencia microcladia or the Sponge Spongia-zimocca of Il-Rogiolo. Helv. Chim. Acta 1991, 74, 47–54. [Google Scholar] [CrossRef]
- Guella, G.; Mancini, I.; Pietra, F. C-15 Acetogenins and Terpenes of the Dictyoceratid Sponge Spongia-zimocca of Il-Rogiolo: A Case of Seaweed-Metabolite Transfer to, and Elaboration within, a Sponge? Comp. Biochem. Phys. B Comp. Biochem. 1992, 103, 1019–1023. [Google Scholar] [CrossRef]
- Brown, J.W.; Kesler, C.T.; Neary, J.T.; Fishman, L.M. Effects of Marine Sponge Extracts on Mitogen-Activated Protein Kinase (MAPK/ERK1,2) Activity in SW-13 Human Adrenal Carcinoma Cells. Toxicon 2001, 39, 1835–1839. [Google Scholar] [CrossRef]
- Brown, J.W.; Cappell, S.; Perez-Stable, C.; Fishman, L.M. Extracts from Two Marine Sponges Lower Cyclin B1 Levels, Cause a G2/M Cell Cycle Block and Trigger Apoptosis in SW-13 Human Adrenal Carcinoma Cells. Toxicon 2004, 43, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Bartolotta, S.A.; Scuteri, M.A.; Hick, A.S.; Palermo, J.; Brasco, M.F.R.; Hajdu, E.; Mothes, B.; Lerner, C.; Campos, M.; Carballo, M.A. Evaluation of Genotoxic Biomarkers in Extracts of Marine Sponges from Argentinean South Sea. J. Exp. Mar. Biol. Ecol. 2009, 369, 144–147. [Google Scholar] [CrossRef]
- Devi, P.; Wahidulla, S.; Kamat, T.; D’Souza, L. Screening Marine Organisms for Antimicrobial Activity against Clinical Pathogens. Indian J. Mar. Sci. 2011, 40, 338–346. [Google Scholar]
- Dellai, A.; Mansour, H.B.; Clary-Laroche, A.; Deghrigue, M.; Bouraoui, A. Anticonvulsant and Analgesic Activities of Crude Extract and Its Fractions of the Defensive Secretion from the Mediterranean Sponge, Spongia officinalis. Cancer Cell Int. 2012, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Dellai, A.; Deghrigue, M.; Laroche-Clary, A.; Masour, H.B.; Chouchane, N.; Robert, J.; Bouraoui, A. Anti-inflammatory and Antiproliferative Activities of Crude Extract and Its Fractions of the Defensive Secretion From the Mediterranean Sponge, Spongia officinalis. Drug Dev. Res. 2010, 71, 412–418. [Google Scholar] [CrossRef]
- Dellai, A.; Deghrigue, M.; Laroche-Clary, A.; Masour, H.B.; Chouchane, N.; Robert, J.; Bouraoui, A. Evaluation of Antiproliferative and Anti-inflammatory Activities of Methanol Extract and Its Fractions from the Mediterranean Sponge. Cancer Cell Int. 2012, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, V.; Ghosal, S. Antiamoebic Activity of Marine Sponge Spongia officinalis var. ceylonensis Dendy. Bangladesh Pharm. J. 2014, 17, 38–42. [Google Scholar]



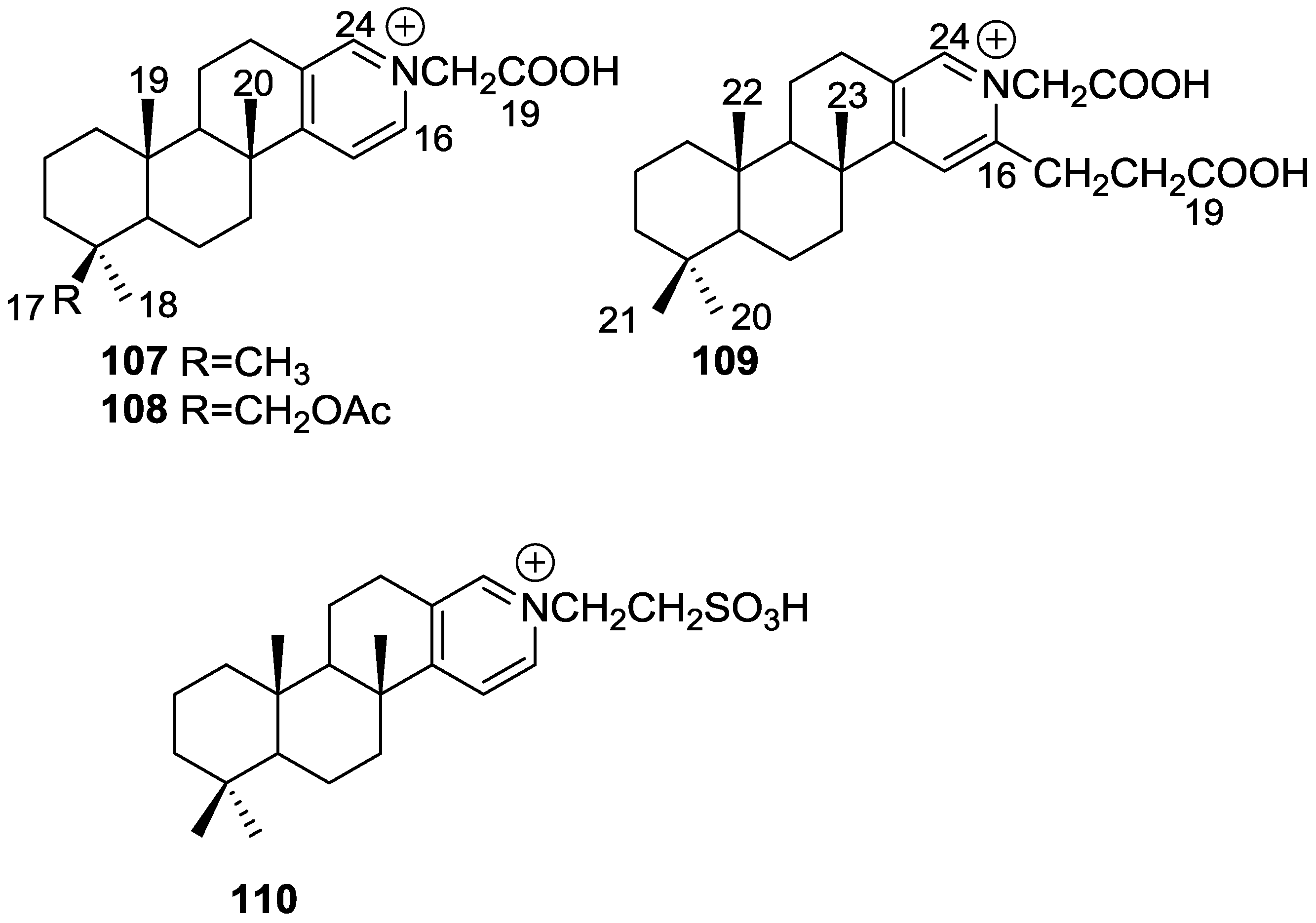
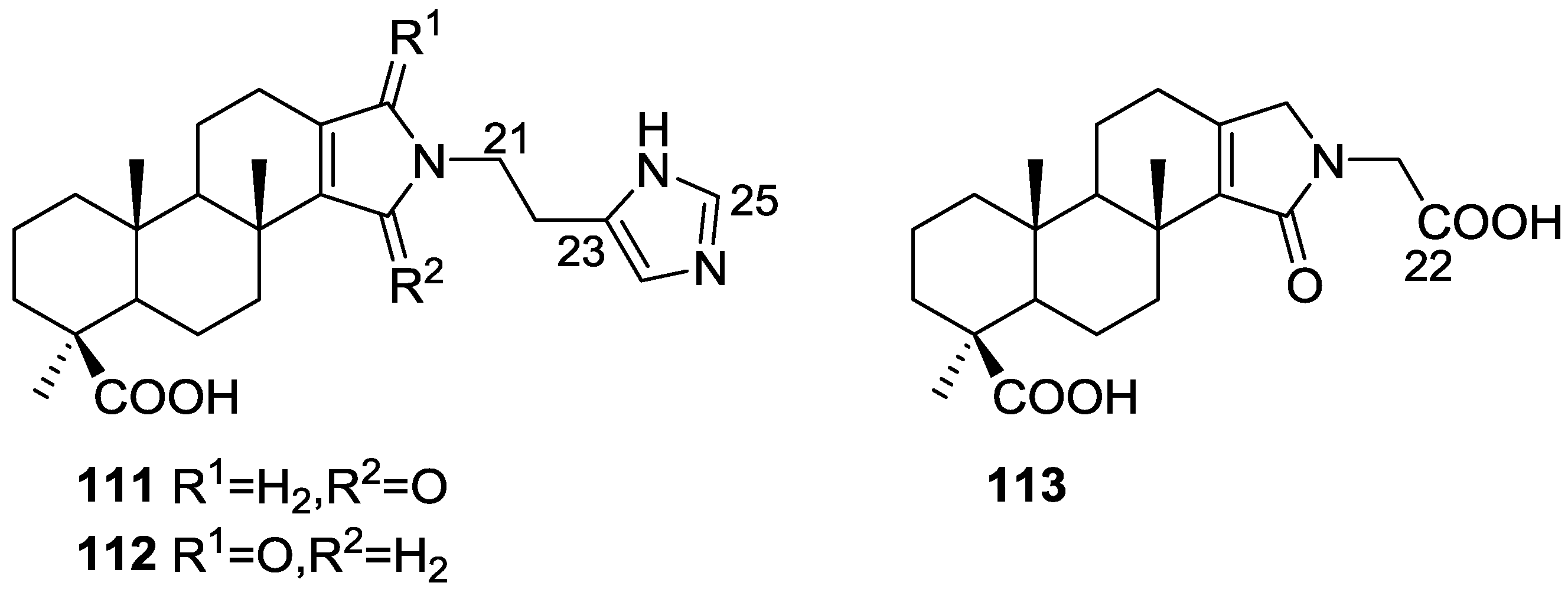
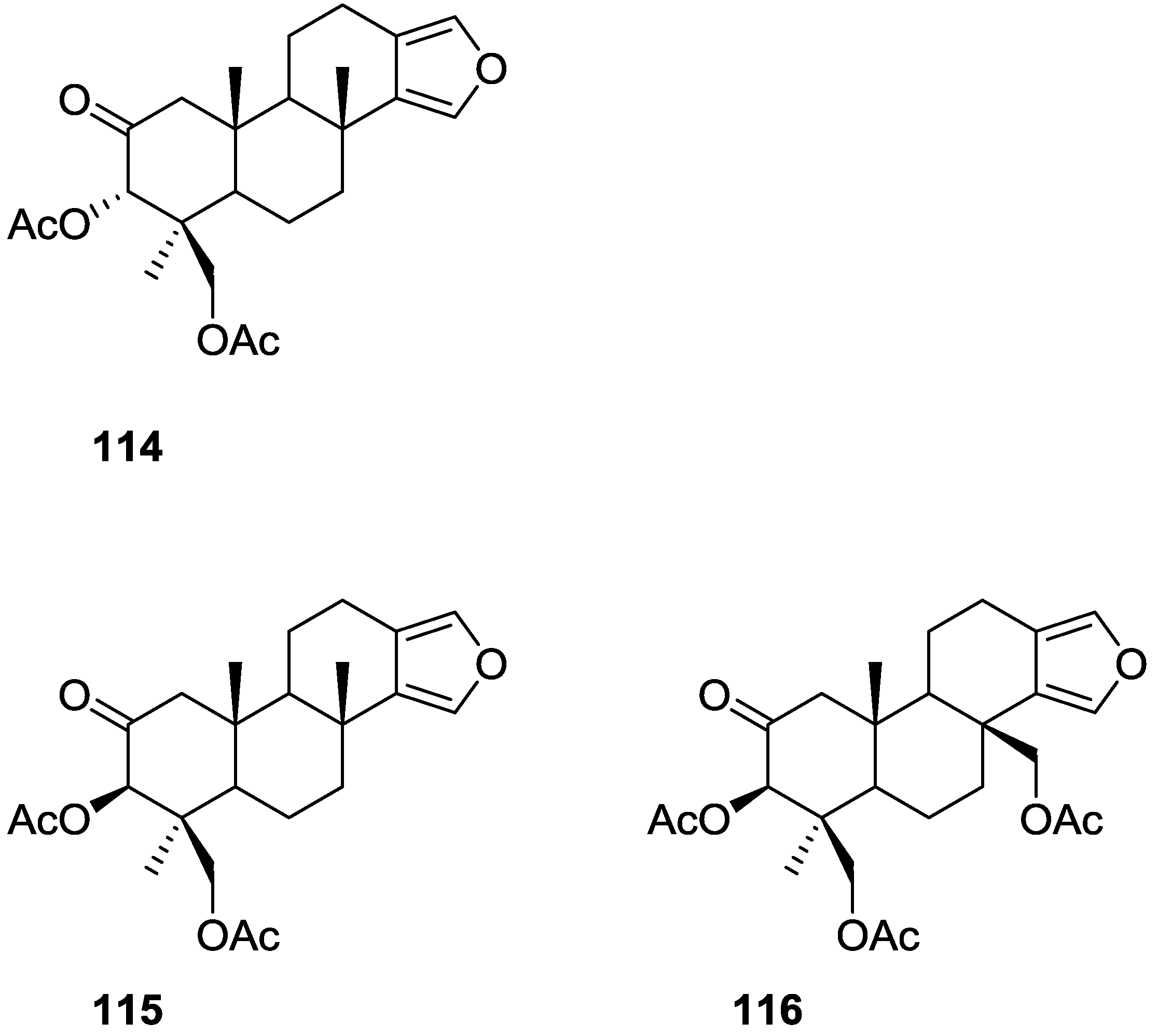
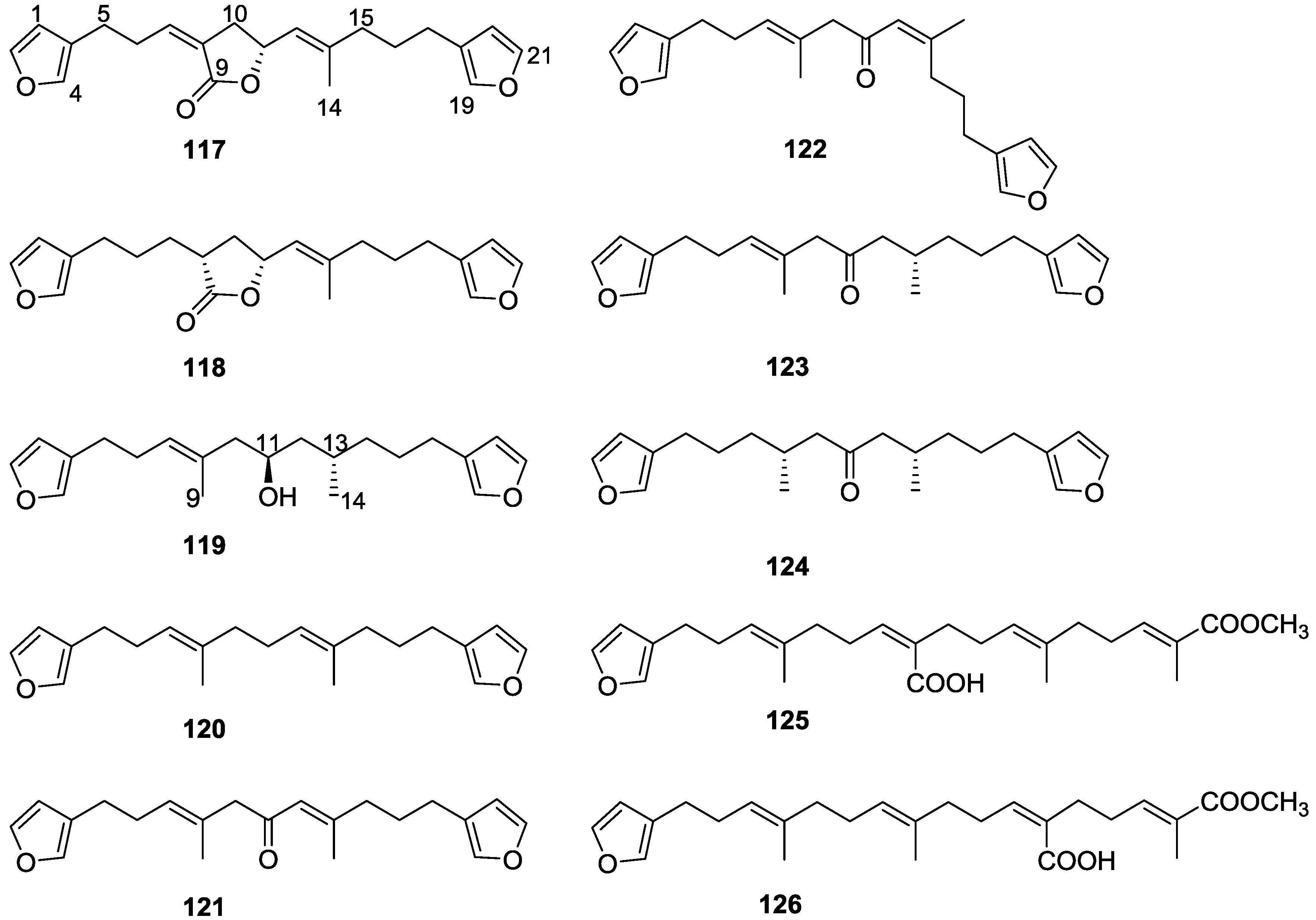


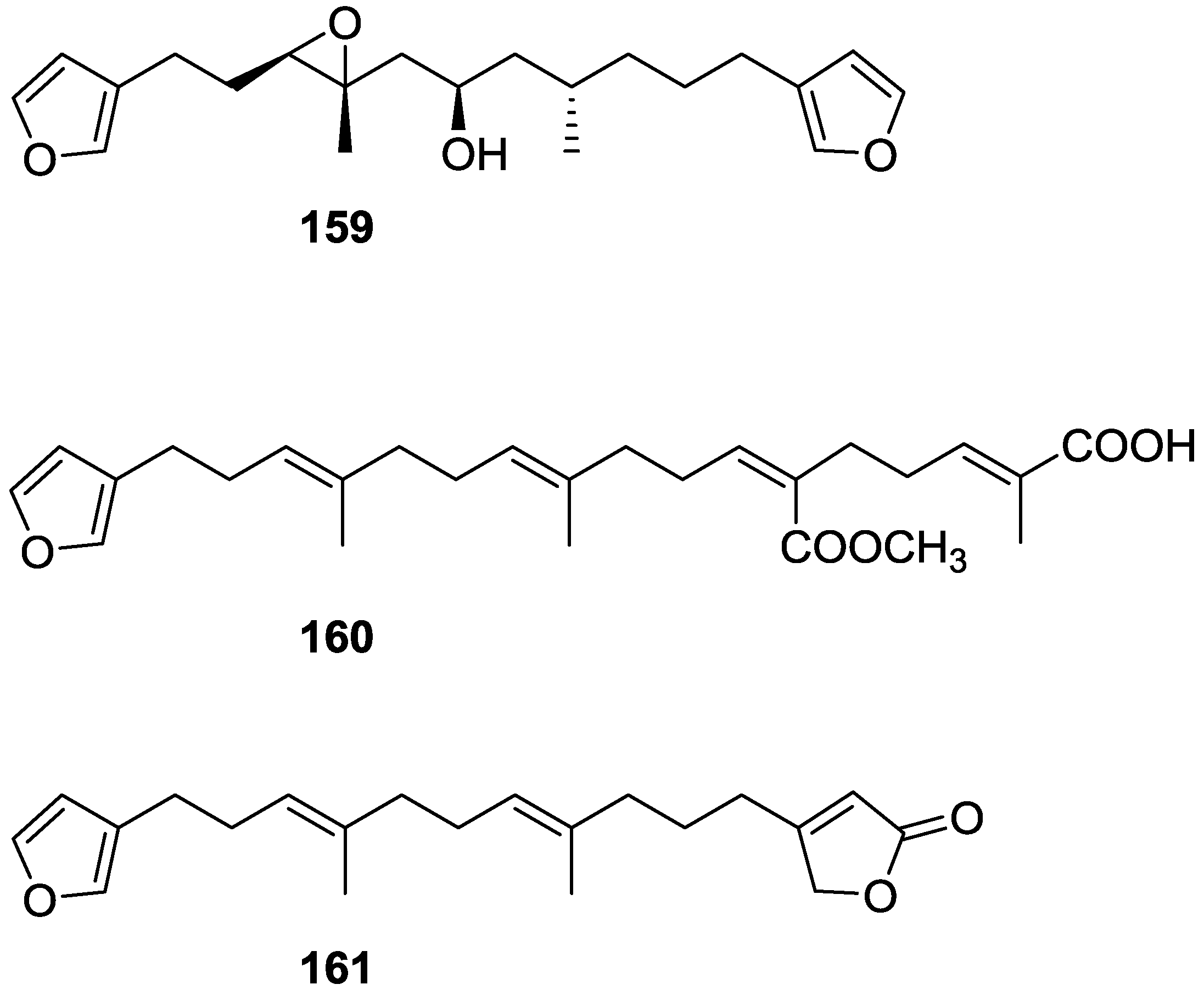
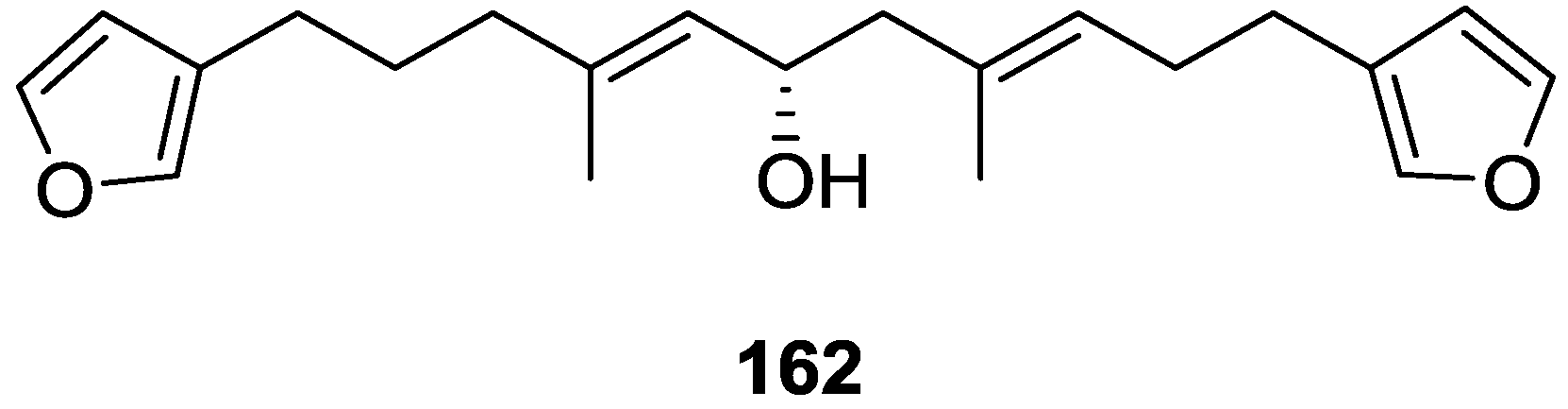
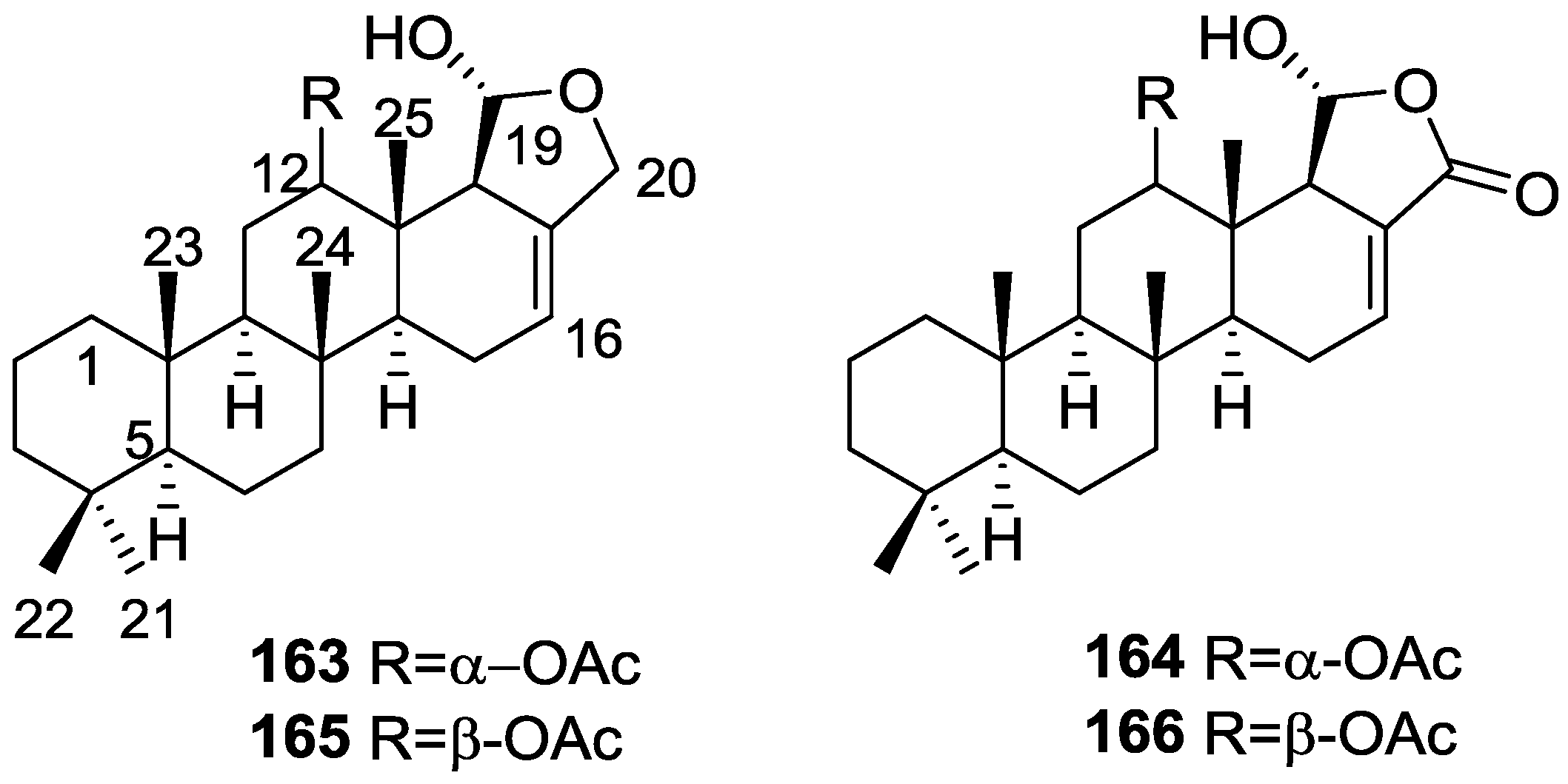
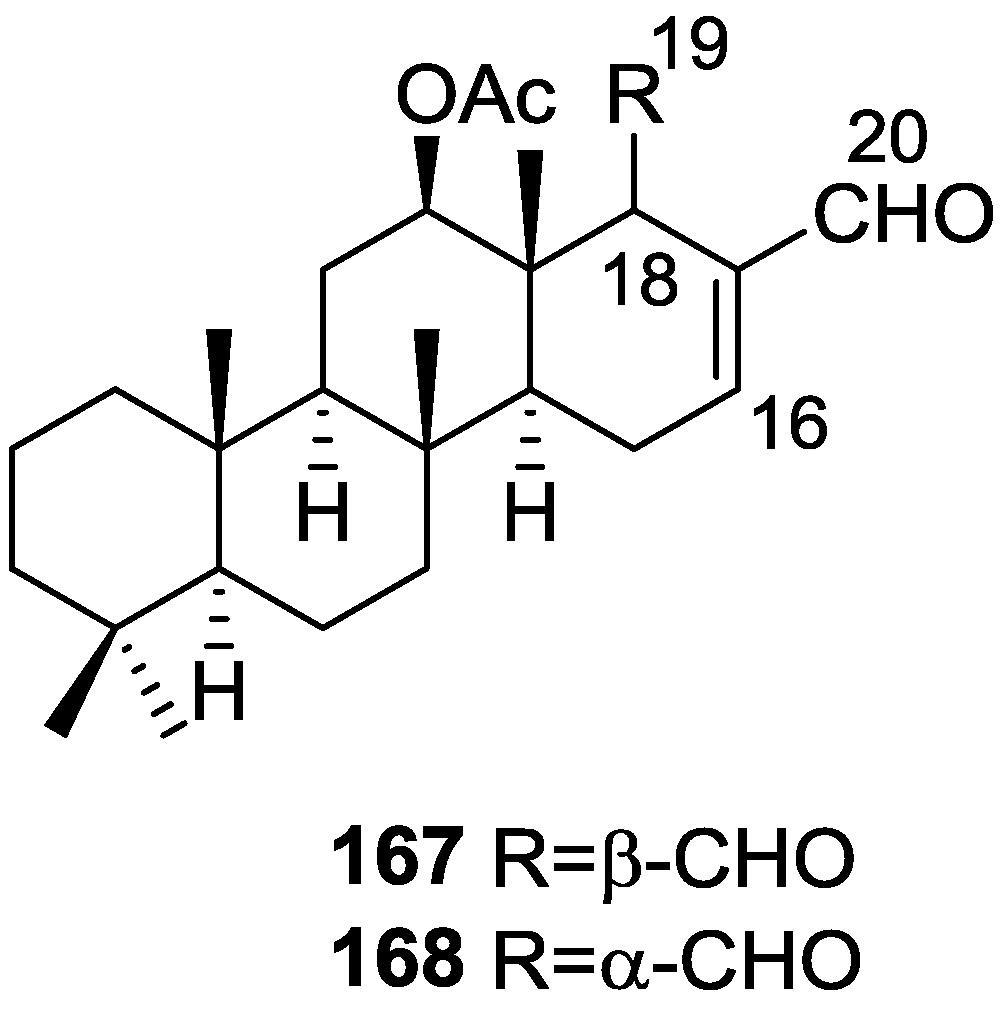
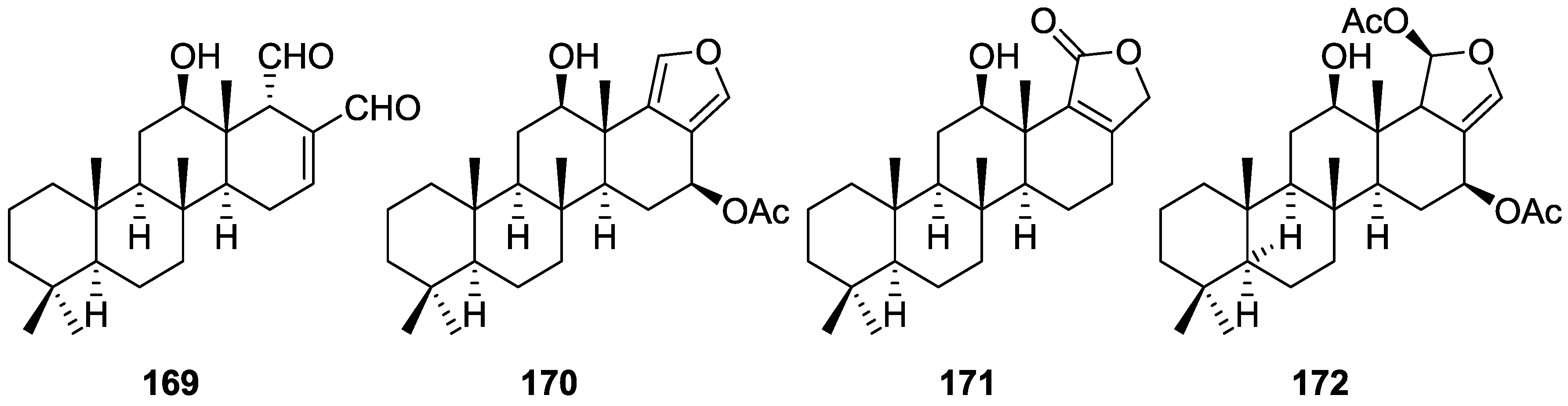
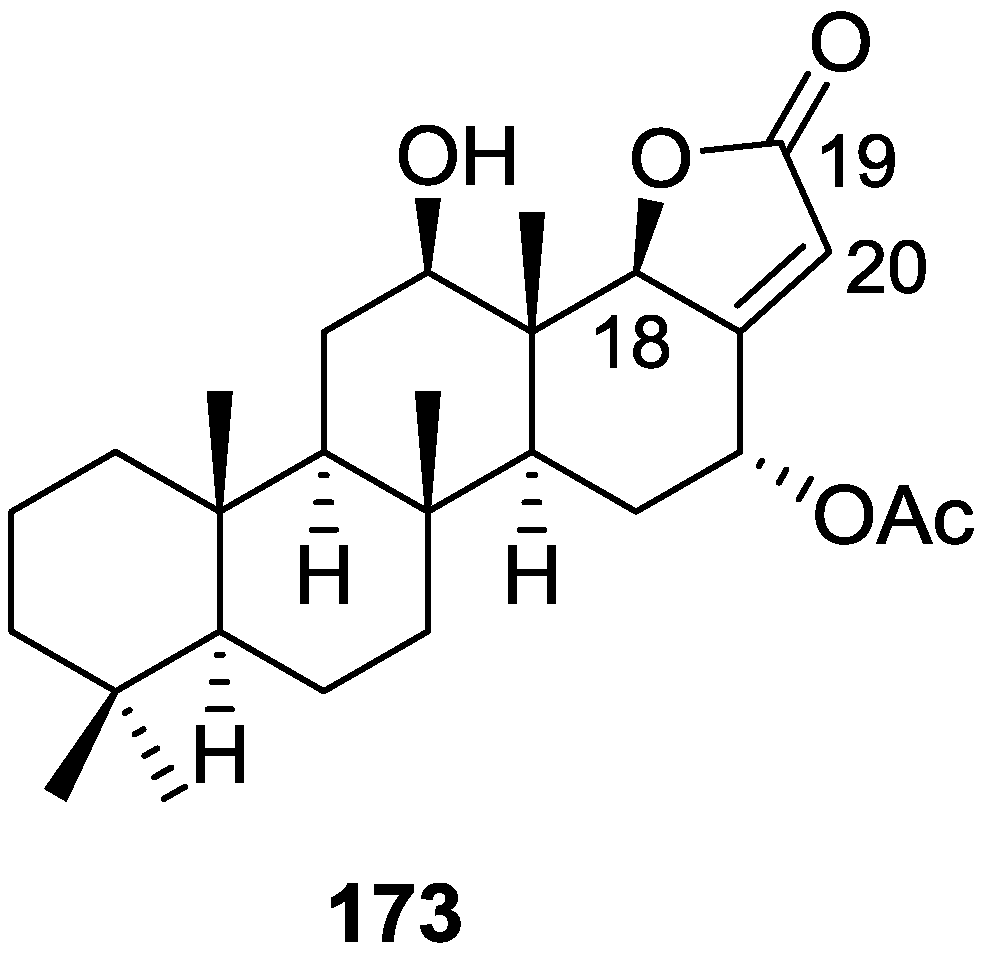

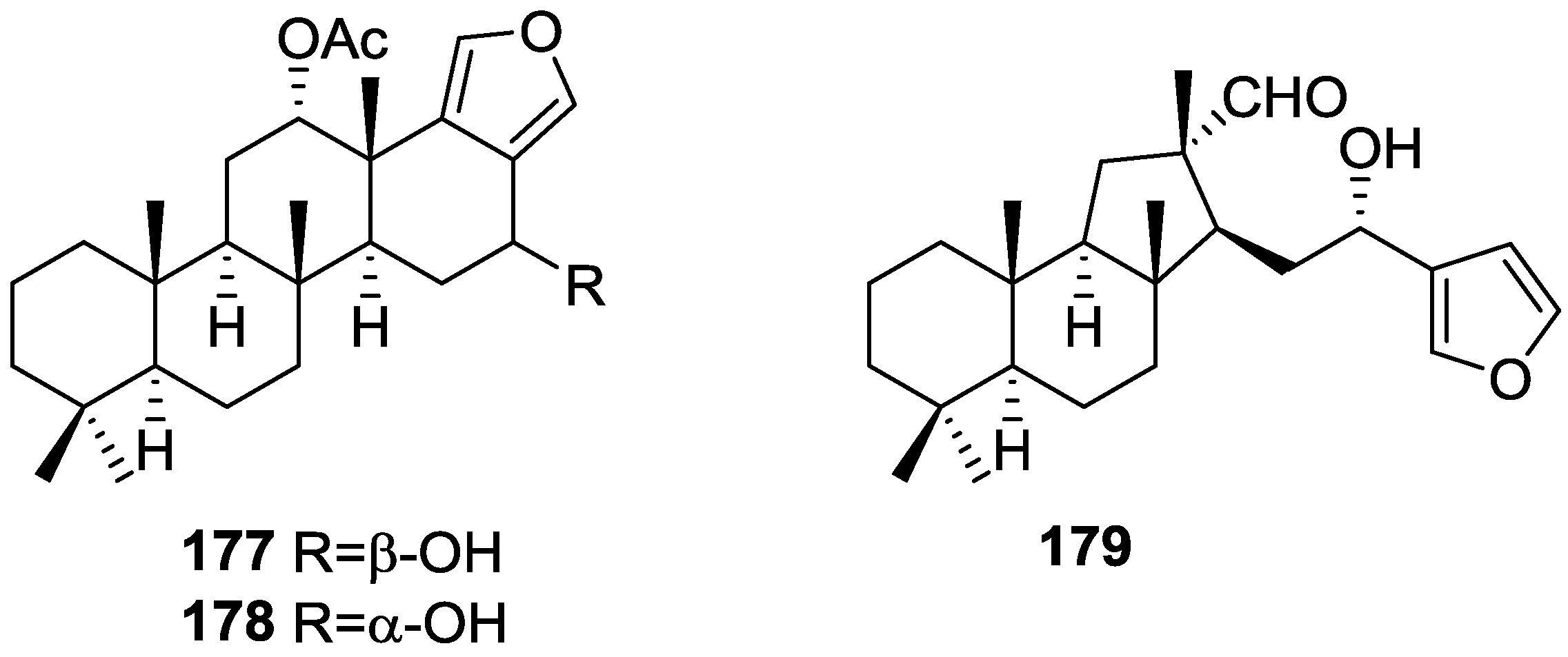
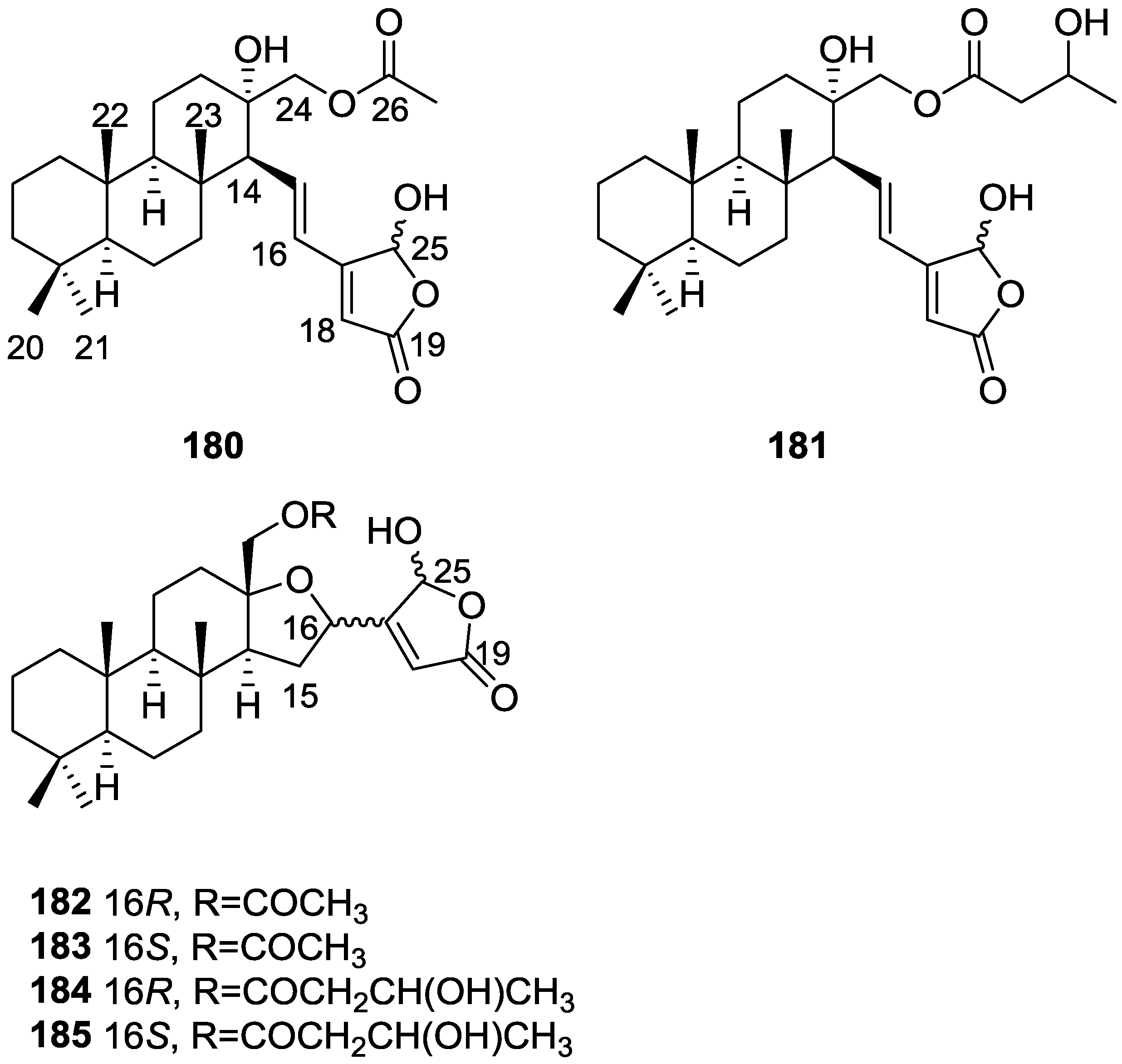



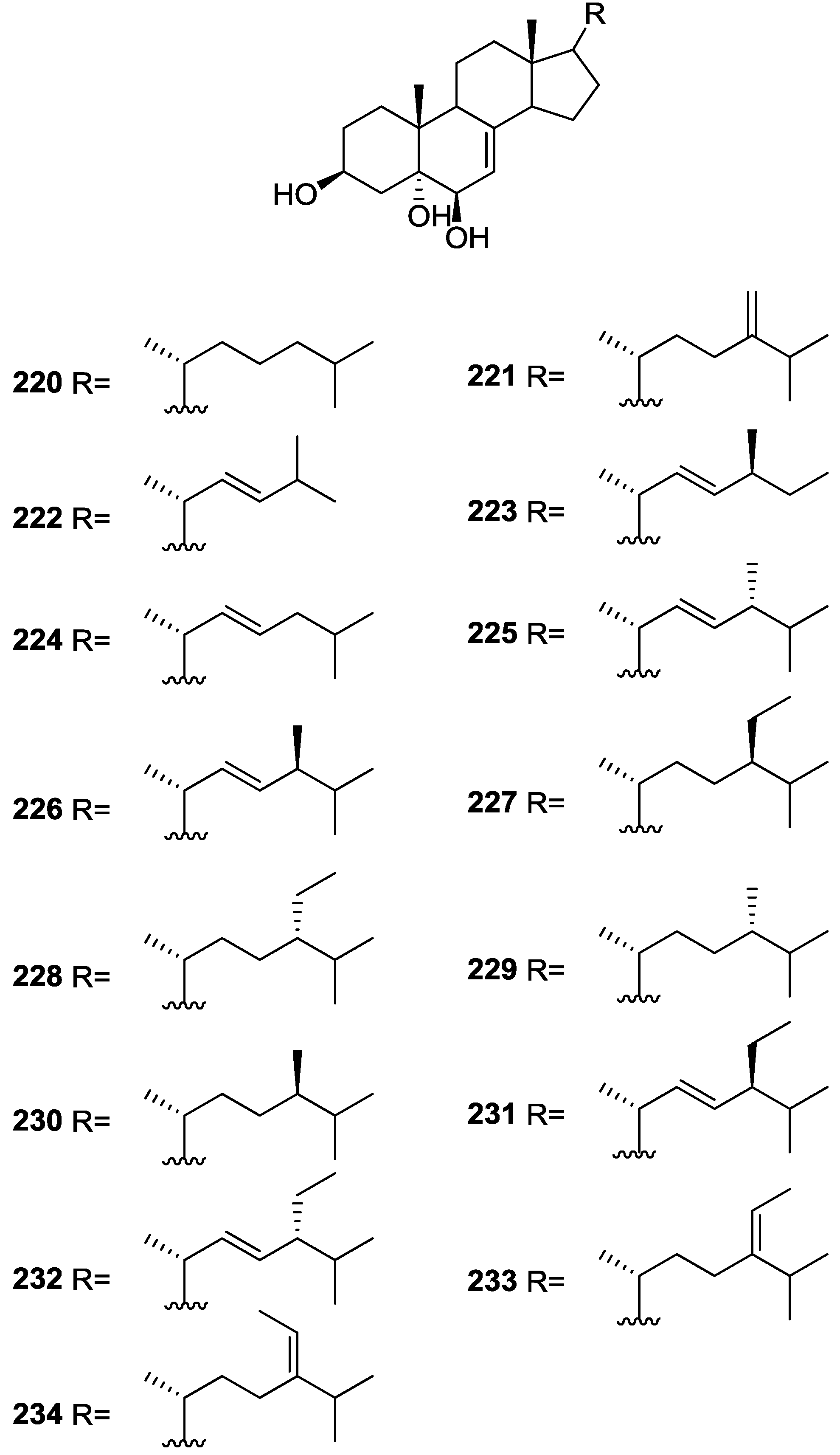
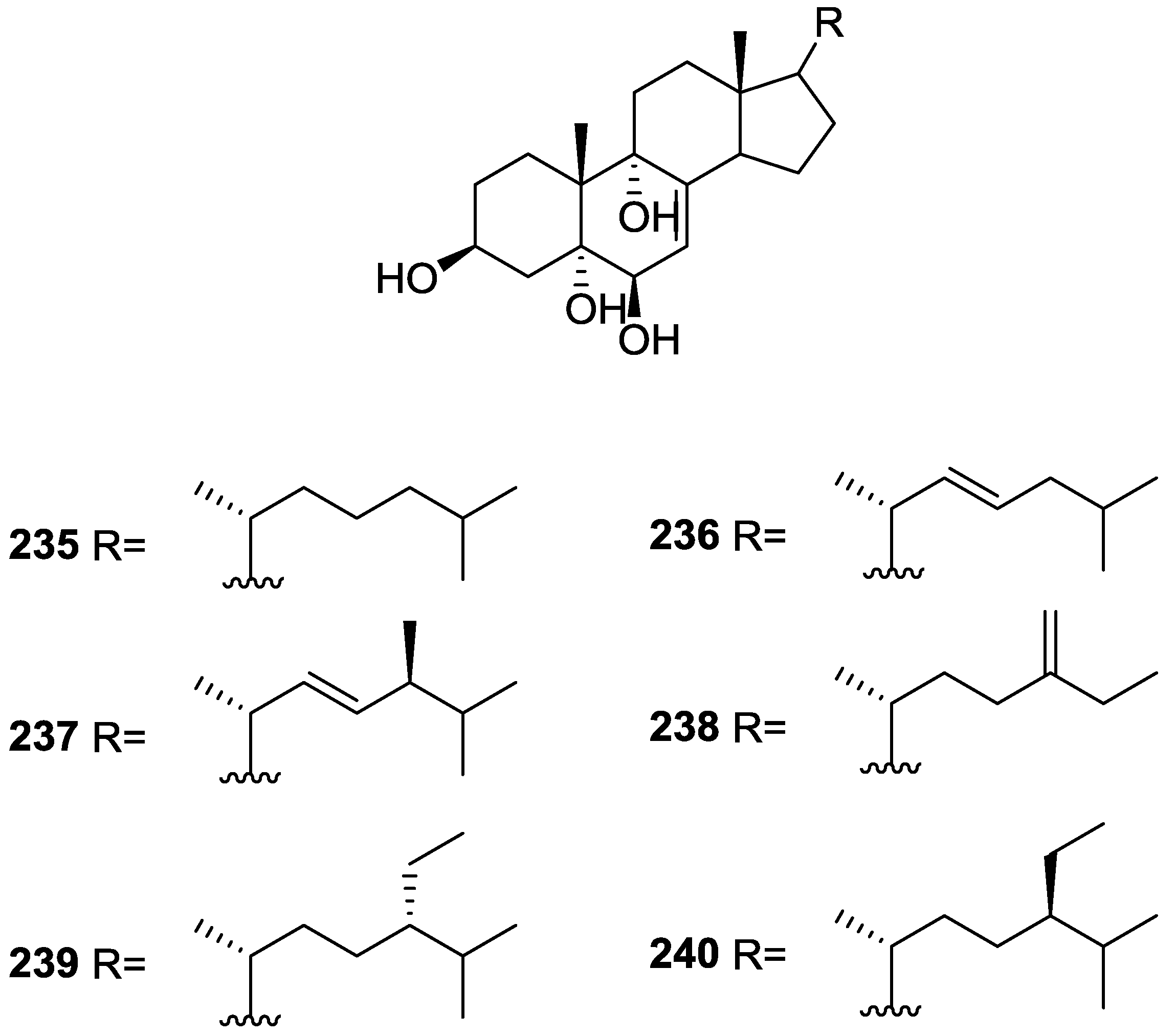
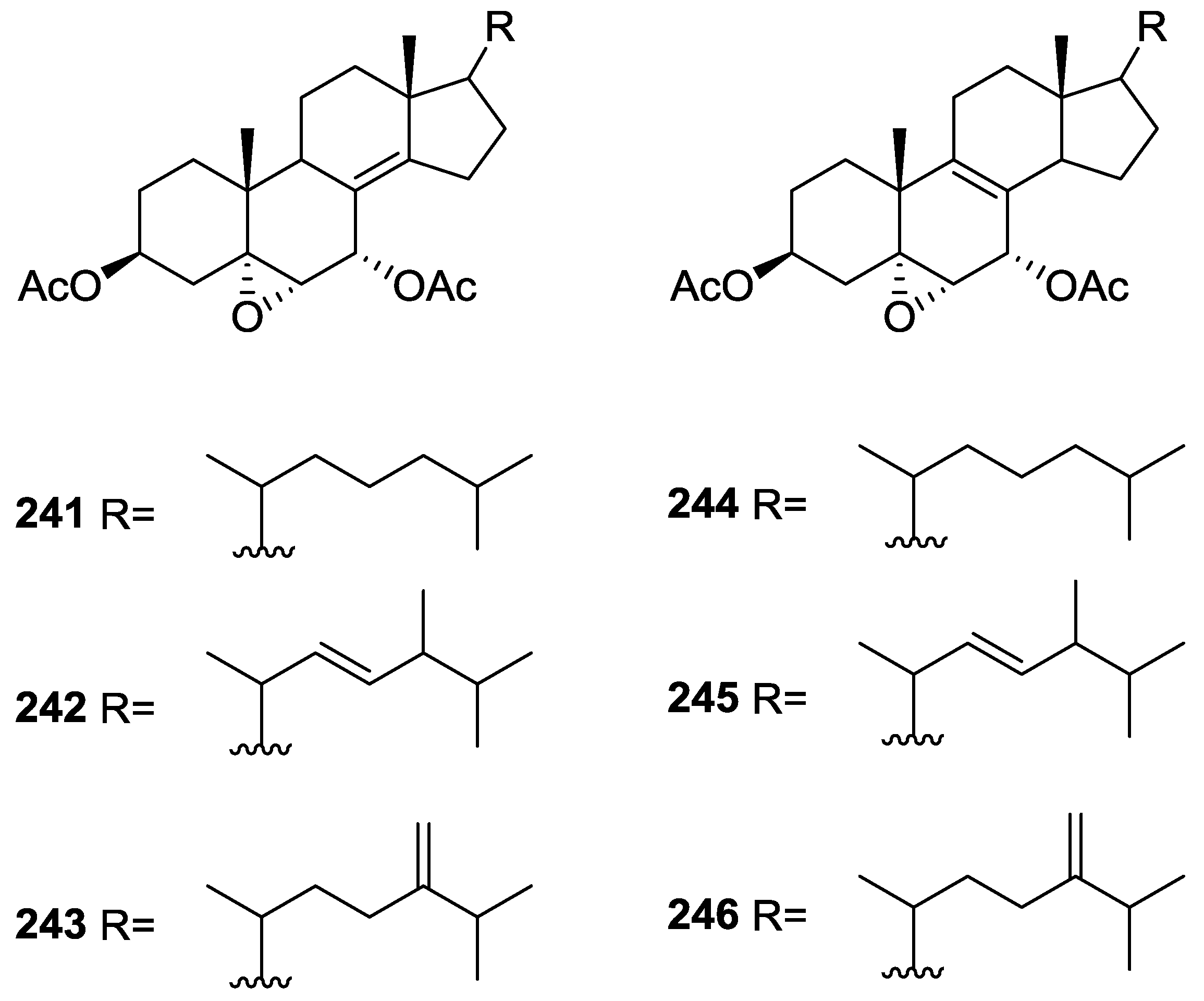
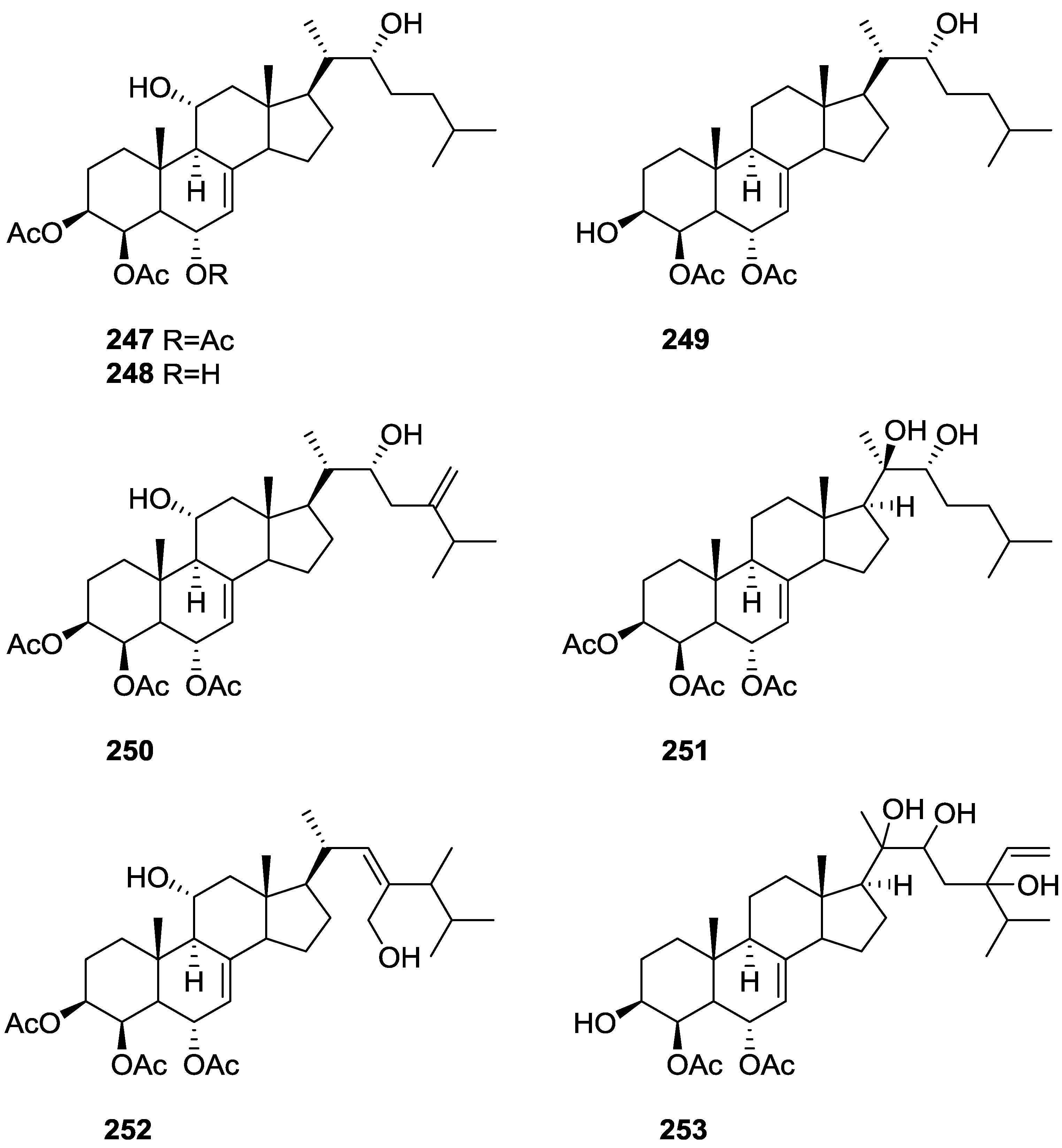




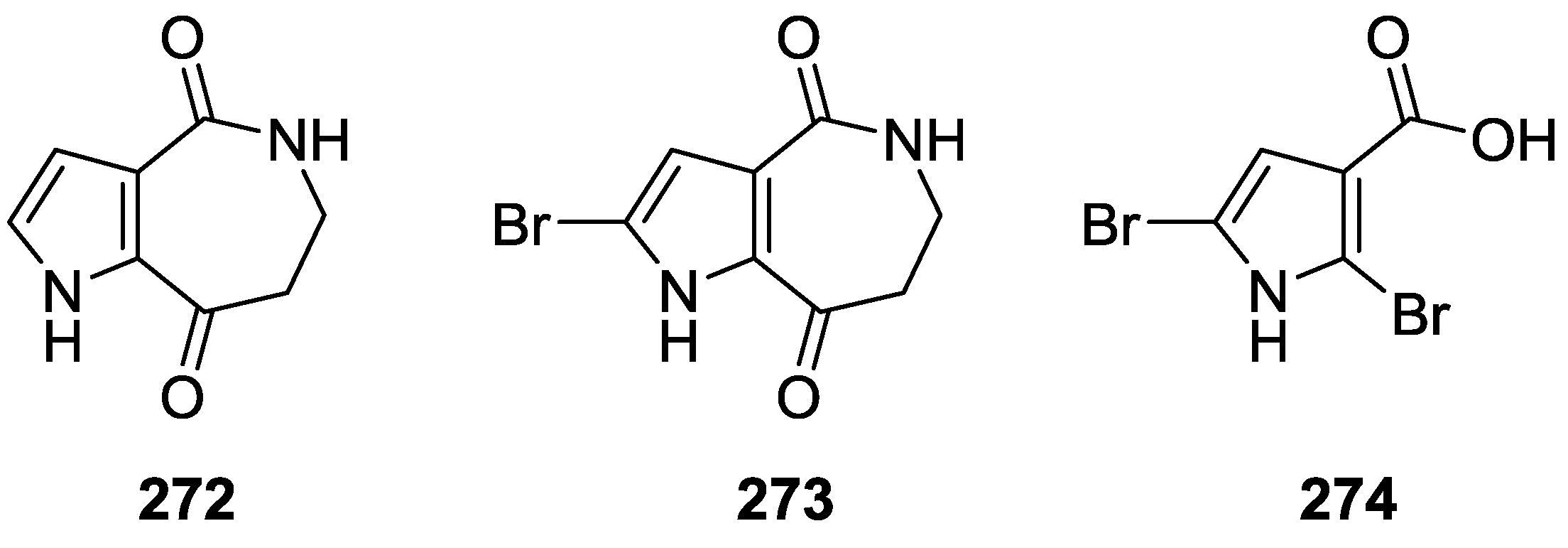
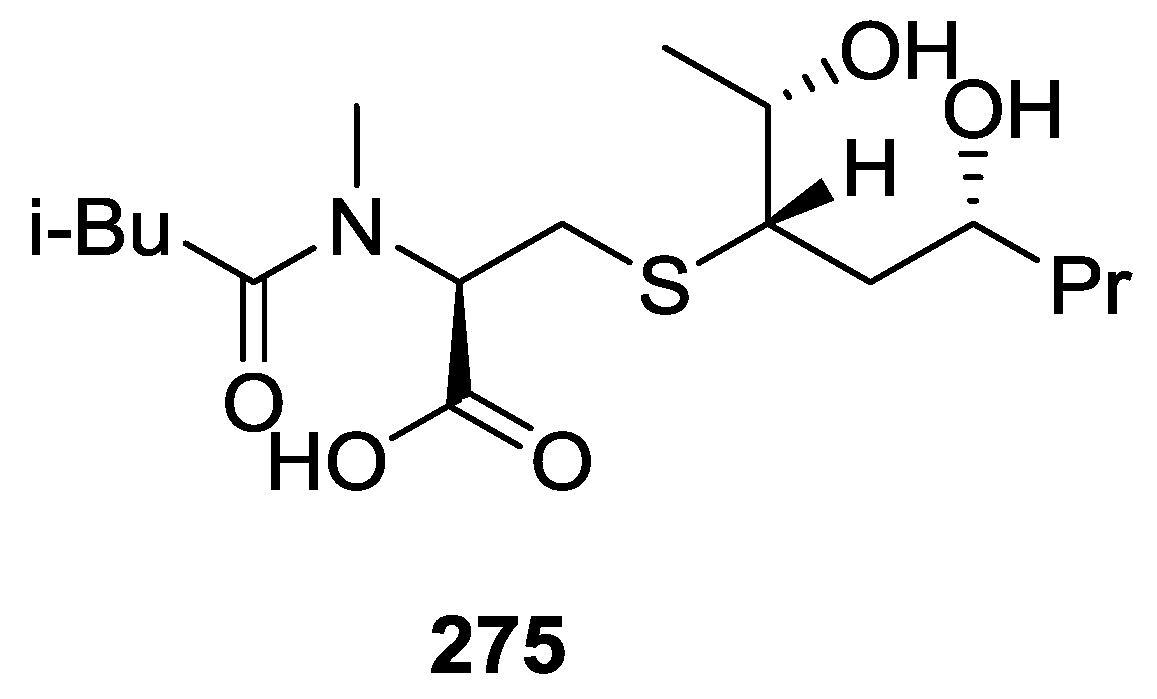
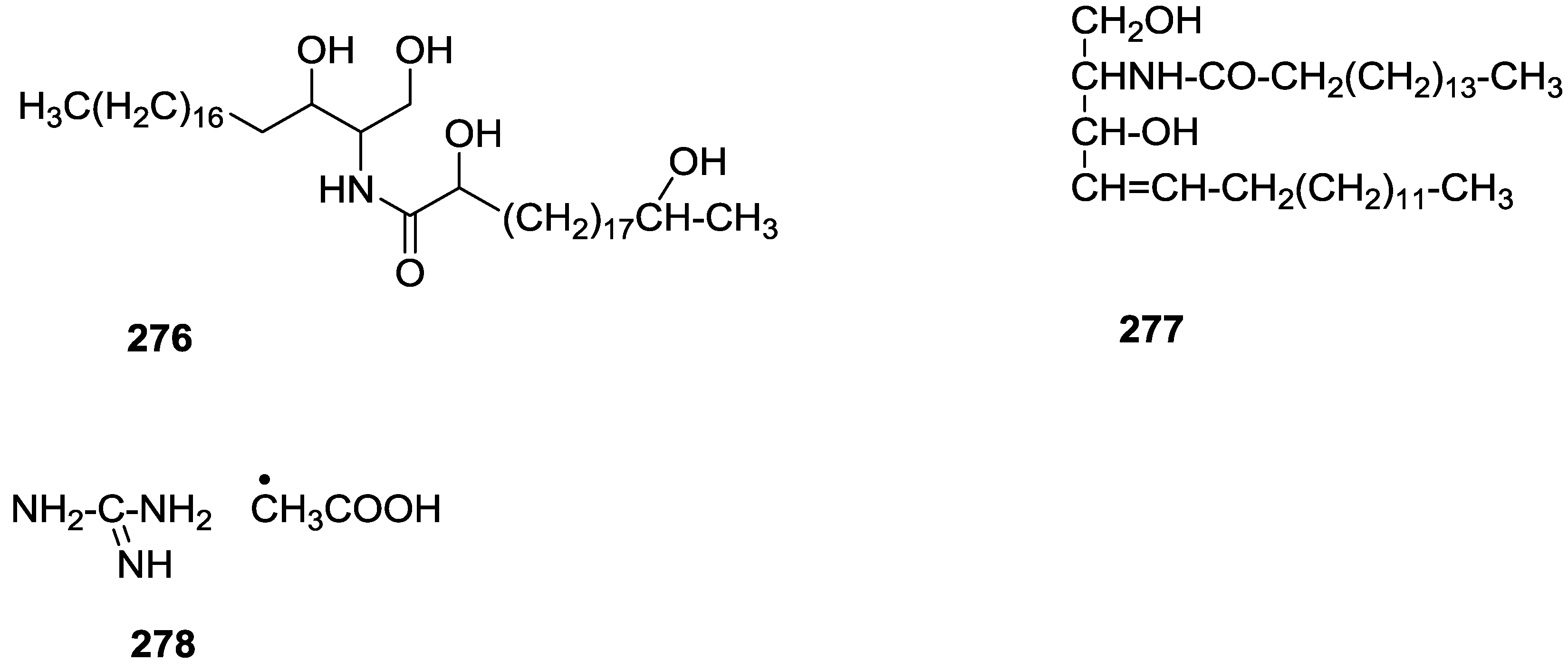
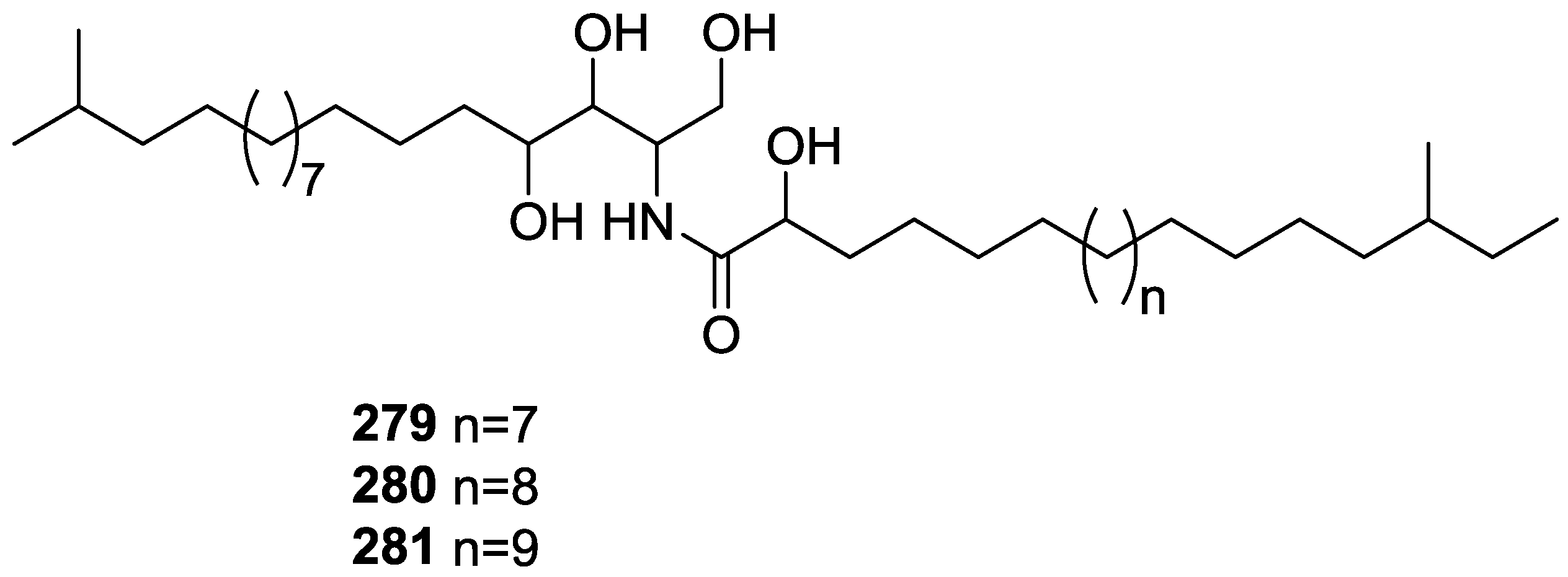
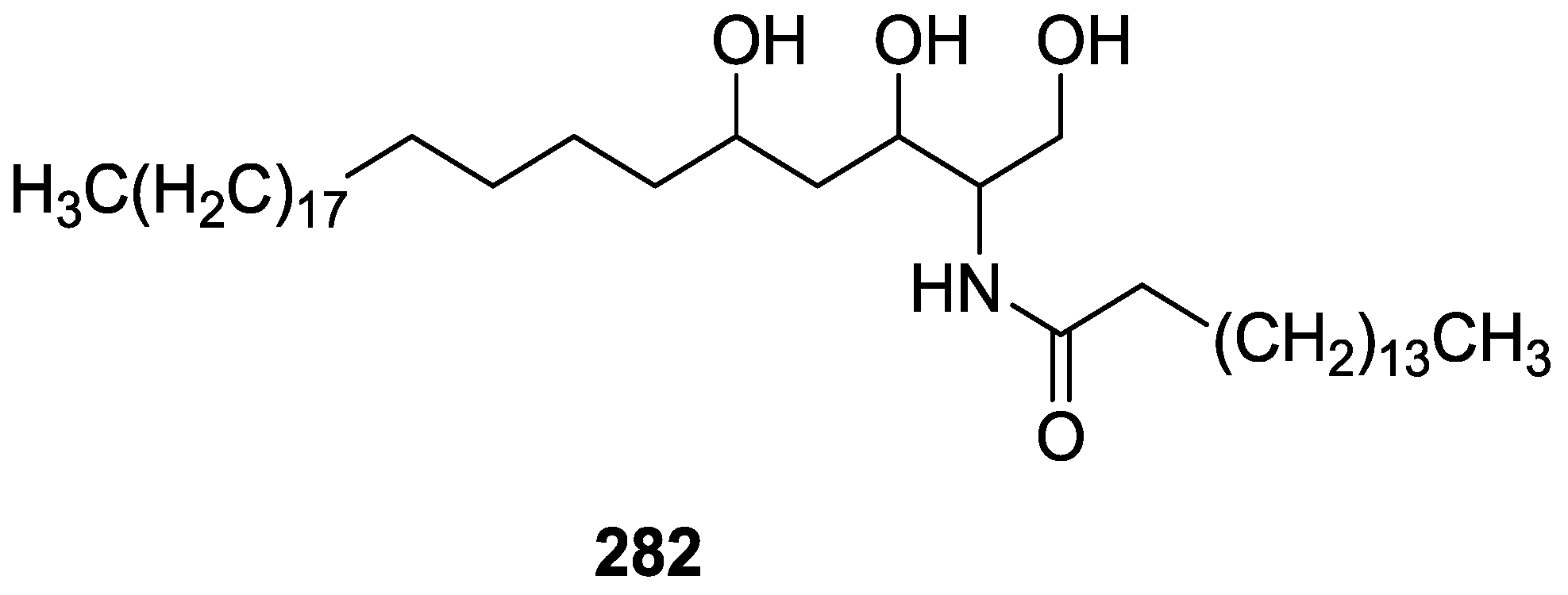
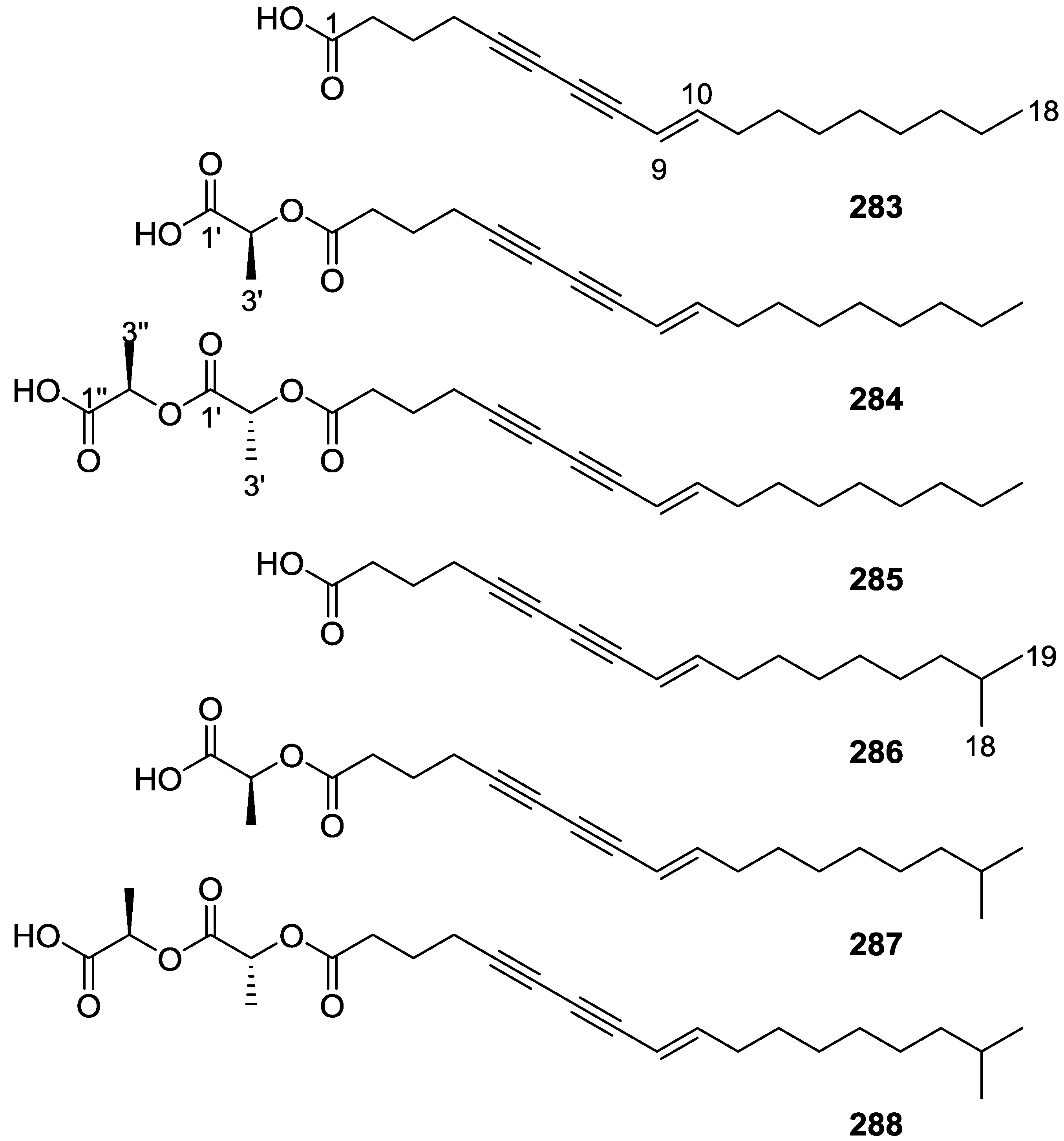
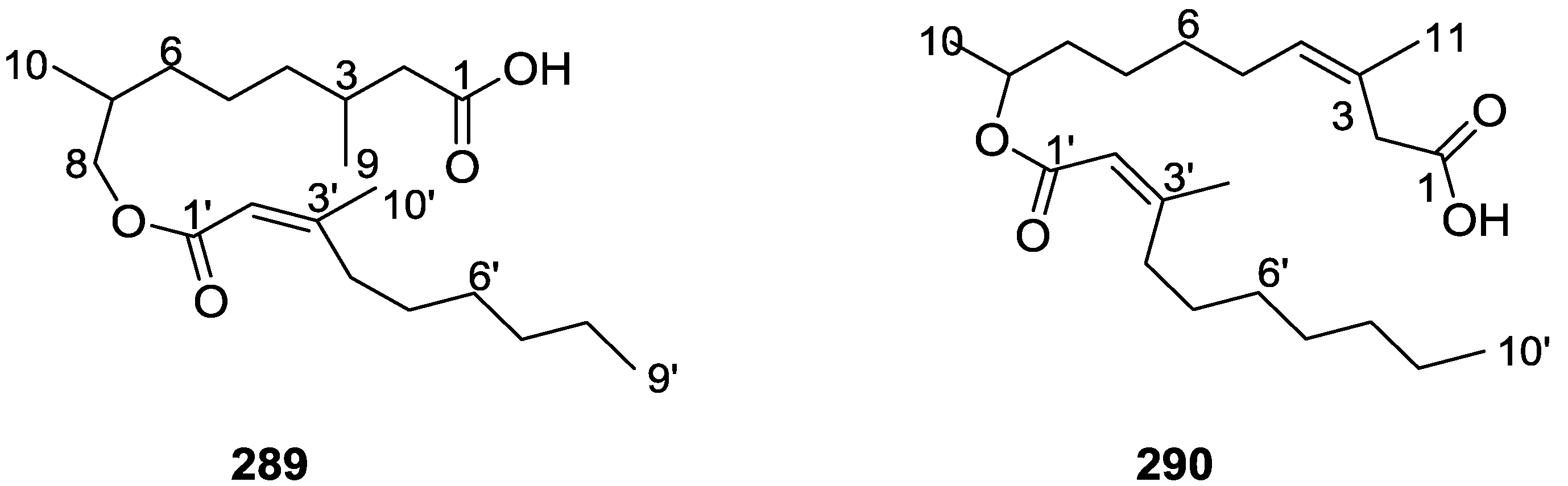
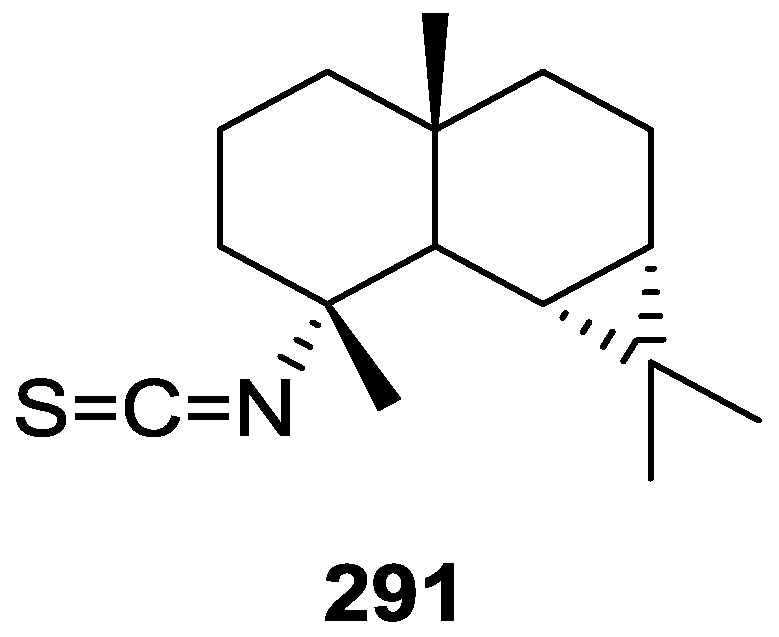
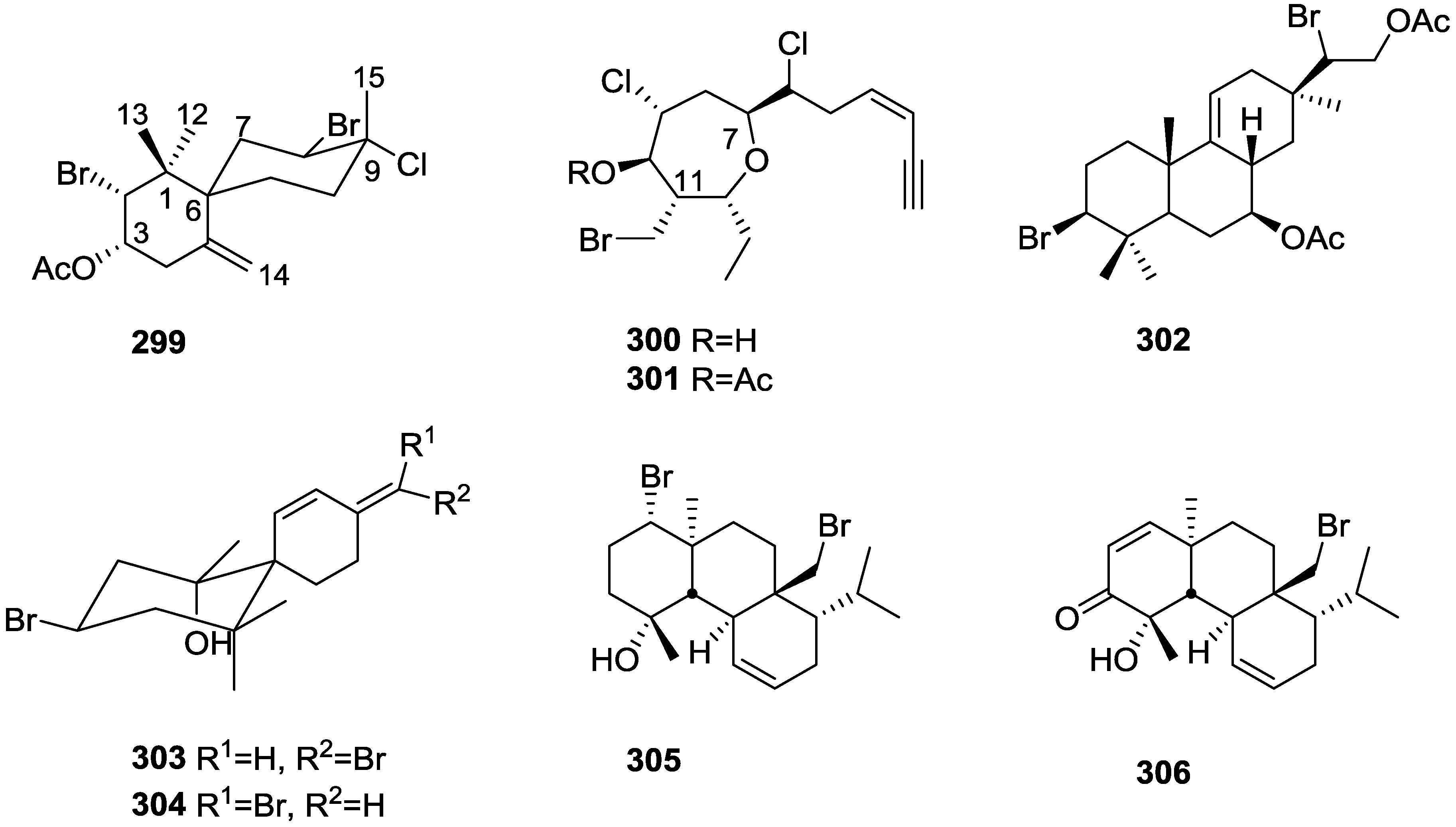

{kind=link}
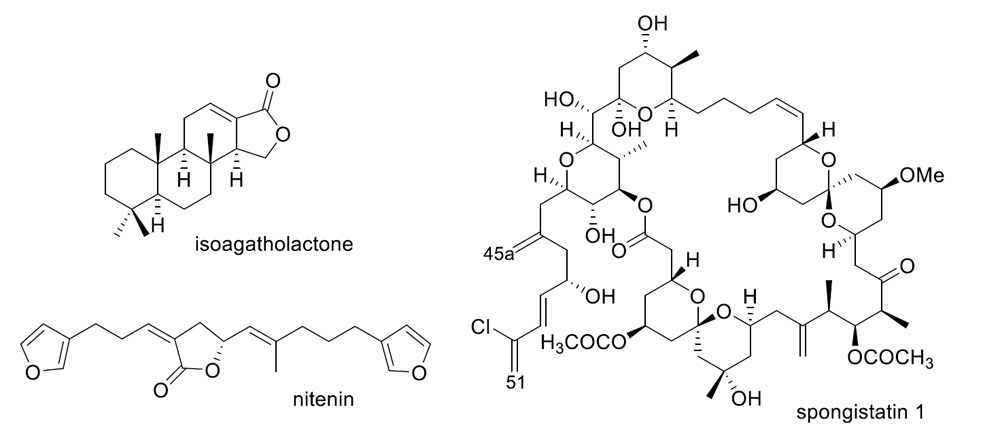
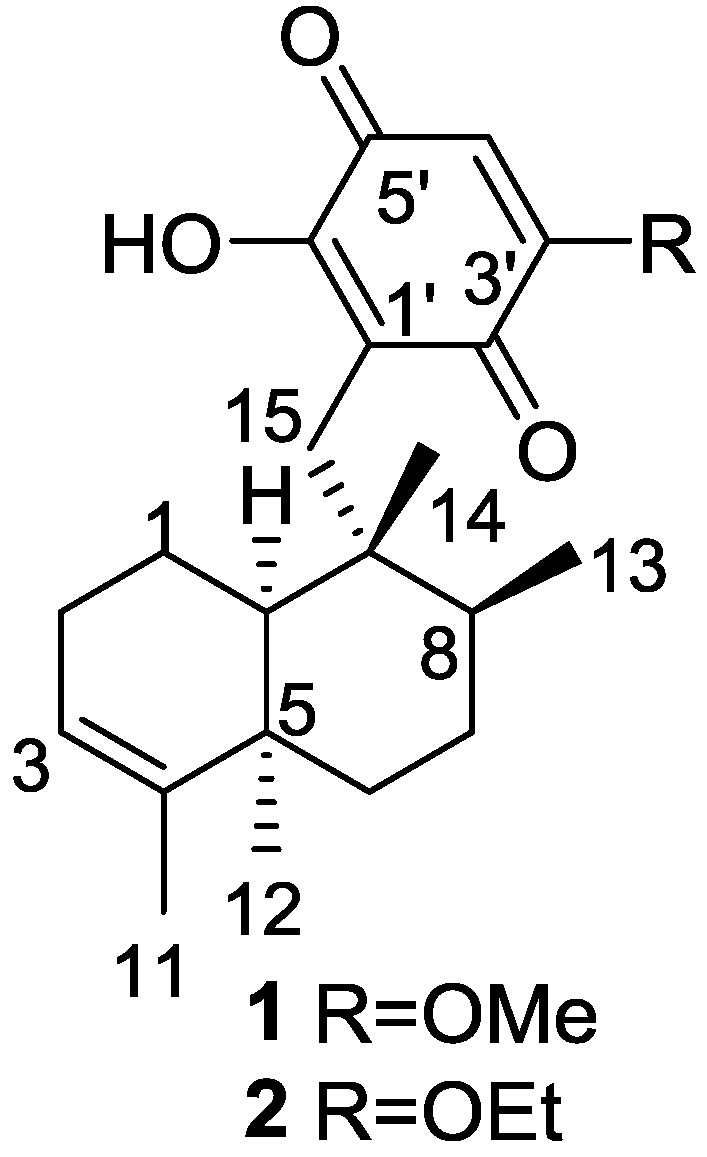{kind=link}
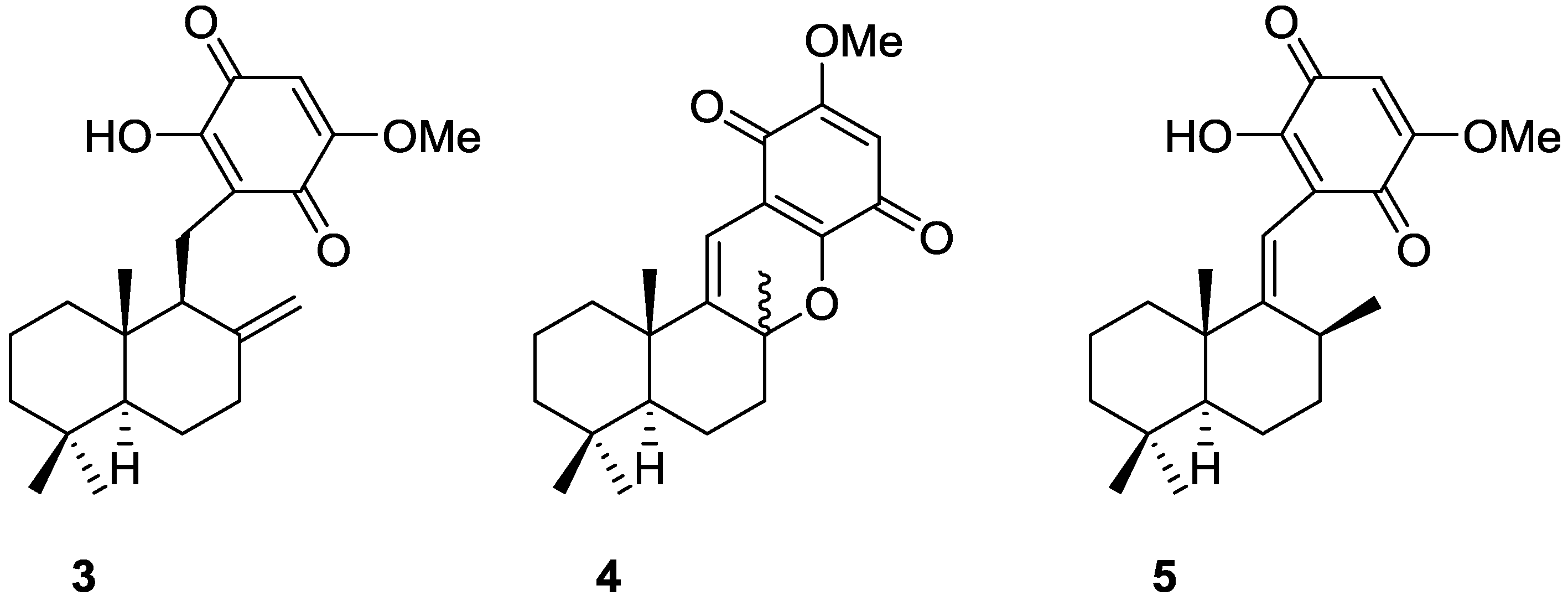{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
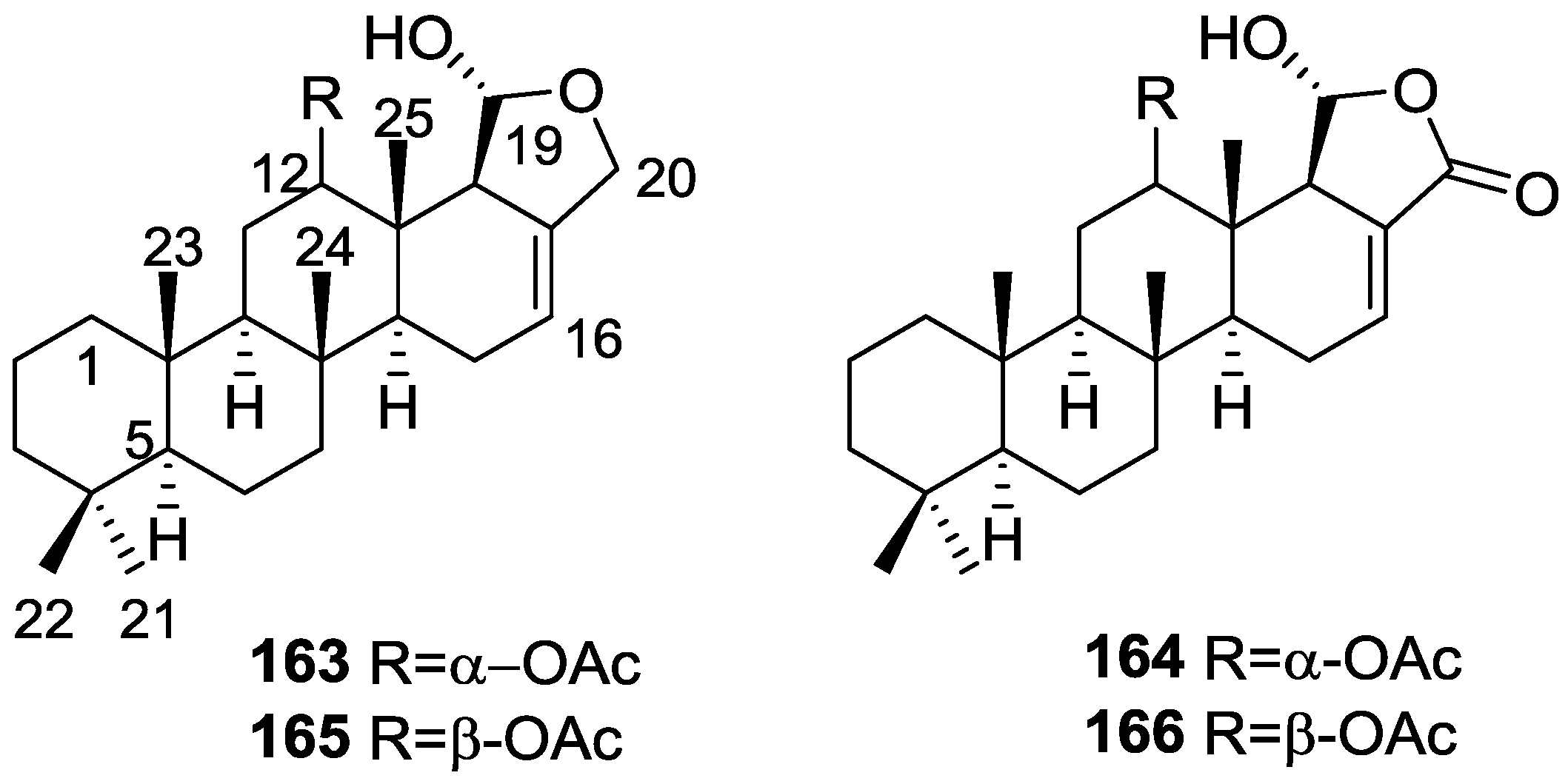{kind=link}
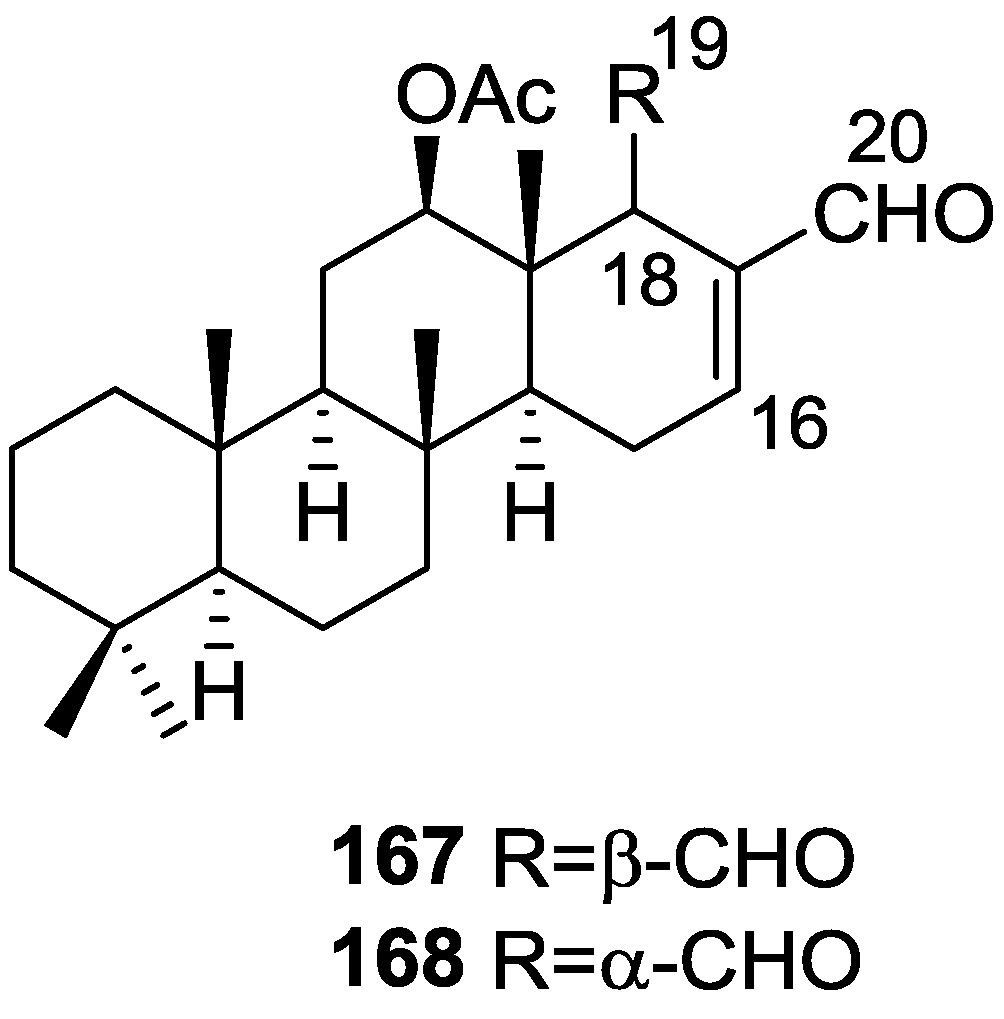{kind=link}
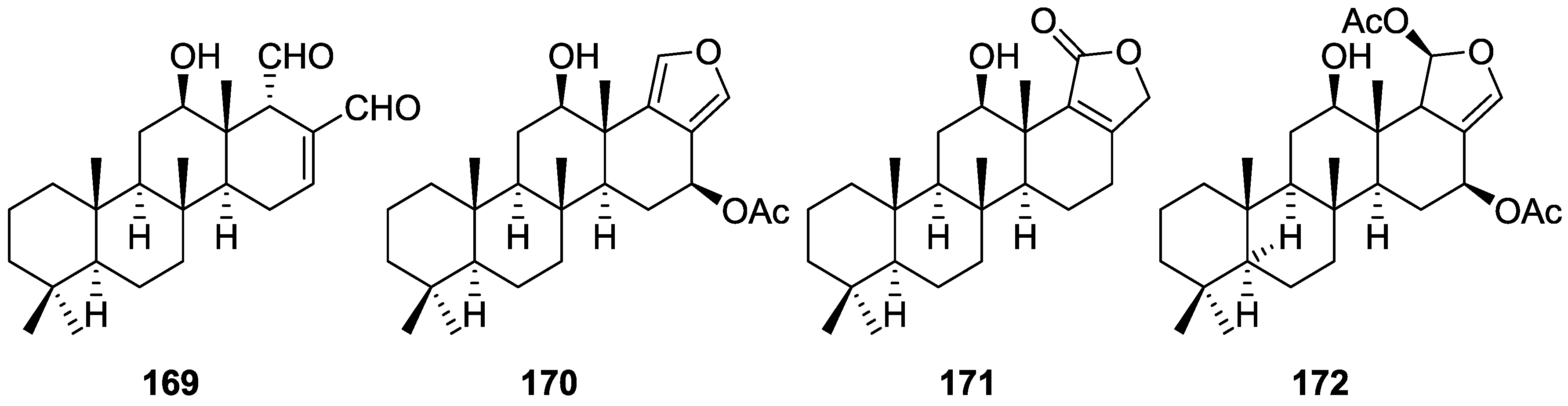{kind=link}
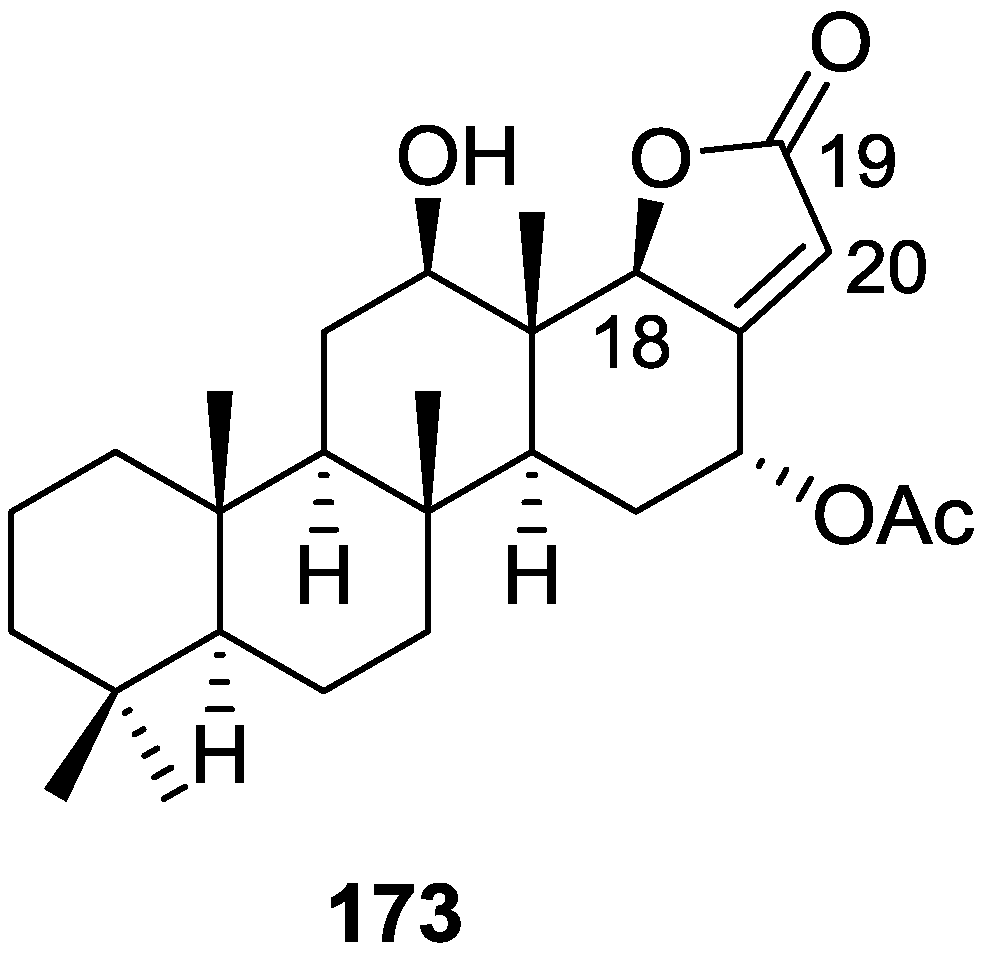{kind=link}
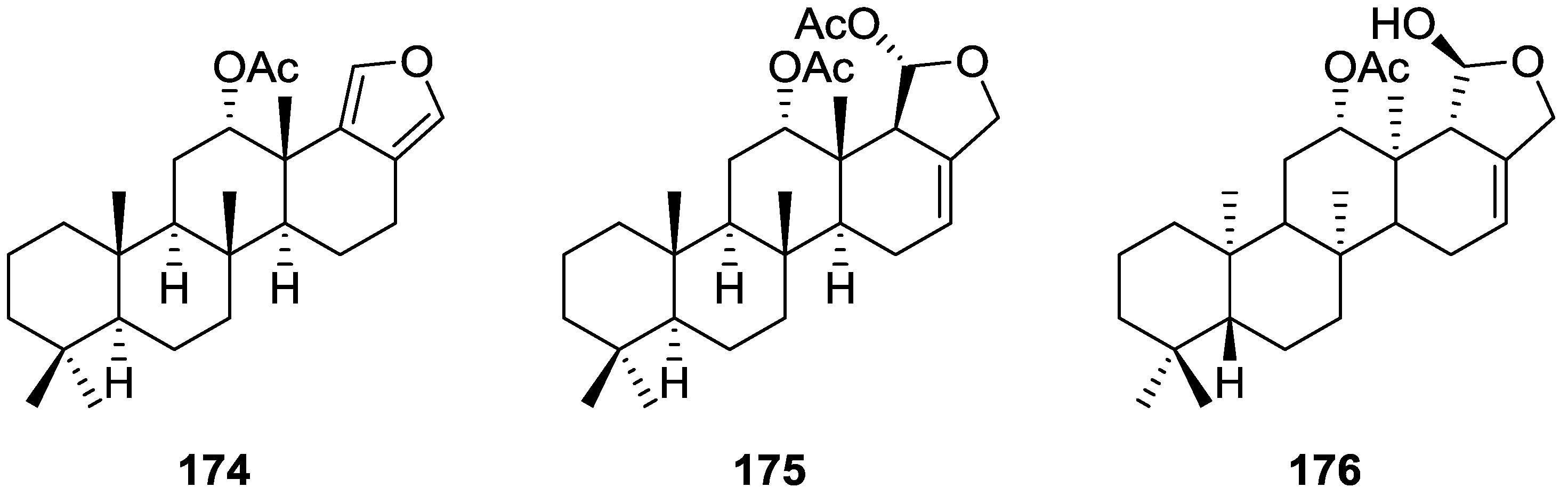{kind=link}
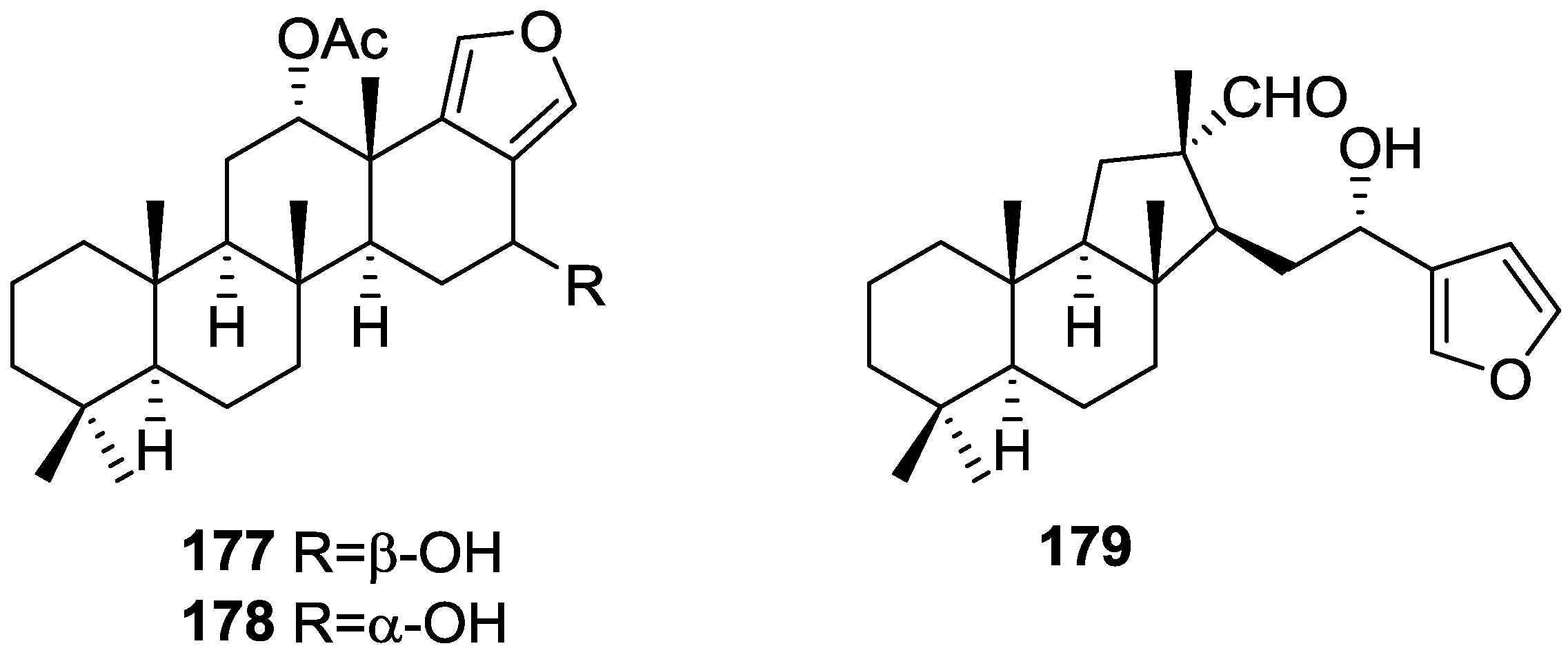{kind=link}
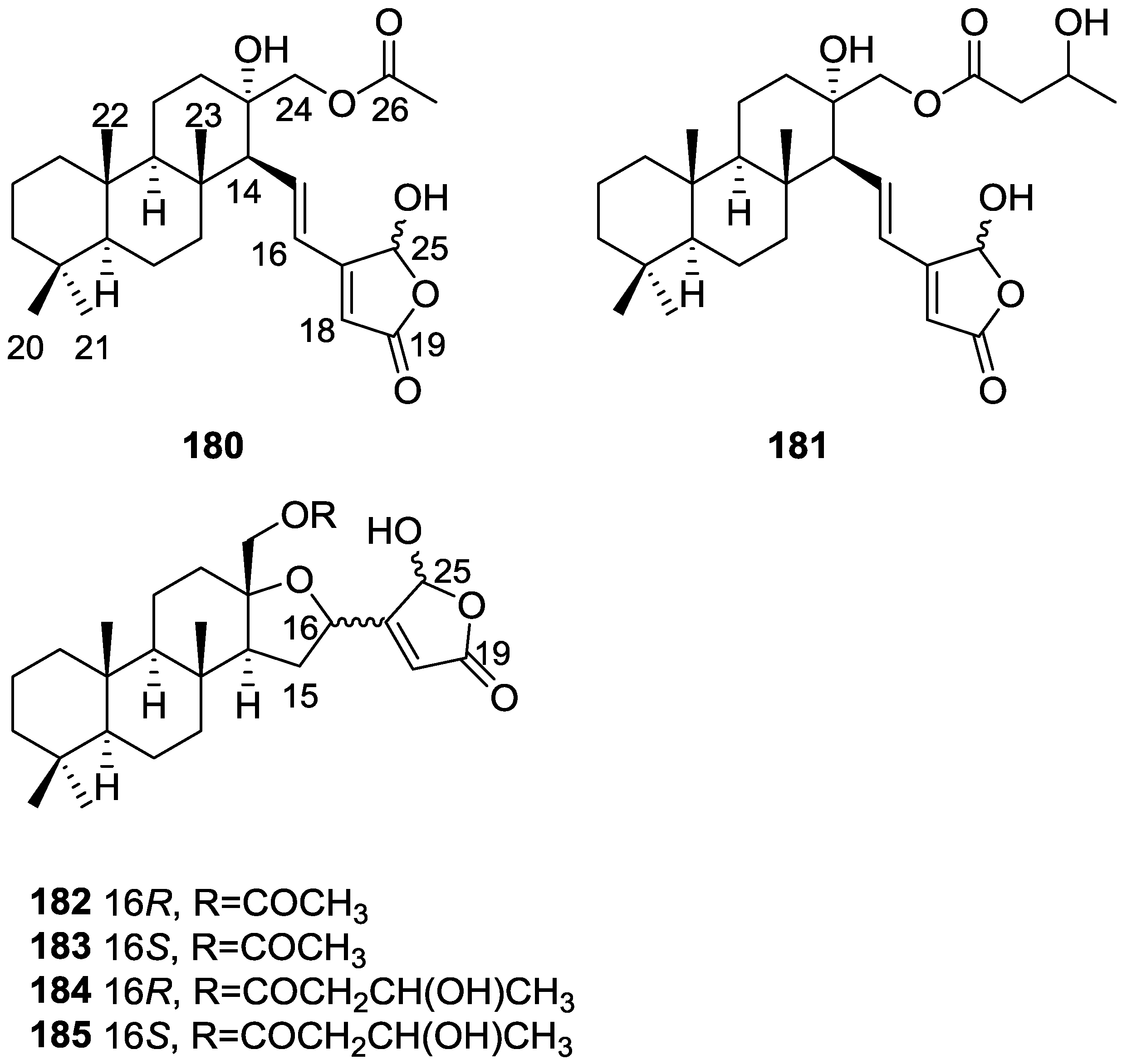{kind=link}
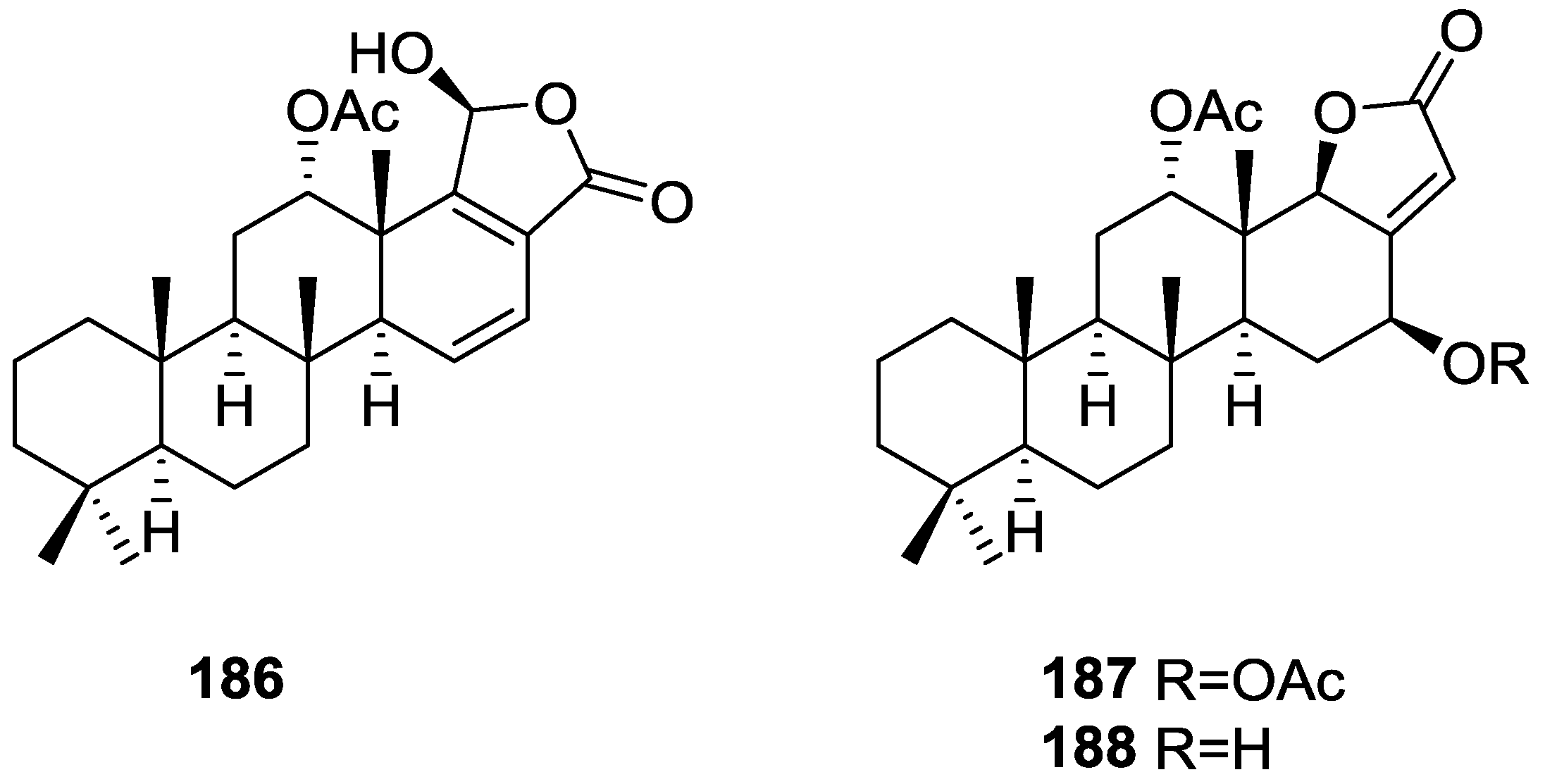{kind=link}
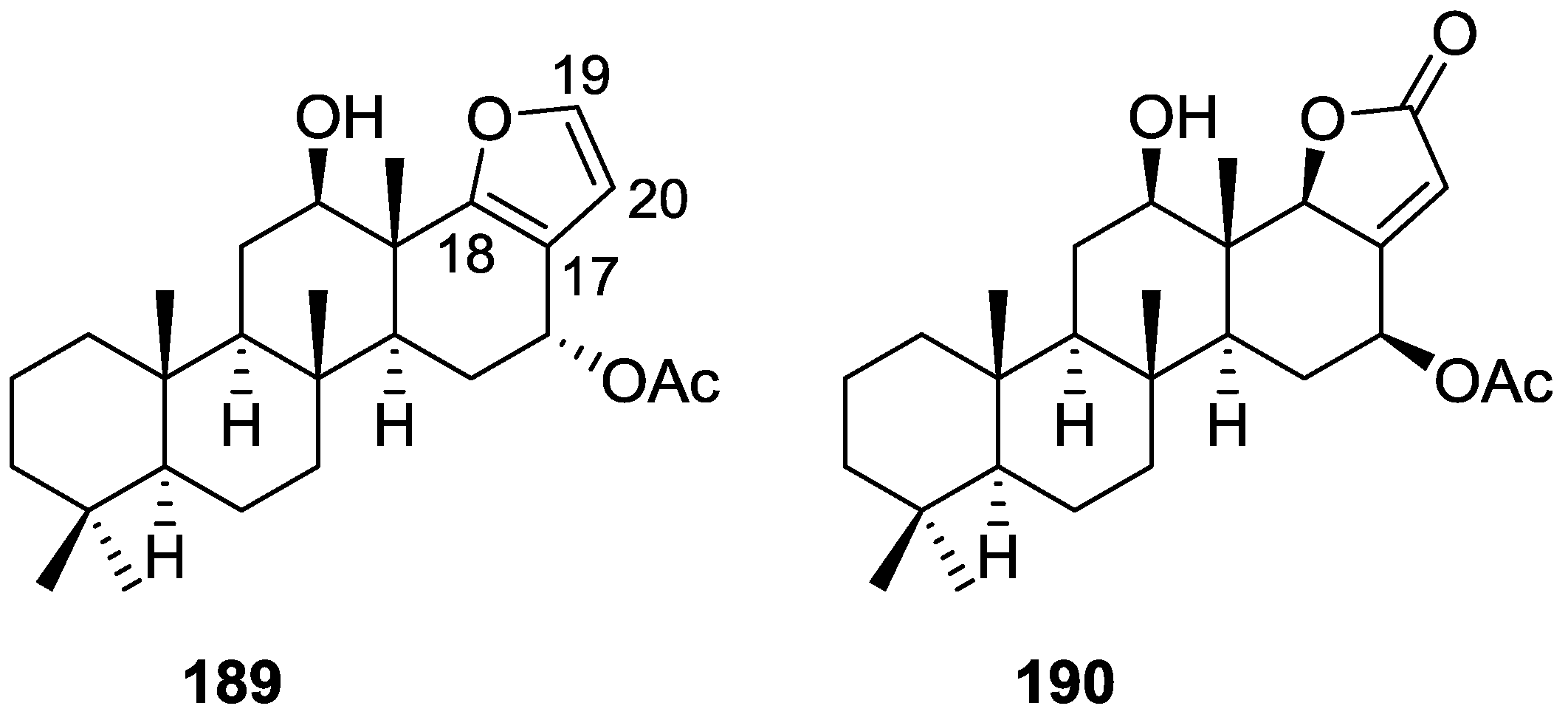{kind=link}
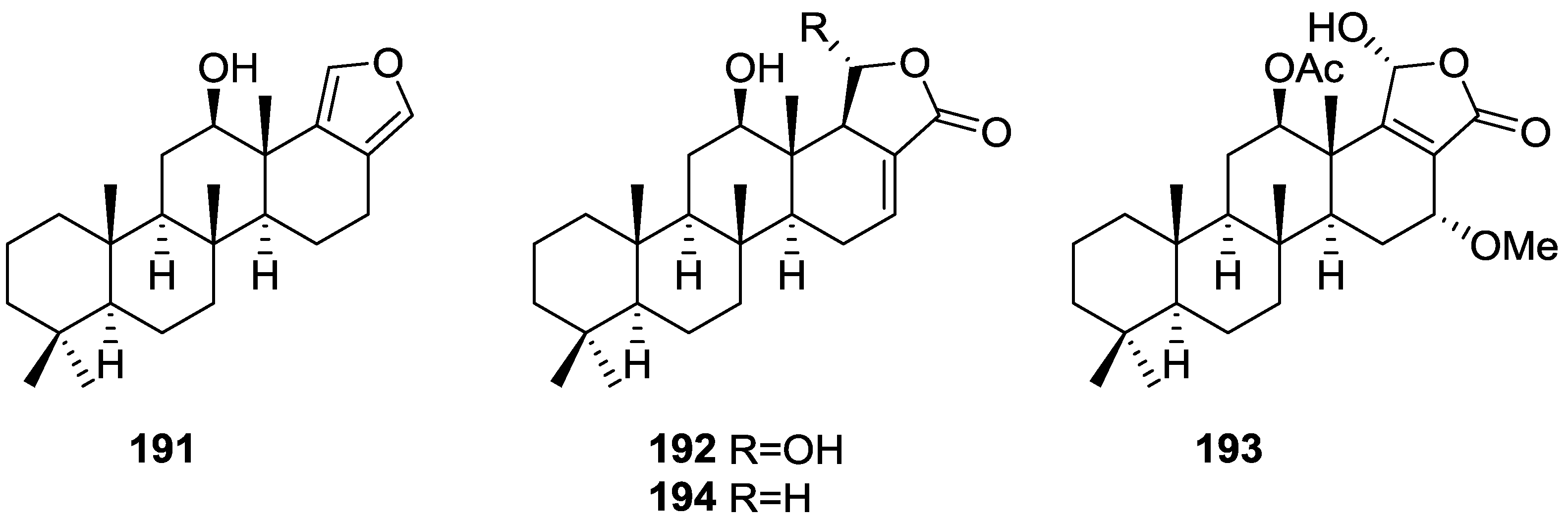{kind=link}
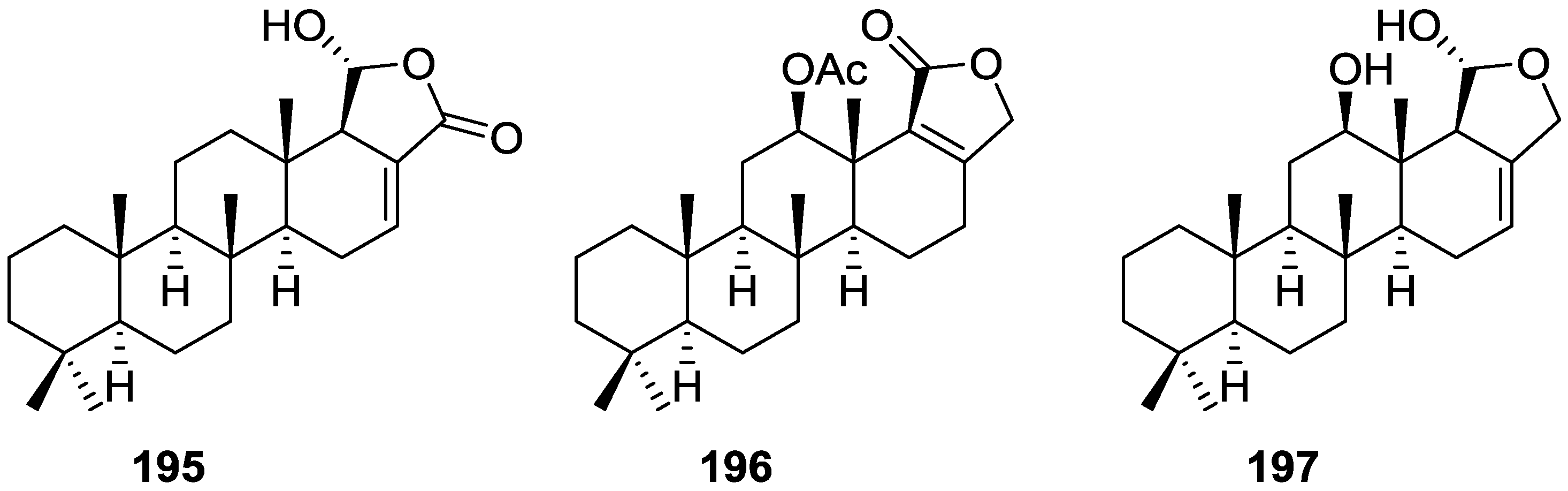{kind=link}
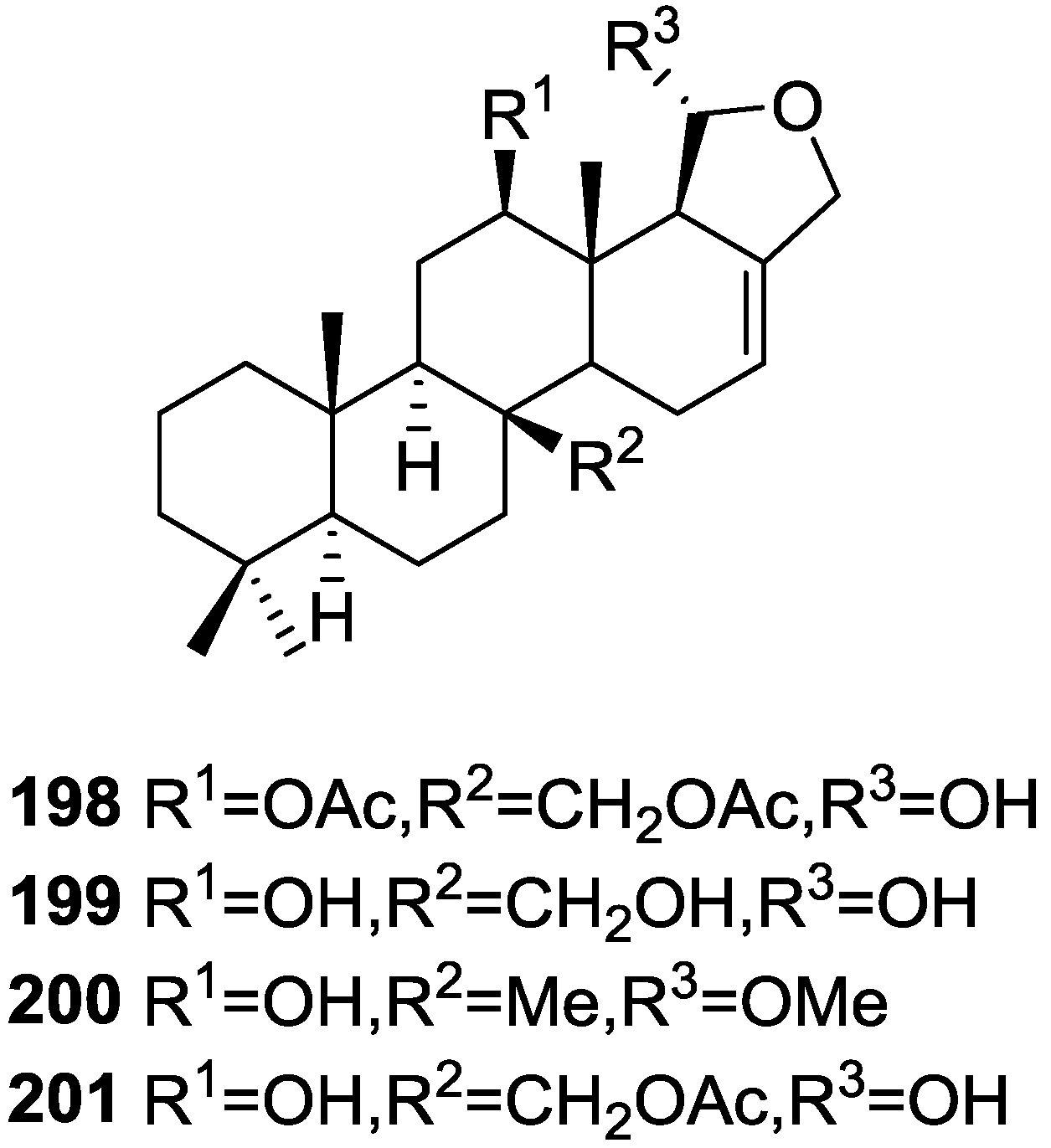{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
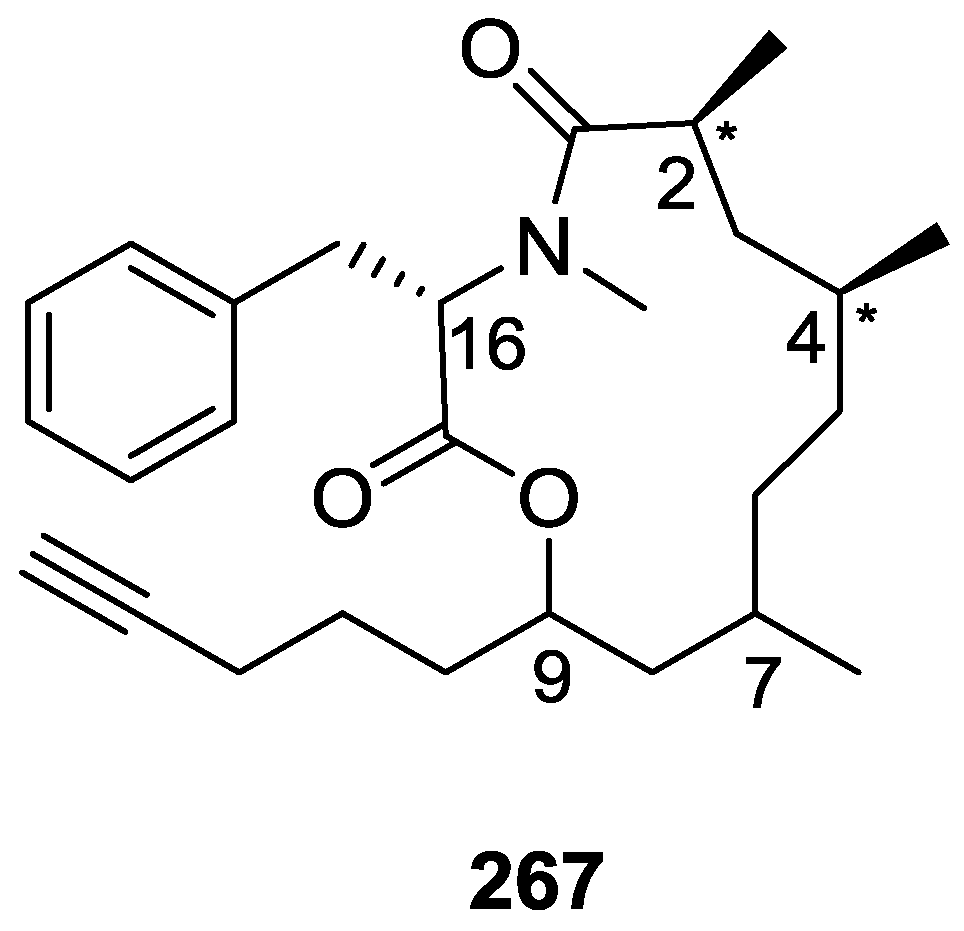{kind=link}
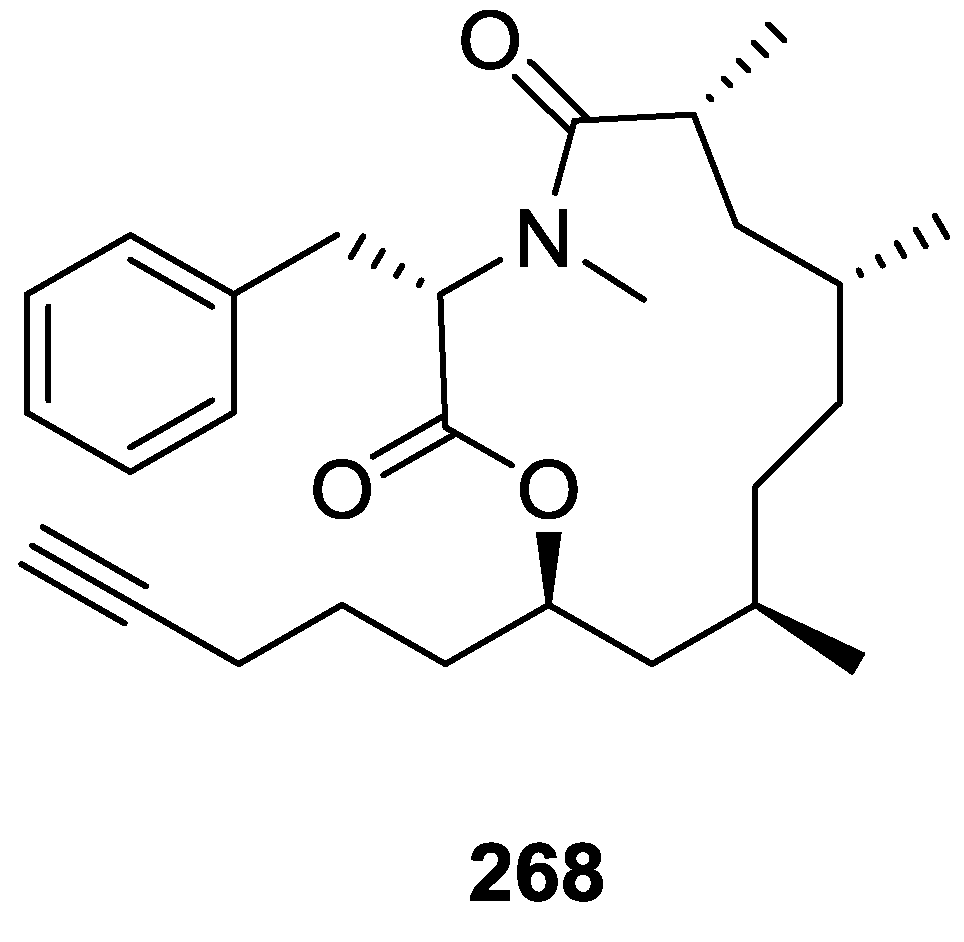{kind=link}
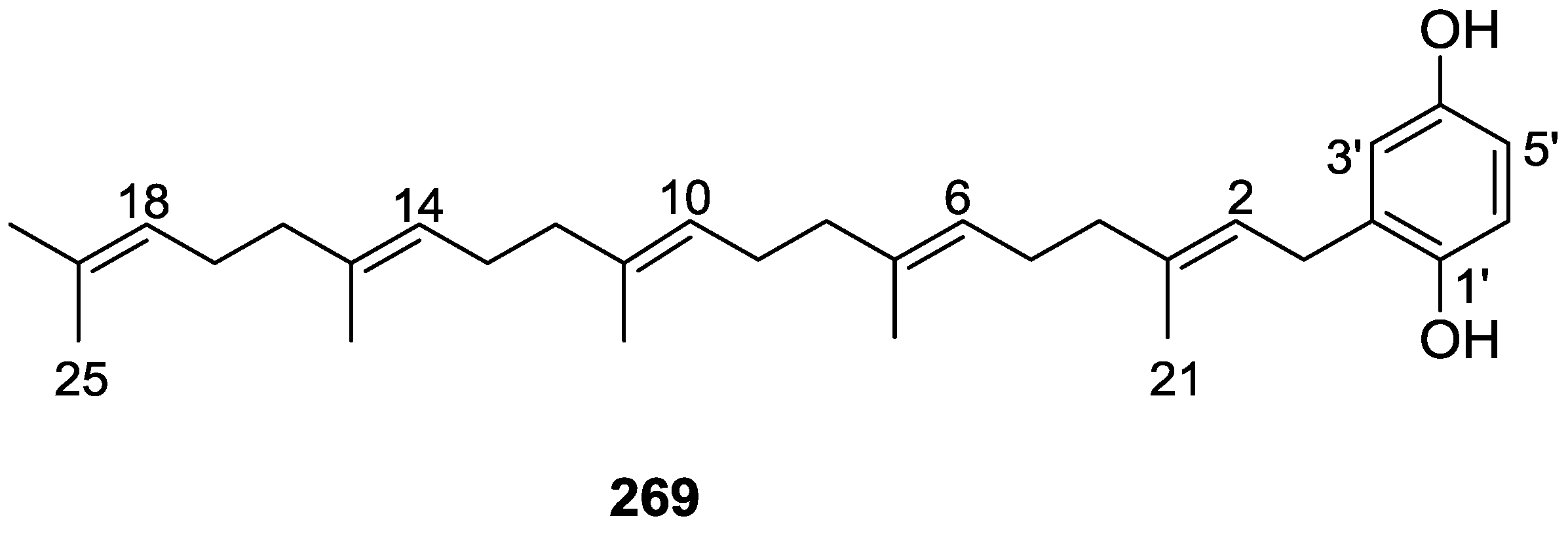{kind=link}
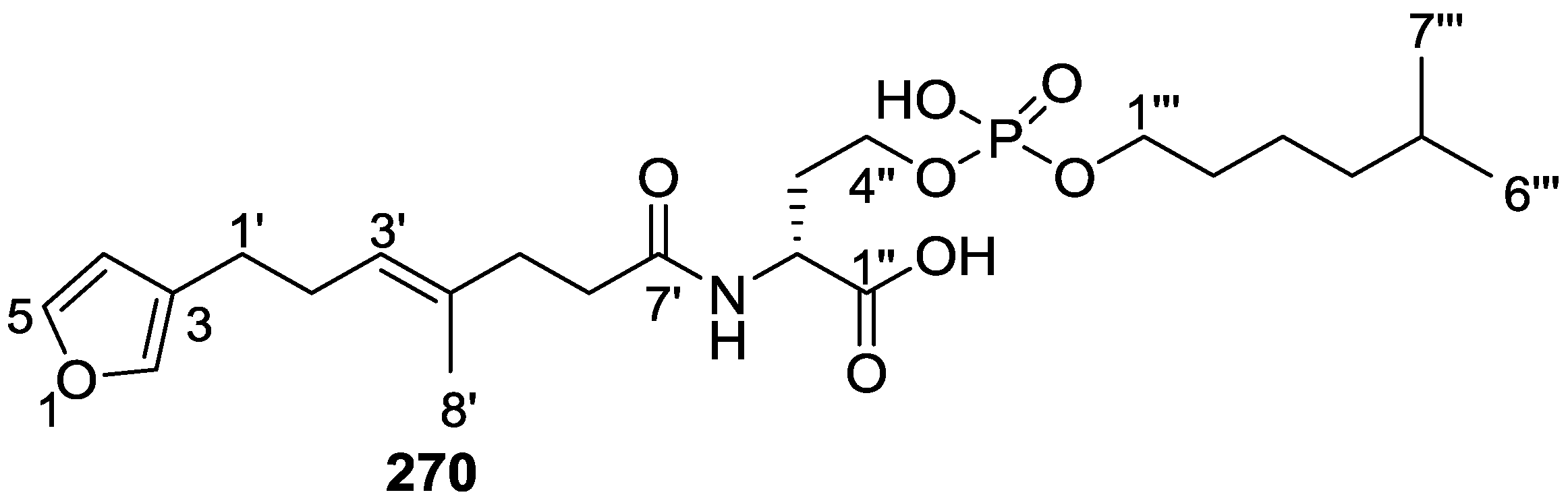{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
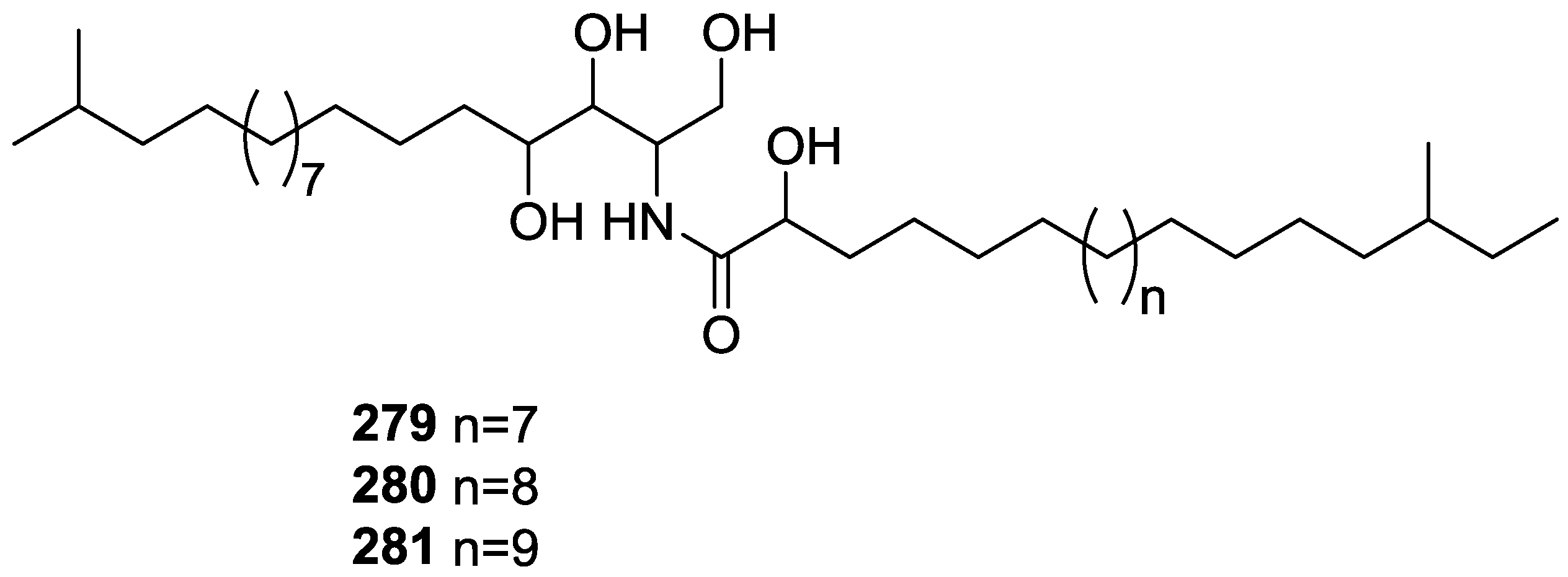{kind=link}
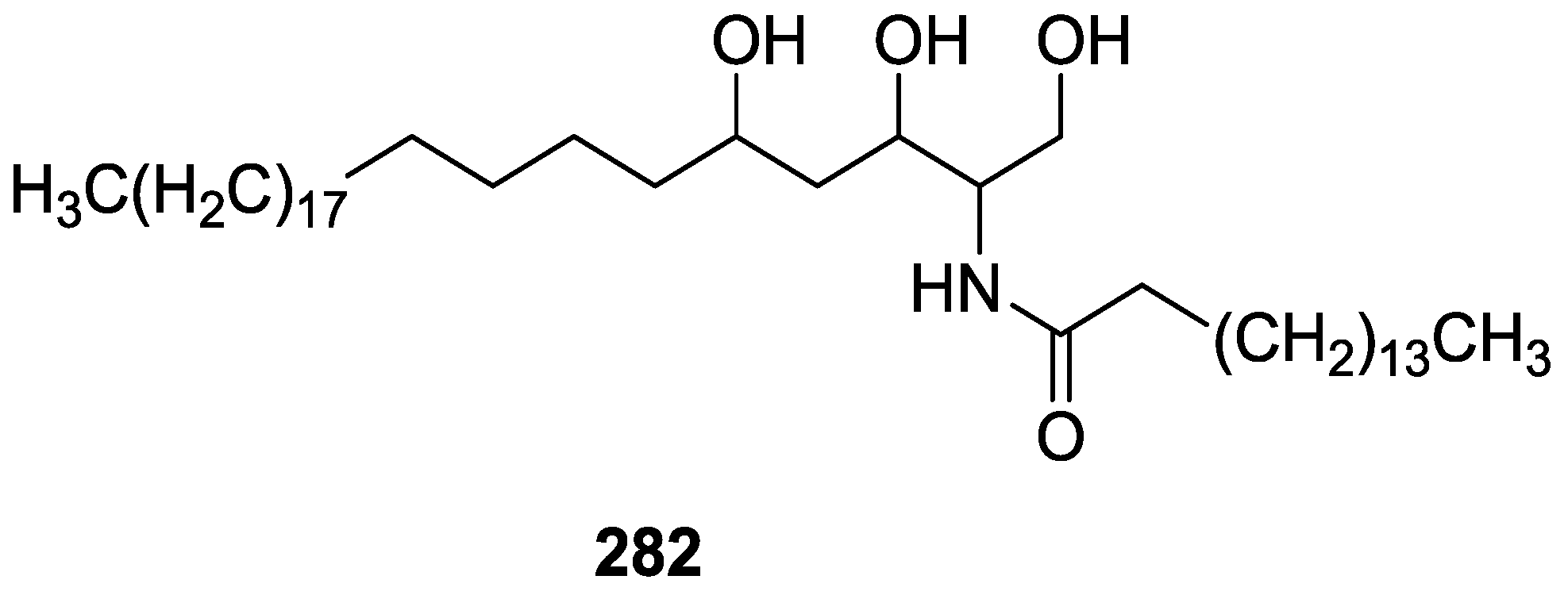{kind=link}
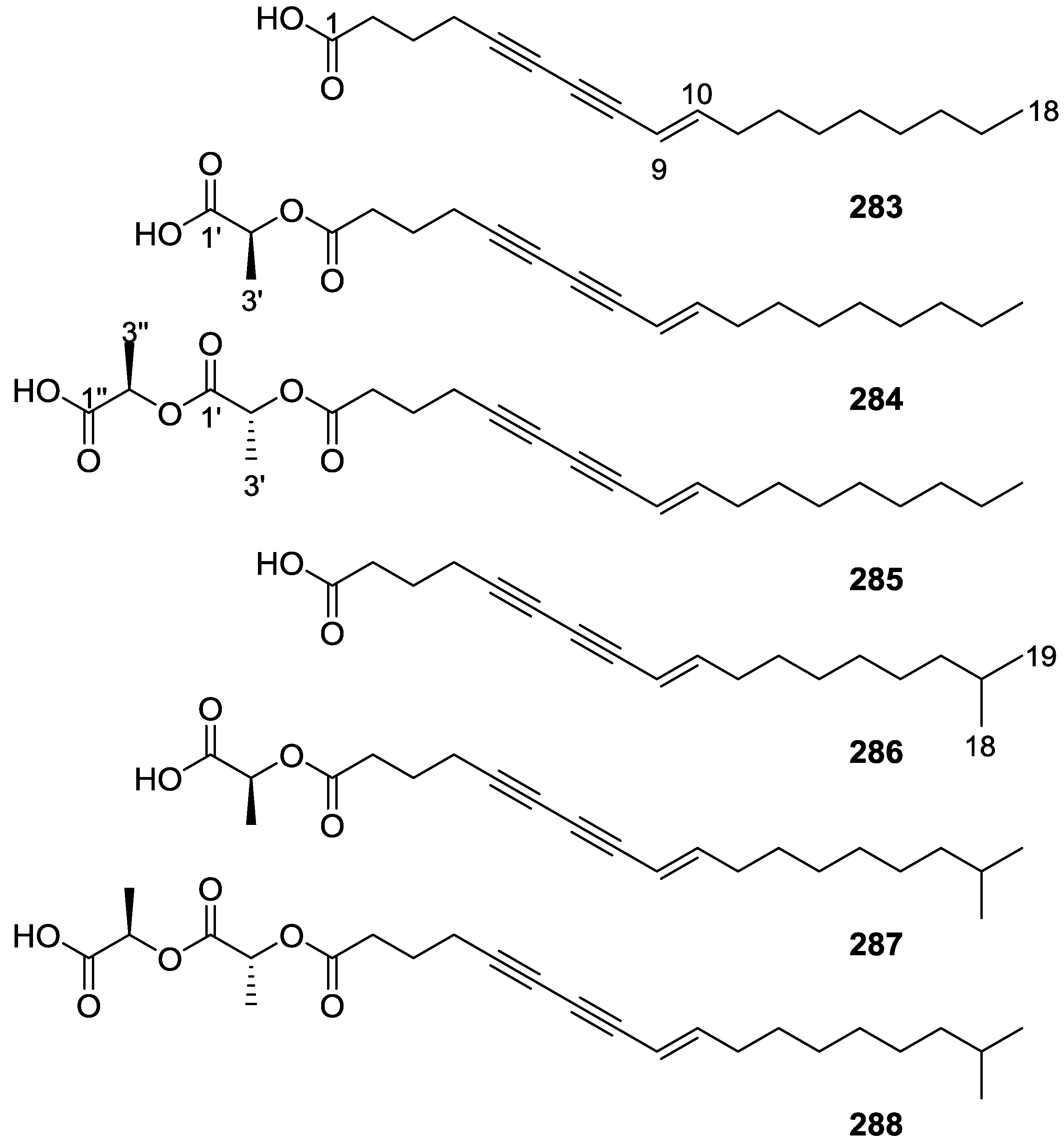{kind=link}
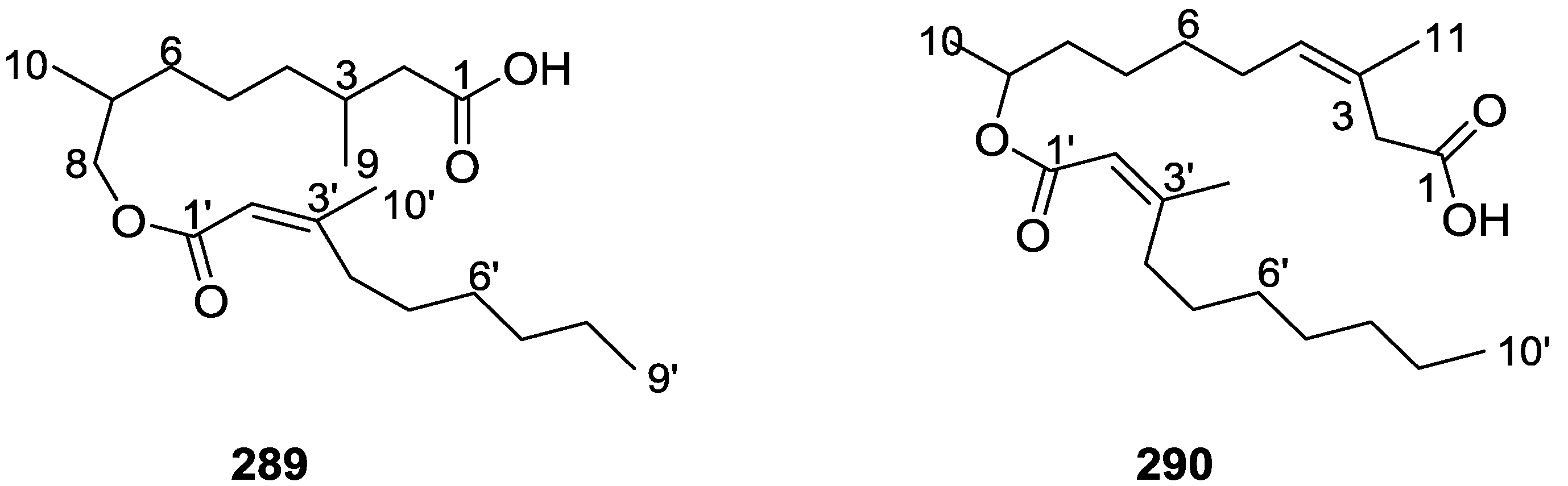{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | N. naja Venom %I (10 μM) | Pancreas %I (10 μM) | Human Synovial %I (10 μM) IC50 (μM) | RAPb + Zymosan %I (10 μM) | Bee Venom %I (10 μM) IC50 (μM) |
|---|---|---|---|---|---|
| 107 | 0.5 ± 0.5 | 18.0 ± 8.1 | 40.1 ± 7.7 d | 17.1 ± 4.6 | 33.1 ± 6.0 c |
| 108 | 0.4 ± 0.4 | 14.2 ± 5.1 | 34.6 ± 5.8 d | 17.9 ± 4.2 | 32.2 ± 6.0 c |
| 109 | 3.1 ± 2.2 | 9.1 ± 3.5 | 40.4 ± 5.7 d | 30.9 ± 5.3 c | 36.2 ± 5.4 d |
| 110 | 0.0 ± 0.0 | 7.6 ± 4.0 | 48.2 ± 3.8 d | 19.6 ± 5.4 | 37.6 ± 6.5 c |
| manoalide | 17.0 ± 1.7 c | 32.3 ± 2.7 d | 93.2 ± 0.2 d 3.9 | 38.4 ± 0.5 d | 62.5 ± 3.8 d 7.5 |
| IC50 (μg/mL) | ||||
|---|---|---|---|---|
| Compound | L1210 | HeLa | A549 | KB |
| 165 | 2.1 | 22.5 | 29.4 | 16.2 |
| 166 | 13.2 | 26.0 | 23.7 | 18.5 |
| 191 | >50 | 19.5 | >50 | >50 |
| 192 | 2.3 | 15.0 | 14.8 | 14.3 |
| 193 | 2.2 | 5.3 | 5.3 | 15.6 |
| 194 | 1.6 | 16.5 | 16.5 | 17.1 |
| Mitomycin C | 0.020 | 0.015 | 0.020 | 0.015 |
| Compound | Inhibition FXR Transactivation IC50 (μM) | Cytotoxicity IC50 (μM) (CV-1 cell) |
|---|---|---|
| 165 | 81.1 | 98.5 |
| 198 | 8.1 | 32.7 |
| 199 | 64.5 | >100 |
| 200 | 24.8 | 86.9 |
| 201 | 25.3 | 29.2 |
| Z-Guggulsterone | 10.0 | Not determined |
| Compound | Inhibition FXR Transactivation IC50 (μM) | Cytotoxicity IC50 (μM) (CV-1 Cell) |
|---|---|---|
| 166 | 60.4 | 75.1 |
| 192 | 75.0 | 77.2 |
| 194 | 31.6 | 41.4 |
| 202 | 2.4 | 49.4 |
| 203 | >100 | >100 |
| 204 | 24.0 | 48.0 |
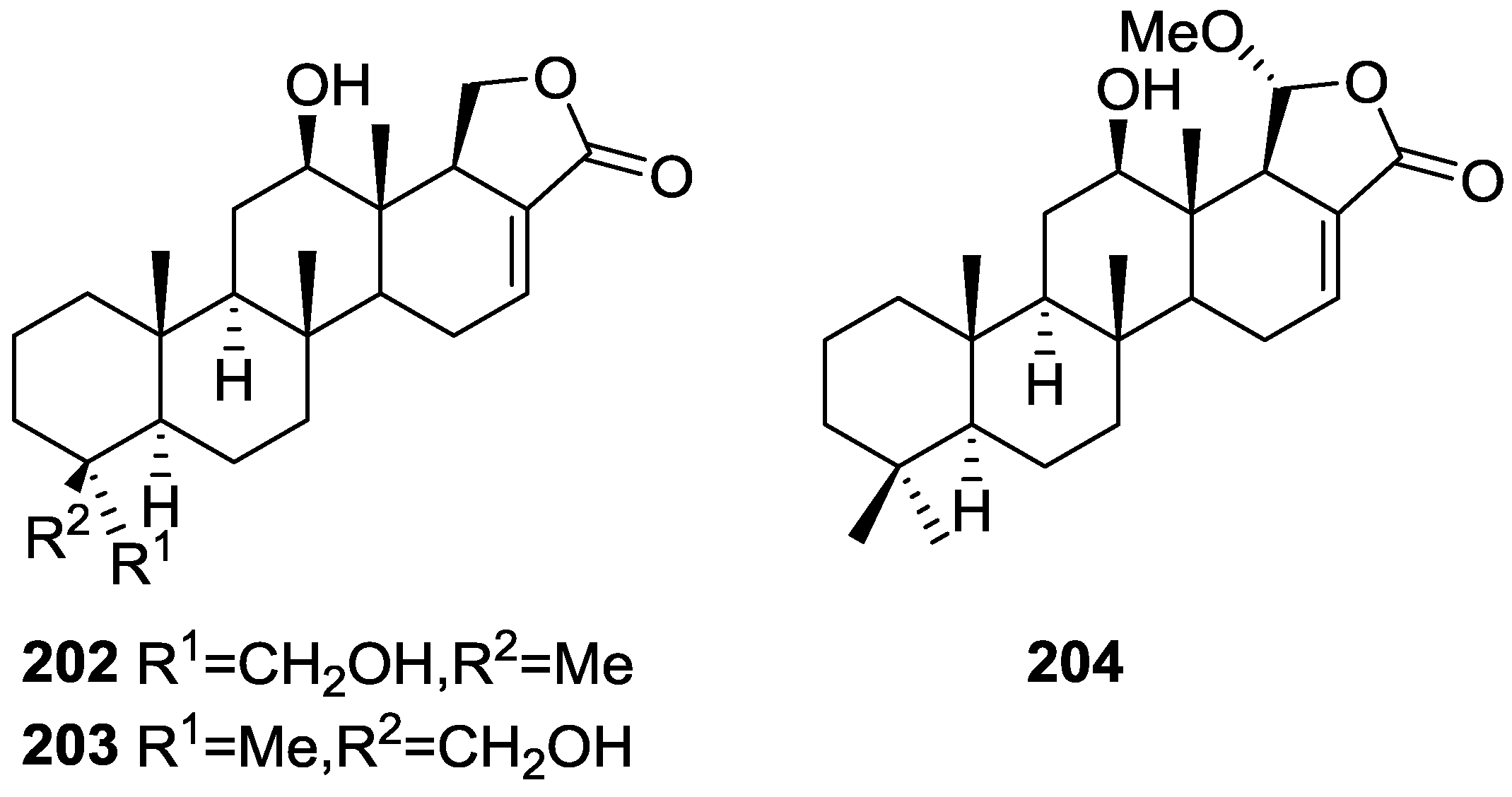
| E-Guggulsterone | 4.1 | Not determined |
| Compound | N. naja Venom %I (10 μM) | Pancreas %I (10 μM) | Human Synovial %I (10 μM) IC50 (μM) | RAPb + Zymosan %I (10 μM) | Bee Venom %I (10 μM) IC50 (μM) |
|---|---|---|---|---|---|
| 205 | 1.3 ± 0.8 | 14.3 ± 6.8 | 34.4 ± 6.5 d | 18.8 ± 3.2 | 37.1 ± 6.3 d |
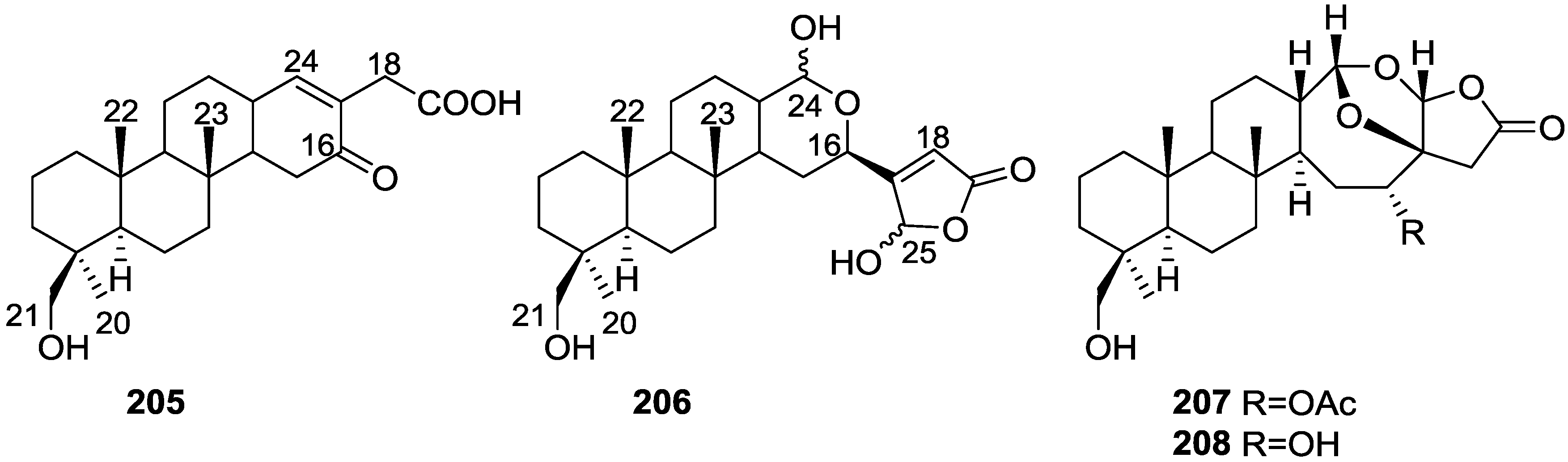
| 206 | 8.7 ± 3.9 | 19.5 ± 3.6 d | 87.2 ± 2.1 d 5.8 | 25.6 ± 1.9 d | 5.4 ± 2.1 |
| manoalide | 17.0 ± 1.7 c | 32.3 ± 2.7 d | 93.2 ± 0.2 d 3.9 | 38.4 ± 0.5 d | 62.5 ± 3.8 d 7.5 |
| Cell Lines | 268 IC50 (μM) | 6-Mercaptopurine IC50 (μM) |
|---|---|---|
| J774.A1 | 0.56 | 0.003 |
| HEK-293 | 0.66 | 0.007 |
| WEHI-164 | 0.42 | 0.017 |
| Activity | Sesquiterpene Quinones | Diterpenes | C21 and Other Furano Terpenes | Sesterterpenes | Sterols | Macrolides | Miscellaneous Compounds |
|---|---|---|---|---|---|---|---|
| Artemia Salina | 53,54,63,74,75 [32] 41 [33] | 139,140 [58] 121,144,145,146 [61] 162 [32] | 165,169,172 [58] | ||||
| Anticancer (cytotoxicity and/or antiproliferative) | 6 [11] 8 [15,22] 12,13 [16] 14,15 [17] 21,22 [18] | 53,54,55 [27] 61 [29] 86 [36] 106 [41] 40,100 [48] | 156,157,158 [65] 296 [121] | 180,181,182,183 [73] 189,190 [67] 165,166,191,192,193,194 [77] | 259,260 [67] | 261,262 [93] 263 [94,95] 267 [82] 297 [121] | 271 [108] 283,284,285,286,287,288 [115] |
| Anticancer (other actions) | 8 [15] 14,15,16,17,18,19 [20] | 111 [43] | 180,181,182, 183,184 [73] 165,192,194 [77] | 263 [103] | |||
| Chemopreventive | 103,104 [40] | ||||||
| DNA repair | 8 [15] | ||||||
| Embryo development | 89,90,91,99,100,101,102 [39] | 291 [39] | |||||
| Immunomodulatory | 90,94 [37] | ||||||
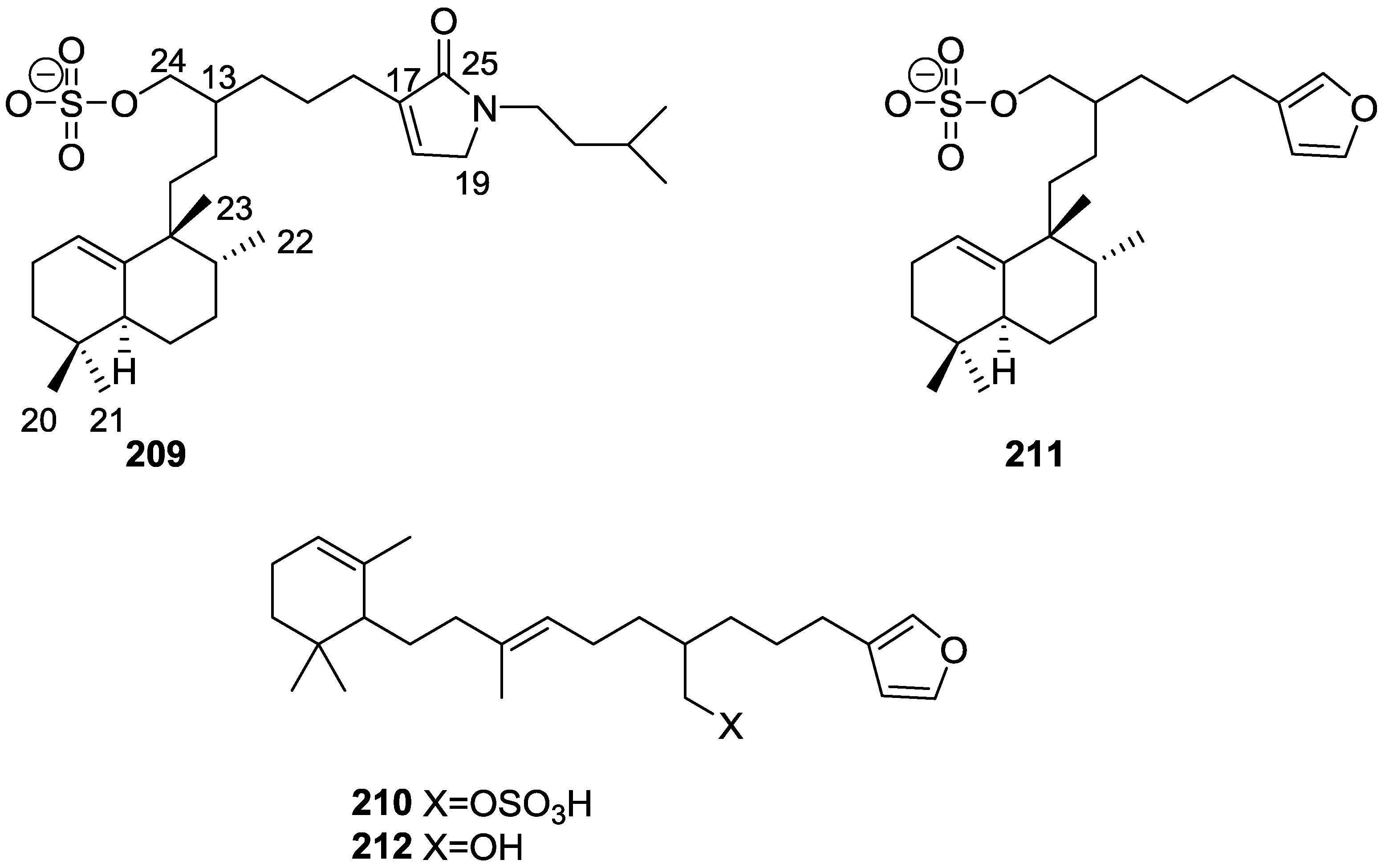
| Immunosuppression | 209,210,211 [81] | ||||||
| P-glycoprotein modulation | 247 [87] 247,248,249,250,251,252,253 [88] | ||||||
| HSV1 or HSV2 | 53,54,55 [27] 43 [48] | ||||||
| HIV | 270 [107] | ||||||
| Microorganisms | 1,2 [9] 3,4,5 [10] | 48,49 [26] | 147 [62] 148 [63] 126 [66] | 271 [108] 275 [110] 283,284,285,286,287,288 [115] | |||
| Antihelminthic | 298 [122] | ||||||
| Predation/Defense | 139 [58] 143 [60] | 169,172 [58] | |||||
| Biofilm induction of E. coli PHL628 | 123,124 [66] | ||||||
| Hypercholesterolemia | 165,198,199,200,201 [79] 166,192,194,202,204 [80] | ||||||
| Lipid metabolism | 283,286 [115] 283 [116] | ||||||
| Antiinflamatory | 205,206 [42] | ||||||
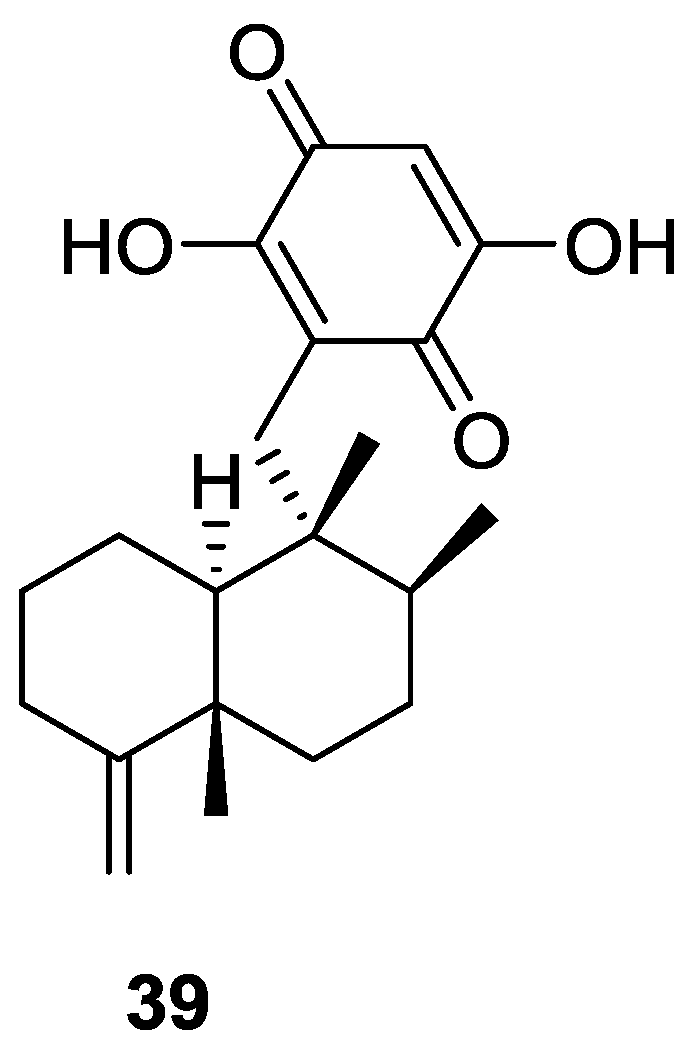
| Antioxidant | 39 [21] | ||||||
| Hemolytic activity | 6 [11] | ||||||
| Smooth muscle paralyzing agent | 147 [62] | ||||||
| Neurotransmission | 43,95,96,97,98 [38] | ||||||
| Neurotrophic | 166,192,194,196,197 [78] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Máximo, P.; Ferreira, L.M.; Branco, P.; Lima, P.; Lourenço, A. The Role of Spongia sp. in the Discovery of Marine Lead Compounds. Mar. Drugs 2016, 14, 139. https://doi.org/10.3390/md14080139
Máximo P, Ferreira LM, Branco P, Lima P, Lourenço A. The Role of Spongia sp. in the Discovery of Marine Lead Compounds. Marine Drugs. 2016; 14(8):139. https://doi.org/10.3390/md14080139
Chicago/Turabian StyleMáximo, Patrícia, Luísa M. Ferreira, Paula Branco, Pedro Lima, and Ana Lourenço. 2016. "The Role of Spongia sp. in the Discovery of Marine Lead Compounds" Marine Drugs 14, no. 8: 139. https://doi.org/10.3390/md14080139
APA StyleMáximo, P., Ferreira, L. M., Branco, P., Lima, P., & Lourenço, A. (2016). The Role of Spongia sp. in the Discovery of Marine Lead Compounds. Marine Drugs, 14(8), 139. https://doi.org/10.3390/md14080139