Cracking the Cytotoxicity Code: Apoptotic Induction of 10-Acetylirciformonin B is Mediated through ROS Generation and Mitochondrial Dysfunction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
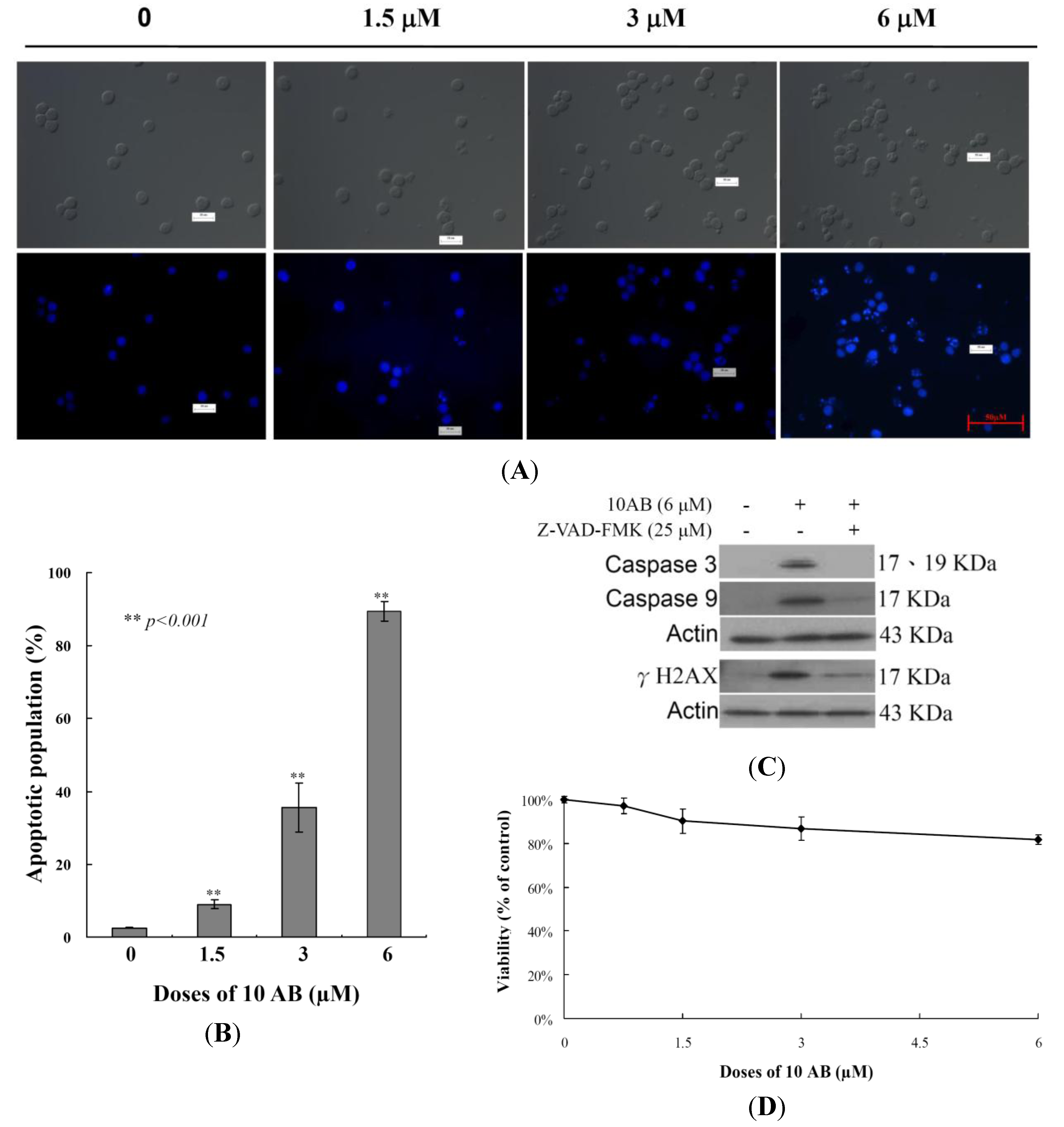
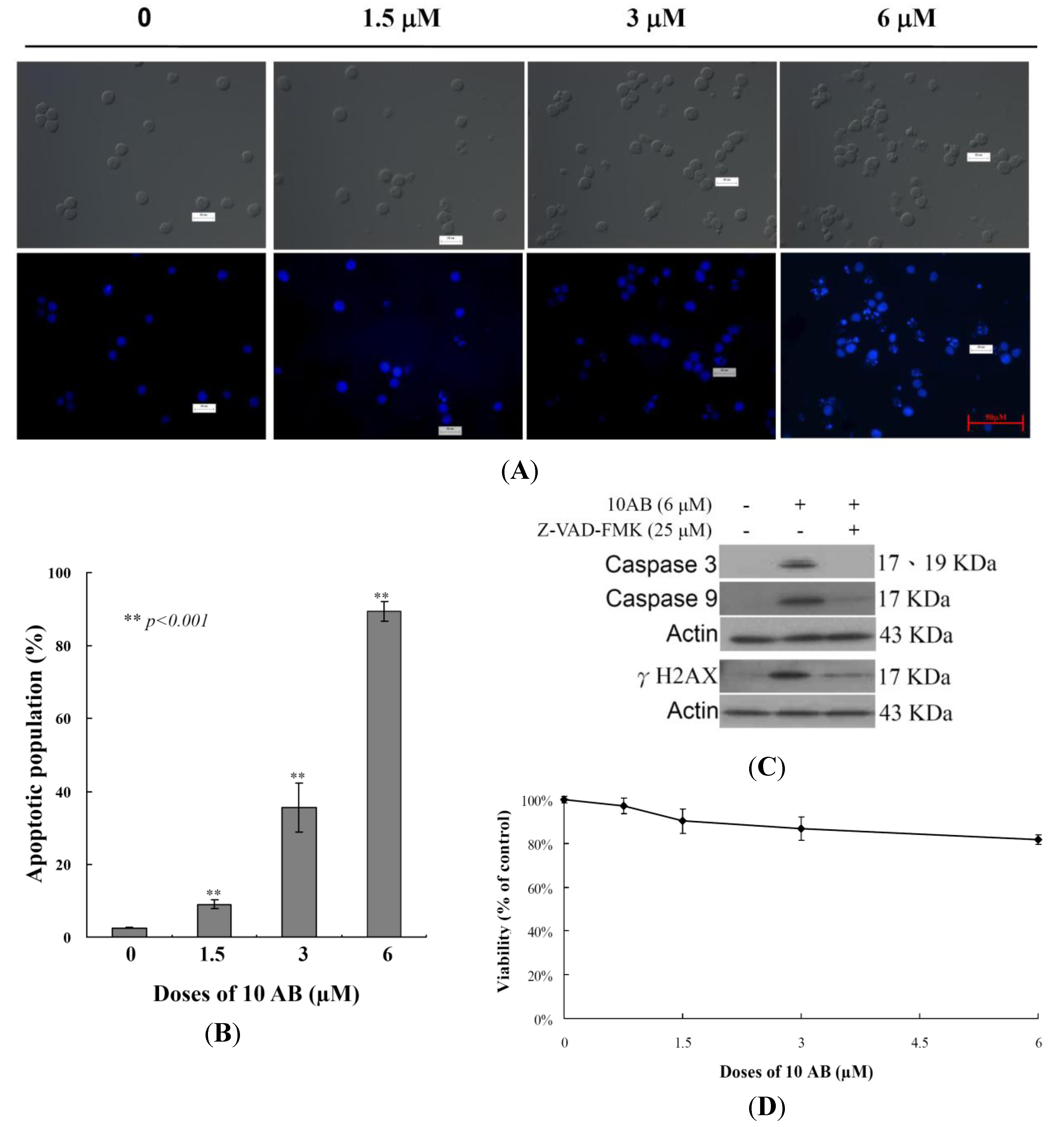
2.1. The Apoptotic Induction Effect of 10AB in HL 60 Cells
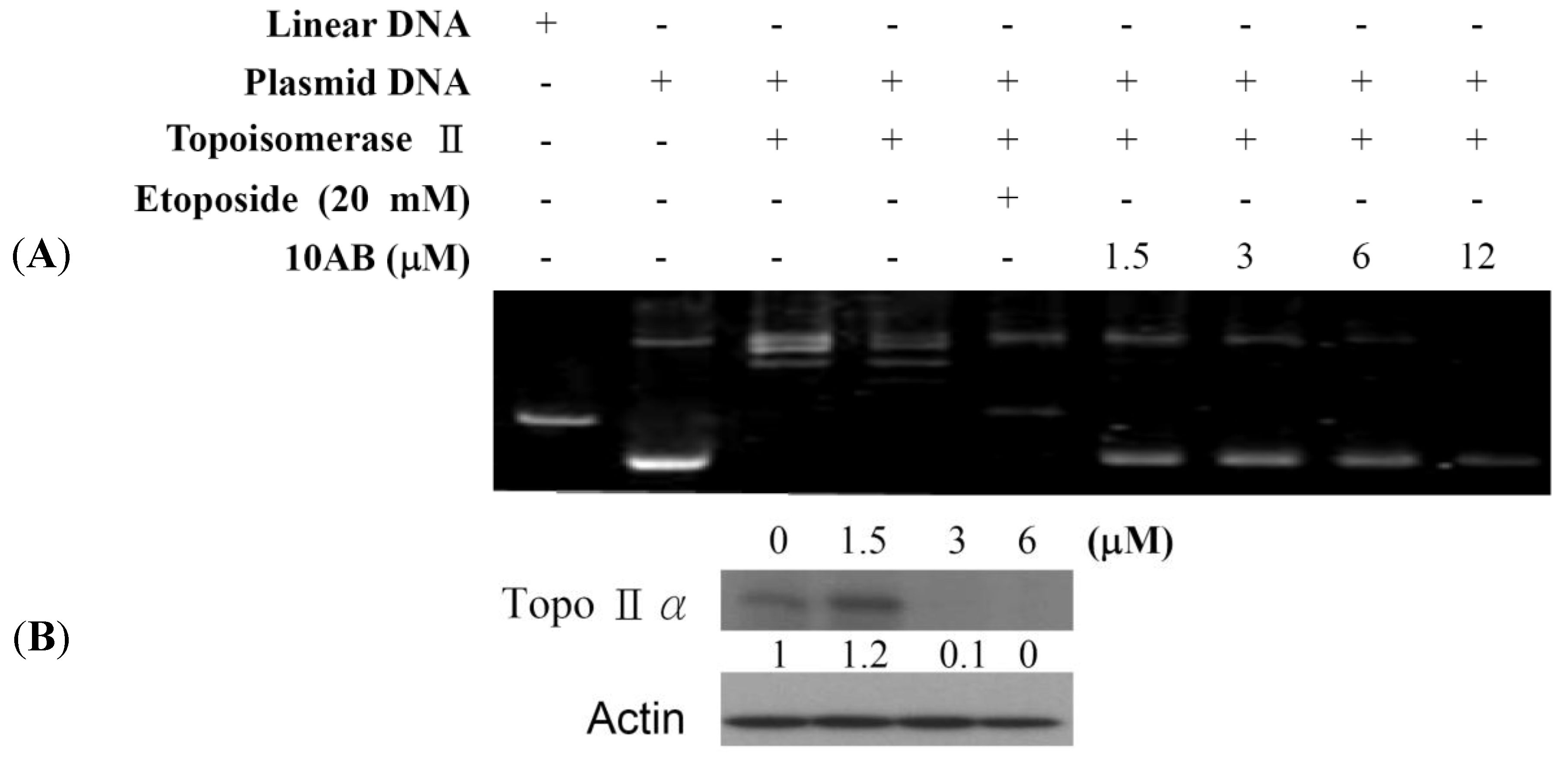
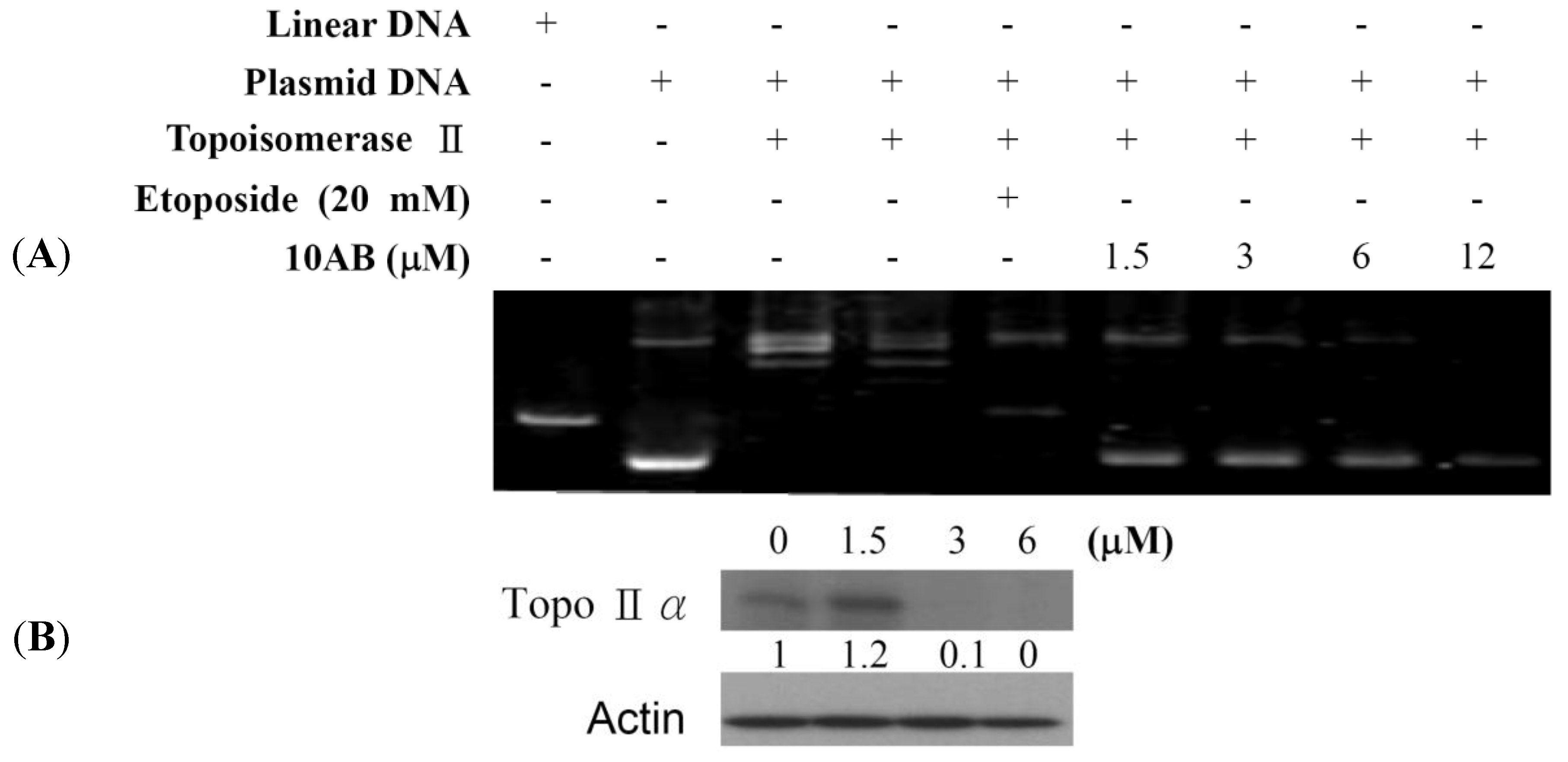
2.2. The Effect of 10AB on Topoisomerase IIα Activity
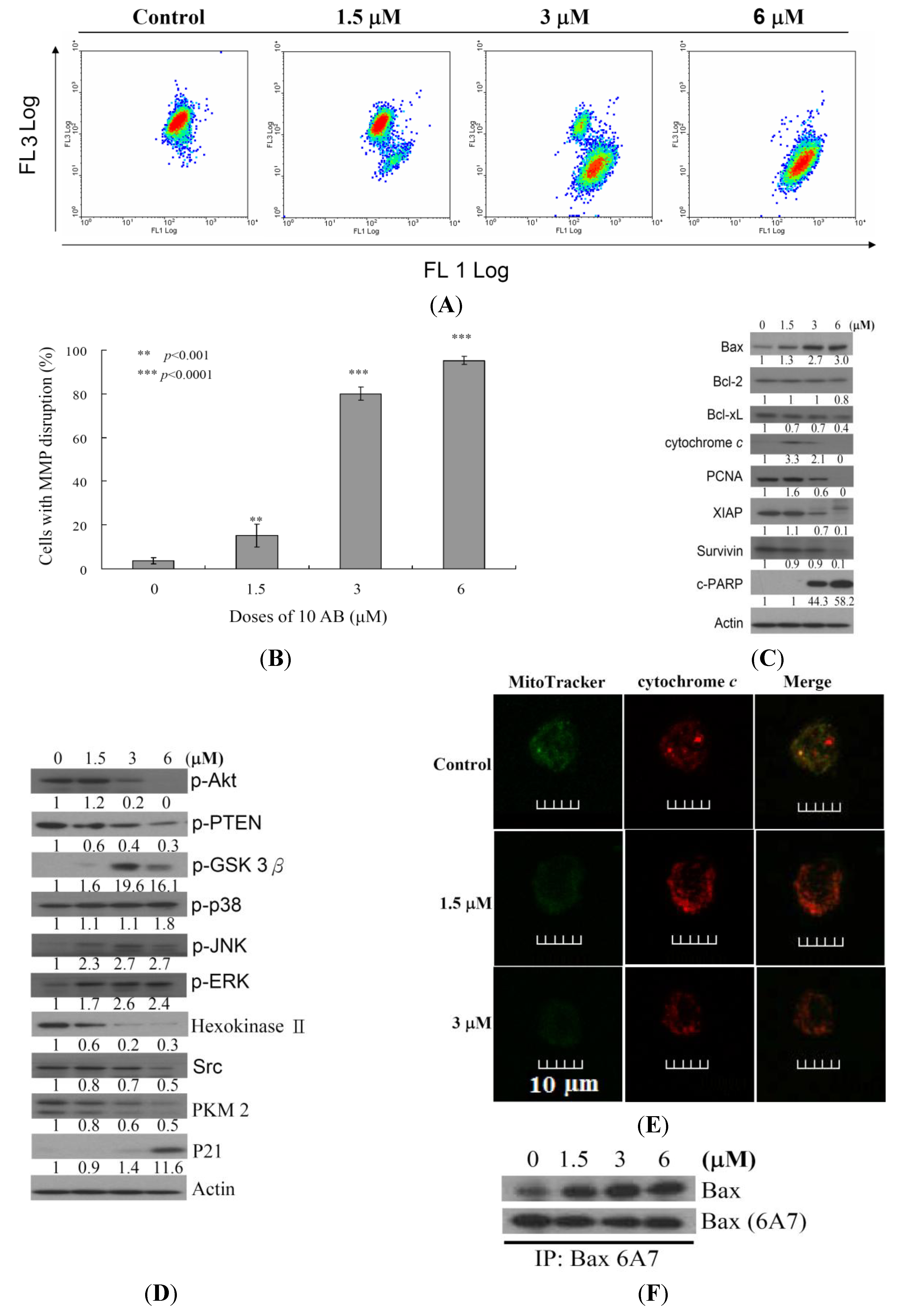
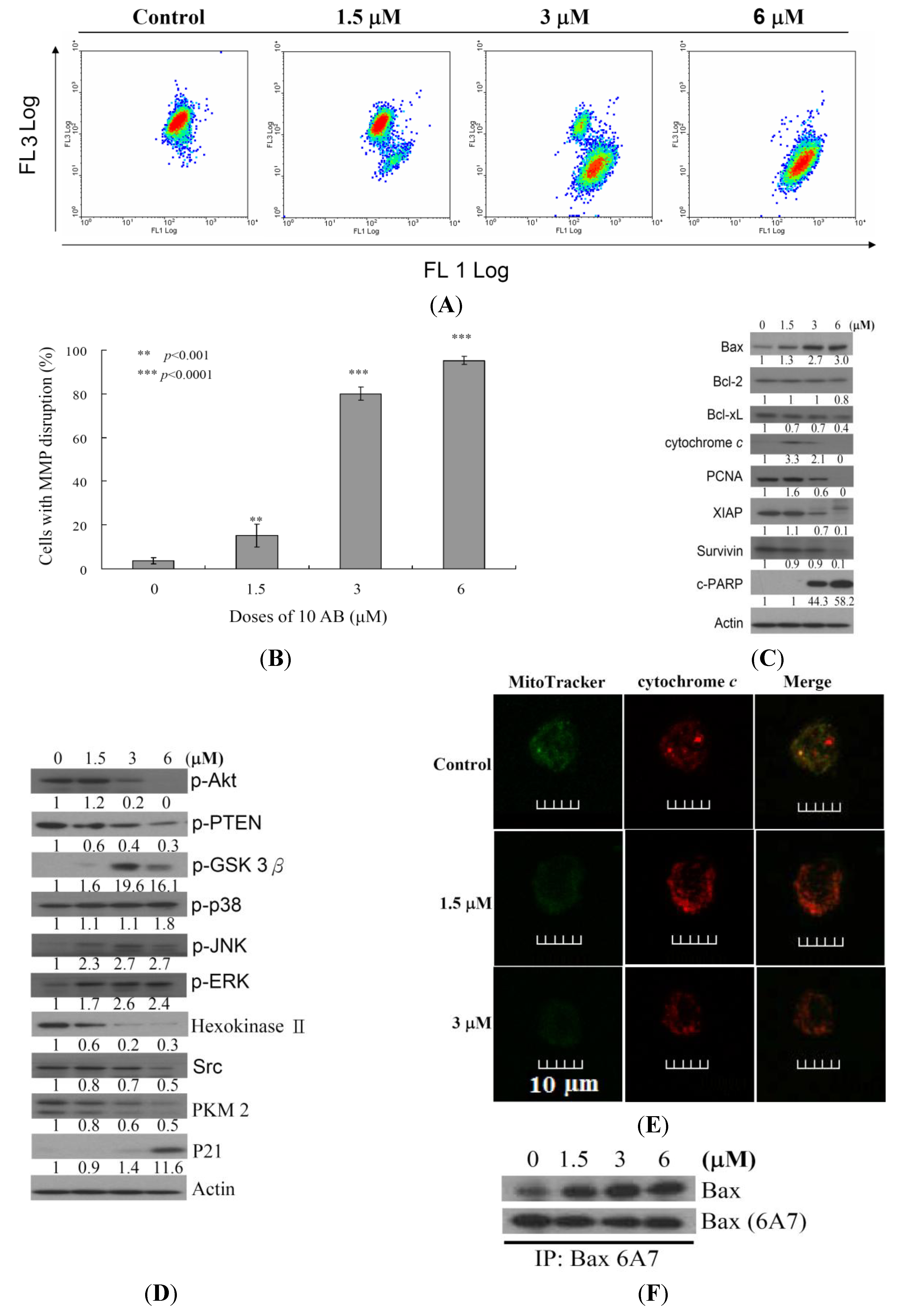
2.3. The Relationship between 10AB-Induced Apoptosis in HL 60 Cells and the Disruption in Mitochondrial Membrane Potential as well as Mitochondrial Metabolism-Related Proteins
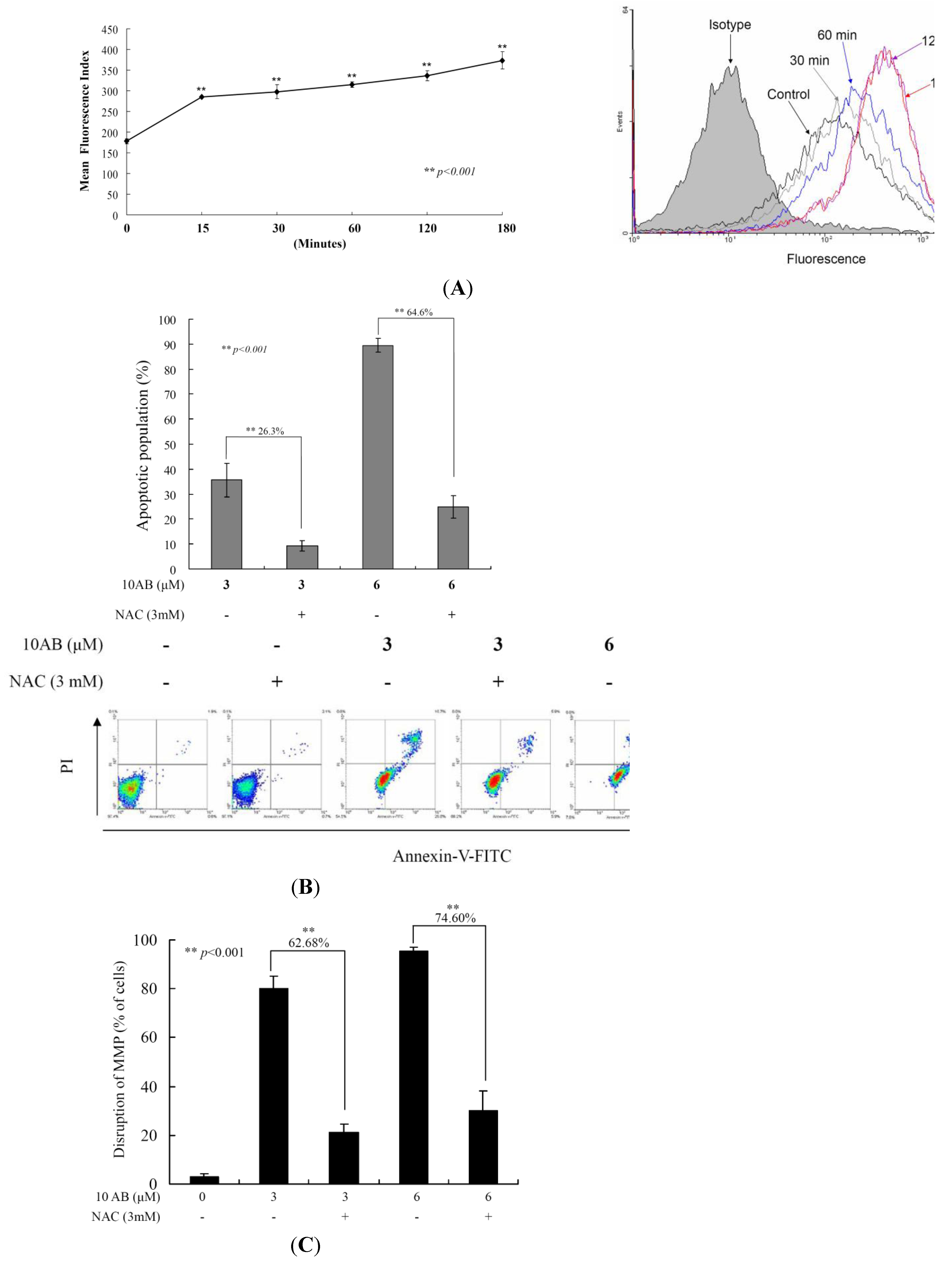
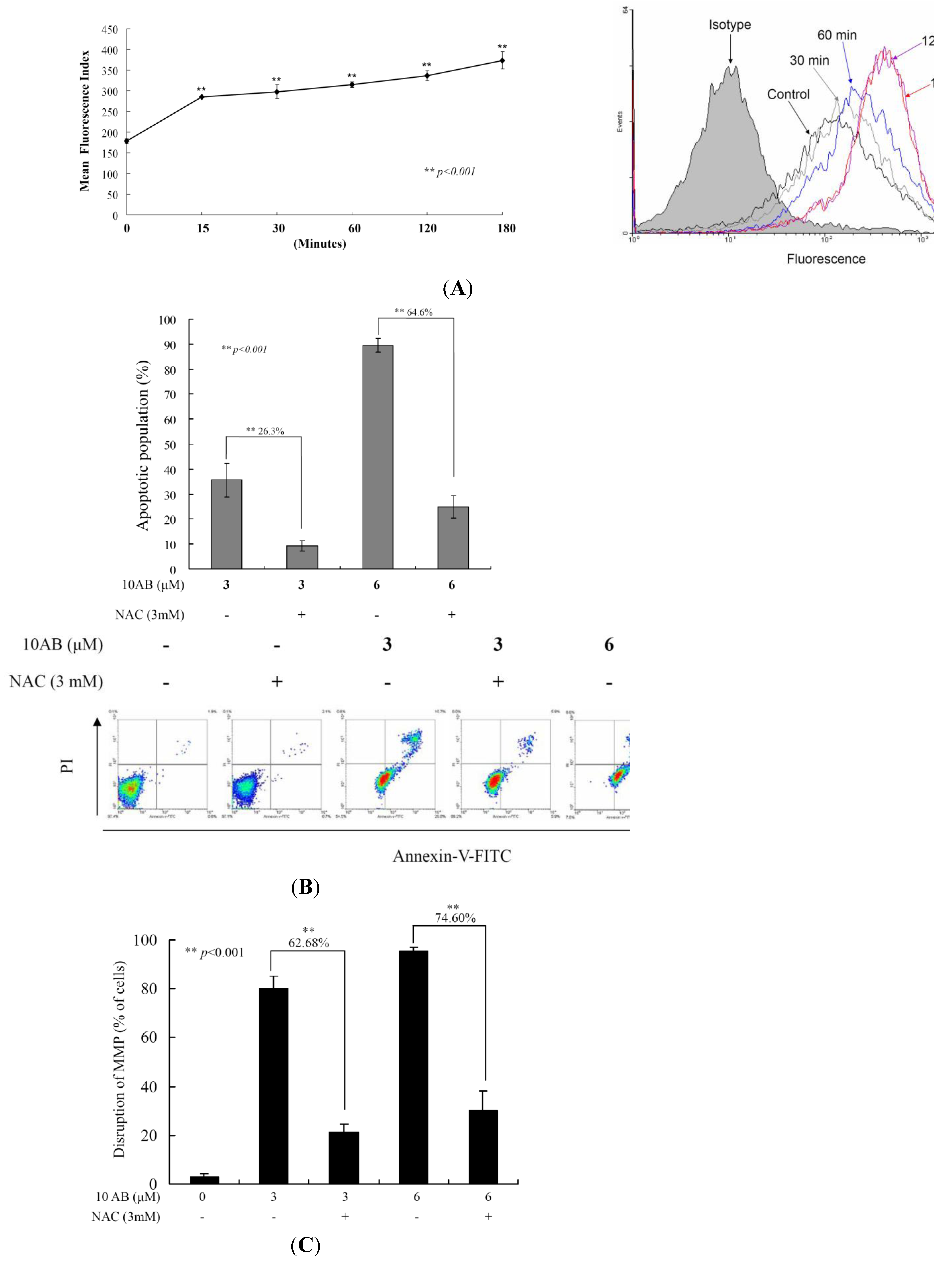
2.4. The Relationship between 10AB-Induced Apoptosis and ROS Generation
3. Discussion
4. Experimental Section
4.1. Bioassays Materials
4.2. Preparation of 10-Acetylirciformonin B (10AB) Stock Solution
4.3. MTT Proliferation Assay
4.4. Annexin V/PI Apoptosis Assay
4.5. Determination of ROS Generation, and MMP Disruption
4.6. Assay of Topoisomerase II Catalytic Inhibitors and Poisons
4.7. Co-Immunoprecipitaion and Western Blotting
4.8. Immunofluorescence Analysis
4.9. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Tatarkova, Z.; Kuka, S.; Petras, M.; Racay, P.; Lehotsky, J.; Dobrota, D.; Kaplan, P. Why mitochondria are excellent targets for cancer therapy. Klin. Onkol. 2012, 25, 421–426. [Google Scholar]
- Gall, J.M.; Wong, V.; Pimental, D.R.; Havasi, A.; Wang, Z.; Pastorino, J.G.; Bonegio, R.G.; Schwartz, J.H.; Borkan, S.C. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int. 2011, 79, 1207–1216. [Google Scholar] [CrossRef]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria as targets for chemotherapy. Apoptosis 2009, 14, 624–640. [Google Scholar]
- Fogg, V.C.; Lanning, N.J.; Mackeigan, J.P. Mitochondria in cancer: At the crossroads of life and death. Chin. J. Cancer 2011, 30, 526–539. [Google Scholar] [CrossRef]
- Gasparre, G.; Porcelli, A.M.; Lenaz, G.; Romeo, G. Relevance of mitochondrial genetics and metabolism in cancer development. Cold Spring Harb. Persp. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- Wenner, C.E. Cell signaling and cancer-possible targets for therapy. J. Cell. Physiol. 2010, 223, 299–308. [Google Scholar]
- Arora, K.K.; Pedersen, P.L. Functional significance of mitochondrial bound hexokinase in tumor cell metabolism. Evidence for preferential phosphorylation of glucose by intramitochondrially generated ATP. J. Biol. Chem. 1988, 263, 17422–17428. [Google Scholar]
- Shulga, N.; Wilson-Smith, R.; Pastorino, J.G. Hexokinase II detachment from the mitochondria potentiates cisplatin induced cytotoxicity through a caspase-2 dependent mechanism. Cell Cycle 2009, 8, 3355–3364. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta 2010, 1797, 1225–1230. [Google Scholar] [CrossRef]
- Murphy, M.P.; Holmgren, A.; Larsson, N.G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell. Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef]
- Kim, K.Y.; Yu, S.N.; Lee, S.Y.; Chun, S.S.; Choi, Y.L.; Park, Y.M.; Song, C.S.; Chatterjee, B.; Ahn, S.C. Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochem. Biophys. Res. Commun. 2011, 413, 80–86. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Bauer, G. Tumor cell-protective catalase as a novel target for rational therapeutic approaches based on specific intercellular ROS signaling. Anticancer Res. 2012, 32, 2599–2624. [Google Scholar]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol 2012, 86, 1649–1665. [Google Scholar] [CrossRef]
- Azad, N.; Iyer, A.; Vallyathan, V.; Wang, L.; Castranova, V.; Stehlik, C.; Rojanasakul, Y. Role of oxidative/nitrosative stress-mediated Bcl-2 regulation in apoptosis and malignant transformation. Ann. N. Y. Acad. Sci. 2010, 1203, 1–6. [Google Scholar] [CrossRef]
- Clement, M.V.; Hirpara, J.L.; Pervaiz, S. Decrease in intracellular superoxide sensitizes Bcl-2-overexpressing tumor cells to receptor and drug-induced apoptosis independent of the mitochondria. Cell Death Differ. 2003, 10, 1273–1285. [Google Scholar] [CrossRef]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef]
- Llovet, J.M.; Di Bisceglie, A.M.; Bruix, J.; Kramer, B.S.; Lencioni, R.; Zhu, A.X.; Sherman, M.; Schwartz, M.; Lotze, M.; Talwalkar, J.; et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J. Natl. Cancer Inst. 2008, 100, 698–711. [Google Scholar] [CrossRef]
- Yap, T.A.; Workman, P. Exploiting the cancer genome: Strategies for the discovery and clinical development of targeted molecular therapeutics. Ann. Rev. Pharmacol. Toxicol. 2012, 52, 549–573. [Google Scholar] [CrossRef]
- Su, J.H.; Tseng, S.W.; Lu, M.C.; Liu, L.L.; Chou, Y.; Sung, P.J. Cytotoxic C21 and C22 terpenoid-derived metabolites from the sponge Ircinia sp. J. Nat. Prod. 2011, 74, 2005–2009. [Google Scholar] [CrossRef]
- Su, J.H.; Chang, W.B.; Chen, H.M.; El-Shazly, M.; Du, Y.C.; Kung, T.H.; Chen, Y.C.; Sung, P.J.; Ho, Y.S.; Kuo, F.W.; et al. 10-Acetylirciformonin B, a sponge furanoterpenoid, induces DNA damage and apoptosis in leukemia cells. Molecules 2012, 17, 11839–11848. [Google Scholar] [CrossRef]
- Du, Y.C.; Chang, F.R.; Wu, T.Y.; Hsu, Y.M.; El-Shazly, M.; Chen, C.F.; Sung, P.J.; Lin, Y.Y.; Lin, Y.H.; Wu, Y.C.; et al. Antileukemia component, dehydroeburicoic acid from Antrodia camphorata induces DNA damage and apoptosis in vitro and in vivo models. Phytomedicine 2012, 19, 788–796. [Google Scholar] [CrossRef]
- Su, J.H.; Chen, Y.C.; El-Shazly, M.; Du, Y.C.; Su, C.W.; Tsao, C.W.; Liu, L.L.; Chou, Y.; Chang, W.B.; Su, Y.D.; et al. Towards the Small and the Beautiful: A Small Dibromotyrosine Derivative from Pseudoceratina sp. Sponge Exhibits Potent Apoptotic Effect through Targeting IKK/NFkappaB Signaling Pathway. Mar. Drugs 2013, 11, 3168–3185. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Zheng, Y.; Yamaguchi, H.; Tian, C.; Lee, M.W.; Tang, H.; Wang, H.-G.; Chen, Q. Arsenic trioxide (As2O3) induces apoptosis through activation of Bax in hematopoietic cells. Oncogene 2005, 24, 3339–3347. [Google Scholar] [CrossRef]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 2004, 279, 21085–21095. [Google Scholar]
- Zhou, H.; Xu, M.; Gao, Y.; Deng, Z.; Cao, H.; Zhang, W.; Wang, Q.; Zhang, B.; Song, G.; Zhan, Y. Matrine induces caspase-independent program cell death in hepatocellular carcinoma through bid-mediated nuclear translocation of apoptosis inducing factor. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Ratain, M.J.; Rowley, J.D. Therapy-related acute myeloid leukemia secondary to inhibitors of topoisomerase II: From the bedside to the target genes. Ann. Oncol. 1992, 3, 107–111. [Google Scholar] [CrossRef]
- Roulston, D.; Anastasi, J.; Rudinsky, R.; Nucifora, G.; Zeleznik-Le, N.; Rowley, J.D.; McGavran, L.; Tsuchida, M.; Hayashi, Y. Therapy-related acute leukemia associated with t(11q23) after primary acute myeloid leukemia with t(8;21): A report of two cases. Blood 1995, 86, 3613–3614. [Google Scholar]
- Kudo, K.; Yoshida, H.; Kiyoi, H.; Numata, S.; Horibe, K.; Naoe, T. Etoposide-related acute promyelocytic leukemia. Leukemia 1998, 12, 1171–1175. [Google Scholar]
- Pedersen-Bjergaard, J.; Philip, P.; Larsen, S.O.; Andersson, M.; Daugaard, G.; Ersboll, J.; Hansen, S.W.; Hou-Jensen, K.; Nielsen, D.; Sigsgaard, T.C.; et al. Therapy-related myelodysplasia and acute myeloid leukemia. Cytogenetic characteristics of 115 consecutive cases and risk in seven cohorts of patients treated intensively for malignant diseases in the Copenhagen series. Leukemia 1993, 7, 1975–1986. [Google Scholar]
- Chiara, A.D.; Pederzoli-Ribeil, M.; Burgel, P.R.; Danel, C.; Witko-Sarsat, V. Targeting cytosolic proliferating cell nuclear antigen in neutrophil-dominated inflammation. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Hoffman, B. Gadd45 in the response of hematopoietic cells to genotoxic stress. Blood Cells Mol. Dis. 2007, 39, 329–335. [Google Scholar] [CrossRef]
- Liebermann, D.A.; Tront, J.S.; Sha, X.; Mukherjee, K.; Mohamed-Hadley, A.; Hoffman, B. Gadd45 stress sensors in malignancy and leukemia. Crit. Rev. Oncog. 2011, 16, 129–140. [Google Scholar] [CrossRef]
- Barreto, G.; Schafer, A.; Marhold, J.; Stach, D.; Swaminathan, S.K.; Handa, V.; Doderlein, G.; Maltry, N.; Wu, W.; Lyko, F.; et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007, 445, 671–675. [Google Scholar] [CrossRef]
- Wadhwa, S.; Mumper, R.J. d-Penicillamine and other low molecular weight thiols: Review of anticancer effects and related mechanisms. Cancer Lett. 2013, 337, 8–21. [Google Scholar] [CrossRef]
- Gillies, L.A.; Kuwana, T. Apoptosis regulation at the mitochondrial outer membrane. J. Cell. Biochem. 2014, 115, 632–640. [Google Scholar] [CrossRef]
- Chiara, F.; Gambalunga, A.; Sciacovelli, M.; Nicolli, A.; Ronconi, L.; Fregona, D.; Bernardi, P.; Rasola, A.; Trevisan, A. Chemotherapeutic induction of mitochondrial oxidative stress activates GSK-3alpha/beta and Bax, leading to permeability transition pore opening and tumor cell death. Cell Death Dis. 2012, 3, e444. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, S.-Y.; Khuri, F.; Curran, W.J.; Deng, X. Mono-or double-site phosphorylation distinctly regulates the proapoptotic function of Bax. PLoS One 2010, 5, e13393. [Google Scholar]
- An, J.; Chen, Y.; Huang, Z. Critical upstream signals of cytochrome c release induced by a novel Bcl-2 inhibitor. J. Biol. Chem. 2004, 279, 19133–19140. [Google Scholar] [CrossRef]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef]
- Palorini, R.; Simonetto, T.; Cirulli, C.; Chiaradonna, F. Mitochondrial complex I inhibitors and forced oxidative phosphorylation synergize in inducing cancer cell death. Int. J. Cell. Biol. 2013, 2013. [Google Scholar] [CrossRef]
- Kim, J.S.; Ahn, K.J.; Kim, J.A.; Kim, H.M.; Lee, J.D.; Lee, J.M.; Kim, S.J.; Park, J.H. Role of reactive oxygen species-mediated mitochondrial dysregulation in 3-bromopyruvate induced cell death in hepatoma cells : ROS-mediated cell death by 3-BrPA. J. Bioenerg. Biomembr. 2008, 40, 607–618. [Google Scholar] [CrossRef]
- Wei, L.; Dai, Q.; Zhou, Y.; Zou, M.; Li, Z.; Lu, N.; Guo, Q. Oroxylin A sensitizes non-small cell lung cancer cells to anoikis via glucose-deprivation-like mechanisms: c-Src and hexokinase II. Biochim. Biophys. Acta 2013, 1830, 3835–3845. [Google Scholar] [CrossRef]
- Sebastian, S.; Kenkare, U.W. Expression of two type II-like tumor hexokinase RNA transcripts in cancer cell lines. Tumor Biol. 1998, 19, 253–260. [Google Scholar] [CrossRef]
- Marin-Hernandez, A.; Rodriguez-Enriquez, S.; Vital-Gonzalez, P.A.; Flores-Rodriguez, F.L.; Macias-Silva, M.; Sosa-Garrocho, M.; Moreno-Sanchez, R. Determining and understanding the control of glycolysis in fast-growth tumor cells. Flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J. 2006, 273, 1975–1988. [Google Scholar] [CrossRef]
- Liu, C.X.; Zhou, H.C.; Yin, Q.Q.; Wu, Y.L.; Chen, G.Q. Targeting peroxiredoxins against leukemia. Exp. Cell Res. 2013, 319, 170–176. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shih, H.-C.; El-Shazly, M.; Juan, Y.-S.; Chang, C.-Y.; Su, J.-H.; Chen, Y.-C.; Shih, S.-P.; Chen, H.-M.; Wu, Y.-C.; Lu, M.-C. Cracking the Cytotoxicity Code: Apoptotic Induction of 10-Acetylirciformonin B is Mediated through ROS Generation and Mitochondrial Dysfunction. Mar. Drugs 2014, 12, 3072-3090. https://doi.org/10.3390/md12053072
Shih H-C, El-Shazly M, Juan Y-S, Chang C-Y, Su J-H, Chen Y-C, Shih S-P, Chen H-M, Wu Y-C, Lu M-C. Cracking the Cytotoxicity Code: Apoptotic Induction of 10-Acetylirciformonin B is Mediated through ROS Generation and Mitochondrial Dysfunction. Marine Drugs. 2014; 12(5):3072-3090. https://doi.org/10.3390/md12053072
Chicago/Turabian StyleShih, Huei-Chuan, Mohamed El-Shazly, Yung-Shun Juan, Chao-Yuan Chang, Jui-Hsin Su, Yu-Cheng Chen, Shou-Ping Shih, Huei-Mei Chen, Yang-Chang Wu, and Mei-Chin Lu. 2014. "Cracking the Cytotoxicity Code: Apoptotic Induction of 10-Acetylirciformonin B is Mediated through ROS Generation and Mitochondrial Dysfunction" Marine Drugs 12, no. 5: 3072-3090. https://doi.org/10.3390/md12053072
APA StyleShih, H.-C., El-Shazly, M., Juan, Y.-S., Chang, C.-Y., Su, J.-H., Chen, Y.-C., Shih, S.-P., Chen, H.-M., Wu, Y.-C., & Lu, M.-C. (2014). Cracking the Cytotoxicity Code: Apoptotic Induction of 10-Acetylirciformonin B is Mediated through ROS Generation and Mitochondrial Dysfunction. Marine Drugs, 12(5), 3072-3090. https://doi.org/10.3390/md12053072