
New Drugs from Marine Organisms in Alzheimer’s Disease


 ,
,
Abstract
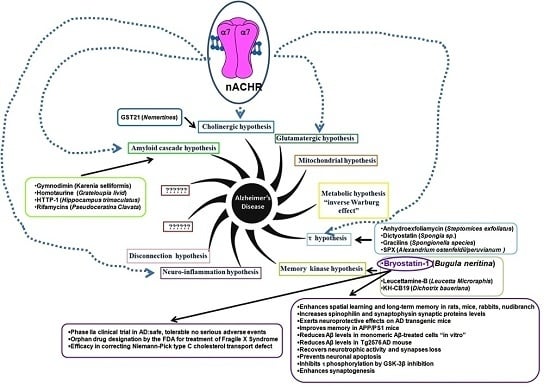
:
1. Introduction
- (i)
- “The amyloid cascade hypothesis”. For decades the hypothesis was the main “framework” for AD research. The pathological accumulation of Aβ, as amyloid plaques, frequently observed in AD brains [6], was considered the main etiopathology cause. The increased Aβ accumulation, according to this hypothesis, triggers a cascade of events leading to synaptic dysfunction, memory loss and structural brain damage in AD advanced stages. The hypothesis that Aβ peptides are the causal factors of AD is now considered an oversimplification. Consequently, a linear toxicity model (increased Aβ deposition which in turn increases a brain damage) is incorrect. However, the possible Aβ role is to trigger other downstream events, such as τ aggregation. The failure of Aβ-targeted clinical trials in AD patients supports the hypothesis that Aβ peptides may be recurrent contributors in the AD process, but it is neither necessary, nor sufficient [6];
- (ii)
- “The cholinergic hypothesis”. The hypothesis is based on the observation ofsignificant loss in cholinergic signaling such as a severe loss of brain white matter with the reduction of cholinergic neurons of the basal forebrain (i.e., Acetylcholine (ACh), nicotine/muscarinic binding sites (nicotinic/muscarinic receptor: nAChR, mAChR)) observed in post-mortem cerebral cortex of AD patients [7]. A significant reduction of the number of nicotine and ACh binding sites in cerebral cortex of AD patients supports a decrease in the number of both nAChR and mAChR. Moreover, the activity of choline acetyltransferase (ChAT) and acetylcholinesterase (AChE) is decreased. The two enzymes are involved in ACh synthesis/degradation: ChAT transfers an acetyl group from the coenzyme (acetyl-CoA) to choline yielding ACh while AChE catalyzes ACh breakdown. Consequently, any failure in the cholinergic system is strictly linked to attention, learning and memory deficit;
- (iii)
- “The glutamatergic hypothesis”. The hypothesis is based on the gradual deterioration of proper synaptic functioning through GluN2A-containing N-methyl-d-aspartate receptors (NMDARs) and the development of excitotoxicity through GluN2B-containing NMDARs. Alteration in NMDARs activity may involve Aβ-induced synaptic impairment, spine loss and neurodegeneration [8];
- (iv)
- (v)
- “The metabolic hypothesis” is based on the assumption that mitochondrial dysregulation up-regulates the oxidative phosphorylation (OXPHOS) activity (known as “inverse Warburg effect”) [11];
- (vi)
- “The τ hypothesis”. The hypothesis is based on the observation that τ dysfunction (abnormal levels, hyperphosphorylation, or ubiquination), in the absence of amyloid pathology, is sufficient to cause synaptic and neuronal loss [12];
- (vii)
- “The memory kinase hypothesis” is based on the involvement of Protein Kinase C (PKC) in acquisition and modification of dendritic spines, in neurite retraction and in synaptic plasticity [13] (For details see Bryostatin-1 section);
- (viii)
- (ix)
- “The clearance systems hypothesis” is based on Aβ clearance failure. In briefly, an excess deposition of Aβ peptides results from an imbalance between their production and clearance; in both EOAD and LOAD, as well as at the prodromal stage [16];
- (x)
- “The Cognitive Reserve (CR) hypothesis”. The hypothesis is proposed to explain the gap between the brain insult and the pathological manifestations. The CR includes two elements: brain (i.e., brain size, synaptic count, and dendritic branching) and cognitive (i.e., neural and compensation reserve) reserve. Two components of the reserve work together and protect the brain from AD [17].
- (xi)
2. Drugs from Marine Organisms
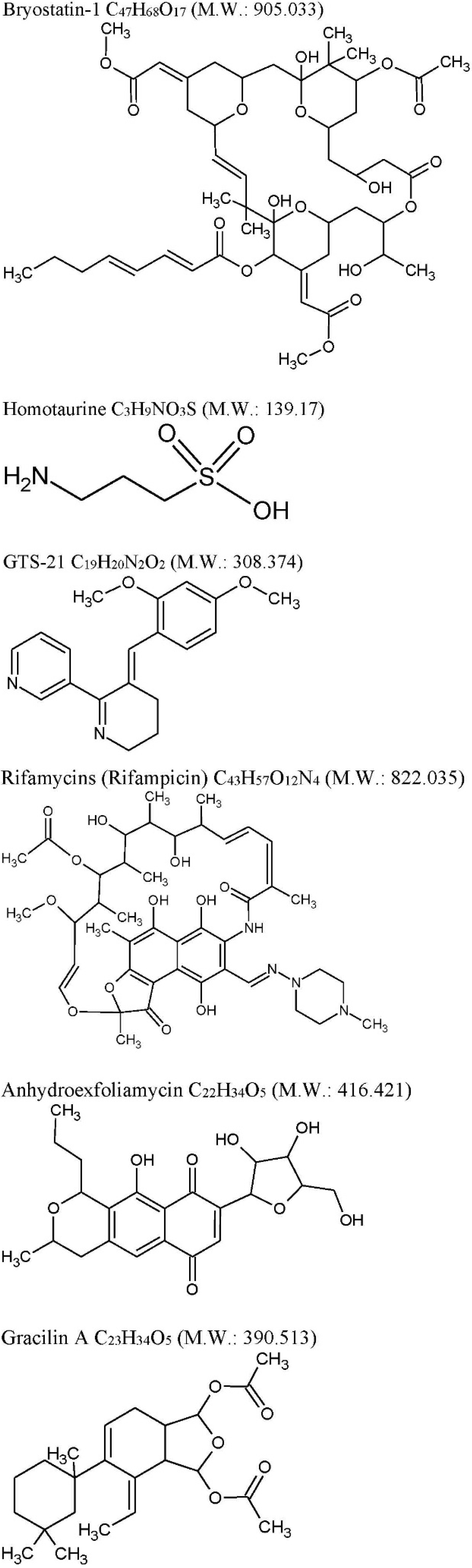
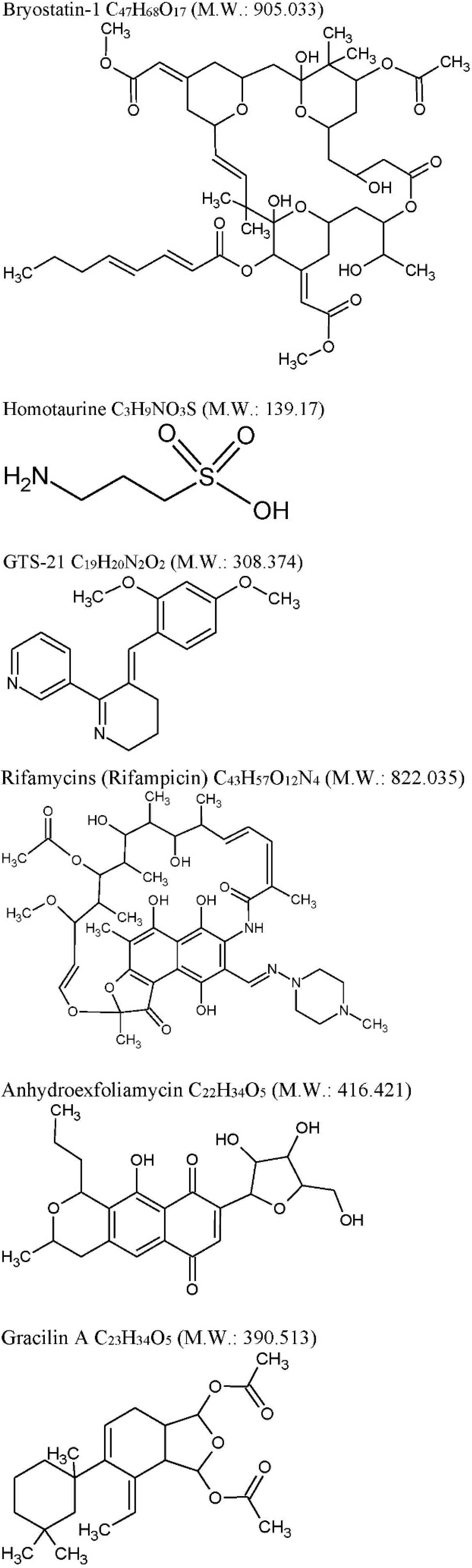
2.1. Bryostatin-1
- (i)
- (ii)
- increase spinophilin (regulatory subunit of protein phosphatase-1 catalytic subunit highly enriched in dendritic spines) and synaptophysin (major synaptic vesicle protein p38), synaptic proteins levels causing synapses structural changes [52];
- (iii)
- exert neuroprotective effects on AD transgenic mice [54];
- (iv)
- improve memory (measured as reduction in latency to escape, after oral Bry-1) in APP/PS1 (mice containing human transgenes for both amyloid precursor protein (APP), bearing the Swedish KM670/671NL (rs63751263, rs63750445) mutation and PSEN1 containing an L166P mutation (rs63750265), both under the control of the Thy1 promoter) transgenic mouse [55];
- (v)
- reduce Aβ levels in monomeric Aβ-treated cells “in vitro” [56];
- (vi)
- reduce Aβ levels in Tg2576 AD mouse (mice overexpressing a mutant form of APP (isoform 695)) and aged rat recovery [57];
- (vii)
- recover neurotrophic activity and synapses loss [57];
- (viii)
- prevent neuronal apoptosis [57];
- (ix)
- inhibit τ phosphorylation by GSK-3β inhibition [57];
- (x)
- enhance synaptogenesis, leading cognitive deficits recovery [57].

{kind=link}
{kind=link}
{kind=link}
| ClinicalTrials.Gov Identifier | Title of the Trial | Study Design/Endpoint Classification | Primary Purpose | Ref |
|---|---|---|---|---|
| Bryostatin-1: C47H68O17 M.W. 905.04 from Bugulaneritina (or brown bryozoans). | ||||
| NCT00606164 Verified: January 2008 by Blanchette Rockefeller Neurosciences Institute. | Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics Study of Bryostatin-1 in Patients With AD. | Randomized Interventional Safety/Efficacy Study Double Blind * | Find out single-dose safety. This study is also being done: (1) to determine how effective a single dose of Bry-1 is in the treatment of AD; (2) to find out what happens to Bry-1 once it enters the body by measuring the levels of Bry-1 in blood; (3) to measure PKC-C in the blood. | [58] |
| NCT02221947 Terminated Verified: April 2015 (not specified) | Study to Evaluate the Preliminary Safety, Efficacy, PK and PD of Bryostatin-1 in Patients With AD. | Randomized Safety/Efficacy Study Double Blind* | Evaluate the safety and tolerability following a single intravenous dose | [59] |
| NCT02431468 Verified: April 2015 by Neurotrope Bioscience, Inc. | A Study Assessing Bryostatin-1 in the Treatment of Moderately Severe to Severe AD. | Randomized Safety/Efficacy Study Double Blind* | To compare different doses for the treatment of moderately severe to severe AD. The study is 28 weeks in duration, including a safety and efficacy 30 days evaluation after the last dose of the study drug. | [60] |
| Homotaurine: (Tramiprosate) C3H9NO3S M.W. 139.17 from a red alga Grateloupia livid | ||||
| NCT00314912 Last verified: July 2007 Bellus Health Inc. | Open-Label Extension of the Phase III Study With Tramiprosate (3APS) in Patients With Mild to Moderate AD. | Randomized, double-blind, placebo-controlled, parallel-group study conducted at 67 study centers across the United States and Canada | Evaluate the long-term safety. Secondary Outcome Measures: To provide additional long-term data on the efficacy of Tramiprosate (3APS). No significant treatment effect | [67,68] |
| -- | Homotaurine induces measurable changes of short latency afferent inhibition in a group of MCI individuals. | 10 MCI patients at 100 mg for 4 weeks | SLAI cortical inhibitory circuit changes, no SICI changes, unable to induce changes of the LTP/LTD mechanisms | [70] |
| GTS-21: C19H20N2O2 M.W. 308.374, anabaseine synthetic derivative from Nemertines (ribbon worms). | ||||
| NCT00414622 Last Updated: April 18, 2007 | A Double Blind, Placebo-Controlled Randomized Study to Compare the Safety and Tolerability of GTS-21 (25 mg TID, 50 mg TID, 75 mg TID and 150 mg TID) When Administered Daily for 28 Days to Participants With Probable AD. | Randomized Double-Blind | Endpoint Classification: Safety/Efficacy Study Primary Purpose: Treatment The study amperes as completed, however no results are present | [72,73] |
| Rifamycins: C43H57O12N4 M.W. 822.036 previously known to be produced only by soil actinobacteria Amycolatopsis is produced by marine bacteria—Salinispora isolated from the marine sponge Pseudoceratinaclavata. | ||||
| -- | A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of AD: the DARAD trial. | DARAD study: multicenter, blinded, randomized, placebo-controlled factorial doxycycline and rifampin | Neither rifampin nor doxycycline provided any benefit to patients with AD. | [74] |
| -- | A randomized, controlled trial of doxycycline and rifampin for patients with AD. | Randomized, triple-blind, controlled trial. | Possible therapeutic role in patients with mild to moderate AD | [75] |
2.2. Drugs in Ongoing Clinical Trials
2.3. Drugs in Preclinical Evaluations
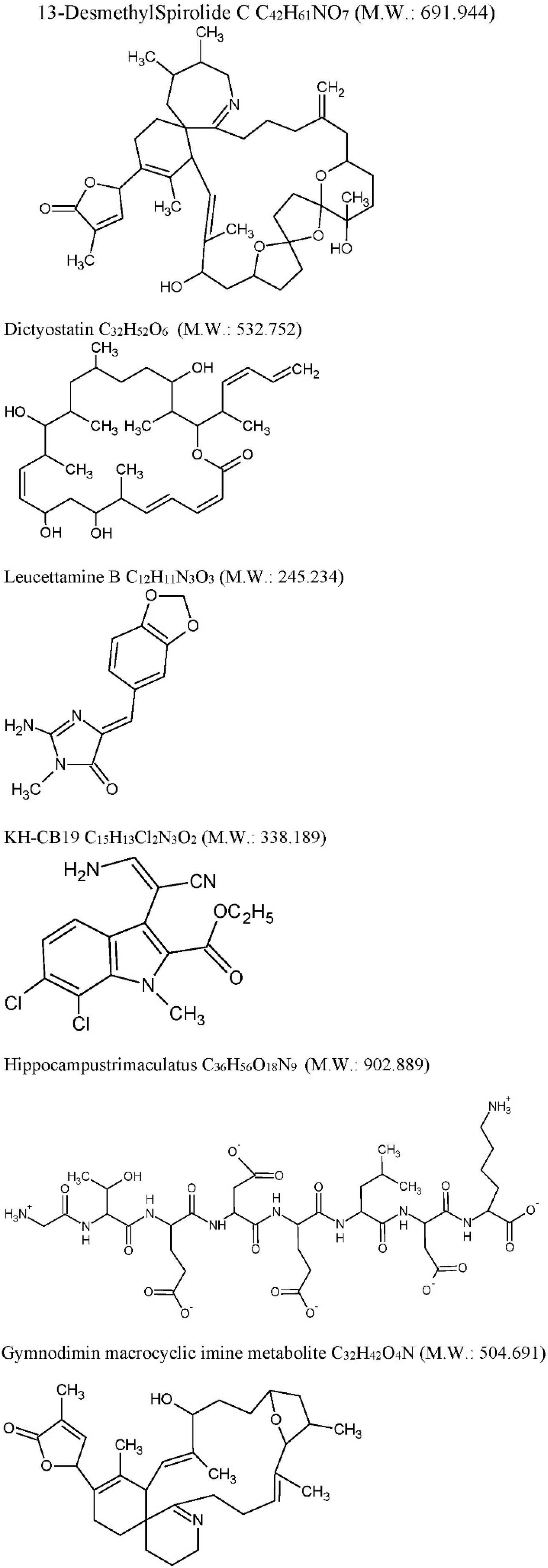
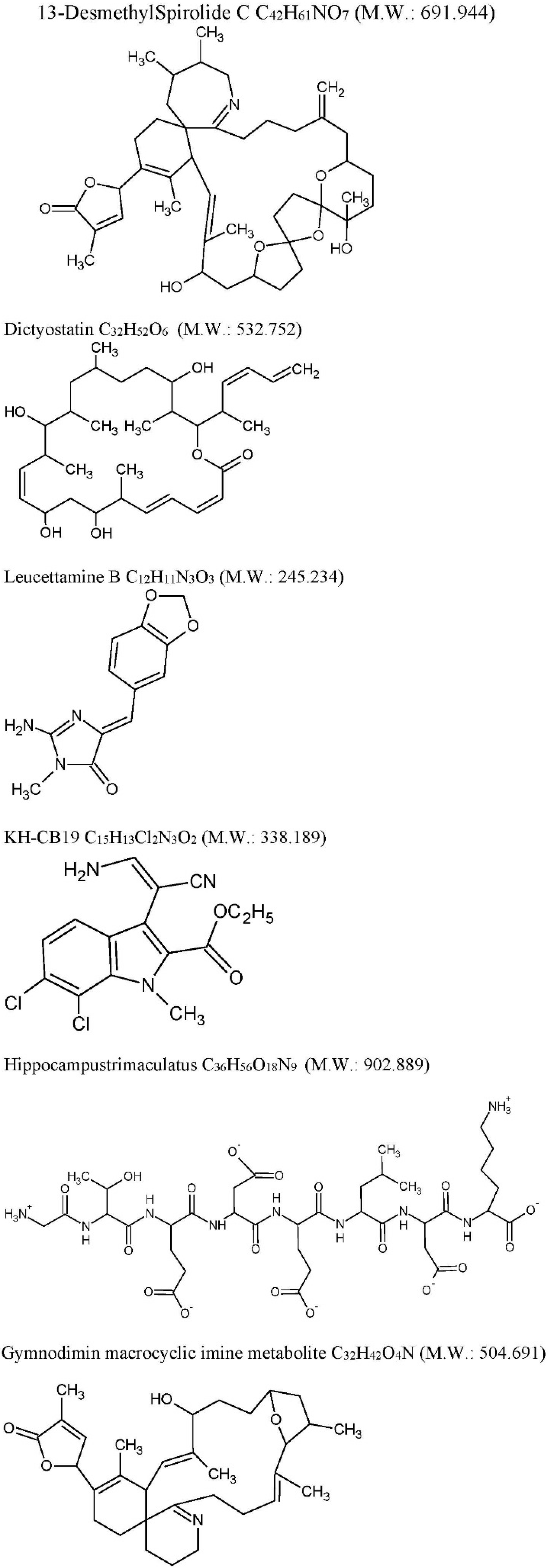
| Drug | Source | Target | Cellular/Animal Model | Effect | Ref |
|---|---|---|---|---|---|
| τ inhibition | |||||
| Anhydroexfoliamycin: C22H24O8 M.W. 416.421 | Streptomyces exfoliatus from marine soil | GSK3β mediated by the JNK pathway | 3xTg-AD mice | GSK3β inhibition τ, phosphorylation reduction | [78] |
| Gracilins: C23H34O5 M.W. 390.513 | Spongionella sp. | Mitochondrial targeting through the induction of Nrf2 translocation. BACE1 and ERK inhibition, τhyperphosphorylation reduction. | 3xTg-AD mice | After chronic intraperitoneal treatments, a preliminary behavioral test pointed a positive trend on learning and spatial memory of mice treated with these compounds. Moreover, in vivo assays confirmed the previous results. Amyloid-β42 and hyperphosphorylated tau levels were decreased after treatments and the ERK inhibition was also observed. | [79] |
| 13-desmethyl spirolide-C (SPX): C42H61NO7 M.W. 691.944 Spirolides* | Alexandriumostenfeldii/peruvianumdinoflagellates | Decrease GSK-3β and ERK. | 3xTg mice cortical neurons | Glutamate-induced neurotoxicity inhibition both in control and 3xTg neurons. | [74] |
| Dictyostatin: C32H52O6 M.W. 532.751 | Spongia sp. and Caribbean sponge family Corallistidae | MT-stabilizing agent | CD1 mice | MT-stabilization in the brain one week after 5 mg/kg i.p. administration | [75] |
| CDC2-like kinase inhibitors | |||||
| Leucettamine B: C12H11N3O3 M.W. 245.234 | Leucettamicroraphis Haeckel (Calcarea) sponge | CLK1, Dyrk1A and Dyrk2 inhibition and CLK3 moderate inhibition. | Human U937 cell membrane | -- | [80] |
| KH-CB19: C15H13Cl2N3O2 M.W. 338.188 dichloroindolylenaminonitrile derived from bauerine C | Dichothrixbaueriana blue-green alga | CLK1 and Dyrk1A potent inhibitor. | Inhibition of human recombinant CLK1 (148 to 484 amino acids) expressed in Escherichia coli BL21. | -- | [80] |
| Amyloid-β Aggregation Inhibitors | |||||
| Trimaculatus-derived neuroprotective peptides HTP-1: Gly-Thr-Glu-Asp-Glu-Leu-Asp-Lys: C36H56O18N9 M.W. 902.889 | Hippocampus trimaculatus (seahorse) | -- | PC12 | Aβ42-induced neuronal death protection. Bcl-2 up-regulation. | [81] |
| Gymnodiminmacrocyclic imine metabolite: C32H45O4N M.W. 504.691 | Kareniaselliformis (formerly named Gymnodiniumselliformis) (dinoflagellate) | Antagonize human α7-nAChR expressed in Xenopus oocytes | 3xTg mice cortical neurons | Aβ intracellular accumulation, τhyperphosphorylation reduction, Glutamate-induced neuronal death prevention | [82] |
3. Concluding Remarks
Author Contributions
Conflicts of Interest
References
- World Alzheimer Report 2015. Available online: http://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf (accessed on 3 September 2015).
- Sachdev, P.S.; Blacker, D.; Blazer, D.G.; Ganguli, M.; Jeste, D.V.; Paulsen, J.S.; Petersen, R.C. Classifying neurocognitive disorders: The DSM-5 approach. Nat. Rev. Neurol. 2014, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ekavali, A.S. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T. On neurodegenerative diseases, models, and treatment strategies: Lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 2000, 163, 495–529. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Toldi, J.; Vécsei, L. Glutamatergicdysfunctioning in Alzheimer’s disease and related therapeutic targets. J. Alzheimers Dis. 2014, 42, 177–187. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Milic, M.; Frustaci, A.; Del Bufalo, A.; Sánchez-Alarcón, J.; Valencia-Quintana, R.; Russo, P.; Bonassi, S. DNA damage in non-communicable diseases: A clinical and epidemiological perspective. Mutat. Res. 2015, 776, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Demetrius, L.A.; Driver, J. Alzheimer’s as a metabolic disease. Biogerontology 2013, 14, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Avila, J. New perspectives on the role of tau in Alzheimer’s disease. Implications for therapy. Biochem. Pharmacol. 2014, 88, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Simpkins, J.W.; Alkon, D.L.; Smith, K.E.; Chen, Y.W.; Tan, Z.; Huber, J.D.; Rosen, C.L. Common mechanisms of Alzheimer’s disease and ischemic stroke: The role of protein kinase C in the progression of age-related neurodegeneration. J. Alzheimers Dis. 2015, 43, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Ofek, K.; Soreq, H. Cholinergic involvement and manipulation approaches in multiple system disorders. Chem. Biol. Interact. 2013, 203, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yu, J.T.; Tan, M.S.; Tan, L. Cognitive reserve and Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 187–208. [Google Scholar] [CrossRef] [PubMed]
- Stam, C.J. Modern network science of neurological disorders. Nat. Rev. Neurosci. 2014, 15, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, F.; Miraglia, F.; Curcio, G.; Altavilla, R.; Scrascia, F.; Giambattistelli, F.; Quattrocchi, C.C.; Bramanti, P.; Vernieri, F.; Rossini, P.M. Cortical brain connectivity evaluated by graph theory in dementia: A correlation study between functional and structural data. J. Alzheimers Dis. 2015, 45, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Zanzoni, A. A Computational Network biology approach to uncover novel genes related to Alzheimer’s disease. Methods Mol. Biol. 2016, 1303, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Kisialiou, A.; Moroni, R.; Prinzi, G.; Fini, M. Effect of genetic polymorphisms (SNPs) in CHRNA7 gene on response to acetylcholinesterase Inhibitors (AChEI) in patients with Alzheimer’s disease. Curr. Drug Targets 2015, in press. [Google Scholar] [CrossRef]
- Parri, H.R.; Hernandez, C.M.; Dineley, K.T. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochem. Pharmacol. 2011, 82, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA. 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [PubMed]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Langa, K.M.; Levine, D.A. The diagnosis and management of mild cognitive impairment: A clinical review. JAMA 2014, 312, 2551–2561. [Google Scholar] [CrossRef] [PubMed]
- Rolinski, M.; Fox, C.; Maidment, I.; McShane, R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst. Rev. 2012, 3, CD006504. [Google Scholar] [CrossRef] [PubMed]
- Qizilbash, N.; Birks, J.; Lopez-Arrieta, J.; Lewington, S.; Szeto, S. WITHDRAWN: Tacrine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2007, 18, CD000202. [Google Scholar]
- FDA Approved Drugs. Available online: http://www.centerwatch.com/drug-information/fda-approved-drugs/year/2015/2014/2013 (accessed on 16 September 2015).
- First WHO Ministerial Conference on Global Action Against Dementia. Available online: http://www.who.int/mediacentre/events/meetings/2015/global-action-against-dementia/en/ (accessed on 16 September 2015).
- Koslow, T. The Silent Deep: The Discovery, Ecology and Conservation of the Deep Sea; University Chicago Press: Chicago, IL, USA, 2007; pp. 1–288. [Google Scholar]
- Russo, P.; Del Bufalo, A.; Fini, A. Deep sea as a source of novel-anticancer drugs: Update on discovery and preclinical/clinical evaluation in a systems medicine perspective. EXCLI J. 2015, 14, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Catassi, A.; Cesario, A.; Arzani, D.; Menichini, P.; Alama, A.; Bruzzo, C.; Imperatori, A.; Rotolo, N.; Granone, P.; Russo, P. Characterization of apoptosis induced by marine natural products in non small cell lung cancer A549 cells. Cell Mol. Life Sci. 2006, 63, 2377–2386. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Nastrucci, C.; Cesario, A. From the sea to anticancer therapy. Curr. Med. Chem. 2011, 18, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Nastrucci, C.; Cesario, A.; Russo, P. Anticancer drug discovery from the marine environment. Recent Pat. Anticancer Drug Discov. 2012, 7, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Cesario, A. New anticancer drugs from marine cyanobacteria. Curr. Drug Targets 2012, 13, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Del Bufalo, A. Conopeptides in addiction disorders treatment. Int. J. Adv. Res. Chem. Sci. IJARCS 2015, in press. [Google Scholar]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Kollár, P.; Rajchard, J.; Balounová, Z.; Pazourek, J. Marine natural products: Bryostatins in preclinical and clinical studies. Pharm Biol. 2014, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.F.; Zhu, H.L. The chemistry and biology of the bryostatins: Potential PKC inhibitors in clinical development. Curr. Med. Chem. 2012, 19, 2652–2664. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cherry Herald, L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Keck, G.E.; Poudel, Y.B.; Cummins, T.J.; Rudra, A.; Covel, J.A. Total synthesis of bryostatin 1. J. Am. Chem. Soc. 2011, 133, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Manaviazar, S.; Hale, K.J. Total synthesis of bryostatin 1: A short route. Angew. Chem. Int. Ed. Engl. 2011, 50, 8786–8789. [Google Scholar] [CrossRef] [PubMed]
- Bryostatin 1 and Cisplatin in Treating Patients With Metastatic or Unresectable Stomach Cancer. ClinicalTrials.gov Identifier: NCT00006389. Available online: https://clinicaltrials.gov/ct2/show/NCT00006389 (accessed on 16 September 2015).
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Díaz, L.; Martínez-Bonet, M.; Sánchez, J.; Fernández-Pineda, A.; Jiménez, J.L.; Muñoz, E.; Moreno, S.; Álvarez, S.; Muñoz-Fernández, M.Á. Bryostatin activates HIV-1 latent expression in human astrocytes through a PKC and NF-κB-dependent mechanism. Sci. Rep. 2015, 5, 12442. [Google Scholar] [CrossRef] [PubMed]
- Bryostatin-1 Effect on HIV-1 Latency and Reservoir in HIV-1 Infected Patients Receiving Antiretroviral Treatment (BRYOLAT). ClinicalTrials.gov Identifier: NCT02269605. Available online: https://clinicaltrials.gov/ct2/show/NCT02269605 (accessed on 16 September 2015).
- Kazanietz, M.G. Novel “nonkinase” phorbol ester receptors: The C1 domain connection. Mol. Pharmacol. 2002, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937–957. [Google Scholar] [CrossRef] [PubMed]
- Keck, G.E.; Poudel, Y.B.; Rudra, A.; Stephens, J.C.; Kedei, N.; Lewin, N.E.; Blumberg, P.M. Role of the C8 gem-dimethyl group of bryostatin 1 on its unique pattern of biological activity. Bioorganic Med. Chem. Lett. 2012, 22, 4084–4088. [Google Scholar] [CrossRef] [PubMed]
- Hongpaisan, J.; Alkon, D.L. A structural basis for enhancement of long-term associative memory in single dendritic spines regulated by PKC. Proc. Natl. Acad. Sci. USA 2007, 104, 19571–19576. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liu, Q.Y.; Alkon, D.L. PKC activators enhance GABAergic neurotransmission and paired-pulse facilitation in hippocampal CA1 pyramidal neurons. Neuroscience 2014, 268, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Etcheberrigaray, R.; Tan, M.; Dewachter, I.; Kuipéri, C.; van der Auwera, I.; Wera, S.; Qiao, L.; Bank, B.; Nelson, T.J.; Kozikowski, A.P.; et al. Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proc. Natl. Acad. Sci. USA. 2004, 101, 11141–11146. [Google Scholar] [CrossRef] [PubMed]
- Schrott, L.M.; Jackson, K.; Yi, P.; Dietz, F.; Johnson, G.S.; Basting, T.F.; Purdum, G.; Tyler, T.; Rios, J.D.; Castor, T.P.; et al. Acute oral Bryostatin-1 administration improves learning deficits in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.S.; Alkon, D.L. PKCε promotes HuD-mediated neprilysin mRNA stability and enhances neprilysin-induced Aβ degradation in brain neurons. PLoS ONE 2014, 9, e97756. [Google Scholar] [CrossRef] [PubMed]
- Hongpaisan, J.; Sun, M.K.; Alkon, D.L. PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci. 2011, 31, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Safety, Efficacy, Pharmacokinetics, and Pharmacodynamics Study of Bryostatin 1 in Patients With Alzheimer’s Disease. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00606164 (accessed on 16 September 2015).
- Study to Evaluate the Preliminary Safety, Efficacy, PK and PD of Bryostatin 1 in Patients With Alzheimer’s Disease. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02221947 (accessed on 16 September 2015).
- A Study Assessing Bryostatin in the Treatment of Moderately Severe to Severe Alzheimer’s Disease. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02431468 (accessed on 16 September 2015).
- Neurotrope Announces Positive Top-Line Results From Its Phase 2a Study of Bryostatin-1 in Alzheimer’s Disease. Available online: http://www.neurotropebioscience.com/Welcome_to_Neurotrope_BioScience/Bryostatin-1.html (accessed on 16 September 2015).
- Search Orphan Drug Designations and Approvals. Available online: http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm (accessed on 16 September 2015).
- Devitt, N.M.; Gallagher, L.; Reilly, R.B. Autism Spectrum Disorder (ASD) and Fragile X Syndrome (FXS): Two Overlapping Disorders Reviewed through Electroencephalography—What Can be Interpreted from the Available Information? Brain Sci. 2015, 5, 92–117. [Google Scholar] [CrossRef] [PubMed]
- Neurotrope to Conduct Study on Niemann-Pick Type C, a Devastating Rare Pediatric Disease. Available online: http://www.prnewswire.com/news-releases/neurotrope-to-conduct-study-on-niemann-pick-type-c-a-devastating-rare-pediatric-disease-300104024.html (accessed on 16 September 2015).
- Brady, R.O.; Filling-Katz, M.R.; Barton, N.W.; Pentchev, P.G. Niemann-Pick disease types C and D. Neurol. Clin. 1989, 7, 75–88. [Google Scholar] [PubMed]
- Malnar, M.; Hecimovic, S.; Mattsson, N.; Zetterberg, H. Bidirectional links between Alzheimer’s disease and Niemann-Pick type C disease. Neurobiol. Dis. 2014, 72, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Open-Label Extension of the Phase III Study With Tramiprosate (3APS) in Patients With Mild to Moderate Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/study/NCT00314912 (accessed on 16 September 2015).
- Aisen, P.S.; Gauthier, S.; Ferris, S.H.; Saumier, D.; Haine, D.; Garceau, D.; Duong, A.; Suhy, J.; Oh, J.; Lau, W.C.; et al. Tramiprosate in mild-to-moderate Alzheimer’s disease—a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 2011, 7, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Caltagirone, C.; Ferrannini, L.; Marchionni, N.; Nappi, G.; Scapagnini, G.; Trabucchi, M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: A review. Aging Clin. Exp. Res. 2012, 24, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; di Lorenzo, F.; Manenti, G.; Semprini, R.; Koch, G. Homotaurine induces measurable changes of short latency afferent inhibition in a group of mild cognitive impairment individuals. Front. Aging Neurosci. 2014, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Tokimura, H.; di Lazzaro, V.; Tokimura, Y.; Oliviero, A.; Profice, P.; Insola, A.; Mazzone, P.; Tonali, P.; Rothwell, J.C. Short latency inhibition of human hand motor cortex by somatosensory input from the hand. J. Physiol. 2000, 523, 503–513. [Google Scholar] [CrossRef] [PubMed]
- GTS21-201 for Alzheimer Disease: GTS-21 Administered Daily for 28 Days to Participants With Probable Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/study/NCT00414622 (accessed on 16 September 2015).
- Russo, P.; Del Bufalo, A.; Frustaci, A.; Fini, M.; Cesario, A. Beyond acetylcholinesterase inhibitors for treating Alzheimer’s disease: α7-nAChR agonists in human clinical trials. Curr. Pharm. Des. 2014, 20, 6014–6021. [Google Scholar] [CrossRef] [PubMed]
- Molloy, D.W.; Standish, T.I.; Zhou, Q.; Guyatt, G. DARAD Study Group. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: The DARAD trial. Int. J. Geriatr. Psychiatry 2013, 28, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yue, Y.; Tian, H.; Tao, L.; Wang, Y.; Xiang, J.; Wang, S.; Ding, H. Tramiprosate protects neurons against ischemic stroke by disrupting the interaction between PSD95 and nNOS. Neuropharmacology 2014, 83, 107–117. [Google Scholar] [CrossRef]
- Ricciardi, L.; de Nigris, F.; Specchia, A.; Fasano, A. Homotaurine in Parkinson’s disease. Neurol. Sci. 2015, 36, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Hewavitharana, A.K.; Shaw, P.N.; Fuerst, J.A. Discovery of a new source of rifamycin antibiotics in marine sponge actinobacteria by phylogenetic prediction. Appl. Environ. Microbiol. 2006, 72, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Yulug, B.; Hanoglu, L.; Kilic, E.; Schabitz, W.R. RIFAMPICIN: An antibiotic with brain protective function. Brain Res. Bull. 2014, 107, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Leirós, M.; Alonso, E.; Rateb, M.E.; Ebel, R.; Jaspars, M.; Alfonso, A.; Botana, L.M. The Streptomyces metabolite anhydroexfoliamycin ameliorates hallmarks of Alzheimer’s disease in vitro and in vivo. Neuroscience 2015, 305, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Leirós, M.; Alonso, E.; Rateb, M.E.; Houssen, W.E.; Ebel, R.; Jaspars, M.; Alfonso, A.; Botana, L.M. Gracilins: Spongionella-derived promising compounds for Alzheimer disease. Neuropharmacology 2015, 93, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Vale, C.; Vieytes, M.R.; Laferla, F.M.; Giménez-Llort, L.; Botana, L.M. 13-Desmethyl spirolide-C is neuroprotective and reduces intracellular Aβ and hyperphosphorylated tau in vitro. Neurochem. Int. 2011, 59, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Karthikeyan, C.; Moorthy, N.S.; Waiker, D.K.; Jain, A.K.; Trivedi, P. Human CDC2-like kinase 1 (CLK1): A novel target for Alzheimer’s disease. Curr. Drug Targets 2014, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Pangestuti, R.; Ryu, B.; Himaya, S.; Kim, S.K. Optimization of hydrolysis conditions, isolation, and identification of neuroprotective peptides derived from seahorse Hippocampus trimaculatus. Amino Acids 2013, 45, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Vale, C.; Vieytes, M.R.; Laferla, F.M.; Gimenez-Llort; Botana, L.M. The cholinergic antagonist gymnodi- mine improves Aβ and tau neuropathology in an in vitro model of Alzheimer disease. Cell Physiol. Biochem. 2001, 27, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Gardner, N.M.; James, M.J.; Yao, Y.; Trojanowski, J.Q.; Lee, V.M.; Paterson, I.; Ballatore, C.; Smith, A.B., 3rd. MT-stabilizer, dictyostatin, exhibits prolonged brain retention and activity: Potential therapeutic implications. ACS Med. Chem. Lett. 2013, 4, 886–889. [Google Scholar] [CrossRef] [PubMed]
- The Nobel Prize in Chemistry 1990. Available online: http//www.nobelprize.org/nobel_prizes/chemistry/laureates/1990/ (accessed on 24 December 2015).
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New Drugs from Marine Organisms in Alzheimer’s Disease. Mar. Drugs 2016, 14, 5. https://doi.org/10.3390/md14010005
Russo P, Kisialiou A, Lamonaca P, Moroni R, Prinzi G, Fini M. New Drugs from Marine Organisms in Alzheimer’s Disease. Marine Drugs. 2016; 14(1):5. https://doi.org/10.3390/md14010005
Chicago/Turabian StyleRusso, Patrizia, Aliaksei Kisialiou, Palma Lamonaca, Rossana Moroni, Giulia Prinzi, and Massimo Fini. 2016. "New Drugs from Marine Organisms in Alzheimer’s Disease" Marine Drugs 14, no. 1: 5. https://doi.org/10.3390/md14010005
APA StyleRusso, P., Kisialiou, A., Lamonaca, P., Moroni, R., Prinzi, G., & Fini, M. (2016). New Drugs from Marine Organisms in Alzheimer’s Disease. Marine Drugs, 14(1), 5. https://doi.org/10.3390/md14010005