
A New Analogue of Echinomycin and a New Cyclic Dipeptide from a Marine-Derived Streptomyces sp. LS298


Abstract
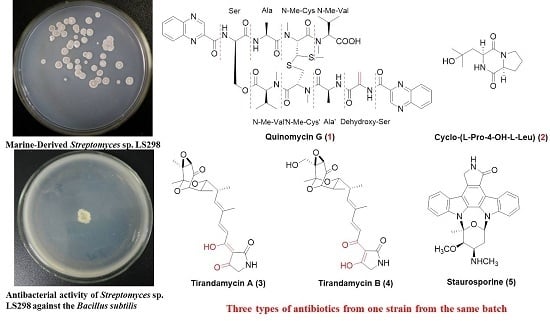
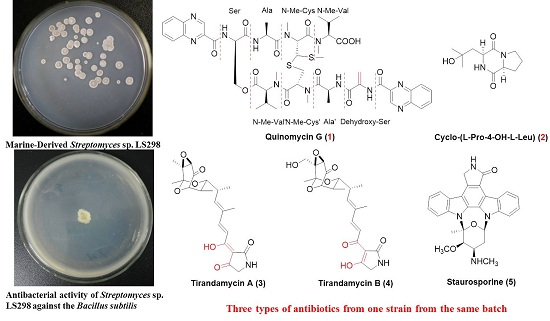
:
1. Introduction

2. Results and Discussion
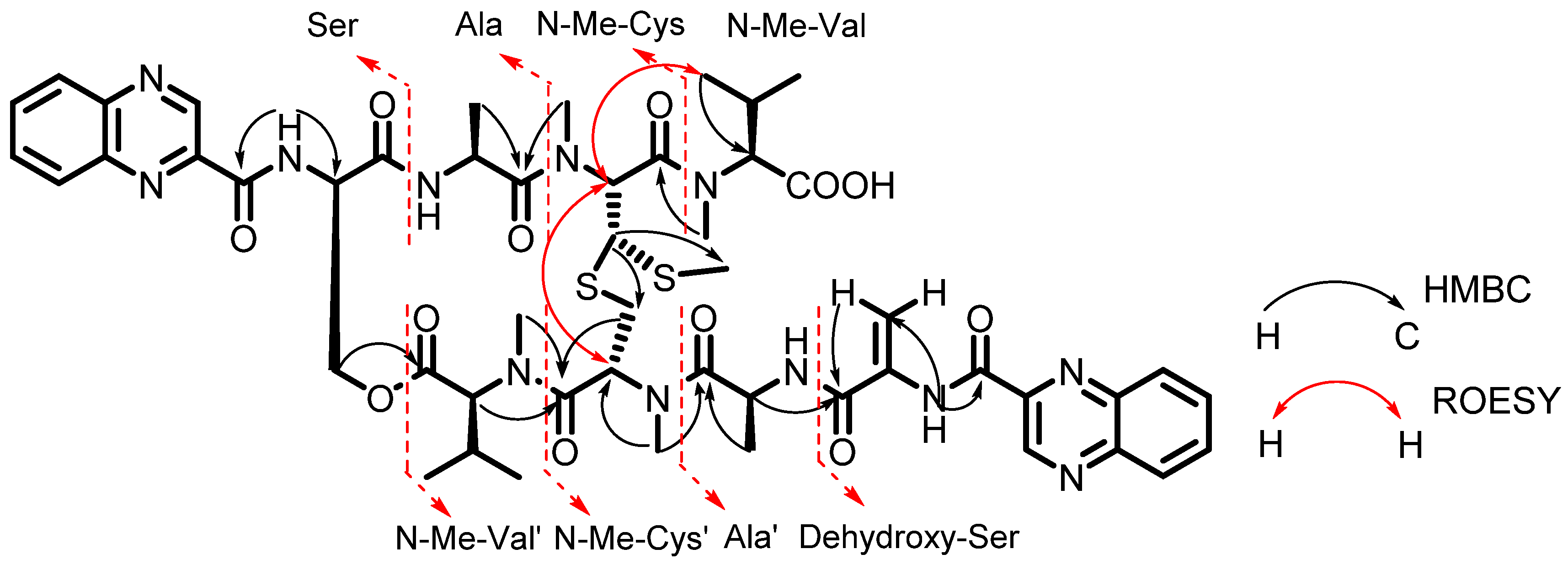
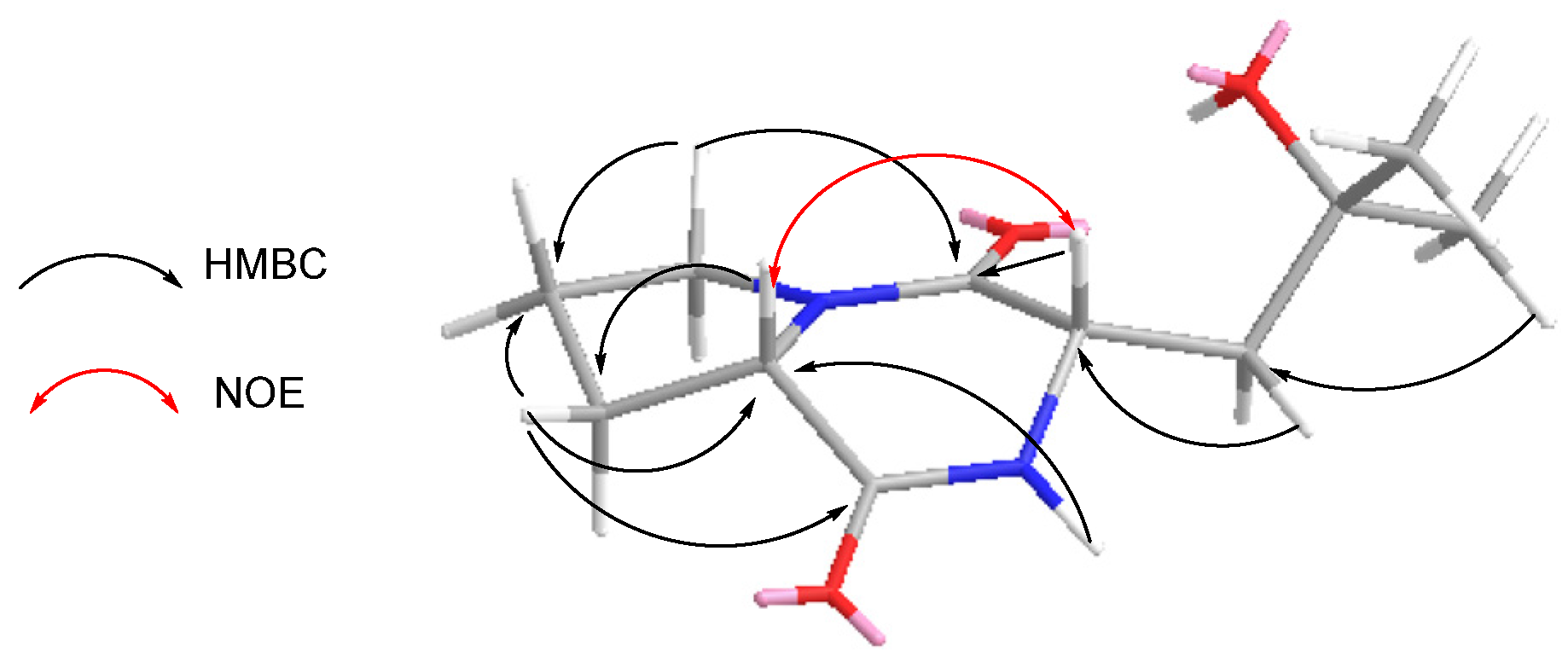
2.1. Structure Elucidation of Compounds 1–5

{kind=link}
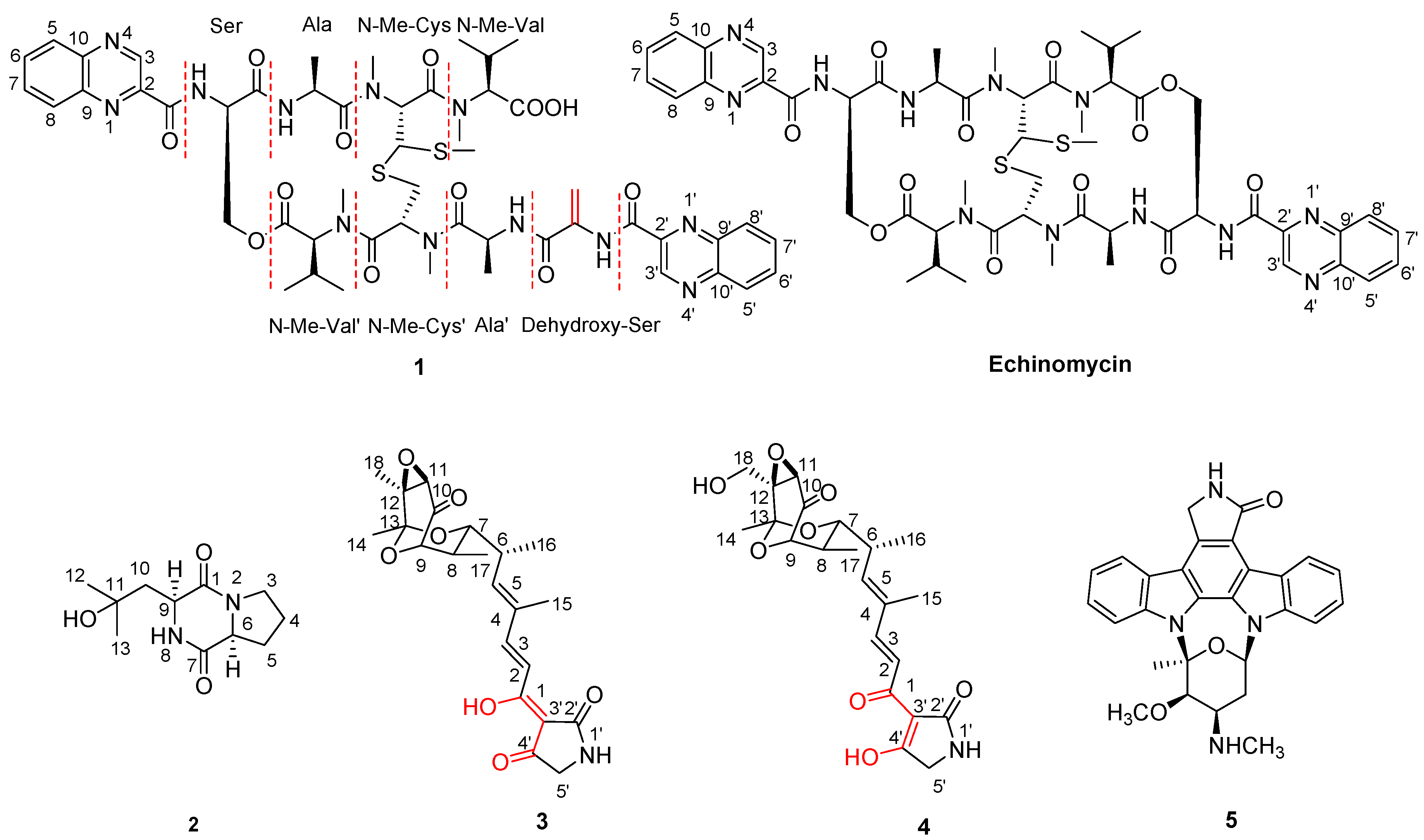{kind=link}
{kind=link}
{kind=link}
| Position | δH, mult (J in Hz) | δC | Position | δH, mult (J in Hz) | δC | Position | δH, mult (J in Hz) | δC |
|---|---|---|---|---|---|---|---|---|
| Quinoxaline | β | 5.02, dd (11.5, 3.0) | 65.8 | α | 5.28, m | 45.6 | ||
| 2 | 143.9 | 4.67, d (11.5) | β | 1.35, d (6.0) | 18.3 | |||
| 2′ | 143.9 | C=O | 169.9 | C=O | 172.2 | |||
| 3 | 9.68, s | 143.5 | Dehydroxy-Ser | Ala′ | ||||
| 3′ | 9.63, s | 143.7 | NH | 10.67, s | NH | 7.83 a | ||
| 5 | 8.20, d (8.0) | 129.4 | α | 133.0 | α | 4.85, m | 44.9 | |
| 5′ | 8.20, d (8.0) | 129.4 | β | 6.90, brs; 6.11, brs | 104.3 | β | 0.19, d (6.0) | 16.4 |
| 6 | 7.84–7.89 a | 132.4 | C=O | 163.2 | C=O | 172.2 | ||
| 6′ | 7.84–7.89 a | 131.9 | N-Me-Cys | N-Me-Val | ||||
| 7 | 7.95–7.97 a | 131.6 | N-Me | 3.15, s | 33.2 | N-Me | 2.97, s | 29.0 |
| 7′ | 7.95–7.97 a | 131.0 | α | 5.70, s | 60.4 | α | 3.42, d (10.5) | 65.8 |
| 8 | 8.27, d (8.0) | 130.0 | β | 3.74, d (2.0) | 52.1 | β | 2.27, m | 27.4 |
| 8′ | 8.20, d (8.0) a | 130.0 | S-Me | 2.07,s | 13.4 | γ | 1.04 a | 19.5 |
| 9 | 140.3 | C=O | 168.3 | γ | 0.97, d (6.0) | 18.5 | ||
| 9′ | 140.3 | N-Me-Cys′ | COOH | 169.8 | ||||
| 10 | 143.4 | N-Me | 3.37, s | 33.8 | N-Me-Val′ | |||
| 10′ | 142.4 | α | 6.03, d (4.0) | 61.1 | N-Me | 3.02, s | 28.8 | |
| C=O | 163.6 | β | 3.47, dd (16.0, 5.0) | 29.2 | α | 4.47, d (11.0) | 64.6 | |
| C=O′ | 161.9 | 2.54, d (16.0) | β | 2.49, m | 28.6 | |||
| Ser | C=O | 171.5 | γ | 1.06, d (7.0) | 19.7 | |||
| NH | 9.01, d (9.5) | Ala | γ | 1.03, d (7.0) | 18.8 | |||
| α | 5.37, d (9.0) | 52.6 | NH | 9.20, d (9.5) | C=O | 169.9 |

| Retention Time (TR) Min | |||
|---|---|---|---|
| Amino Acid Standards | Compound 1 | Compound 2 | |
| l-Ser | 15.787 | - | - |
| d-Ser | 16.422 | 16.649 | - |
| l-Ala | 22.125 | 21.952 | - |
| d-Ala | 23.789 | - | - |
| N-Me-l-Val | 17.499 | 17.528 | - |
| N-Me-d-Val | 19.732 | - | - |
| l-Pro | 14.544 | - | 14.677 |
| d-Pro | 17.528 | - | - |
| FDAA | 19.772 | 19.804 | 19.782 |
| No. | δH, mult (J in Hz) | δC | No. | δH, mult (J in Hz) | δC |
|---|---|---|---|---|---|
| 1 | 166.1 | 7 | 169.7 | ||
| 2 | 8 | 7.66, s | |||
| 3 | 3.56, m | 45.6 | 9 | 4.23, d (10.5) | 53.2 |
| 4α | 1.89, m | 22.6 | 10α | 2.32, dd (14.5, 2.0) | 41.1 |
| 4β | 2.01, m | 10β | 1.79, dd (14.5, 11.0) | ||
| 5α | 2.36, dd (8.5, 2.5) | 28.3 | 11 | 70.9 | |
| 5β | 2.10, m | 12 | 1.31, s | 27.4 | |
| 6 | 4.08, t (8.0) | 58.8 | 13 | 1.35, s | 32.3 |

| No. | Tirandamycin A | Tirandamycin B | ||
|---|---|---|---|---|
| δH, mult (J in Hz) | δC | δH, mult (J in Hz) | δC | |
| 1 | 173.5 | 181.0 | ||
| 2 | 7.05, d (15.5) | 116.2 | 7.55, d (15.5) | 124.8 |
| 3 | 7.47, d (15.5) | 147.9 | 7.14, d (15.5) | 143.2 |
| 4 | 134.1 | 134.2 | ||
| 5 | 6.19, d (9.5) | 143.7 | 5.81, d (10.0) | 137.9 |
| 6 | 2.88, m | 33.9 | 2.83, m | 33.5 |
| 7 | 3.77, dd (9.0, 2.0) | 75.8 | 3.73, m | 76.1 |
| 8 | 1.81, m | 33.9 | 1.77, m | 33.7 |
| 9 | 4.03, d (6.5) | 77.8 | 4.04, d (6.5) | 77.7 |
| 10 | 202.8 | 202.5 | ||
| 11 | 3.47, s | 59.9 | 3.56, s | 56.0 |
| 12 | 56.7 | 59.8 | ||
| 13 | 96.2 | 95.3 | ||
| 14 | 1.44, s | 22.4 | 1.43, s | 23.3 |
| 15 | 1.84, s | 12.0 | 1.79, s | 12.5 |
| 16 | 1.07, d (7.0) | 16.6 | 1.05, d (6.5) | 16.9 |
| 17 | 0.62, d (7.0) | 11.0 | 0.62, d (6.5) | 11.1 |
| 18 | 1.40, s | 15.1 | 3.84, dd (12.5, 3.5) 3.74, dd (12.5, 3.5) | 56.5 |
| 1′ | 8.74, s | 7.58, s | ||
| 2′ | 176.0 | 177.9 | ||
| 3′ | 100.7 | 101.2 | ||
| 4′ | 193.9 | 192.9 | ||
| 5′ | 3.77, dd (9.0, 2.0) 3.75, dd (9.0, 2.0) | 51.4 | 3.47, s | 50.2 |
| 18-OH | 5.02 brs | |||
2.2. Biological Assays
| MIC (μg/mL) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Microorganism | Staphylococcus epidermidis | Staphylococcus aureus | Enterococcu faecium | Enterococcus faecalis | |||||||||||
| Strain No. | ATCC 12228 | 12-6 | 12-8 | ATCC 29213 | ATCC 33591 | 15 | 12-28 | 12-33 | ATCC 700221 | 12-1 | 12-3 | ATCC 29212 | ATCC 51299 | 12-5 | 09-9 |
| Phenotype | MSSE a | MSSE | MRSE b | MSSA c | MRSA d | MSSA | MSSA | MRSA | VRE e | VRE | VSE f | VSE | VRE | VSE | VRE |
| Compound 1 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 64 | 16 | 16 | 16 | 16 |
| Echinomycin | 0.5 | 0.25 | 0.5 | 0.5 | 0.5 | 0.5 | 0.25 | 0.5 | 0.5 | 0.5 | 0.5 | 0.25 | 0.5 | 0.25 | 0.25 |
| Levofloxacin (conrol) | 0.25 | 0.5 | 4 | 0.125 | 0.25 | 0.125 | 0.25 | 64 | 128 | 128 | 64 | 0.5 | 1 | 1 | >128 |
| Compounds | IC50 (μM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACHN | SW1990 | Mia-PaCa-2 | 786-O | U87 MG | SK-N-SH | Jurkat | HCT-116 | NCI-H1650 | HepG2 | BGC-823 | A2780 | |
| Compound 1 | 0.552 | 2.560 | 4.750 | 0.721 | 0.627 | 5.174 | 0.414 | 8.61 | 3.90 | >10 | >10 | >10 |
| Echinomycin | <0.0032 | 0.0026 | <0.0032 | <0.0032 | <0.0032 | 0.027 | <0.0032 | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
3. Experimental Section
3.1. General
3.2. Bacterial Material and Fermentation
3.3. Extraction and Isolation
3.4. Hydrolysis of Compounds 1–2 and HPLC Analysis by Marfey’s Method
3.5. Biological Assays
3.5.1. Antibacterial Activity
3.5.2. Anti-Tumor Assay
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhatnagar, I.; Kim, S.-K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraj, S. Marine Streptomyces as a novel source of bioactive substances. World J. Microb. Biot. 2010, 26, 2123–2139. [Google Scholar] [CrossRef]
- Balachandran, C.; Duraipandiyan, V.; Emi, N.; Ignacimuthu, S. Antimicrobial and cytotoxic properties of Streptomyces sp.(ERINLG-51) isolated from Southern Western Ghats. South Indian J. Biol. Sci. 2015, 1, 7–14. [Google Scholar]
- Sugiyama, R.; Nishimura, S.; Matsumori, N.; Tsunematsu, Y.; Hattori, A.; Kakeya, H. Structure and biological activity of 8-deoxyheronamide C from a marine-derived Streptomyces sp.: Heronamides target saturated hydrocarbon chains in lipid membranes. J. Am. Chem. Soc. 2014, 136, 5209–5212. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.; Carlsson, M.; Uvell, H.; Islam, K.; Edlund, K.; Cullman, I.; Altermark, B.; Mei, Y.F.; Elofsson, M.; Willassen, N.P.; et al. Isolation and characterization of anti-adenoviral secondary metabolites from marine actinobacteria. Mar. Drugs 2014, 12, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Kong, F.D.; Wei, J.J.; Wang, Y.; Wang, W.; Hong, K.; Zhu, W.M. Alkaloids from the Mangrove-Derived Actinomycete Jishengella endophytica 161111. Mar. Drugs 2014, 12, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Ma, L.; Li, S.M.; Liu, Z.; Chen, Y.C.; Zhang, H.B.; Zhang, G.T.; Zhang, Q.B.; Tian, X.P.; Yuan, C.S.; et al. Indimicins A–E, Bisindole Alkaloids from the Deep-Sea-Derived Streptomyces sp. SCSIO 03032. J. Nat. Prod. 2014, 77, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; Liu, W.C.; Zhu, P.; Yang, J.L.; Cheng, K.D. Phylogenetic diversity of bacteria associated with the marine sponge Gelliodes carnosa collected from the Hainan Island coastal waters of the South China Sea. Microb. Ecol. 2011, 62, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.Z.; Gong, T.; Zhu, P. Bioaetive metabolites produced by Streptomyces sp. LS298 isolated from the marine sponge Gelliodes carnosa. Chin. Med. Biotechnol. 2012, 12, 418–425. [Google Scholar]
- Wang, Y.; Liu, Y.; Tang, F.; Bernot, K.M.; Schore, R.; Marcucci, G.; Caligiuri, M.A.; Zheng, P.; Liu, Y. Echinomycin protects mice against relapsed acute myeloid leukemia without adverse effect on hematopoietic stem cells. Blood 2014, 124, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Ryang, Y.S.; Kim, J.B.; Chang, J.-H.; Cho, H.C.; Kim, S.-K. DNA bis-intercalating agent, echinomycin-induced apoptosis via Bcl-2 dependence pathway in human colon cancer cells. Mol. Cell. Toxicol. 2008, 4, 144–149. [Google Scholar]
- Park, Y.S.; Shin, W.S.; Kim, S.K. In vitro and in vivo activities of echinomycin against clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 2008, 61, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Lee, G.S.; Kim, Y.B.; Kim, S.K.; Kim, Y.H. In vitro antibacterial activity of echinomycin and a novel analogue, YK2000, against vancomycin-resistant enterococci. Int. J. Antimicrob. Agents 2004, 24, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikai, Y.; Mayumi, T.; Oka, H.; Suzuki, M.; Harada, K.-I. A Nonempirical Method Using LC/MS for Determination of the Absolute Configuration of Constituent Amino Acids in a Peptide: Elucidation of Limitations of Marfey’s Method and of Its Separation Mechanism. Anal. Chem. 1997, 69, 3346–3352. [Google Scholar] [CrossRef]
- Song, Y.X.; Li, Q.L.; Liu, X.; Chen, Y.C.; Zhang, Y.; Sun, A.J.; Zhang, W.M.; Zhang, J.R.; Ju, J.H. Cyclic Hexapeptides from the Deep South China Sea-Derived Streptomyces scopuliridis SCSIO ZJ46 Active Against Pathogenic Gram-Positive Bacteria. J. Nat. Prod. 2014, 77, 1937–1941. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.J.; Rinehart, K.L., Jr. C NMR spectra of streptolydigin, tirandamycin, and related degradation products. J. Antibiot. (Tokyo) 1980, 33, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.C.; Li, S.; Burr, D.A.; Sherman, D.H. Isolation and characterization of tirandamycins from a marine-derived Streptomyces sp. J. Nat. Prod. 2009, 72, 2076–2079. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Yu, Z.G.; Yan, Y.J.; Yang, D.; Huang, T.T.; Vodanovic-Jankovic, S.; Kron, M.A.; Shen, B. Medium optimization of Streptomyces sp. 17944 for tirandamycin B production and isolation and structural elucidation of tirandamycins H, I and J. J. Antibiot. (Tokyo) 2014, 67, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.G.; Vodanovic-Jankovic, S.; Ledeboer, N.; Huang, S.X.; Rajski, S.R.; Kron, M.; Shen, B. Tirandamycins from Streptomyces sp. 17944 inhibiting the parasite Brugia malayi asparagine tRNA synthetase. Org. Lett. 2011, 13, 2034–2037. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.R.; Yao, Y.L.; Wang, Z.W.; Tian, X.P.; Zhang, S.; Zhang, C. Fermentation optimization, isolation and identification of tirandamycim A and B from marine-derived Streptomyces sp. SCSIO 1666. Chin. J. Mar. Drugs 2010, 29, 12–20. [Google Scholar]
- MacKellar, F.A.; Grostic, M.F.; Olson, E.C.; Wnuk, R.J. Tirandamycin. I. Structure assignment. J. Am. Chem. Soc. 1971, 93, 4943–4945. [Google Scholar] [PubMed]
- Feng, Z.M.; He, J.; Jiang, J.S.; Chen, Z.; Yang, Y.N.; Zhang, P.C. NMR solution structure study of the representative component hydroxysafflor yellow A and other quinochalcone C-glycosides from Carthamus tinctorius. J. Nat. Prod. 2013, 76, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Brill, G.M.; McAlpine, J.B.; Whittern, D. Tirandalydigin, a novel tetramic acid of the tirandamycin-streptolydigin type. II. Isolation and structural characterization. J. Antibiot. (Tokyo) 1988, 41, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Asano, K.; Kawamoto, I.; Tamaoki, T.; Nakano, H. UCN-01 and UCN-02, new selective inhibitors of protein kinase C.I. Screening, producing organism and fermentation. J .Antibiot. (Tokyo) 1989, 42, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Wikler, M.A. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard, 8th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2009. [Google Scholar]
- Wu, C.Y.; Tan, Y.; Gan, M.L.; Wang, Y.G.; Guan, Y.; Hu, X.X.; Zhou, H.X.; Shang, X.Y.; You, X.F.; Yang, Z.Y.; et al. Identification of elaiophylin derivatives from the marine-derived actinomycete Streptomyces sp. 7-145 using PCR-based screening. J. Nat. Prod. 2013, 76, 2153–2157. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- He, L.; Liu, Y.M.; Shi, J.G.; Pei, Q. Synthesis and antitumor activity of cholest-4α-methyl-7-en-3β-ol derivatives. Steroids 2006, 71, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Wang, D.X.; Chen, R.Y.; Liu, P.; Yu, D.Q. Novel benzil and isoflavone derivatives from Millettia dielsiana. Planta Med. 2009, 75, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.E. Tirandamycin, a new antibiotic isolation and characterization. J. Antibiot. (Tokyo) 1971, 24, 558–560. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhen, X.; Gong, T.; Liu, F.; Zhang, P.-C.; Zhou, W.-Q.; Li, Y.; Zhu, P. A New Analogue of Echinomycin and a New Cyclic Dipeptide from a Marine-Derived Streptomyces sp. LS298. Mar. Drugs 2015, 13, 6947-6961. https://doi.org/10.3390/md13116947
Zhen X, Gong T, Liu F, Zhang P-C, Zhou W-Q, Li Y, Zhu P. A New Analogue of Echinomycin and a New Cyclic Dipeptide from a Marine-Derived Streptomyces sp. LS298. Marine Drugs. 2015; 13(11):6947-6961. https://doi.org/10.3390/md13116947
Chicago/Turabian StyleZhen, Xin, Ting Gong, Fu Liu, Pei-Cheng Zhang, Wan-Qi Zhou, Yan Li, and Ping Zhu. 2015. "A New Analogue of Echinomycin and a New Cyclic Dipeptide from a Marine-Derived Streptomyces sp. LS298" Marine Drugs 13, no. 11: 6947-6961. https://doi.org/10.3390/md13116947
APA StyleZhen, X., Gong, T., Liu, F., Zhang, P.-C., Zhou, W.-Q., Li, Y., & Zhu, P. (2015). A New Analogue of Echinomycin and a New Cyclic Dipeptide from a Marine-Derived Streptomyces sp. LS298. Marine Drugs, 13(11), 6947-6961. https://doi.org/10.3390/md13116947