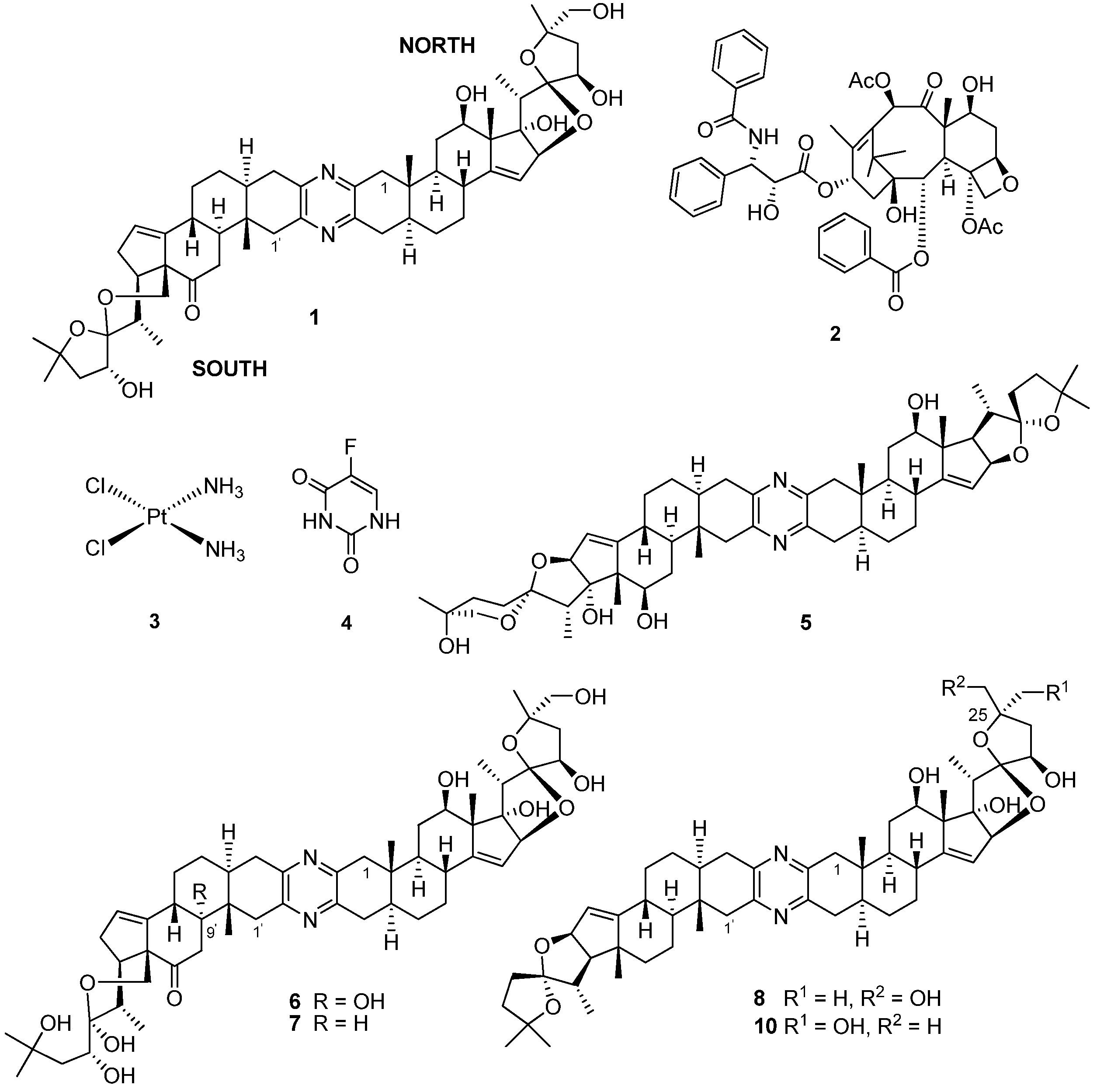
2. Tumor Growth Inhibiting Cephalostatins from the South African Marine Tube Worm Cephalodiscus gilchristi
The first large-scale collections of African marine invertebrates solely for the purpose of new drug discovery were coordinated by Professor G. R. Pettit of Arizona State University, USA, over three decades ago off South Africa’s temperate southern coast [
5]. Cephalostatin 1 (
1,
Figure 2) was isolated in low yield (
ca. 2.3 × 10
−7%) from two separate and substantial SCUBA collections (166 and 450 kg (wet weight) collected in 1981 and 1990, respectively) of the hemichordate marine tube worm
Cephalodiscus gilchristi (
Figure 1a) [
6]. Cephalostatin 1 has emerged as one of the most potent cell growth-inhibiting secondary metabolites ever screened by the U.S. National Cancer Institute (NCI) (ED
50 0.1–0.0001 pM in a P338 leukemia cell line) [
6,
7].
Of immediate interest to those exploring this compound’s tumor growth inhibitory activities was, first, the comparative GI
50 values (quantification of the concentration required to inhibit cellular growth by 50%) of
1 (GI
50 1.2 nM) with commercially available anticancer drugs, e.g., taxol (
2, GI
50 29 nM), cisplatin (
3, GI
50 2000 nM) and 5-fluorouracil (
4, 24,000 nM), and, second, the 275-times higher concentration of
1 required to kill 50% of cancer cells (LC
50 330 nM) relative to the amount required for 50% cell growth inhibition [
6]. In addition, the application of the NCI’s COMPARE algorithm [
8] to the GI
50 data acquired for
1 indicated that this novel bis-steroidal pyrazine alkaloid possesses a unique mechanism of action against the proliferation of cancer cells in the NCI’s
in vitro 60 cancer cell line screen, and therefore, not surprisingly,
1 is increasingly proving to be a valuable tool for the discovery of new apoptosis signaling pathways [
9]. Vollmar and co-workers’ early studies into cephalostatin’s apoptotic mechanism of action established that
1 promotes the release of Smac (second mitochondria-derived activator of caspase) through the dissipation of mitochondrial membrane potential [
6,
9,
10] as part of a novel apoptosome-independent, caspase-9-mediated apoptotic pathway [
6]. Furthermore, Shair and co-workers have shown that
1 also selectively binds to oxysterol binding protein (OSBP) and OSBP-related protein 4L (ORP4L) [
11] and drew attention to these proteins, whose role in cancer cell survival was little known at the time. A further eighteen naturally-occurring and semi-synthetic analogues of
1 have subsequently been reported (1988–2012) in the chemical and patent literature (e.g., U.S. Patents 4873245, 5047532, 5583224 and WO 8908655). The isolation, structure elucidation, synthesis and bioactivity of this cohort of cephalostatins has been comprehensively reviewed along with the closely-related bis-steroidal pyrazine alkaloids, the ritterazines, e.g., ritterazine G, (
5) from the Japanese ascidian (tunicate),
Ritterella tokioka [
6]. Since the publication of Iglesias-Arteaga and Morzycki’s extensive review [
6], the chemical structure of the twentieth member of the cephalostatin series, cephalostatin 20 (
6), has recently been reported by Pettit
et al. [
12]. Compound
6, the 9′-α-hydroxy analog of cephalostatin 9 (
7), was isolated in low yield (1 × 10
7%) from the combined bioactive (cytotoxic to P338 murine lymphocyte cells) fractions from the original extract of
C. gilchristi [
5] nearly a quarter of a century ago. Interestingly, the cell growth inhibitory activities of
6 and
7 against six human tumor cell lines was 100–1000-times less active than
1 in the same tumor cell panel, thus underlining the importance of an intact spirostanol structure in the southern unit of cephalostatins to the growth inhibition activities of these compounds [
12].
Figure 2.
Chemical structures of compounds 1–8 and 10.
Figure 2.
Chemical structures of compounds 1–8 and 10.
Significant effort [
6,
13,
14] has been directed towards the total enantioselective syntheses of
1 over the last two decades. Following on from their first 65-step convergent total synthesis of
1 and potently active cephalostatin/ritterazine hybrids [
15], Fuchs and co-workers have recently reported the first convergent total synthesis of 25-
epi ritterostatin G
N1
N 8 [
16] from commercially available dihydroxyhecogenin acetate (
9,
Figure 3). Fuchs and co-workers identified the key step in their synthesis as a chiral ligand ((DHQ)
2PHAL)-mediated dihydroxylation reaction, which introduced the 25-
epi functionality into the north segment (analogous to the north unit of cephalostatin) [
16]. Compound
8, structurally incorporating the north units of both
1 and
5, exhibited a mean GI
50 (0.48 nM) in a panel of eight cancer cell lines and was 30-fold more active than ritterostatin (
10), also screened in the same cell line panel [
16].
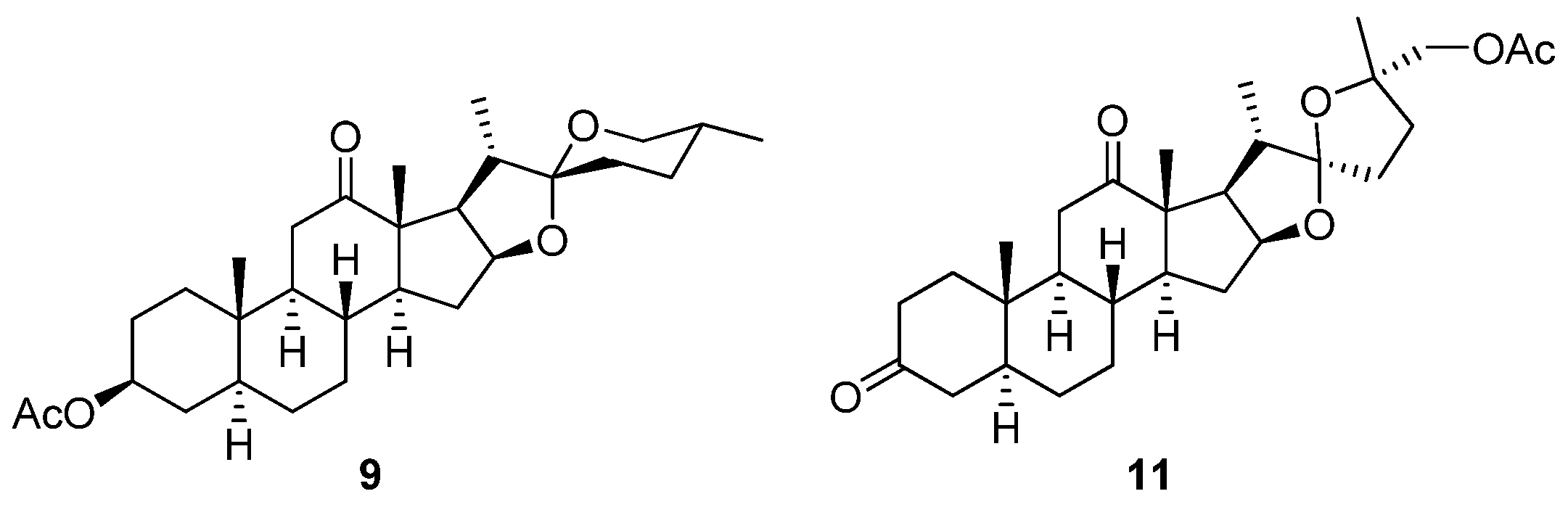
Figure 3.
Chemical structures of synthetic intermediates 9 and 11.
Figure 3.
Chemical structures of synthetic intermediates 9 and 11.
The daunting synthetic challenges of cephalostatin molecular architecture continue to inspire the synthesis of simpler analogs with similar bioactivities to
1 [
6]. The latest target in this series, [5.5]-spiroketal (
11,
Figure 3), which shares the steroidal scaffold of the northern hemisphere of
1 with an intact 1,6-dioxaspiro[5.5]nonane side chain, but with a diminished oxygenation pattern, was synthesized by Pettit
et al. in seven steps from
9 in an overall 4.6% yield [
17]. Although
11 and several synthetic precursors of this compound were not cytotoxic to P388 leukemia cells, Pettit
et al. suggested that
9 was potentially useful as a synthetically-accessible starting point for further synthetic modification into both symmetrical and asymmetrical trisdecacyclic bis-steroidal pyrazine congeners of
1 [
17] through well-established pyrazine ring construction protocols [
16].
3. Synthesis and Revision of the Absolute Configuration of the Cytotoxic Mandelalides from the South African Marine Ascidian, Lissoclinum sp.
The encrusting colonial didemnid ascidian
Lissoclinum sp. (
Figure 1b) collected by SCUBA from Algoa Bay, on the southeast coast of South Africa, afforded sub-milligram (0.5–0.8 mg) quantities of the glycosylated, polyketide macrolides, the mandelalides A–D (
12a,
13–
15,
Figure 4). Mandelalides A and B exhibited potent low nanomolar cytotoxicity (IC
50 12 and 44 nM, respectively) against NCI-H460 lung cancer cells [
18]. The relative configuration of the macrolide rings in
12a,
13–
15 was established through integration of ROESY data with homonuclear (
3JHH) and heteronuclear (
2,3JCH) coupling constants, while the absolute configurations of
12a and
13 were extrapolated from the hydrolysis and subsequent chiral GC-MS analysis of the respective monosaccharide residues (2-
O-methyl-6-dehydro-α-L-rhamnose and 2-
O-methyl-6-dehydro-α-L-talose). The paucity of
12a,
13–
15 isolated from the MeOH-CH
2Cl
2 extract of
Lissoclinum sp. (
i.e., 0.8 mg of
12a) and difficulties encountered in the further supply of these compounds from their natural source implied that the synthesis of
12a (the most active compound in the mandelalide series) would provide sufficient quantities of
12a to explore the mechanism of
in vitro cytotoxicity exhibited by this compound. As described below, the synthesis of
12a and the diastereomer
16 by Willwacher
et al. and Xu, Ye and co-workers revealed errors in the original assignment of the absolute configuration at positions C17, C18 C20, C21 and C23 and resulted in the correction of the chemical structure of mandelalide A (
12a) to
16 [
19,
20].
In 2014, two years after the isolation of the mandelalides was first reported [
18], Willwacher and Fürstner reported the first total synthesis of
12a in a 4.5% overall yield [
21]. They also noted the structural similarities between
12a and madeirolide A (
17,
Figure 4), an equally scarce metabolite previously isolated from a marine sponge,
Leiodermatium sp. [
22,
23], and ascribed the absence of anti-proliferative activity against pancreatic cancer cells reported for
17 to the structural differences between these two compounds. Anticipating in their proposed synthesis of
12a that final closure of the macrolide ring, concomitant with insertion of the ∆
14 Z-olefin, could be achieved with ring closing alkyne metathesis (RCAM), Willwacher and Fürstner successfully synthesized the two main building blocks (
18) and (
19) emerging from their retrosynthetic analysis of
12a. Cobalt-catalyzed carbonylative epoxide opening and iridium-catalyzed two-directional Krische allylation were identified as key synthetic steps required for the synthesis of
18 and
19, respectively, while the RCAM protocol would not have been possible without the use of a highly-selective molybdenum alkylidene complex catalyst; the first time that this catalyst has been successfully incorporated into a natural product total synthesis [
21]. Finally, regioselective trimethylsilyl trifluoromethanesulfonate (TESOTf)-catalyzed rhamnosylation of the mandelalide aglycone proceeded smoothly to afford mandelalide A with the chemical structure
12a, originally proposed by McPhail and co-workers [
18].
Figure 4.
Chemical structures of compounds 12a–19.
Figure 4.
Chemical structures of compounds 12a–19.
Comparing the spectroscopic data acquired for their synthetic product with those of naturally-occurring mandelalide A (
16), Willwacher and Fürstner noted significant chemical shift and coupling constant differences between the NMR datasets of synthetic
12a and the natural product (
Table 1 and
Table 2). Initially, given the relative magnitude of the observed differences, their attention was focused on differences in the
13C chemical shift assigned to the C11 methine and the C25 methyl carbon atoms (
Table 1) and discrepancies in the
3J11,12 coupling constants (
Table 2;
J11,12 = 9.7 and 7.6 Hz, respectively, for naturally-occurring mandelalide A and
12a, respectively) associated with the C11 stereogenic center. However, synthesis of the 11-
epi-diasteromer of
12a (
12b) did not provide clarity on the source of spectroscopic differences between the synthetic and natural products, and Willwacher and Fürstner were at a loss to explain where the anomalies resided in the proposed structure of
12a [
21]. Reddy
et al. [
24] also tackled the synthesis of the aglycone of
12a, reflected in the original structure proposed by McPhail and co-workers [
18]. Their 32-step synthesis afforded the putative mandelalide A aglycone in a 6.3% overall yield, and the spectroscopic data acquired for the synthetic product was consistent with the analogous data for the aglycone of
12a synthesized by Willwacher and Fürstner.
Table 1.
Comparative
13C NMR data (CDCl
3, 175
† and 150
‡ MHz) reported by McPhail and co-workers [
18] for naturally-occurring
16; by Willwacher
et al. [
20,
21] for synthetic
12a,
12b and
16; and by Xu, Ye and co-workers [
19] for synthetic
12a and
16.
Table 1.
Comparative 13C NMR data (CDCl3, 175 † and 150 ‡ MHz) reported by McPhail and co-workers [18] for naturally-occurring 16; by Willwacher et al. [20,21] for synthetic 12a, 12b and 16; and by Xu, Ye and co-workers [19] for synthetic 12a and 16.
| Carbon | Naturally-Occurring 16 [18] † | Synthetic 12a [21] ‡ | Synthetic 12b [21] ‡ | Synthetic 12a [19] ‡ | Synthetic 16 [19] ‡ | Synthetic 16 [20] ‡ |
|---|
| 1 | 167.4 | 167.3 | 166.8 | 167.1 | 167.4 | 167.4 |
| 2 | 123.1 | 123.1 | 123.6 | 122.9 | 123.0 | 123.1 |
| 3 | 147.1 | 146.3 | 146.1 | 146.2 | 147.2 | 147.1 |
| 4 | 38.8 | 38.5 | 39.5 | 38.2 | 38.8 | 38.8 |
| 5 | 73.9 | 73.4 | 73.9 | 73.2 | 73.9 | 73.9 |
| 6 | 37.6 | 36.7 | 38.2 | 36.4 | 37.6 | 37.6 |
| 7 | 73.1 | 72.8 | 72.7 | 72.6 | 73.1 | 73.1 |
| 8 | 39.7 | 39.3 | 39.2 | 39.0 | 39.7 | 39.7 |
| 9 | 72.5 | 73.1 | 73.2 | 72.9 | 72.5 | 72.5 |
| 10 | 43.1 | 42.9 | 43.5 | 42.6 | 43.1 | 43.1 |
| 11 | 34.2 | 32.8 | 34.1 | 32.6 | 34.2 | 34.2 |
| 12 | 141.5 | 140.9 | 141.3 | 140.6 | 141.6 | 141.5 |
| 13 | 123.9 | 123.8 | 124.9 | 123.6 | 123.9 | 123.9 |
| 14 | 131.3 | 130.5 | 130.6 | 130.3 | 131.3 | 131.3 |
| 15 | 126.9 | 126.5 | 126.2 | 126.3 | 127.0 | 126.9 |
| 16 | 31.1 | 31.2 | 31.0 | 31.0 | 31.1 | 31.1 |
| 17 | 81.0 | 81.3 | 81.8 | 81.1 | 81.0 | 81.0 |
| 18 | 37.3 | 37.1 | 36.9 | 36.9 | 37.4 | 37.4 |
| 19 | 36.8 | 36.0 | 36.4 | 35.8 | 36.8 | 36.8 |
| 20 | 83.2 | 82.7 | 82.1 | 82.5 | 83.2 | 83.2 |
| 21 | 73.0 | 73.4 | 73.3 | 72.3 | 73.0 | 73.1 |
| 22 | 34.1 | 34.1 | 34.7 | 33.9 | 34.1 | 34.1 |
| 23 | 72.3 | 72.5 | 74.0 | 72.3 | 72.3 | 72.3 |
| 24 | 66.1 | 65.7 | 65.7 | 65.6 | 66.1 | 66.1 |
| 25 | 18.3 | 20.1 | 22.0 | 19.9 | 18.3 | 18.3 |
| 26 | 14.5 | 14.7 | 14.9 | 14.6 | 14.6 | 14.5 |
| 1′ | 94.2 | 94.0 | 94.1 | 93.7 | 94.2 | 94.2 |
| 2′ | 80.8 | 80.9 | 80.9 | 80.6 | 80.8 | 80.8 |
| 3′ | 71.7 | 71.7 | 71.6 | 71.4 | 71.7 | 71.7 |
| 4′ | 74.3 | 74.2 | 74.2 | 74.0 | 74.3 | 74.3 |
| 5′ | 68.1 | 68.2 | 68.2 | 67.9 | 68.1 | 68.1 |
| 6′ | 17.7 | 17.7 | 17.7 | 17.5 | 17.7 | 17.7 |
| 7′ | 59.1 | 59.2 | 59.1 | 59.0 | 59.2 | 59.1 |
Table 2.
Comparative
1H NMR data (CDCl
3, 700
† and 600
‡ MHz) reported by McPhail and co-workers [
18] for naturally-occurring
16 and by Willwacher
et al. [
20,
21] for synthetic
12a and
16.
Table 2.
Comparative 1H NMR data (CDCl3, 700 † and 600 ‡ MHz) reported by McPhail and co-workers [18] for naturally-occurring 16 and by Willwacher et al. [20,21] for synthetic 12a and 16.
| Naturally-Occurring 16 [18] † | Synthetic 12a [21] ‡ | Synthetic 16 [20] ‡ |
|---|
| No. | δ (ppm) | mult. | J (Hz) | δ (ppm) | mult. | J (Hz) | δ (ppm) | mult. | J (Hz) |
| 1 | | | | | | | | | |
| 2 | 6.01 | dd | 15.5, 1.2 | 5.92 | dt | 15.6, 1.5 | 6.01 | dt | 15.5, 0.8 |
| 3 | 6.97 | ddd | 15.2, 10.4, 4.6 | 7.02 | ddd | 15.5, 8.6, 5.5 | 6.96 | ddd | 15.3, 10.4, 4.9 |
| 4a | 2.36 | m | | 2.34 | dddd | 15.2, 6.5, 5.6, 1.8 | 2.36 | m | |
| 4b | 2.39 | ddd | 14.1, 10.6, 10.6 | 2.46 | dddd | 15.2, 8.6, 3.7, 1.2 | 2.39 | ddd | 13.9, 10.8, 10.7 |
| 5 | 3.36 | dddd | 11.4, 11.4, 2.3, 2.3 | 3.42 | m | | 3.37 | m | |
| 6a | 1.20 | m | | 1.26 | m | | 1.20 | m | |
| 6b | 2.02 | dddd | 12.6, 4.4, 2.3, 1.6 | 1.94 | ddt | 12.0, 4.6, 1.9 | 2.02 | dddd | 12.1, 5.6, 2.3, 1.6 |
| 7 | 3.82 | dddd | 11.1, 10.5, 4.4, 4.4 | 3.77 | m | | 3.82 | dddd | 11.3, 10.6, 4.8, 4.5 |
| 8a | 1.22 | m | | 1.22 | m | | 1.22 | m | |
| 8b | 1.87 | m | | 1.84 | dddd | 12.5, 4.2, 1.9, 1.9 | 1.87 | dddt | 13.2, 7.8, 5.3, 1.9 |
| 9 | 3.32 | dddd | 11.2, 11.2, 2.2, 2.2 | 3.33 | m | | 3.31 | tt | 10.7, 2.1 |
| 10a | 1.21 | ddd | 15.2, 9.6, 2.2 | 1.27 | m | | 1.21 | m | |
| 10b | 1.51 | ddd | 15.2, 11.2, 3.7 | 1.69 | ddd | 14.1, 9.1, 5.1 | 1.52 | ddd | 14.1, 11.1, 3.3 |
| 11 | 2.37 | dqd | 9.6, 6.5, 3.7 | 2.44 | m | | 2.37 | m | |
| 12 | 5.45 | dd | 14.8, 9.7 | 5.61 | dd | 15.2, 7.6 | 5.44 | dd | 14.9, 9.9 |
| 13 | 6.28 | dd | 14.8, 9.7 | 6.22 | ddt | 15.2, 10.8, 1.0 | 6.27 | dd | 14.8, 11.1 |
| 14 | 6.05 | dd | 10.9, 10.9 | 6.01 | tt | 10.8, 1.8 | 6.05 | dd | 10.9, 10.9 |
| 15 | 5.28 | ddd | 10.8, 10.8, 5.6 | 5.27 | ddd | 10.8, 8.3, 7.5 | 5.28 | dt | 10.8, 5.6 |
| 16a | 1.88 | m | | 2.14 | dddd | 14.8, 6.8, 5.1, 1.9 | 1.88 | m | |
| 16b | 2.28 | ddd | 13.1, 11.4, 11.4 | 2.29 | dtd | 14.8, 8.5, 1.6 | 2.25 | m | |
| 17 | 3.98 | ddd | 11.1, 8.1, 1.8 | 4.03 | ddd | 8.6, 7.2, 4.9 | 3.98 | ddd | 10.9, 8.5, 1.7 |
| 18 | 2.52 | dddq | 12.0, 7.0, 7.0 | 2.43 | m | | 2.52 | dddq | 12.3, 7.0, 7.0, 6.9 |
| 19a | 1.17 | ddd | 11.9, 11.9, 10.3 | 1.28 | m | | 1.17 | ddd | 12.2, 12.1, 10.2 |
| 19b | 2.01 | ddd | 12.2, 7.0, 5.6 | 2.04 | dt | 12.3, 6.7 | 2.01 | ddd | 11.8, 7.1, 6.0 |
| 20 | 3.63 | m | | 3.71 | ddd | 8.4, 8.2, 6.7 | 3.63 | m | |
| 21 | 3.42 | ddd | 11.1, 8.8, 1.8 | 3.45 | m | | 3.42 | ddd | 11.2, 8.9, 1.8 |
| 22a | 1.46 | ddd | 14.1, 11.1, 1.9 | 1.54 | ddd | 14.4, 10.5, 2.5 | 1.46 | ddd | 14.2, 11.3, 1.9 |
| 22b | 1.76 | ddd | 13.9, 11.7, 1.8 | 1.77 | ddd | 14.4, 10.8, 2.0 | 1.76 | ddt | 12.8, 12.6, 1.5 |
| 23 | 5.23 | dddd | 11.7, 4.9, 2.9, 1.9 | 5.24 | m | | 5.23 | dddd | 11.6, 5.1, 3.1, 2.0 |
| 24a | 3.61 | m | | 3.65 | m | | 3.61 | m | |
| 24b | 3.81 | dd | 12.1, 2.9 | 3.78 | dd | 12.1, 3.3 | 3.79 | m | |
| 25 | 0.85 | d | 6.6 | 1.00 | d | 6.7 | 0.85 | d | 6.6 |
| 26 | 1.03 | d | 6.9 | 0.98 | d | 7.0 | 1.02 | d | 7.0 |
| 1′ | 5.02 | d | 1.1 | 5.02 | d | 1.5 | 5.02 | d | 1.1 |
| 2′ | 3.40 | dd | 3.8, 1.4 | 3.40 | dd | 3.8, 1.5 | 3.40 | dd | 3.4, 1.5 |
| 3′ | 3.68 | m | | 3.69 | m | | 3.68 | td | 9.8, 3.7 |
| 4′ | 3.34 | dd | 9.4, 9.4 | 3.34 | t | 9.4 | 3.34 | dd | 10.5, 9.3 |
| 5′ | 3.62 | m | | 3.63 | dd | 9.4, 6.1 | 3.62 | dd | 9.9, 5.9 |
| 6′ | 1.27 | d | 6.3 | 1.28 | d | 6.3 | 1.26 | d | 6.3 |
| 7′ | 3.45 | s | | 3.46 | s | | 3.45 | s | |
| OH-1′ | | | | 2.45–2.33 | | | 2.69 | br s | |
| OH-2′ | | | | 2.56–2.33 | | | 2.31 | br s | |
| OH-3′ | 2.24 | s | | 2.44–2.34 | | | 2.35 | m | |
| OH-4′ | 1.54 | s | | 2.78–2.64 | br s | | 2.53 | br s | |
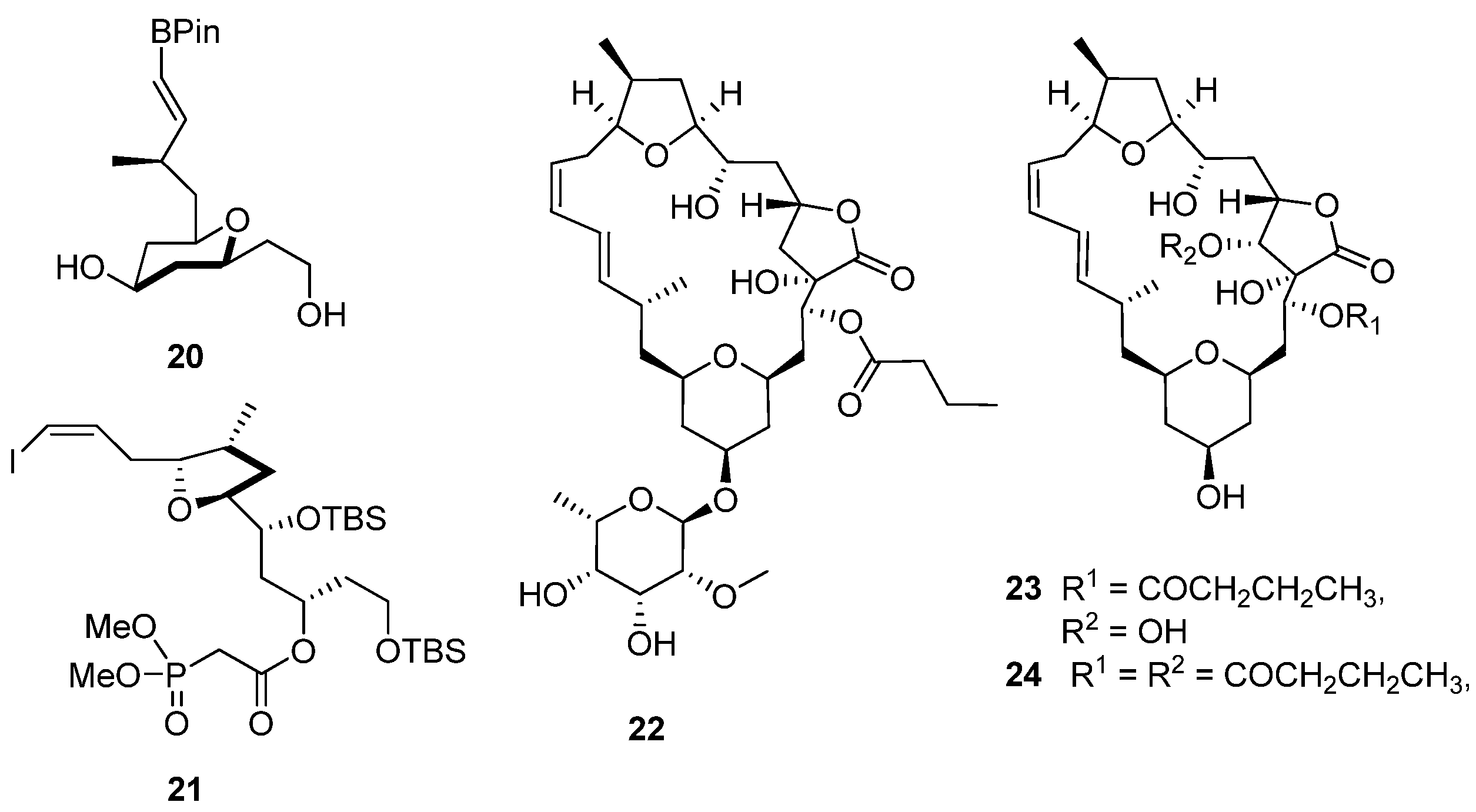
A second synthesis of
12a by Xu, Ye and co-workers [
19] was published in Angewandte Chemie International Edition shortly after Willwacher and Fürstner’s synthetic communication appeared in the same volume of the journal. Approaching the synthesis of the 24-membered macrocycle via a different route to that used by Willwacher and Fürstner; Xu, Ye and co-workers initially constructed the two sub-units (
20 and
21,
Figure 5) with Prins cyclization, providing diastereoselective access to the tetrahydropyran moiety in
20 and a Rychnovsky–Bartlett cyclization generating the tetrahydrofuran ring in
21. As anticipated from the retrosynthetic analysis that guided this synthetic approach, both subunits were successfully assembled into the aglycone via Suzuki coupling and Horner–Wadsworth–Emmons macrocyclization. The synthesis of
12a was concluded with the addition of a protected rhamnose moiety to the mandelalide aglycone via a Kahne glycosylation reaction followed by a single-step collective removal of the silyl protecting groups. Xu, Ye and co-workers also identified the incompatibility of the NMR datasets acquired for their synthetic product and naturally-occurring mandelalide A, including the significant chemical shift differences associated with the C11 and C25 carbon atoms (
Table 1 and
Table 2). However, from direct comparison of the opposite configurations of the stereogenic centers in the northern hemisphere of
12a and
17, they correctly postulated that, assuming
12a and
17 share a common biogenesis, the differences in absolute configuration would probably be confined to this part of the molecule and not C11, to which an
S configuration was assigned in both
12a and
17. The convergent synthetic approach to
12a by Xu, Ye and co-workers enabled them to synthesize the diastereomer,
16, with opposite configurations at positions C17, C18 C20, C21 and C23 to those initially reported for
12a [
18] and consistent with the configurations assigned to the analogous chiral centers in
17 [
19]. Willwacher
et al. also recently reported a further synthesis of
16 [
20] and confidently postulated revised structures for mandelalide B–D
(22–
24,
Figure 5). Comparison of the NMR data of naturally-occurring mandelalide A with those of
16 (
Table 1 and
Table 2) confirmed that these two compounds were identical and that any uncertainties around the correct structure of mandelalide A had been successfully resolved.
Figure 5.
Chemical structures of compounds 20–25.
Figure 5.
Chemical structures of compounds 20–25.
Although the chemical structure of mandelalide A has now been unequivocally established as
16, the inconsistency in the cytotoxicity data reported for this compound remains unresolved. McPhail and co-workers reported that the naturally-occurring mandelalides A and B possessed potent cytotoxicity against human NCI-H460 lung cancer cells (IC
50 12 and 44 nM, respectively) and Neuro-2A neuroblastoma cells (IC
50 29 and 84 nM, respectively) [
18]. These results are at variance with the reported lack of cytotoxicity exhibited by synthetic
16 when screened against a panel of ten cancer cell lines of different histological origin by Xu, Ye and co-workers [
19] (
Table 3). Willwacher
et al. also noted the negligible cytotoxicity of
16 against cancer cell lines with the exception of a single human breast carcinoma cell line [
20] (
Table 3).
Table 3.
Comparative IC
50 data (μM) reported by McPhail and co-workers [
18] for naturally-occurring
16 and by Xu, Ye and co-workers [
19] and Willwacher
et al. [
20] for synthetic
16.
Table 3.
Comparative IC50 data (μM) reported by McPhail and co-workers [18] for naturally-occurring 16 and by Xu, Ye and co-workers [19] and Willwacher et al. [20] for synthetic 16.
| Cell Line | Histological Origin | Naturally-Occurring 16 [18] | Synthetic 16 [19] | Synthetic 16 [20] |
|---|
| Neuro-2A | Neuro | 0.044 | | |
| NCI-H460 | Lung | 0.012 | | |
| H1299 | Lung | | 25.5 | |
| PC-3 | Prostate | | 108.7 | |
| PLC/PRF/5 | Liver | | 140.6 | |
| MHCC97L | Liver | | >500 | |
| HeLa | Cervix | | 249.0 | |
| SH-SY5Y | Brain | | >500 | |
| HCT 116 | Colon | | >500 | |
| HT-29 | Colon | | >500 | >1000 |
| MCF7 | Breast | | 271.5 | |
| MDA-MB-361-DYT2 | Breast | | | 0.041 |
| N87 | Stomach | | | 0.206 |
4. Bisindole Alkaloid Inhibitors of Methicillin-Resistant Staphylococcus aureus Pyruvate Kinase from the South African Marine Sponge Topsentia sp.
Methicillin-resistant
Staphylococcus aureus (MRSA), euphemistically also referred to as the “super bug”, was initially encountered in public healthcare facilities and remains a significant cause of mortality in these facilities. MRSA is no longer confined to healthcare facilities and has been increasingly reported from the general population and domestic livestock worldwide [
25,
26,
27]. Annually, MRSA accounts for
ca. 94,000 infections and 18,000 deaths in the USA and 150,000 infections in the EU [
27]. There are no available MRSA mortality data from Southern Africa. However, a 2015 study conducted in three South African academic hospitals reported a MRSA prevalence rate (MRSA infections as a % of all recorded
S. aureus infections) of 36%, which is comparable to Israel (33.5%) Ireland (38.1%) and the U.K. (35.5%) [
28]. The escalating infection and mortality rates associated with the ongoing spread of drug-resistant pathogenic bacteria, e.g., MRSA, are further exacerbated by the dearth of new antibiotics entering the clinic [
29].
Paradoxically, the targeting of bacteria-specific proteins in new antibacterial drug development programs is problematic given the concomitant selective pressure that drugs, emerging from this classic drug discovery approach, exert on the pathogens, leading to the proliferation of drug-resistant bacterial strains [
30]. Protein target-based antibiotic drug discovery is, however, not redundant. Contemporary genomic and proteomic studies of MRSA [
31,
32] have increased our understanding of the complex protein-protein interaction networks (interactomes) in this organism. The detailed mapping of interactomes has led to the identification of highly-connected hub proteins, which, given their centrality within the interactomes, are essential for mediating key cellular processes and sustaining MRSA viability [
30,
31]. Out of necessity, hub proteins are evolutionarily-conserved proteins, given the deleterious effect that mutations of hub proteins would have on the complex interactomes in which they play a key role [
30]. Therefore, targeting hub proteins within the MRSA interactomes will minimize the potential for the emergence of drug resistance in MRSA and is a novel strategy for developing much needed new chemotherapeutic interventions against this drug-resistant pathogen [
31]. Amongst the suite of hub proteins in a 608-protein interactome network (comprising 23% of the proteome in a hospital-acquired strain of MRSA), Zoraghi
et al. identified pyruvate kinase (PK) as a suitable target for possible antibiotic drug discovery [
30]. Catalyzing the rate-limiting irreversible conversion of phosphoenolpyruvate into pyruvate during glycolysis, pyruvate kinases are, not surprisingly, ubiquitous in both prokaryotes and eukaryotes. Fortuitously, the MRSA PK homotetramer (
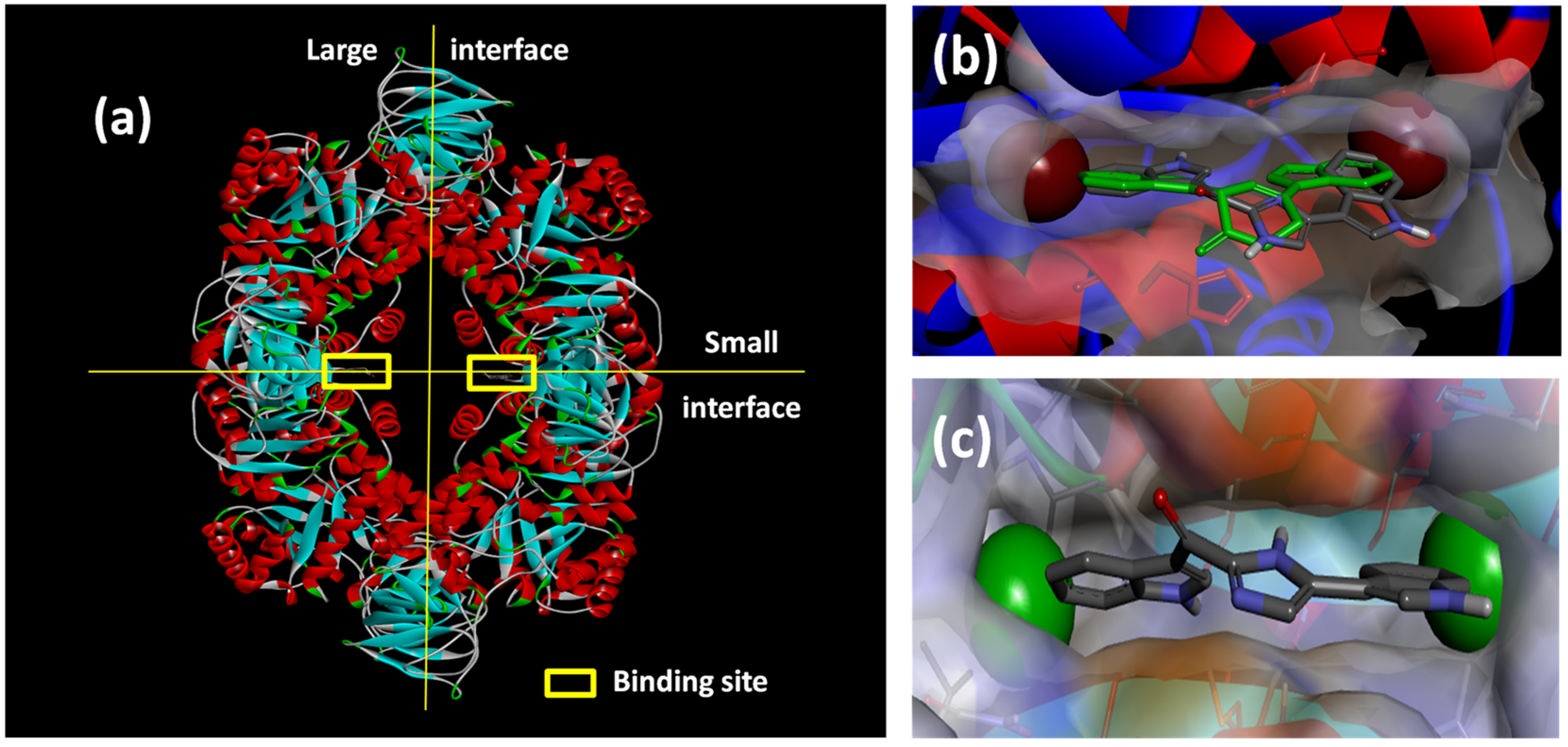
Figure 6a) has several possible lipophilic binding pockets that are absent in human PK orthologs, allowing potential selective inhibition of this enzyme target [
33]. Initially, two parallel strategies were used to generate lead compounds to exploit the inhibition of this key enzyme. The first strategy involved the random screening of >900 marine invertebrate extracts, including those from South African marine invertebrates, for selective MRSA PK inhibition. The second rational drug design strategy coupled knowledge of the detailed structure of the MRSA PK enzyme binding site with contemporary computer-aided drug design techniques to generate new synthetic MRSA PK inhibitors. Both strategies are reviewed in more detail below.
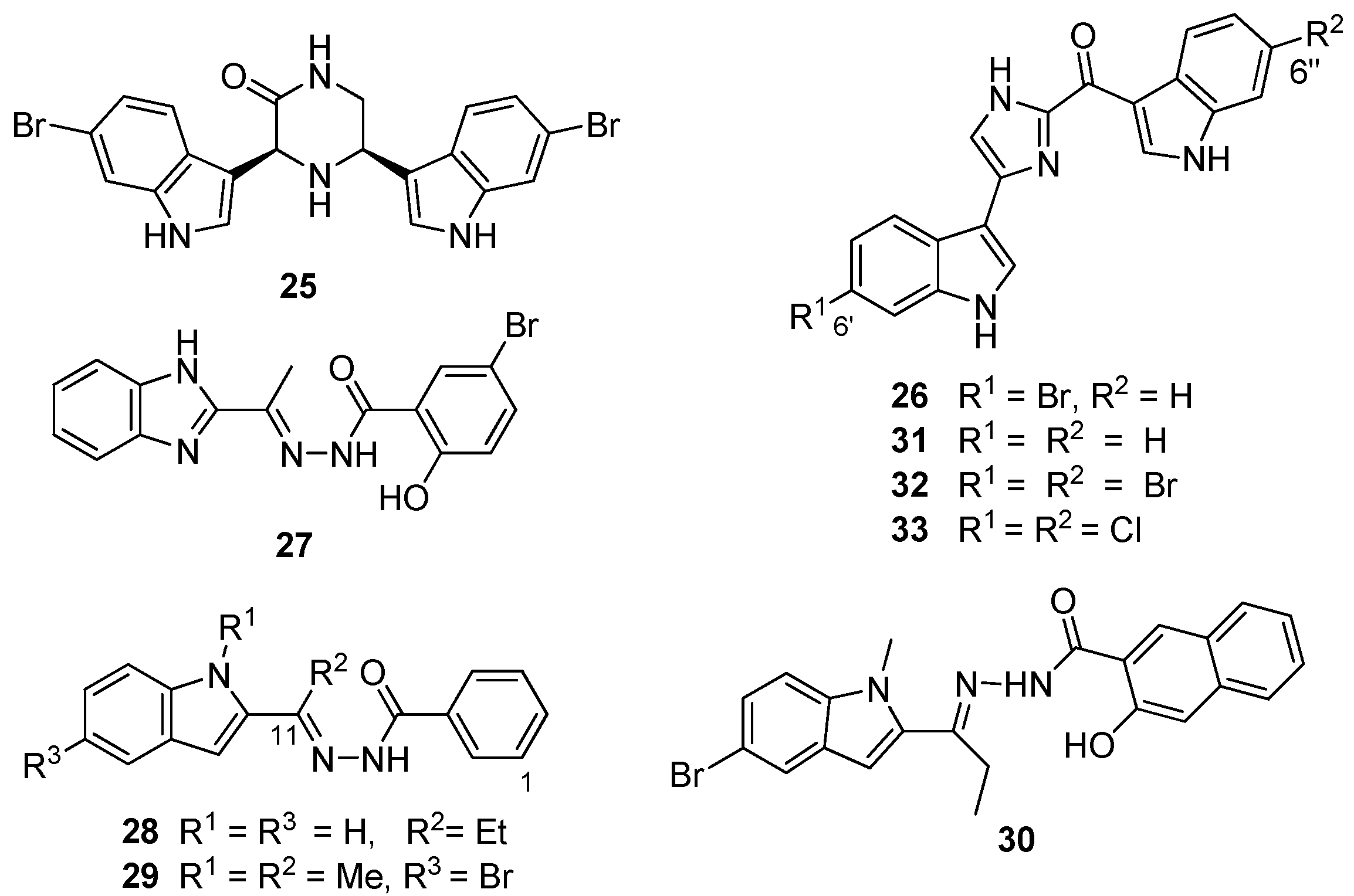
The random screening of 968 marine invertebrate extracts, collected from seven different benthic marine environments around the world, [
34], afforded only one extract that was active in the MRSA PK inhibition assay. The methanolic extract of the South African sponge,
Topsentia pachastrelloides (
Figure 1c), showed significant activity in the MRSA PK inhibition assay, and subsequent bioassay-guided fractionation of this extract yielded a cohort of four bisindole alkaloids of which, the two known metabolites
cis-3,4-dihydrohamacanthin B (
25,
Figure 7) and bromodeoxytopsentin (
26) proved to be the most active compounds (IC
50 16 and 60 nM, respectively). These two compounds also exhibited between 166- and 600-fold selectivity for MRSA PK when compared to similar inhibition data acquired from screening
25 and
26 against four human PK orthologs. X-ray crystallographic analysis of the co-crystallized
cis-3,4-dihydrohamacanthin B-MRSA PK complex revealed that
25 was neither bound to the recognized activation nor allosteric effector binding sites on this enzyme, but was instead unexpectedly bound to two identical lipophilic binding sites on the small interface of the MRSA PK homotetramer [
34].
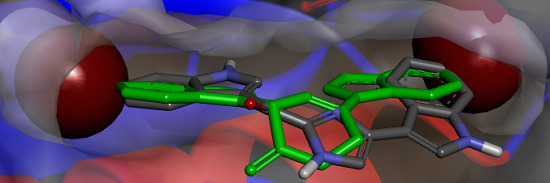
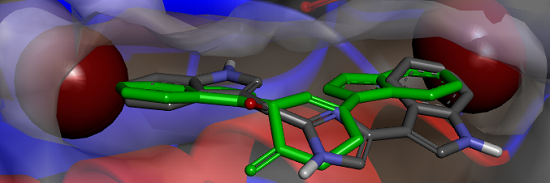
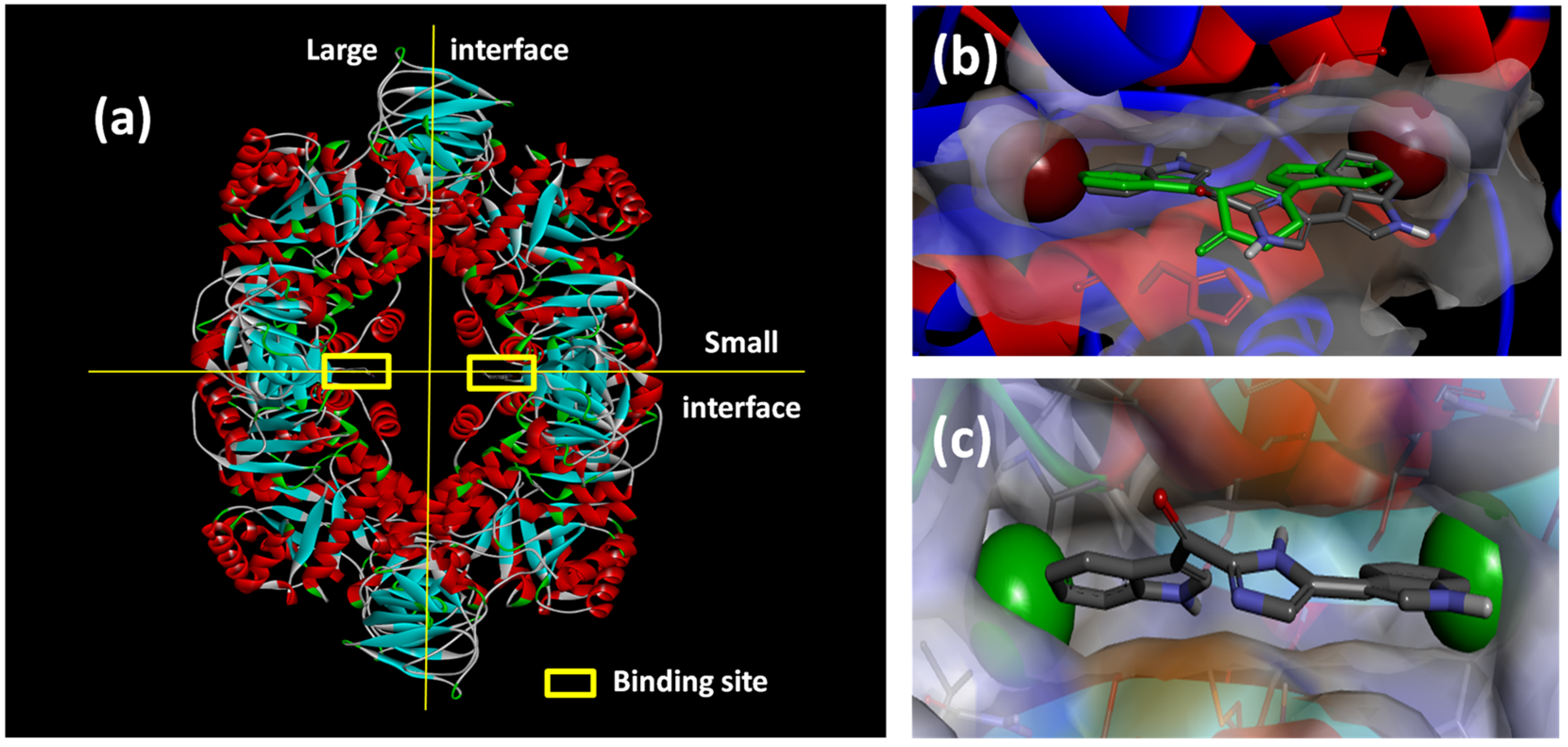
Figure 6.
(a) X-ray structure of MRSA PK (PDB Accession Number 3T07) with the large and small interfaces and the cis-3,4-dihydrohamacanthin B binding sites indicated; (b) X-ray co-crystal generated diagram of 25 (green) in the cis-3,4-dihydrohamacanthin B binding site of MRSA PK with 26 (grey) overlaid in its highest scoring docked conformation with its bromines displayed as CPK models; (c) highest scoring docked conformation of the synthetic compound 33, with its chlorines displayed as CPK models, in the MRSA PK cis-3,4-dihydrohamacanthin B binding site.
Figure 6.
(a) X-ray structure of MRSA PK (PDB Accession Number 3T07) with the large and small interfaces and the cis-3,4-dihydrohamacanthin B binding sites indicated; (b) X-ray co-crystal generated diagram of 25 (green) in the cis-3,4-dihydrohamacanthin B binding site of MRSA PK with 26 (grey) overlaid in its highest scoring docked conformation with its bromines displayed as CPK models; (c) highest scoring docked conformation of the synthetic compound 33, with its chlorines displayed as CPK models, in the MRSA PK cis-3,4-dihydrohamacanthin B binding site.
The small interface in MRSA PK is postulated to be crucial for establishing the rigidity of MRSA PK necessary for catalytic activity, and the binding of either
25 or
26 to this region of the protein is therefore thought to induce flexibility and, subsequently, to reduce enzyme activity [
33]. The symmetrical
cis-3,4-dihydrohamacanthin B binding sites are characterized by two lipophilic pockets with an appropriate spatial arrangement to readily accommodate the bromine substituents of
25 and
26 (
Figure 6b). In addition, the histidine residues (HIS365) on the neighboring parallel MRSA PK α-helices rearrange to anchor the indole rings through π-interactions [
34,
35]. Interestingly, sequence alignment between MRSA and human PK isoforms indicated that access to the analogous binding sites in human PK orthologs is hindered by a group of amino acids that effectively shield these sites from potential ligands [
33]. This structural difference around the entrance to the binding sites is consequently thought to account for the greater selectivity of
25 and
26, and related synthetic inhibitors
vide infra, for MRSA PK over human PK orthologs [
33,
35].
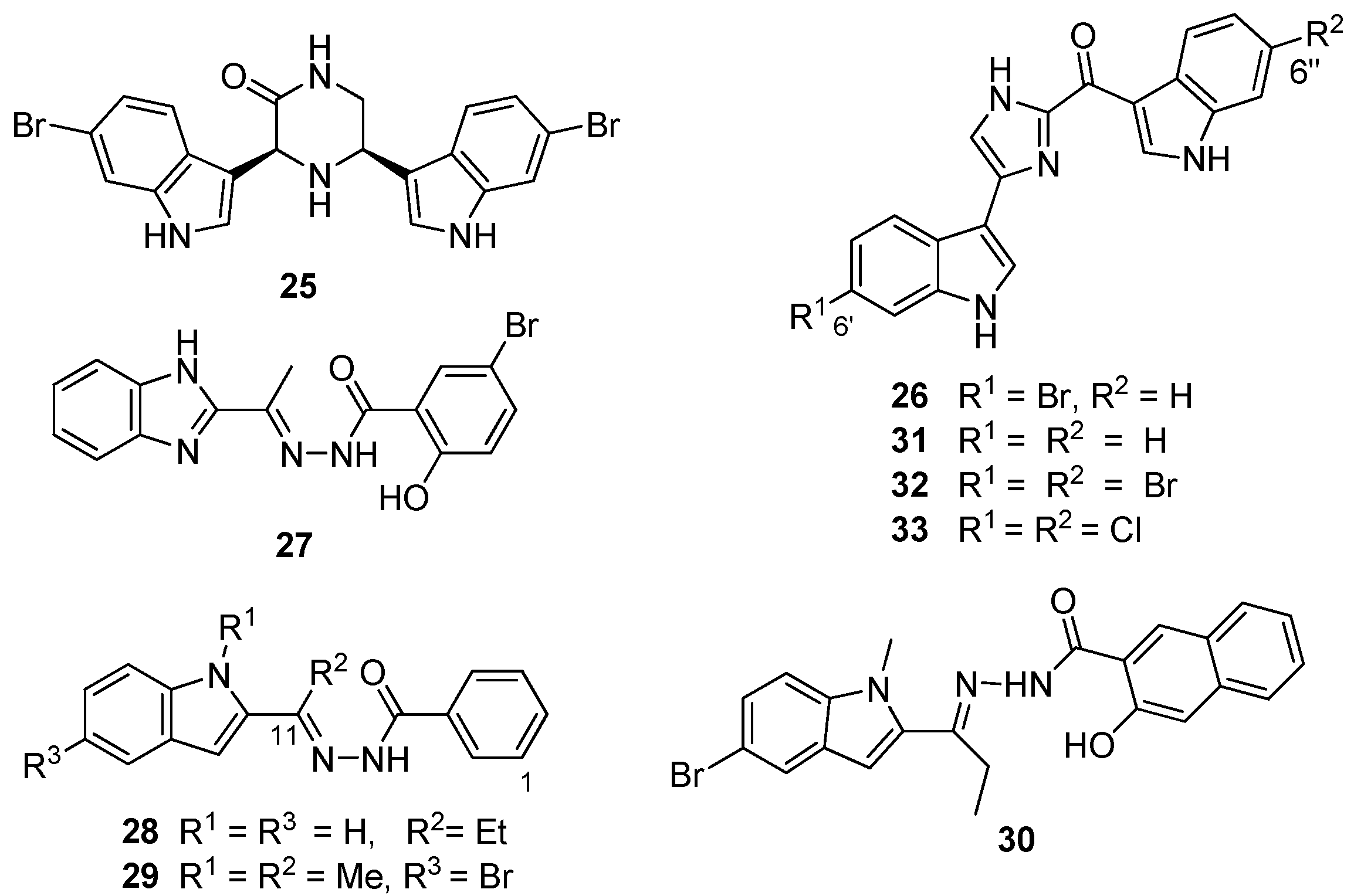
Figure 7.
Chemical structures of compounds 25–33.
Figure 7.
Chemical structures of compounds 25–33.
From the preliminary screening of an
in silico library, coupled with functional enzyme assays, Zoraghi
et al. identified the benzimidazole compound IS-130 (
27,
Figure 7) as a potent MRSA PK inhibitor (IC
50 91 nM) with good specificity (>1000-fold) for MRSA PK over human isoforms, but with poor
in vivo antibacterial activity at the cellular level against a methicillin-susceptible strain of
S. aureus (MSSA) (minimum inhibitory concentration (MIC) >187 μg/mL) [
30]. Nevertheless, the structural motif of
27 provided the starting point for a medicinal chemistry program aimed at improving selectivity, potency and antibacterial activity of potential MRSA PK inhibitors. Compound AM-168 (
28) exhibited only slightly reduced potency (IC
50 126 nM) and substantially increased antibacterial activity (MIC 9.7 μg/mL), which was attributed by Zoraghi
et al. to increased cell membrane penetration due to the increased lipophilicity imparted by the C11 ethyl substituent to this compound [
30]. Accordingly, additional alkyl substitution, e.g., NSK5-15 (
29), further enhanced antibacterial activity against several strains of MSSA and MRSA (MIC 1.4–5.8 μg/mL) with a further decrease in MRSA PK inhibitory potency (185 nM). Kumar
et al. [
33] extended Zoraghi
et al.’s preliminary study and prepared a series of >70 compounds in which systematic structural changes were made to the heteroaromatic ring, the phenolic moiety and the central linker unit of the hit compound
27. This series of compounds was screened against MRSA PK and methicillin-susceptible
S. aureus, with Kumar
et al. reporting varying levels of potency (IC
50 15–380 nM) and antibacterial activity (MIC 1–>194 μg/mL), respectively. Interestingly, co-crystallization of
27 and
28 with MRSA PK followed by X-ray analysis revealed that these compounds were also bound to the
cis-3,4-dihydrohamacanthin B binding sites of the MRSA PK enzyme (
Figure 6a) [
33]. Unfortunately, Kumar
et al.’s synthetic program did not shed any light on the structure activity relationships that might conclusively link potency (IC
50) with antibacterial activity (MIC). Ultimately,
N-methylindole (
30) provided the best combination of
in vitro MRSA PK inhibition (IC
50 79 nM) and antibacterial activity (MIC 1 μg/mL), possibly warranting further exploitation of
30 and analogous compounds as potential antibiotics effective against MRSA [
33].
Veale
et al. [
35] used the chemical structure of the naturally-occurring hit compound
26 identified in the South African sponge extract as a starting point for an extensive ligand-receptor docking study of various analogs of
26 with the
cis-3,4-dihydrohamacanthin B binding site. Postulating that a dihalogenated analog of
26 would better exploit the opportunities offered by the symmetrical
cis-3,4-dihydrohamacanthin B binding site, in particular the two terminal lipophilic binding pockets, Veale
et al. prepared the 6′, 6″dihalogenated (F, Cl, Br and I,
Figure 7) analogues of
26 and the debrominated compound, deoxytopsentin (
31), for a comparative MRSA PK inhibition study. The target halogenated synthetic compounds were readily accessed in reasonable overall yield (10%–32% over five steps) via the dehydrative cyclocondensation of the respective
N-Boc-protected 6-halo-indolyl-3-glyoxals with ammonium acetate in ethanol, followed by thermolytic cleavage of the
N-Boc groups. As expected, the MRSA inhibition activity of the non-halogenated compound
31 (IC
50 240 nM) was less active than the naturally-occurring monobrominated compound
26 (IC
50 60 nM), while both the dibrominated and dichlorinated (
Figure 6c) analogs (
32 and
33) were an order of magnitude more potent (IC
50 2 and 1.5 nM, respectively) than
26 coupled to improved selectivity for MRSA PK over the four human orthologs assayed [
35].
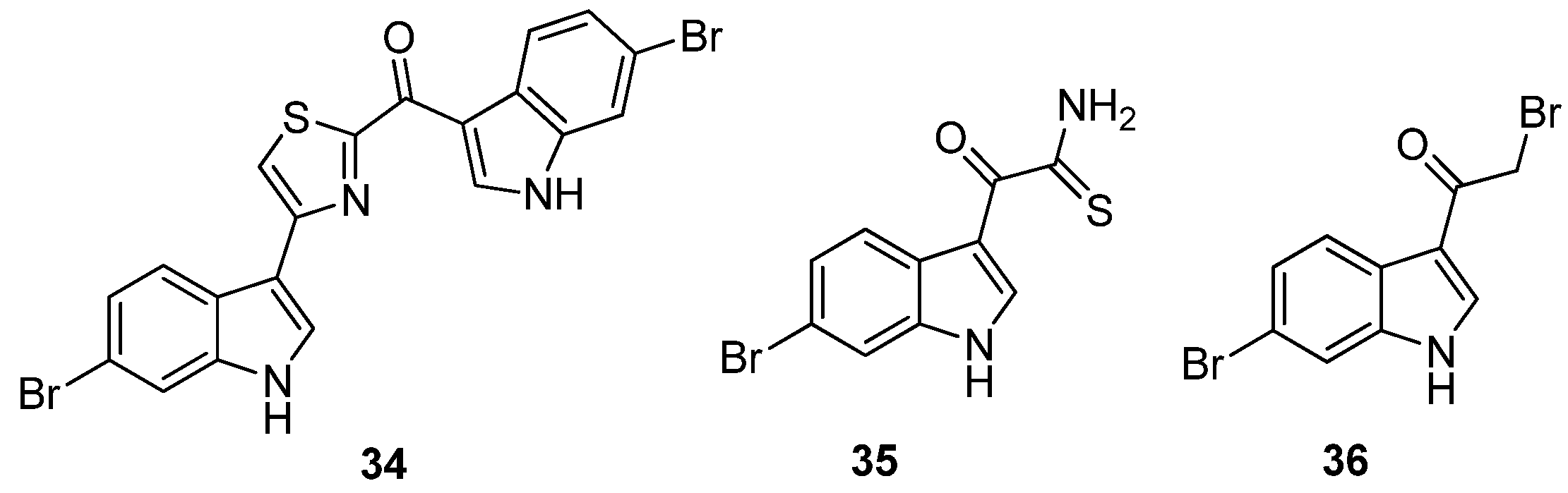
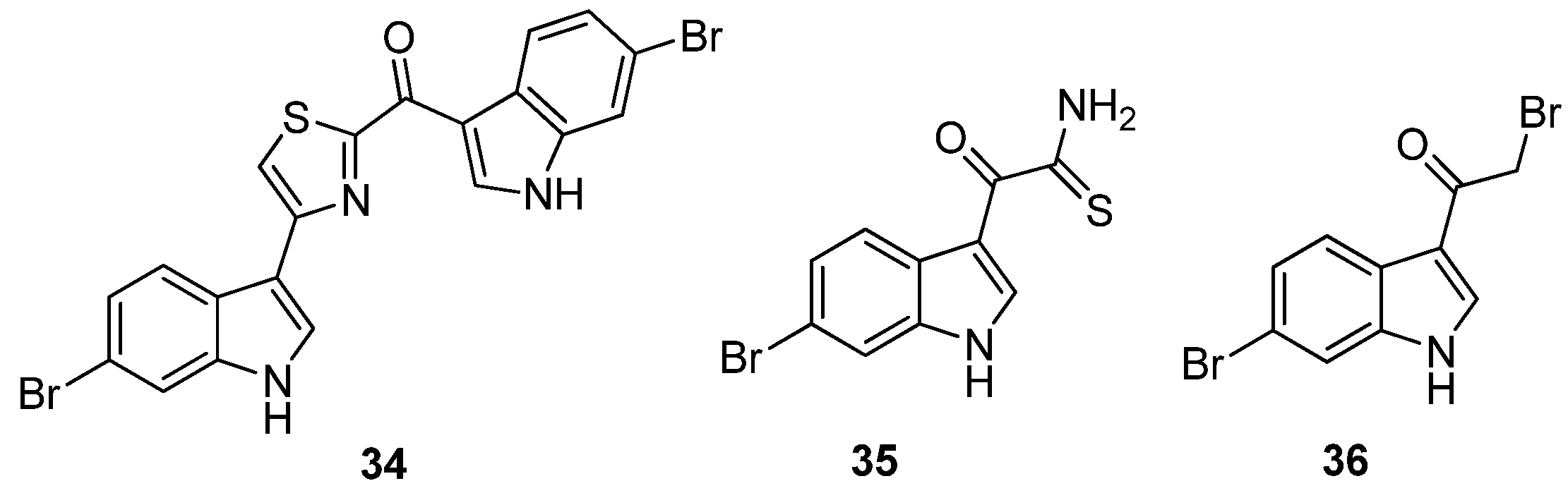
Veale
et al. further evaluated the importance of the imidazole ring to MRSA PK inhibition in dihalogenated bisindole alkaloids by preparing a similar series of dihalogenated bisindoles in which the imidazole ring was replaced by a thiazole moiety, e.g.,
34 [
36] (
Figure 8). Coupling of α-oxo-1
H-indole-3-thioacetamide (
35) with the α-bromoketone (
36) in a regiospecific Hantzsch thiazole ring formation reaction afforded the targeted halogenated bisindole thiazoles. The μM activity of the synthetic bisindole thiazoles (e.g., IC
50 5 μM for
35) indicated that bioisosteric replacement of the imidazole ring with a thiazole had a negative impact on MRSA PK potency [
36]. The antibacterial activity of both the synthetic bisindole imidazoles and bisindole thiazoles was not recorded.
Figure 8.
Chemical structures of compounds 34–36.
Figure 8.
Chemical structures of compounds 34–36.
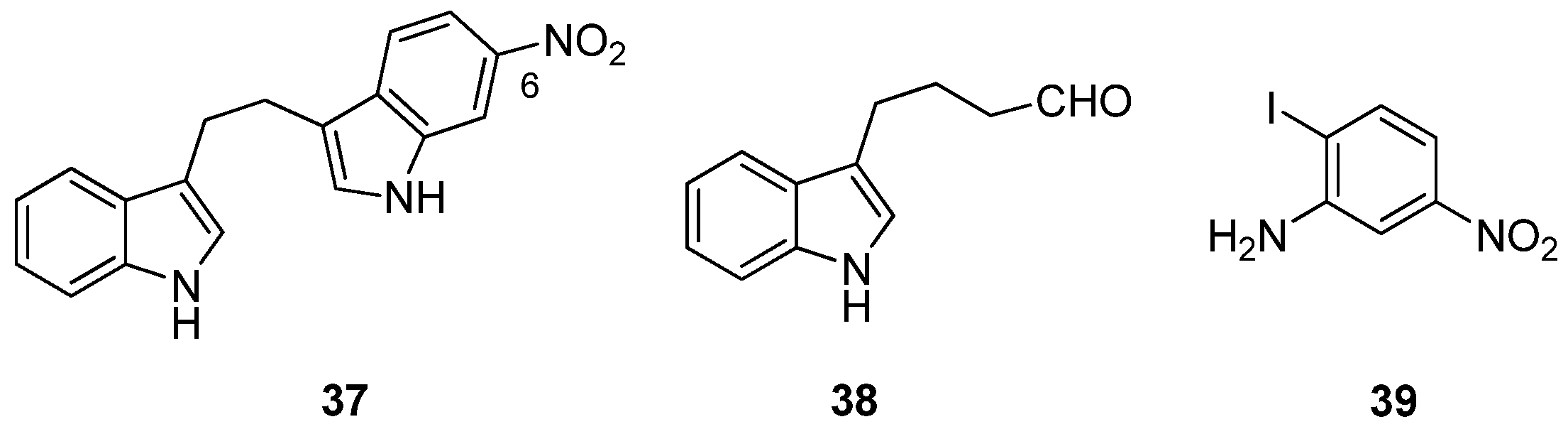
Furthermore, acknowledging the significance of a bisindole motif to increased inhibition of MRSA PK, Sperry and co-workers have recently described the selective MRSA PK inhibition of a cohort of eleven, variously-substituted, synthetic 1,2-bis(3-indolyly)ethane compounds, e.g.,
37 [
37] (
Figure 9). Compound
37, accessed via palladium catalyzed heteroannulation of the aldehyde (
38) and 1-iodo-2-amino-4-nitrobenzene (
39) [
38], was the most potent of the series (IC
50 0.9 μM) and exhibited a 20–106-fold selectivity for MRSA PK over four human PK isoforms. Replacing the C6 nitro substituent with chloro, nitrile, methoxy and methyl functionalities had a deleterious effect on MRSA PK inhibition, with the nitrile and methoxy analogs inactive and the chloro and methyl analogs two orders of magnitude less active (IC
50 272 and 294 μM, respectively) [
37].
Figure 9.
Chemical structures of compounds 37–39.
Figure 9.
Chemical structures of compounds 37–39.
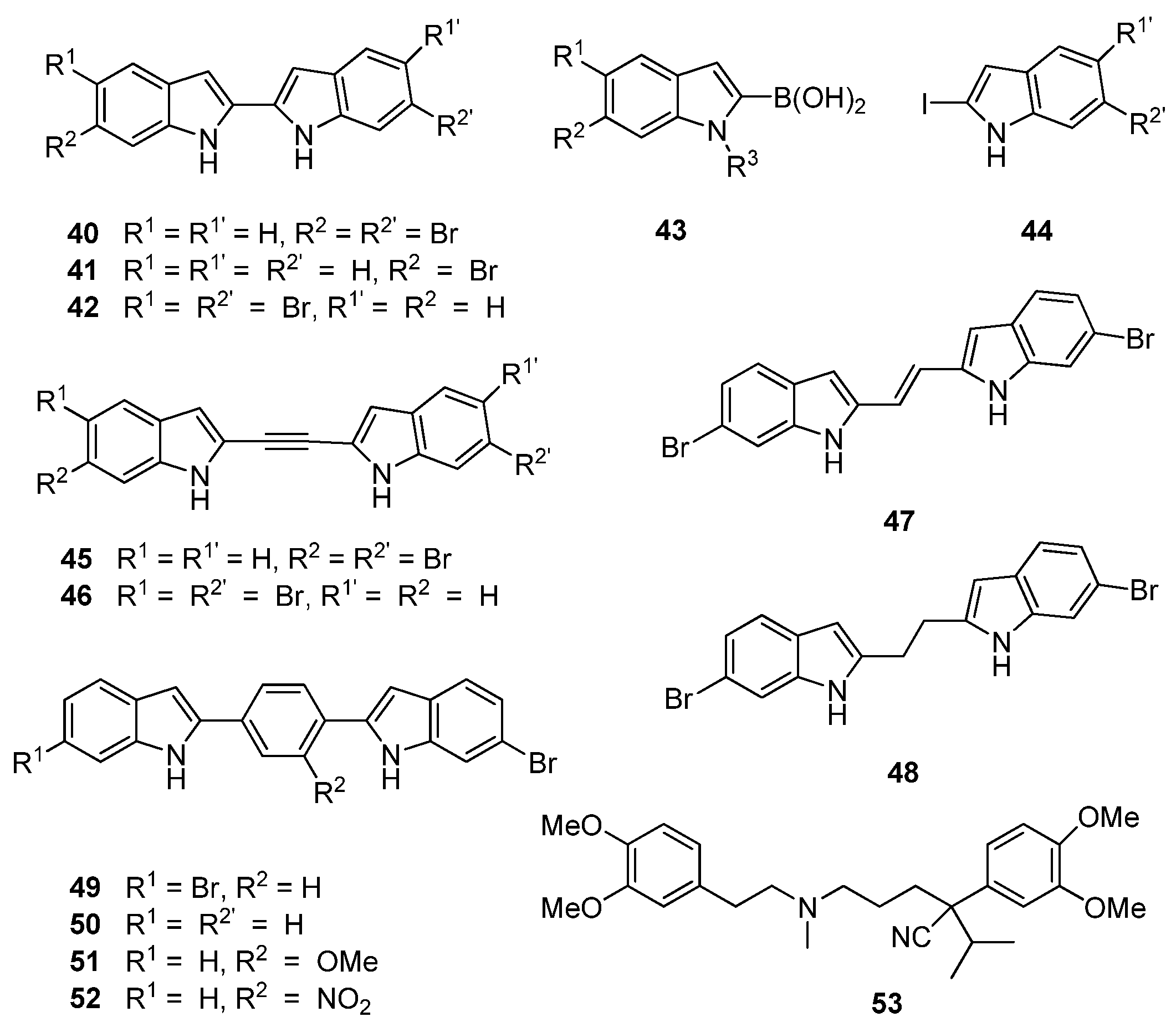
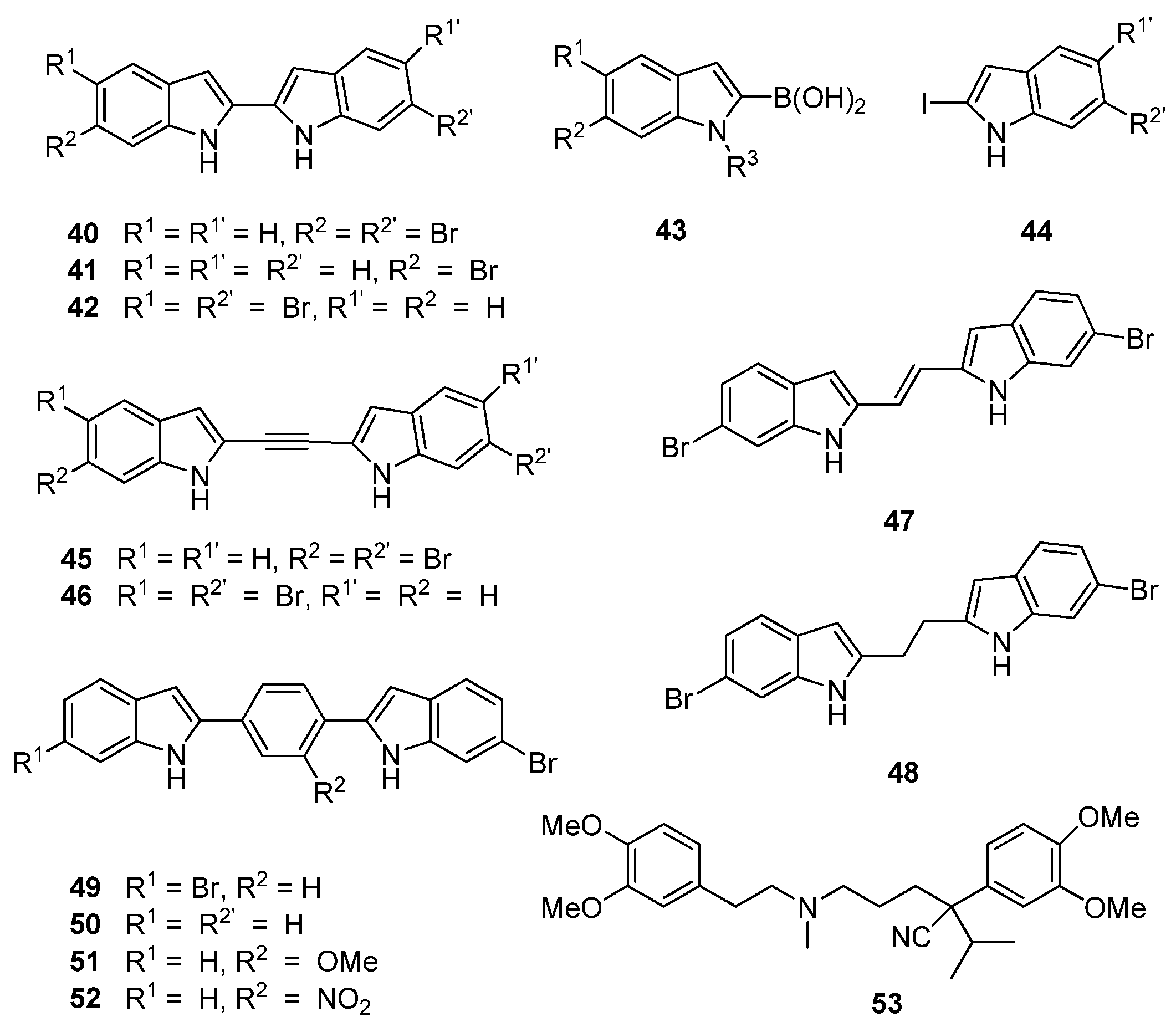
Similarly, Kumar
et al. [
39] changed direction from their earlier medicinal chemistry program based on the putative MRSA PK inhibitor,
27, to focus on potential bisindole inhibitors of MRSA PK in line with the chemical structures of the potent naturally-occurring bisindole MRSA PK inhibitors,
25 and
26. Central to their strategy was varying the linker units between the indole rings in order to uncover the relationship between activity and indole orientation relative to the linker unit in addition to providing further insight into size constraints within the MRSA PK binding site. Their initial cohort of directly-linked 2,2′-biindoles (
40–
42,
Figure 10) was synthesized through a Suzuki–Miyaura coupling of boronic acid precursor
43 and iodinated indole
44. This cohort of compounds were generally found to potently inhibit MRSA PK at concentrations as low as 1 nM. Initial inhibitory data obtained by Kumar
et al. supported previous observations made between deoxytopsentin analogues
26 and
32 that 6-6′ dibrominated bisindoles, such as 40 (IC
50 7 nM), display superior MRSA PK inhibition than a corresponding monobrominated analogue, e.g.,
41 (IC
50 21 nM). However, the opposite trend was observed with regard to MIC values (16 and 2 μg/mL, respectively) against MSSA strains [
39]. Interestingly, the 6,5′ dibrominated analogue (
42) also displayed potent MRSA PK inhibition (IC
50 2.2 nM) coupled to a significantly improved MIC against
S. aureus (0.3 μg/mL). The structure activity relationship of the linker group between indoles was further explored through insertion of acetylene, ethylene and ethyl moieties between the two substituted indole rings (
45–
48,
Figure 10) using standard synthetic protocols. Similar MPSA PK inhibitory activity was observed between this group and the 2,2′-biindoles. However, MIC activity against
S. aureus was generally lost, with the single exception of the 6,5′ dibrominated analogue
46. An additional set of aryl linked bisindole analogues (
49–
52) were prepared with a view toward exploiting possible interactions with the aromatic histidine residues present in the binding site. The dibrominated compound (
49) was found to be comparatively less active than compound
40, while activity was restored with the mono-brominated compound (
50), leading the authors to suggest that Compound
50 defines the maximum permissible length within the MRSA PK binding site. Interestingly, analogues of
50 featuring substitutions on the aryl ring (
51,
52) were found to be the most active in the series against MRSA PK (IC
50 ca. 2 nM) while the nitro-containing compound
52 showed encouraging activity against
S. aureus (MIC 2.0 μg/mL). While no conclusive rationale for the differences between MRSA PK inhibitory activity and MIC values was postulated, Kumar
et al. determined, through co-administration of their synthetic compounds with the calcium channel blocker verapamil (
53), that several bisindoles were actively removed from the cells via cellular efflux mechanisms, possibly accounting for the contrasting antibacterial activities observed in their study [
39].
Figure 10.
Chemical structures of compounds 40–53.
Figure 10.
Chemical structures of compounds 40–53.



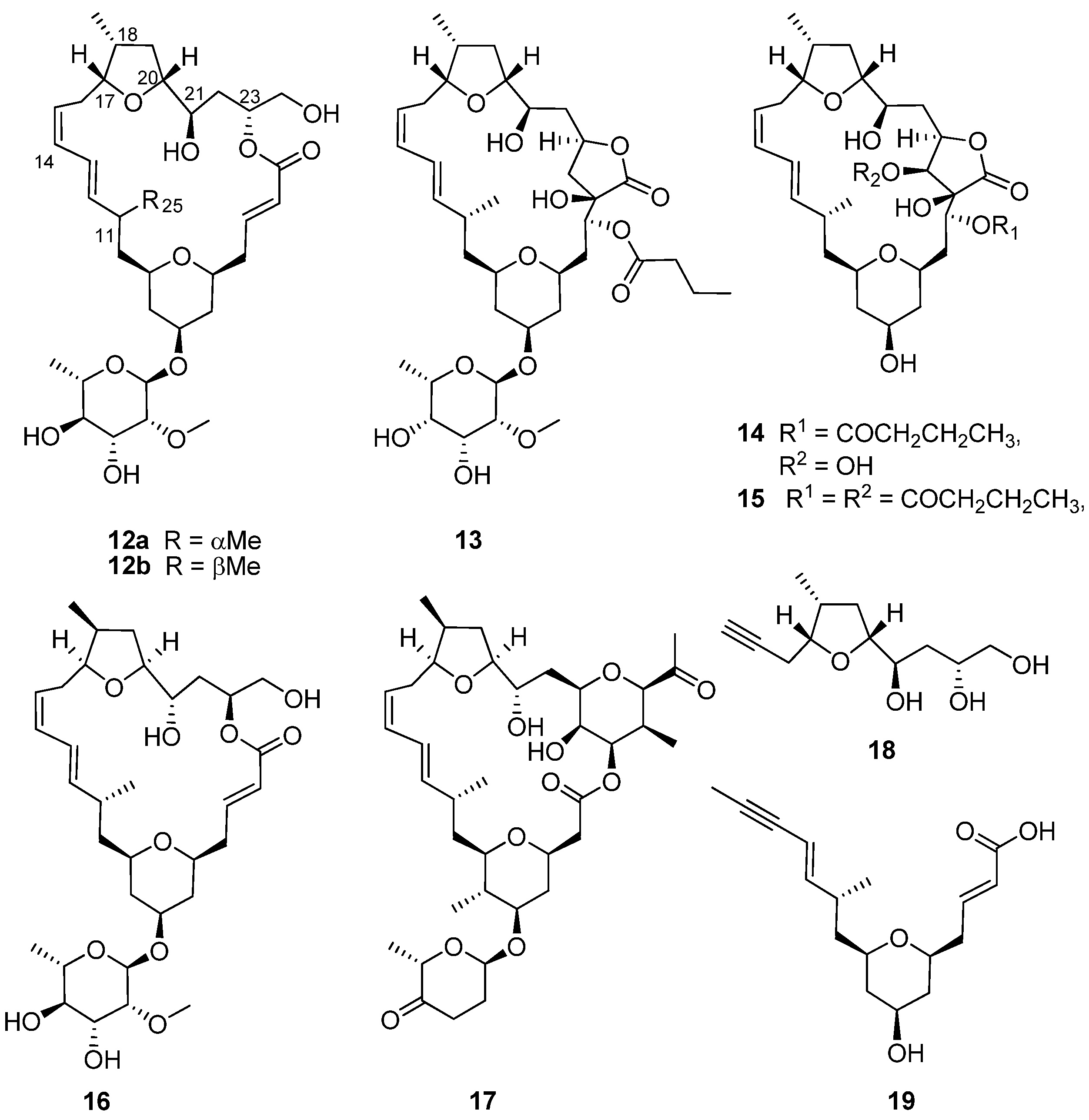

{kind=link}
{kind=link}
{kind=link}
{kind=link}
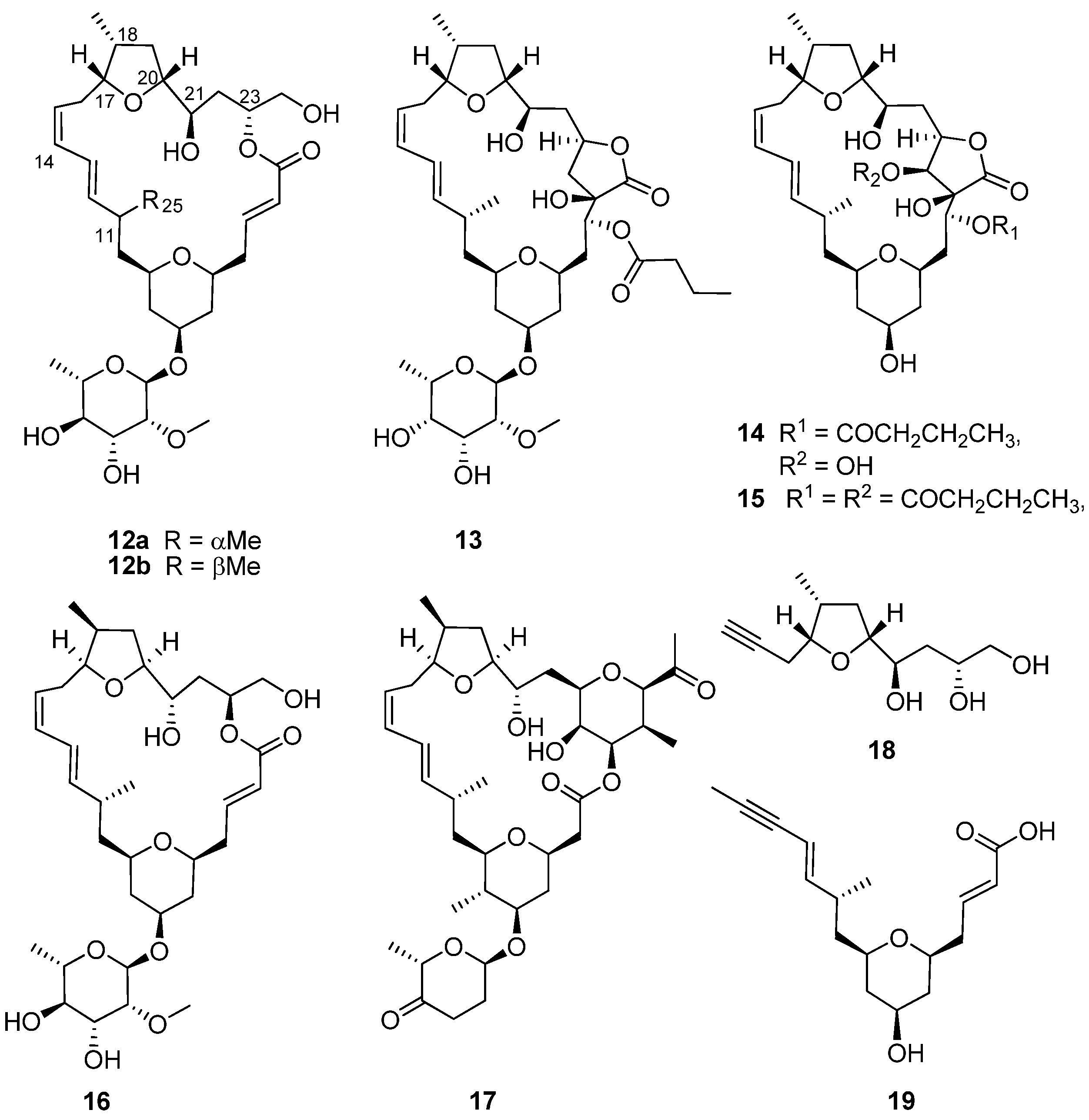{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}