Marine and Semi-Synthetic Hydroxysteroids as New Scaffolds for Pregnane X Receptor Modulation




Abstract
:
{kind=link}
{kind=link}
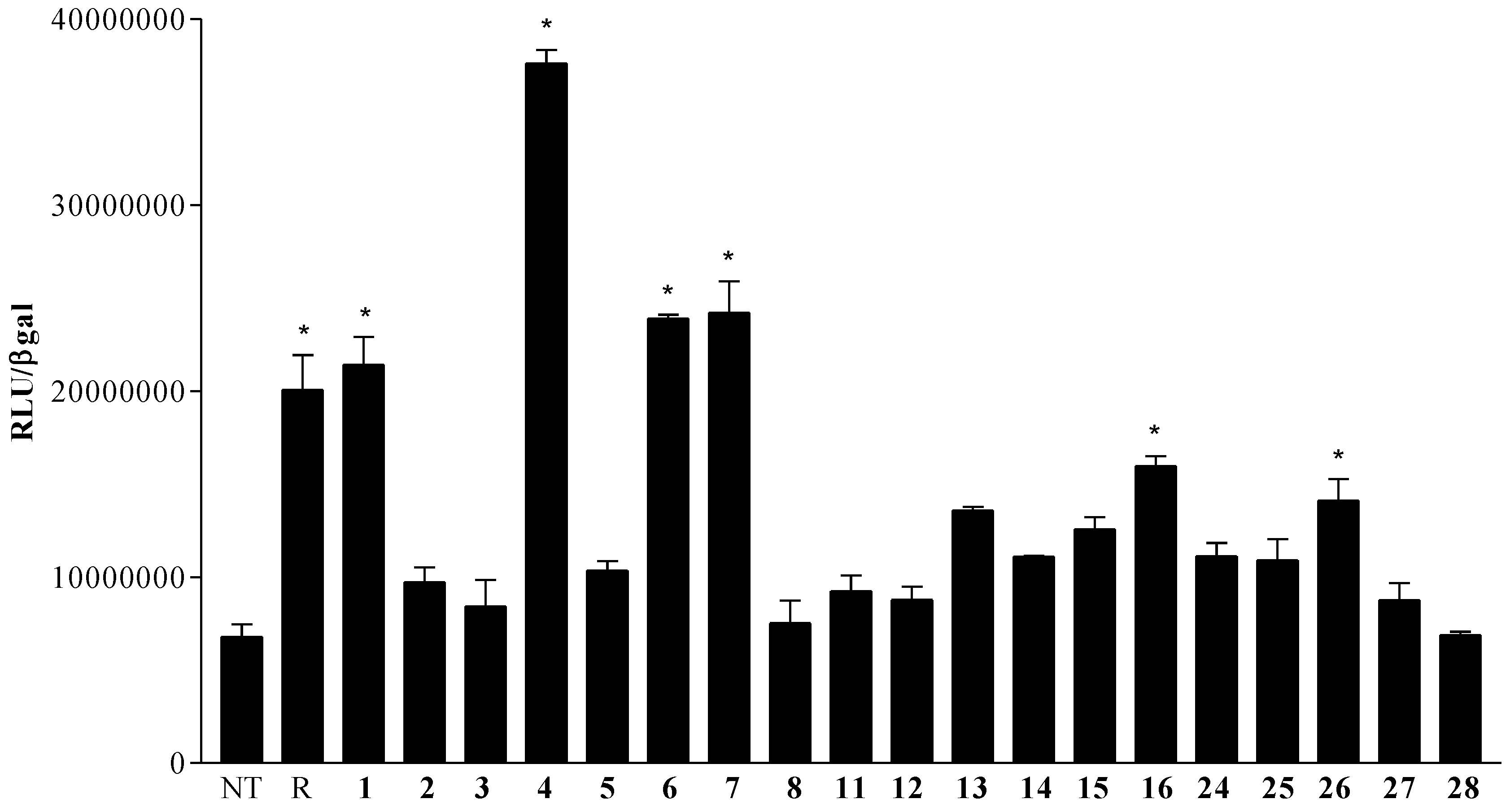{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
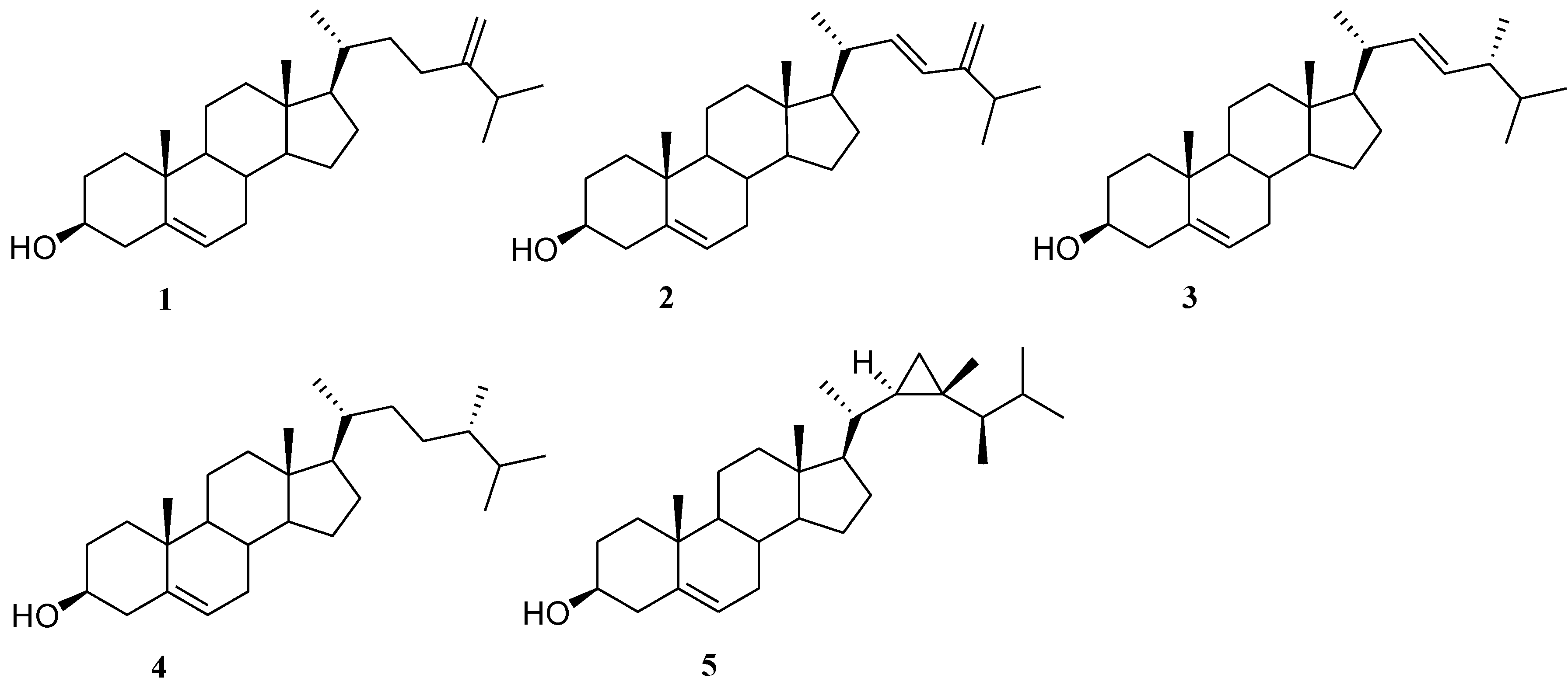
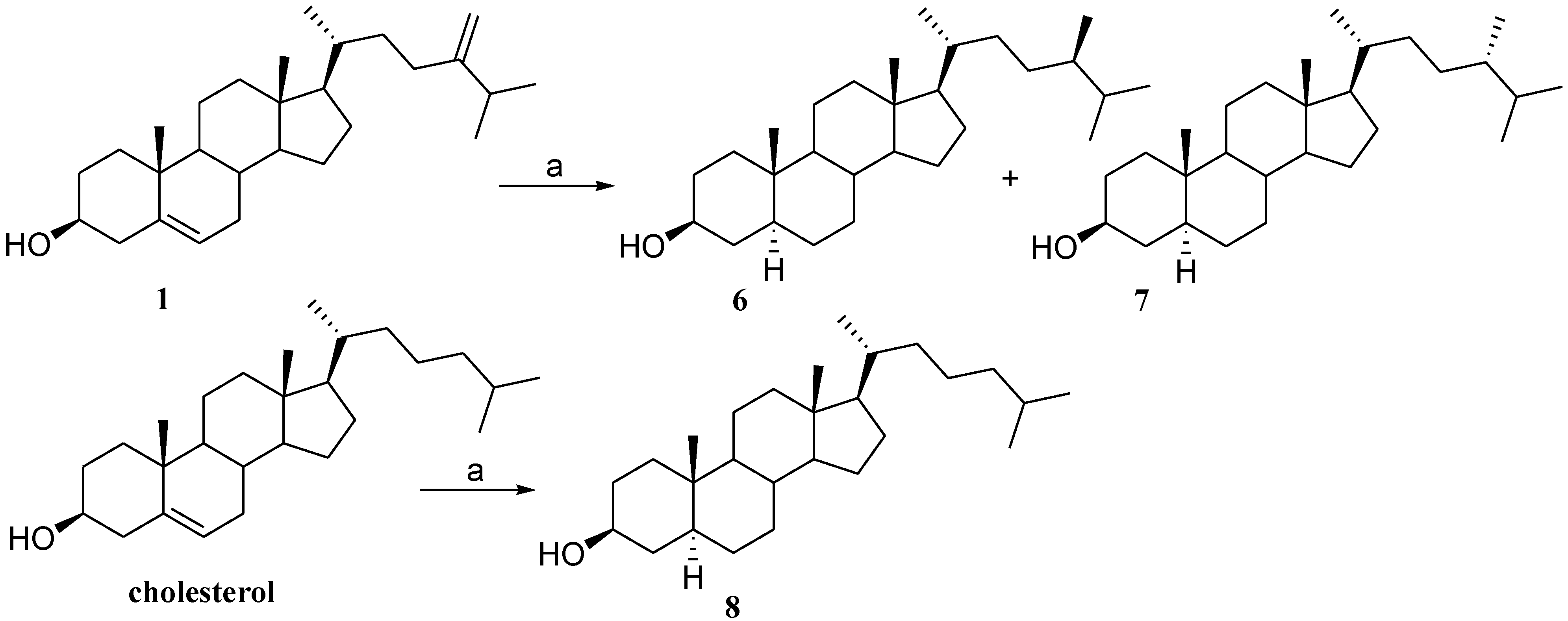
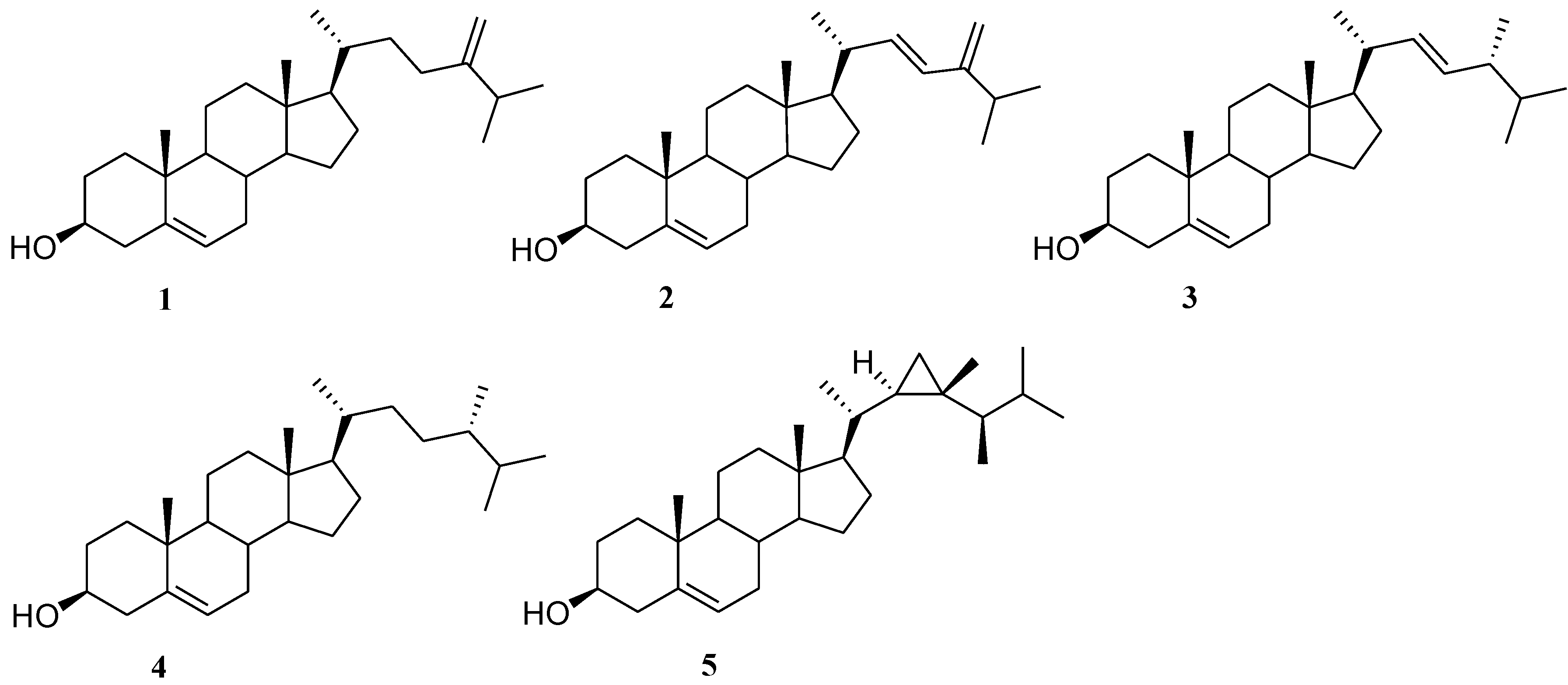
2.1. Isolation of Cholesterol Derivatives with C-24 Alkylation from Indian Ocean Collection of Sinularia
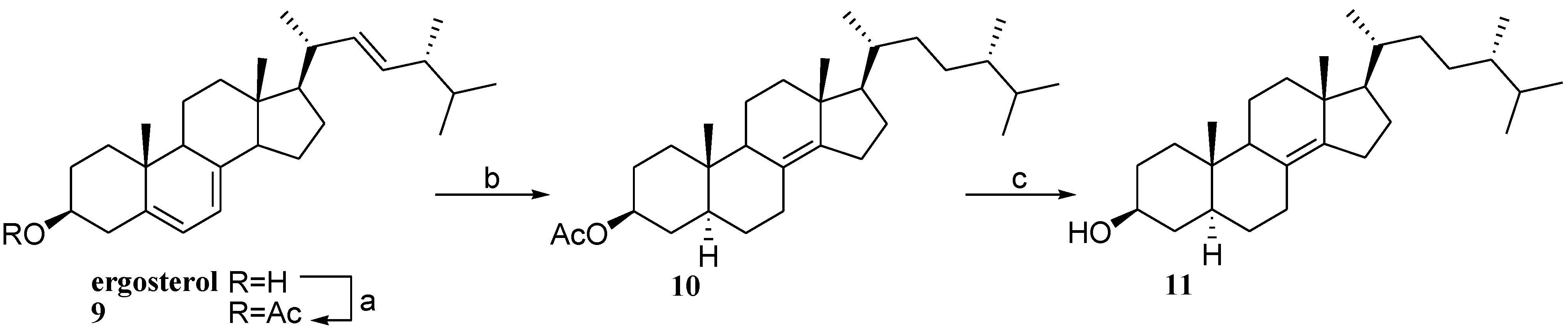
2.2. Preparation of 24-Methyl Stanols
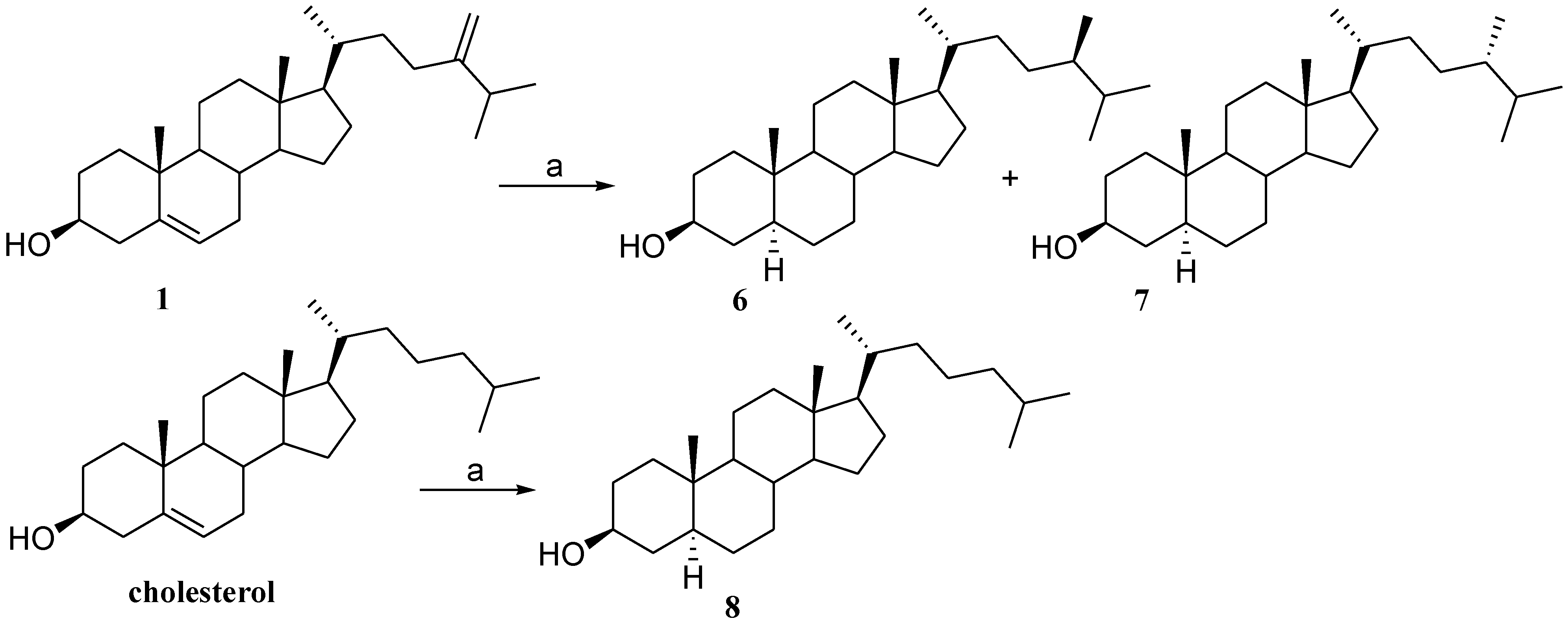
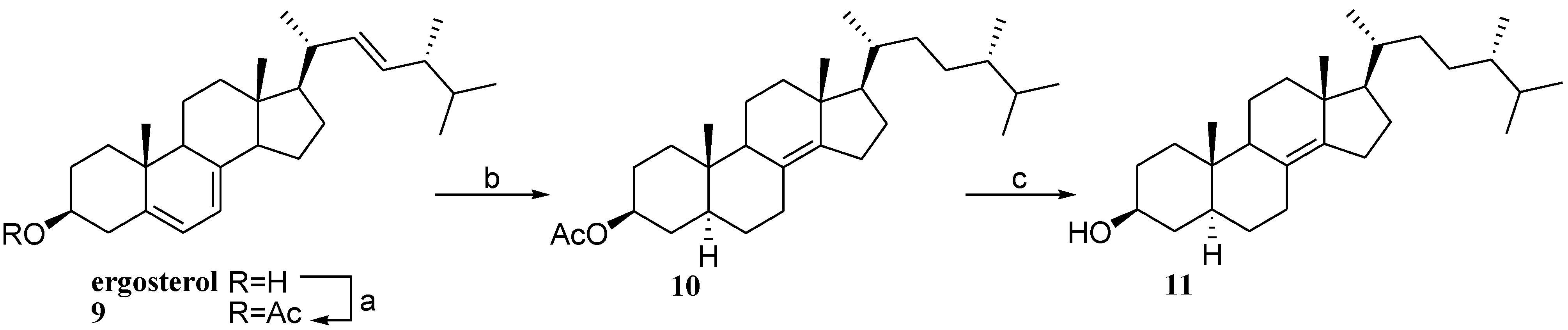
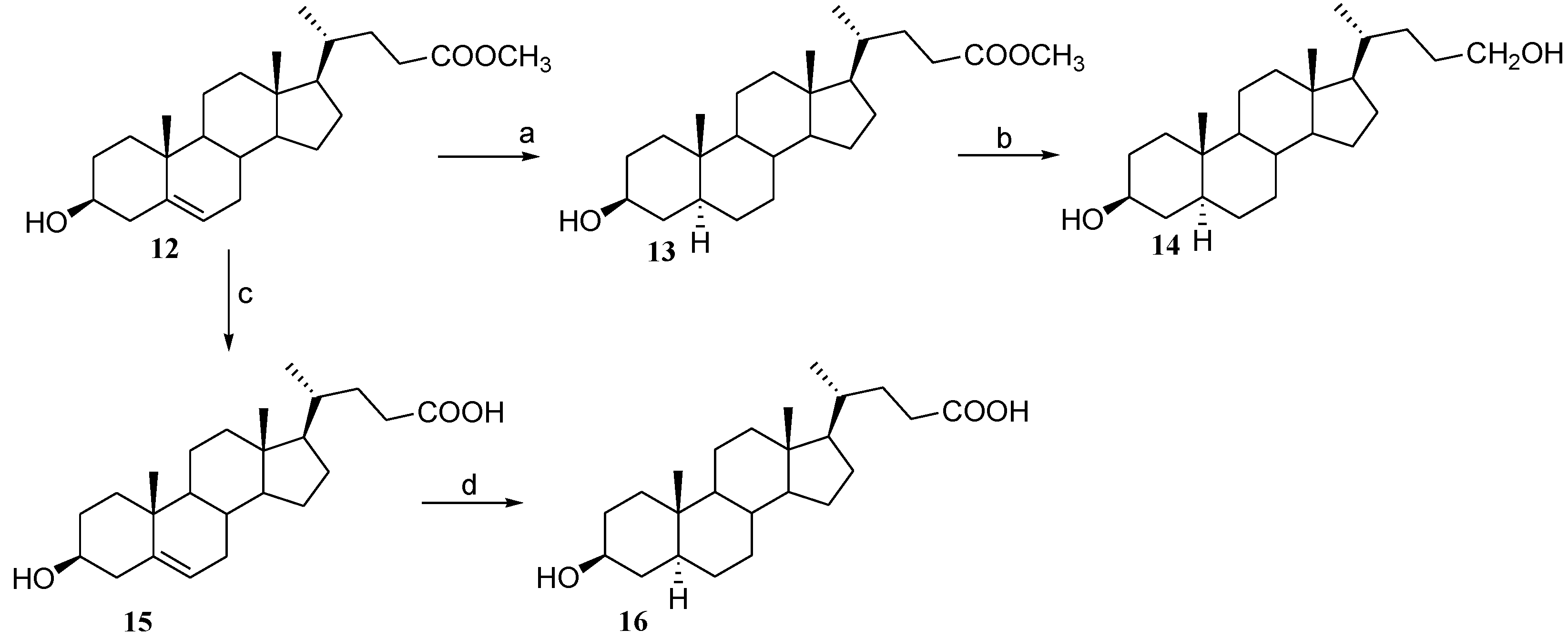
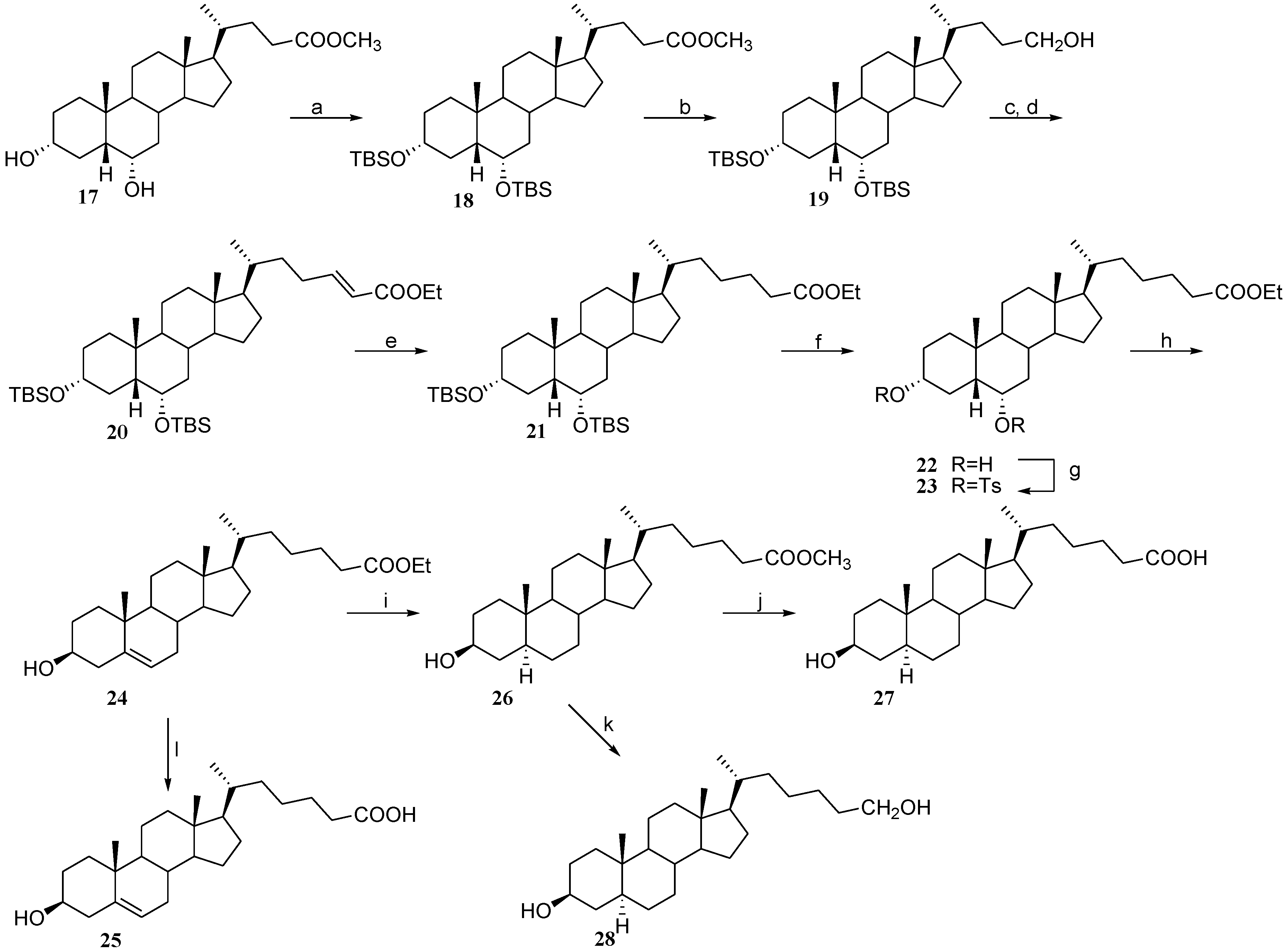
2.3. Preparation of Polar Side Chain Modified 3β-Hydroxy Steroids
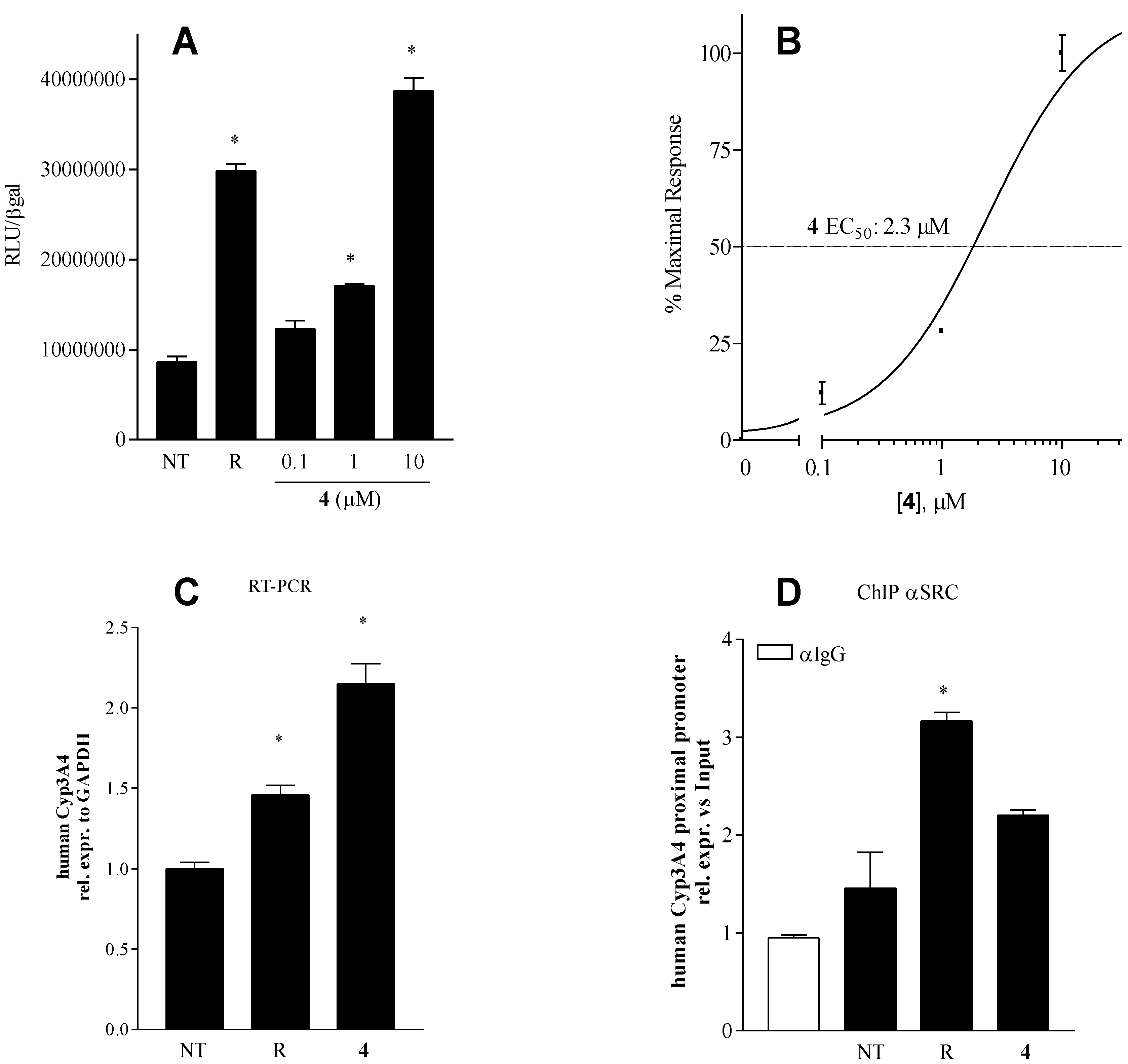
2.4. Pharmacological Evaluation
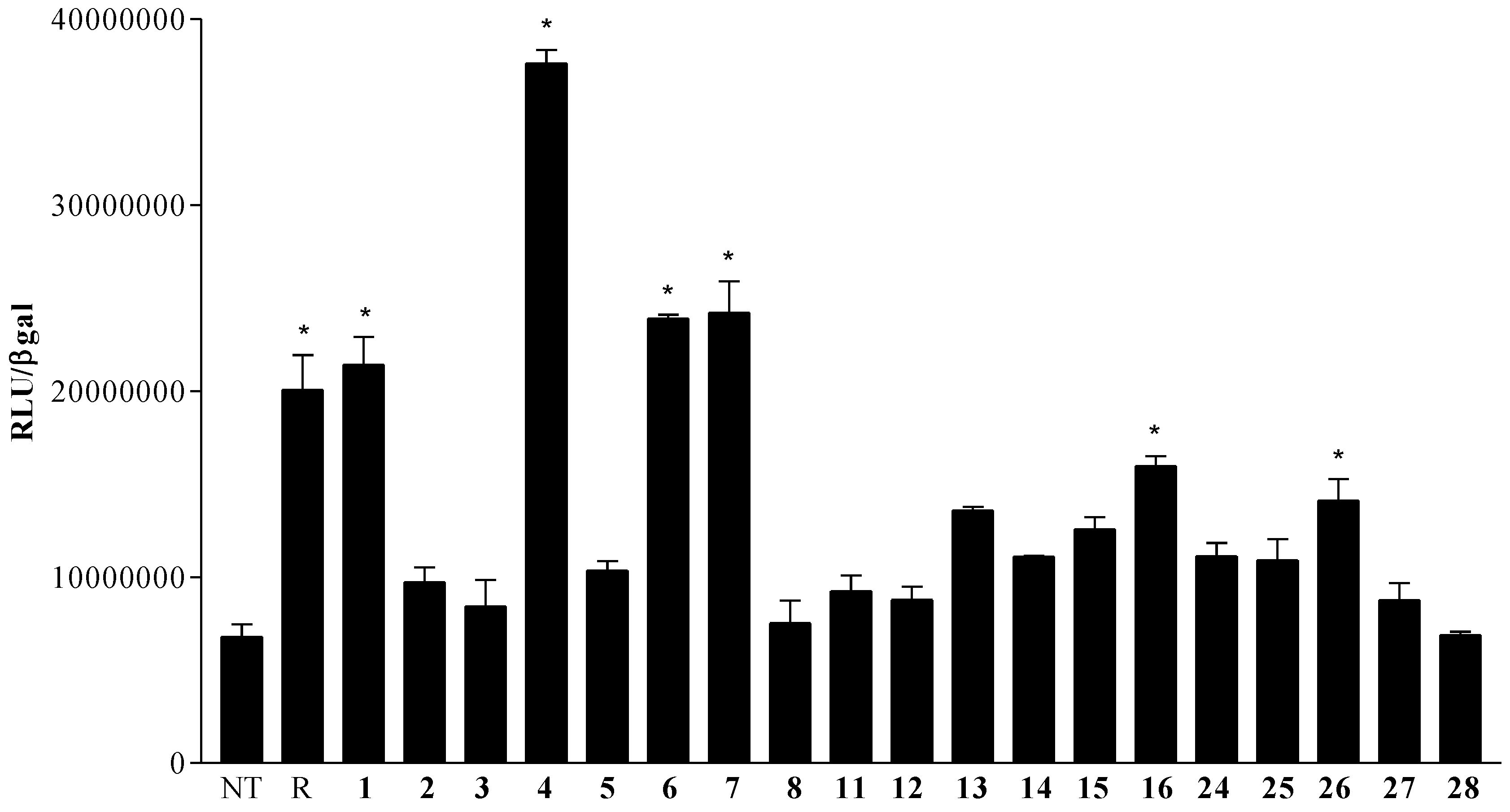
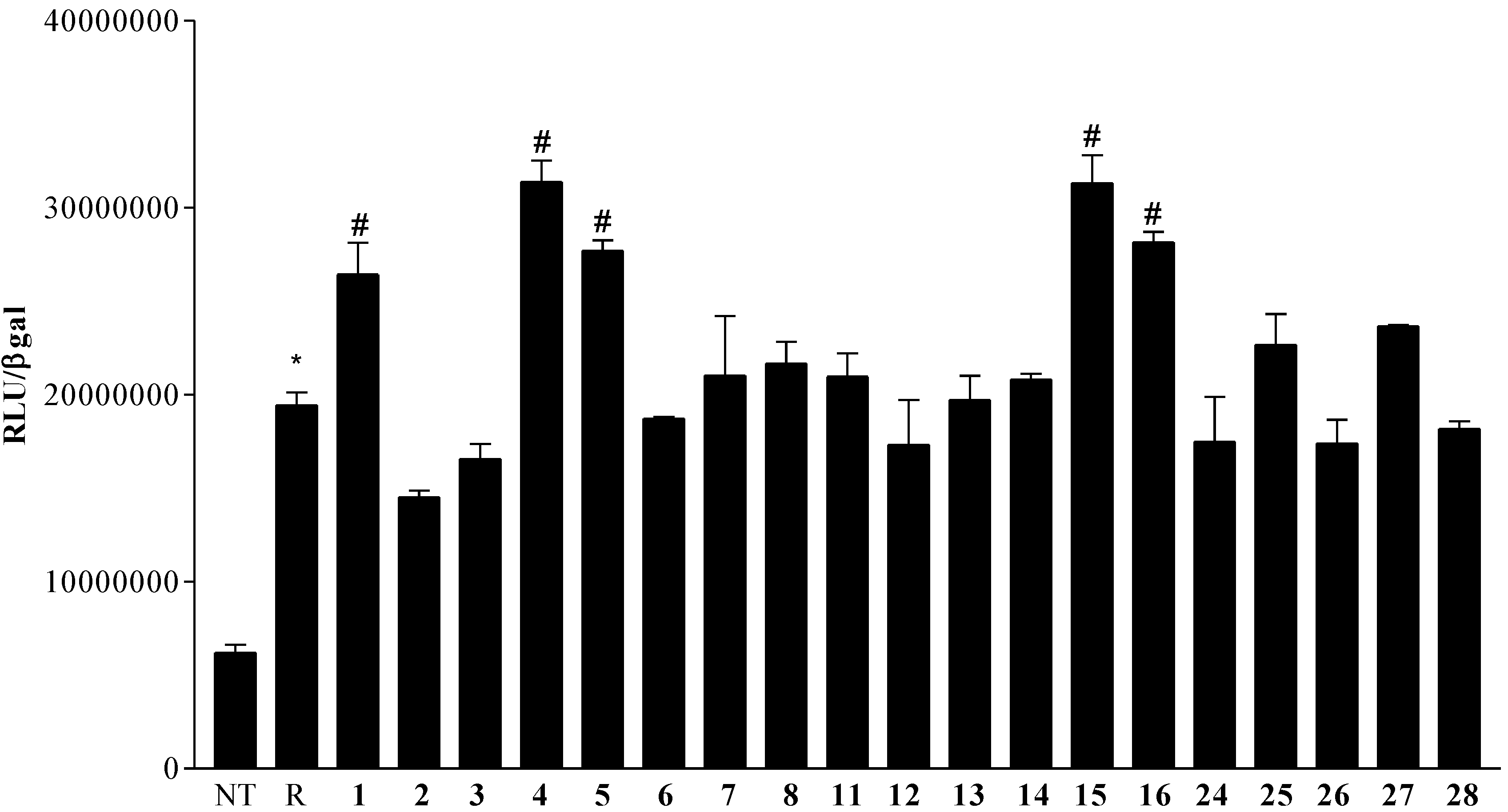
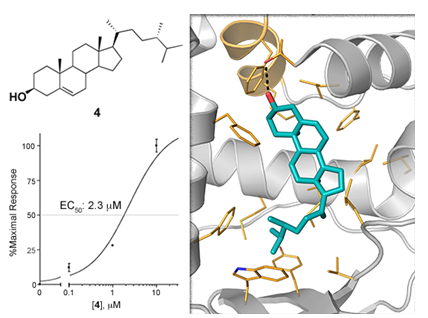
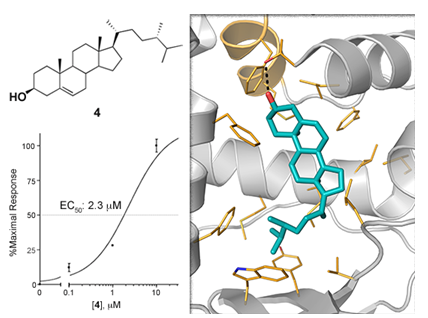
Pharmacologial evaluation on 4
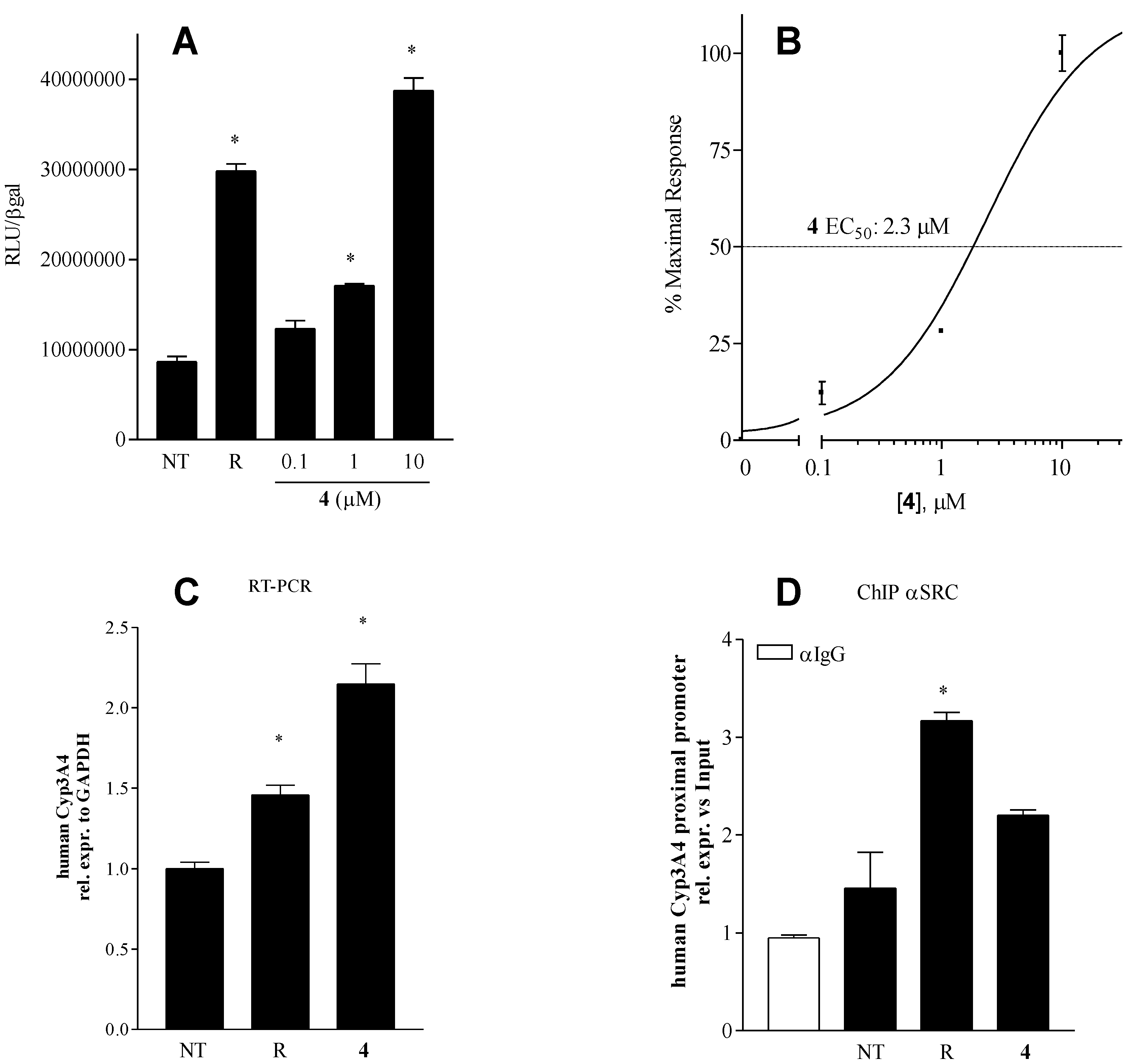
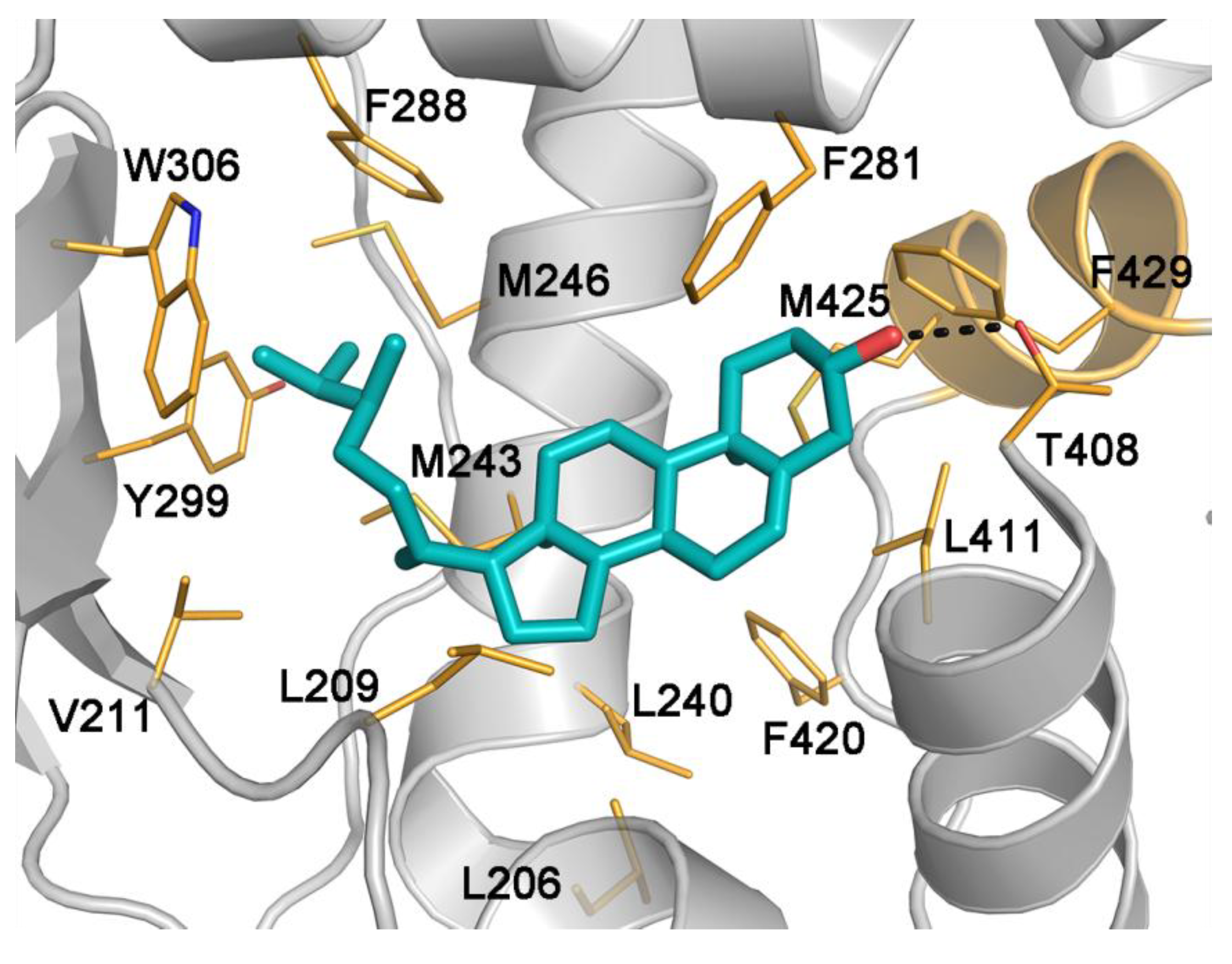
2.5. Binding Mode of Compound 4
3. Experimental Section
3.1. Chemistry
3.1.1. General Procedures
3.1.2. Isolation Procedures
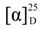 −16.6 (c 0.04, CHCl3); HRMS-ESI m/z 399.3625 [M + H]+, C28H47O requires 399.3627. Selected 1H NMR (C6D6): δH 5.36 (br d, J = 5.0 Hz, 1H), 4.92 (s, 1H), 4.90 (s, 1H), 3.40 (m, 1H), 1.09 (d, J = 6.8 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H), 0.95 (s, 3H), 0.66 (s, 3H). −38.7 (c 0.05, CHCl3); HRMS-ESI m/z 397.3464 [M + H]+, C28H45O requires 397.3470. Selected 1H NMR (C6D6): δH 6.07 (d, J = 15.6 Hz, 1H), 5.63 (dd, J = 8.7, 15.6 Hz, 1H), 5.36 (br d, J = 3.9 Hz, 1H), 5.03 (s, 1H), 4.96 (s, 1H), 3.40 (m, 1H), 1.14 (d, J = 7.0 Hz, 6H), 1.10 (d, J = 7.0 Hz, 3H), 0.95 (s, 3H), 0.66 (s, 3H). −23.6 (c 0.04, CHCl3); HRMS-ESI m/z 399.3622 [M + H+], C28H47O requires 399.3627. Selected 1H NMR (C6D6): δH 5.36 (br d, J = 5.0 Hz, 1H), 5.29 (ovl, 1H), 5.27 (ovl, 1H), 3.39 (m, 1H), 1.12 (d, J = 7.0 Hz, 3H), 1.01 (d, J = 7.0 Hz, 3H), 0.95 (s, 3H), 0.92 (d, J = 6.8 Hz, 6H), 0.68 (s, 3H). −42.5 (c 0.04, CHCl3); HRMS-ESI m/z 401.3778 [M + H]+, C28H49O requires 401.3783. NMR data as previously reported [19]. −29.5 (c 0.06, CHCl3); HRMS-ESI m/z 443.4249 [M + H]+, C31H55O requires 443.4253. NMR data as previously reported [20].
−16.6 (c 0.04, CHCl3); HRMS-ESI m/z 399.3625 [M + H]+, C28H47O requires 399.3627. Selected 1H NMR (C6D6): δH 5.36 (br d, J = 5.0 Hz, 1H), 4.92 (s, 1H), 4.90 (s, 1H), 3.40 (m, 1H), 1.09 (d, J = 6.8 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H), 0.95 (s, 3H), 0.66 (s, 3H). −38.7 (c 0.05, CHCl3); HRMS-ESI m/z 397.3464 [M + H]+, C28H45O requires 397.3470. Selected 1H NMR (C6D6): δH 6.07 (d, J = 15.6 Hz, 1H), 5.63 (dd, J = 8.7, 15.6 Hz, 1H), 5.36 (br d, J = 3.9 Hz, 1H), 5.03 (s, 1H), 4.96 (s, 1H), 3.40 (m, 1H), 1.14 (d, J = 7.0 Hz, 6H), 1.10 (d, J = 7.0 Hz, 3H), 0.95 (s, 3H), 0.66 (s, 3H). −23.6 (c 0.04, CHCl3); HRMS-ESI m/z 399.3622 [M + H+], C28H47O requires 399.3627. Selected 1H NMR (C6D6): δH 5.36 (br d, J = 5.0 Hz, 1H), 5.29 (ovl, 1H), 5.27 (ovl, 1H), 3.39 (m, 1H), 1.12 (d, J = 7.0 Hz, 3H), 1.01 (d, J = 7.0 Hz, 3H), 0.95 (s, 3H), 0.92 (d, J = 6.8 Hz, 6H), 0.68 (s, 3H). −42.5 (c 0.04, CHCl3); HRMS-ESI m/z 401.3778 [M + H]+, C28H49O requires 401.3783. NMR data as previously reported [19]. −29.5 (c 0.06, CHCl3); HRMS-ESI m/z 443.4249 [M + H]+, C31H55O requires 443.4253. NMR data as previously reported [20].3.1.3. Synthesis
+0.4 (c 0.09, CH3OH); selected 1H NMR (700 MHz, C6D6): δH 3.37 (m, 1H), 2.99 (d, J = 5.0 Hz, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H), 0.70 (s, 3H), 0.66 (s, 3H). HRMS-ESI m/z 403.3937 [M + H]+, C28H51O requires 403.3940. +0.6 (c 0.12, CH3OH); selected 1H NMR (700 MHz, C6D6): δH 3.40 (m, 1H), 3.01 (d, J = 5.0 Hz, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H), 0.70 (s, 3H), 0.66 (s, 3H). HRMS-ESI m/z 403.3935 [M + H]+, C28H51O requires 403.3940. +11.8 (c 0.27, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 3.58 (m, 1H), 0.89 (d, J = 6.0 Hz, 3H), 0.85 (d, J = 6.0 Hz, 6H), 0.79 (s, 3H), 0.64 (s, 3H). 13C NMR (100 MHz, CDCl3): δC 71.7, 56.8, 56.5, 54.6, 45.1, 42.8, 40.3, 39.8, 38.5, 37.2, 36.4, 36.1, 35.8, 35.7, 32.4, 31.8, 29.0, 28.5, 28.2, 24.5, 24.0, 23.0, 22.8, 21.5, 18.9, 12.6, 12.3; HRMS-ESI m/z 389.3781 [M + H]+, C27H49O requires 389.3783. −86.0 (c 0.07, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 5.56 (m, 1H), 5.37 (m, 1H), 4.70 (m, 1H), 2.50 (m, 1H), 2.36 (m, 1H), 2.04 (s, 3H), 1.03 (d, J = 6.8 Hz, 3H), 0.95 (s, 3H), 0.91 (d, J = 6.8 Hz, 3H), 0.84 (d, J = 6.4 Hz, 3H), 0.82 (d, J = 6.4 Hz, 3H), 0.62 (s, 3H). 13C NMR (100 MHz, CDCl3): δC 170.5, 141.7, 138.8, 135.6, 132.0, 120.2, 116.2, 72.7, 55.7, 54.5, 46.0, 42.7 (2C), 40.4, 38.9, 37.8, 37.0, 36.6, 33.0, 28.2, 28.0, 22.9, 21.3, 21.0, 20.9, 19.8, 19.6, 17.5, 16.0, 12.0; HRMS-ESI m/z 439.6920 [M + H]+, C30H47O2 requires 439.6924. +1.23 (c 0.19, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 4.71 (m, 1H), 2.02 (s, 3H), 0.92 (d, J = 6.7 Hz, 3H), 0.84 (d, J = 6.7 Hz, 3H), 0.83 (s, 3H), 0.77 (d, J = 6.5 Hz, 6H), 0.69 (s, 3H). 13C NMR (100 MHz, CDCl3): δC 170.7, 143.0, 126.2, 73.7, 56.7, 49.2, 44.1, 42.7, 39.1, 37.3, 36.7, 36.3, 34.8, 34.1, 33.5, 31.5, 30.4, 29.5, 28.7, 27.5, 27.0, 25.8, 21.5, 20.5, 19.9, 19.3, 18.2, 17.6, 15.4, 12.7. HRMS-ESI m/z 443.3885 [M + H]+, C30H51O2 requires 443.3889. +10.2 (c 0.13, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 3.62 (m, 1H), 0.93 (d, J = 6.5 Hz, 3H), 0.85 (d, J = 6.4 Hz, 3H), 0.84 (s, 3H), 0.78 (d, J = 6.5 Hz, 6H), 0.63 (s, 3H); 13C NMR (100 MHz, CDCl3): δC 142.6, 126.3, 71.2, 56.6, 49.2, 44.2, 42.7, 39.0, 38.2, 37.2, 36.7, 36.5, 34.8, 33.5, 31.5, 30.3, 29.6, 28.8, 27.0 (2C), 25.8, 20.5, 19.9, 19.2, 18.2, 17.5, 15.4, 12.8; HRMS-ESI m/z 401.3780 [M + H]+, C28H49O requires 401.3783. −9.0 (c 0.73, CHCl3); selected 1H NMR (400 MHz CDCl3): δH 5.34 (d, J = 5.0 Hz, 1H), 3.66 (s, 3H), 3.50 (m, 1H), 1.00 (s, 3H), 0.92 (d, J = 6.5 Hz, 3H), 0.67 (s, 3H); 13C NMR (100 MHz CDCl3): δC 175.1, 141.0, 121.8, 71.9, 57.0, 56.0, 51.7, 50.4, 42.4 (2C), 39.9, 37.5, 35.6 (2C), 32.1 (2C), 31.7, 31.3 (2C), 28.3, 24.5, 21.3, 19.7, 18.5, 12.1; HRMS-ESI m/z 389.3053 [M + H]+, C25H41O3 requires 389.3056. +3.4 (c 0.54, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 3.64 (s, 3H), 3.56 (m, 1H), 0.89 (d, J = 6.0 Hz, 3H), 0.78 (s, 3H), 0.63 (s, 3H).13C NMR (100 MHz, CDCl3): δC 175.3, 71.5, 56.7, 56.1, 54.6, 51.8, 45.1, 42.9, 40.3, 38.3, 37.2, 35.7 (2C), 35.6, 32.3, 31.6, 31.3, 31.2, 28.9, 28.4, 24.4, 21.5, 18.5, 12.5, 12.3. HRMS-ESI m/z 391.3210 [M + H]+, C25H43O3 requires 391.3212. +21 (c 0.08, CH3OH); selected 1H NMR (400 MHz, CD3OD): δH 3.50 (m, ovl, 2H), 3.50 (m, ovl, 1H), 0.94 (d, J = 6.4 Hz, 3H), 0.82 (s, 3H), 0.69 (s, 3H). 13C NMR (100 MHz, CDCl3): δC 71.4, 63.7, 56.4, 56.1, 54.3, 44.8, 42.3, 40.0, 38.2 (2C), 36.9 (2C), 35.5, 35.4, 32.1, 32.0, 31.7, 29.3, 28.6, 24.2, 21.2, 18.6, 12.3, 12.0; HRMS-ESI m/z 363.3260 [M + H]+, C24H43O2 requires 362.3263. −13.6 (c 0.1, CH3OH); selected 1H NMR (400 MHz, CD3OD): δH 5.35 (d, J = 5.0 Hz, 1H), 3.50 (m, 1H), 1.00 (s, 3H), 0.96 (d, J = 6.4 Hz, 3H), 0.71 (s, 3H). 13C NMR (100 MHz, CD3OD): δC 177.0, 142.5, 122.5, 72.5, 58.2, 57.3, 51.7, 43.5, 43.0, 41.1, 38.6, 36.7, 33.3, 33.0 (2C), 32.3 (2C), 32.0, 29.1, 25.3, 22.2, 19.8, 18.8, 12.3. HRMS-ESI m/z 375.2895 [M + H]+, C24H39O3 requires 375.2899. +4.16 (c 1.6, CH3OH); selected 1H NMR (400 MHz, CDCl3): δH 3.57 (m, 1H), 0.94 (d, J = 6.4 Hz, 3H), 0.81 (s, 3H), 0.66 (s, 3H). 13C NMR (100 MHz, CD3OD): δC 178.1, 72.0, 57.9, 57.5, 55.8, 46.2, 43.8, 41.4, 38.9, 38.3, 36.9, 36.7, 36.6, 33.3, 32.3, 32.2, 32.0, 30.0, 29.1, 25.3, 22.4, 18.7, 12.8, 12.6. HRMS-ESI m/z 377.3053 [M + H]+, C24H41O3 requires 419.3161. +2.2 (c 0.55, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 3.98 (dt, J = 3.8, 8.0 Hz, 1H), 3.67 (s, 3H), 3.52 (m, 1H), 0.90 (d, J = 6.4 Hz, 3H), 0.88 (s, 9H), 0.87 (s, 9H), 0.86 (s, 3H), 0.62 (s, 3H), 0.04 (s, 6H), 0.02 (s, 6H). 13C NMR (100 MHz, CDCl3): δC 175.0, 73.0, 68.6, 56.0, 55.8, 51.4, 49.5, 42.8, 39.9, 39.5 (2C), 35.9 (2C), 35.3 (2C), 34.8, 31.0 (2C), 30.9, 30.8, 29.7, 28.0, 25.9 (3C), 25.5 (3C), 24.2, 23.5, 20.7, 18.2, 12.0, −4.5, −4.7, −4.8, −4.9; HRMS-ESI m/z 635.4893 [M + H]+, C37H71O4Si2 requires 635.4891. +6.3 (c 0.4, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 3.95 (dt, J = 3.4, 11.2 Hz, 1H), 3.57 (t, J = 6.6 Hz, 2H), 3.50 (m, 1H), 1.92 (m, 1H), 1.83 (m, 1H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (s, 9H), 0.85 (s, 9H), 0.85 (s, 3H), 0.60 (s, 3H), 0.03 (s, 6H), 0.01 (s, 6H). 13C NMR (100 MHz, CDCl3): δC 73.0, 68.5, 63.3, 56.0 (2C), 49.5, 42.7, 39.9, 39.5 (2C), 35.8 (2C), 35.5, 35.3, 34.7, 31.7, 30.8 (2C), 29.8, 29.3, 28.1, 25.9 (3C), 25.8 (3C), 24.1, 23.4, 20.7, 18.5, 12.0, −4.5, −4.7, −4.8, −4.9; HRMS-ESI m/z 607.4940 [M + H]+, C36H71O3Si2 requires 607.4942. −0.46 (c 0.3, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH 6.97 (dt, J = 5.1, 15.6 Hz, 1H), 5.81 (d, J = 15.6 Hz, 1H), 4.19 (q, J = 7.5 Hz, 2H), 3.99 (m, 1H), 3.53 (m, 1H), 1.29 (t, J = 7.6 Hz, 3H), 0.93 (d, J = 6.5 Hz, 3H), 0.90 (s, 9H), 0.90 (s, 3H), 0.89 (s, 9H), 0.64 (s, 3H), 0.06 (s, 6H), 0.04 (s, 6H). 13C NMR (100 MHz, CDCl3): δC 166.8, 149.8, 121.0, 72.9, 68.6, 60.0, 56.0 (2C), 49.5, 42.8, 39.9, 39.6, 35.9, 35.8, 35.4 (2C), 34.8, 34.2, 31.0 (3C), 29.8, 28.9, 28.1, 25.9 (3C), 25.8 (3C), 24.2, 23.5, 20.7, 18.4, 14.3, 12.0, −4.5, −4.6, −4.7, −4.8; HRMS-ESI m/z 675.5200 [M + H]+, C40H75O4Si2 requires 675.5204. −9.0 (c 0.05, CHCl3); selected 1H NMR (400 MHz, CDCl3): δH4.14 (q, J = 7.1 Hz, 2H), 4.01 (m, 1H), 3.55 (m, 1H), 2.31 (t, J = 7.6 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H), 0.93 (d, J = 7.0 Hz, 3H), 0.91 (s, 9H), 0.89 (s, 9H), 0.90 (s, 3H), 0.64 (s, 3H), 0.07 (s, 6H), 0.05 (s, 6H). 13C NMR (100 MHz, CD3OD): δC 174.0, 73.0, 68.6, 60.2, 56.2 (2C), 49.6, 42.8, 40.0, 39.8, 39.6, 35.9, 35.6, 35.5, 34.9, 34.4, 31.0 (2C), 30.4, 29.8, 29.6, 28.9, 28.2, 26.0, 25.9 (3C), 25.8 (3C), 24.2, 23.5, 20.8, 18.6, 14.3, 12.0, −4.5, −4.6, −4.7, −4.8. HRMS-ESI m/z 677.5357 [M + H]+, C40H77O4Si2 requires 677.5360. −3.78 (c 0.38, CH3OH); selected 1H NMR (400 MHz, CDCl3): δH 4.12 (q, J = 7.3 Hz, 2H), 4.04 (m, 1H), 3.63 (m, 1H), 2.10 (t, J = 7.3 Hz, 2H), 1.25 (t, J = 7.3 Hz, 3H), 0.90 (s, 3H), 0.89 (d, J = 6.6 Hz, 3H), 0.62 (s, 3H).13C NMR (100 MHz, CDCl3): δC 174.2, 71.0, 67.5, 60.2, 56.1, 56.0, 48.4, 42.6, 39.8 (2C), 39.7, 35.7, 35.5, 35.4, 35.3, 34.7, 34.6, 33.9, 29.8, 29.2, 28.1, 25.5, 24.0, 23.5, 20.6, 18.4, 14.3, 12.0. HRMS-ESI m/z 449.3629 [M + H]+, C28H49O4 requires 449.3631. +6.36 (c 0.34, CH3OH); 1H NMR (400 MHz, CDCl3): δH 7.78 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 4.78 (m, 1H), 4.30 (m, 1H), 4.12 (q, J = 7.1 Hz, 2H), 2.28 (t, J = 7.5 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H), 0.80 (s, 3H), 0.60 (s, 3H). 13C NMR (100 MHz, CDCl3): δC 173.7, 149.0, 144.2, 135.8, 134.0, 129.3 (2C), 128.6, 127.0, 126.9, 124.9, 123.4, 123.3, 81.4, 79.3, 59.7, 55.4, 55.3, 49.7, 45.8, 42.2, 39.1, 38.9, 35.6, 34.9, 34.8, 34.3, 33.9, 31.6, 27.5, 26.8, 25.9, 25.0, 24.8, 23.4 (2C), 22.3, 21.1, 19.9, 18.0, 13.8, 11.4; HRMS-ESI m/z 757.3805 [M + H]+, C42H61O8S2 requires 757.3808. −8.5 (c 0.28, CH3OH); selected 1H NMR (400 MHz, CDCl3): δH 5.31 (d, J = 4.2 Hz, 1H), 4.09 (q, J = 7.0 Hz, 2H), 3.48 (m, 1H), 2.26 (t, J = 7.5 Hz, 2H), 1.23 (t, J = 7.0 Hz, 3H), 0.98 (s, 3H), 0.88 (d, J = 6.2 Hz, 3H), 0.65 (s, 3H); 13C NMR (100 MHz CDCl3): δC 174.0, 140.8, 121.6, 71.7, 60.2, 56.7, 55.9, 50.0, 42.2 (2C), 39.7, 37.2, 36.4, 35.5, 35.4, 34.3, 31.8 (3C), 28.1, 25.5, 25.4, 24.2, 21.0, 19.3, 18.6, 14.2, 11.8; HRMS-ESI m/z 431.3250 [M + H]+, C28H47O requires 431.3252. −8.0 (c 0.10, CH3OH); selected 1H NMR (400 MHz, CD3OD): δH 5.33 (d, J = 4.3 Hz, 1H), 3.40 (m, 1H), 2.20 (t, J = 7.5 Hz, 1H), 1.02 (s, 3H), 0.94 (d, J = 6.3 Hz, 3H), 0.71 (s, 3H). 13C NMR (100 MHz, CD3OD): δH 177.8, 142.3, 122.5, 72.6, 58.2, 57.6, 51.8, 43.5, 43.0, 41.2, 38.6, 37.7, 37.0, 36.8, 35.0, 33.3, 33.1, 32.3, 29.3, 26.7, 26.5, 25.3, 22.2, 19.9, 19.2, 12.3; HRMS-ESI m/z 403.3210 [M + H]+, C26H49O3 requires 403.3212. +0.6 (c 0.1, CHCl3); 1H NMR (400 MHz, CDCl3): δH 3.61 (s, 3H), 3.52 (m, 1H), 2.25 (t, J = 7.5 Hz, 2H), 0.84 (d, J = 6.3 Hz, 3H), 0.75 (s, 3H), 0.59 (s, 3H). 13C NMR (100 MHz, CDCl3): δC 174.5, 71.0, 56.3, 56.0, 54.2, 51.3, 44.8, 42.5, 39.9, 38.0, 36.9, 35.4 (2C), 35.3 (2C), 34.0, 31.9, 31.3, 31.0, 28.6, 25.5, 25.3, 24.1, 21.1, 18.5, 12.2, 12.0; HRMS-ESI m/z 419.3520 [M + H]+, C27H47O3 requires 419.3525. +4.2 (c 0.2, CHCl3); selected 1H NMR (400 MHz, CD3OD): δH 3.50 (m, 1H), 2.28 (t, J = 7.6 Hz, 2H), 0.93 (d, J = 6.7 Hz, 3H), 0.83 (s, 3H), 0.70 (s, 3H). 13C NMR (100 MHz, CD3OD): δC 177.8, 71.9, 57.9, 57.6, 55.8, 46.2, 43.8, 41.4, 38.9, 38.3, 37.0, 36.9, 36.8, 36.6, 35.0, 33.3, 32.2, 30.0, 29.3, 26.7, 26.5, 25.2, 22.4, 19.2, 12.8, 12.6. HRMS-ESI m/z 405.3367 [M + H]+, C26H45O3 requires 405.3369. +3.6 (c 0.85, CHCl3); 1H NMR (400 MHz CD3OD): δH 3.53 (t, J = 7.2 Hz, 2H), 3.48 (m, 1H), 0.92 (d, J = 6.1 Hz, 3H), 0.83 (s, 3H), 0.69 (s, 3H); HRMS-ESI m/z 391.3573 [M + H]+, C26H47O2 requires 391.3576.3.2. Transfection and Luciferase Assays
3.3. Real Time PCR
3.4. CHiP
3.5. Statistical Analysis
3.6. Computational Methods
3.6.1. Ligand and Protein Preparation
3.6.2. Docking Calculations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef]
- Chang, T.K.H.; Waxman, D.J. Synthetic drugs and natural products as modulators of constitutive androstane receptor (CAR) and pregnane X receptor (PXR). Drug Metab. Rev. 2006, 38, 51–73. [Google Scholar] [CrossRef]
- D’Auria, M.V.; Sepe, V.; Zampella, A. Natural ligands for nuclear receptors: Biology and potential therapeutic applications. Curr. Top. Med. Chem. 2012, 12, 637–669. [Google Scholar] [CrossRef]
- Kittayaruksakul, S.; Zhao, W.; Xu, M.; Ren, S.; Lu, J.; Wang, J.; Downes, M.; Evans, R.M.; Venkataramanan, R.; Chatsudthipong, V.; et al. Identification of three novel natural product compounds that activate PXR and CAR and inhibit inflammation. Pharm. Res. 2013, 30, 2199–2208. [Google Scholar] [CrossRef]
- Cheng, J.; Shah, Y.M.; Gonzalez, F.J. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol. Sci. 2012, 33, 323–330. [Google Scholar] [CrossRef]
- Gu, X.; Ke, S.; Liu, D.; Sheng, T.; Thomas, P.E.; Rabson, A.B.; Gallo, M.A.; Xie, W.; Tian, Y. Role of NF-kappaB in regulation of PXR-mediated gene expression: A mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J. Biol. Chem. 2006, 281, 17882–17889. [Google Scholar]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Mencarelli, A.; D’Amore, C.; Renga, B.; Zampella, A.; Fiorucci, S. Total synthesis and pharmacological characterization of solomonsterol A, a potent marine pregnane-X-receptor agonist endowed with anti-inflammatory activity. J. Med. Chem. 2011, 54, 4590–4599. [Google Scholar] [CrossRef]
- Dou, W.; Zhang, J.; Zhang, E.; Sun, A.; Ding, L.; Chou, G.; Wang, Z.; Mani, S. Chrysin ameliorates chemically induced colitis in the mouse through modulation of a PXR/NF-κB signaling pathway. J. Pharmacol. Exp. Ther. 2013, 345, 473–482. [Google Scholar] [CrossRef]
- Cheng, J.; Shah, Y.M.; Ma, X.; Pang, X.; Tanaka, T.; Kodama, T.; Krausz, K.W.; Gonzalez, F.J. Therapeutic role of rifaximin in inflammatory bowel disease: Clinical implication of human pregnane X receptor activation. J. Pharmacol. Exp. Ther. 2010, 335, 32–41. [Google Scholar] [CrossRef]
- Mencarelli, A.; Renga, B.; Palladino, G.; D’Amore, C.; Ricci, P.; Distrutti, E.; Barbanti, M.; Baldelli, F.; Fiorucci, S. Inhibition of NF-κB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur. J. Pharmacol. 2011, 668, 317–324. [Google Scholar]
- Fiorucci, S.; Distrutti, E.; Bifulco, G.; D’Auria, M.V.; Zampella, A. Marine sponge steroids as nuclear receptor ligands. Trends Pharmacol. Sci. 2012, 33, 591–601. [Google Scholar] [CrossRef]
- Fiorucci, S.; Zampella, A.; Distrutti, E. Development of FXR, PXR and CAR agonists and antagonists for treatment of liver disorders. Curr. Top. Med. Chem. 2012, 12, 605–624. [Google Scholar] [CrossRef]
- Festa, C.; Lauro, G.; de Marino, S.; D’Auria, M.V.; Monti, M.C.; Casapullo, A.; D’Amore, C.; Renga, B.; Mencarelli, A.; Petek, S.; et al. Plakilactones from the marine sponge Plakinastrella mamillaris. Discovery of a new class of marine ligands of peroxisome proliferator-activated receptor γ. J. Med. Chem. 2012, 55, 8303–8317. [Google Scholar] [CrossRef]
- Festa, C.; de Marino, S.; D’Auria, M.V.; Bifulco, G.; Renga, B.; Fiorucci, S.; Petek, S.; Zampella, A. Solomonsterols A and B from Theonella swinhoei. The first example of C-24 and C-23 sulfated sterols from a marine source endowed with a PXR agonistic activity. J. Med. Chem. 2011, 54, 401–405. [Google Scholar] [CrossRef]
- De Marino, S.; Sepe, V.; D’Auria, M.V.; Bifulco, G.; Renga, B.; Petek, S.; Fiorucci, S.; Zampella, A. Towards new ligands of nuclear receptors. Discovery of malaitasterol A, an unique bis-secosterol from marine sponge Theonella swinhoei. Org. Biomol. Chem. 2011, 9, 4856–4862. [Google Scholar] [CrossRef]
- Festa, C.; D’Amore, C.; Renga, B.; Lauro, G.; de Marino, S.; D’Auria, M.V.; Bifulco, G.; Zampella, A.; Fiorucci, S. Oxygenated polyketides from Plakinastrella mamillaris as a new chemotype of PXR agonists. Mar. Drugs 2013, 11, 2314–2327. [Google Scholar] [CrossRef]
- Chini, M.G.; Jones, C.R.; Zampella, A.; D’Auria, M.V.; Renga, B.; Fiorucci, S.; Butts, C.P.; Bifulco, G. Quantitative NMR-derived interproton distances combined with quantum mechanical calculations of 13C chemical shifts in the stereochemical determination of conicasterol F, a nuclear receptor ligand from Theonella swinhoei. J. Org. Chem. 2012, 77, 1489–1496. [Google Scholar] [CrossRef]
- De Marino, S.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; Renga, B.; D’Amore, C.; Fiorucci, S.; Debitus, C.; Zampella, A. Theonellasterols and conicasterols from Theonella swinhoei. Novel marine natural ligands for human nuclear receptors. J. Med. Chem. 2011, 54, 3065–3075. [Google Scholar] [CrossRef]
- De Marino, S.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; D’Amore, C.; Renga, B.; Mencarelli, A.; Petek, S.; Fiorucci, S.; et al. 4-Methylenesterols from Theonella swinhoei sponge are natural pregnane-X-receptor agonists and farnesoid-X-receptor antagonists that modulate innate immunity. Steroids 2012, 77, 484–495. [Google Scholar] [CrossRef]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; Renga, B.; D’Amore, C.; Debitus, C.; Fiorucci, S.; Zampella, A. Conicasterol E, a small heterodimer partner sparing farnesoid X receptor modulator endowed with a pregnane X receptor agonistic activity, from the marine sponge Theonella swinhoei. J. Med. Chem. 2012, 55, 84–93. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; D’Amore, C.; Cipriani, S.; D’Auria, M.V.; Sepe, V.; Chini, M.G.; Monti, M.C.; Bifulco, G.; Zampella, A.; et al. Discovery that theonellasterol a marine sponge sterol is a highly selective FXR antagonist that protects against liver injury in cholestasis. PLoS One 2012, 7, e30443. [Google Scholar] [CrossRef]
- Koizumi, N.; Fujimoto, Y.; Takeshita, T.; Ikekawa, N. Studies on steroids. Part 49. Carbon-13 nuclear magnetic resonance of 24-substituted steroids. Chem. Pharm. Bull. 1979, 27, 38–42. [Google Scholar] [CrossRef]
- Ling, N.C.; Hale, R.L.; Djerassi, C. The structure and absolute configuration of the marine sterol gorgosterol. J. Am. Chem. Soc. 1970, 92, 5281–5282. [Google Scholar] [CrossRef]
- Kho, E.; Imagawa, D.K.; Rohmer, M.; Kashman, Y.; Djerassi, C. Sterols in marine invertebrates. 22. Isolation and structure elucidation of conicasterol and theonellasterol, two new 4-methylene sterols from the Red Sea sponges Theonella conica and Theonella swinhoei. J. Org. Chem. 1981, 46, 1836–1839. [Google Scholar] [CrossRef]
- Rubinstein, I.; Goad, L.J.; Clague, A.D.H.; Mulheirn, L.J. The 220 MHz NMR spectra of phytosterols. Phytochemistry 1976, 15, 195–200. [Google Scholar] [CrossRef]
- Lee, W.-H.; Lutsky, B.N.; Schroepfcr, G.J. 5 Alpha-cholest-8(14)-en-3 beta-ol, a possible intermediate in the biosynthesis of cholesterol. Enzymatic conversion to cholesterol and isolation from rat skin. J. Biol. Chem. 1969, 244, 5440–5448. [Google Scholar]
- Trottier, J.; Milkiewicz, P.; Kaeding, J.; Verreault, M.; Barbier, O. Coordinate regulation of hepatic bile acid oxidation and conjugation by nuclear receptors. Mol. Pharm. 2006, 3, 212–222. [Google Scholar]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Lauro, G.; Bifulco, G.; D’Amore, C.; Renga, B.; Fiorucci, S.; Zampella, A. Modification in the side chain of solomonsterol A: Discovery of cholestan disulfate as a potent pregnane-X-receptor agonist. Org. Biomol. Chem. 2012, 10, 6350–6362. [Google Scholar] [CrossRef]
- Iida, T.; Kakiyama, G.; Hibiya, Y.; Miyata, S.; Inoue, T.; Ohno, K.; Goto, T.; Mano, N.; Junichi Goto, J.; Nambara, T.; et al. Chemical synthesis of the 3-sulfooxy-7-N-acetylglucosaminyl-24-amidated conjugates of 3β,7β-dihydroxy-5-cholen-24-oic acid, and related compounds: Unusual, major metabolites of bile acid in a patient with Niemann-Pick disease type C1. Steroids 2006, 71, 18–29. [Google Scholar] [CrossRef]
- Iida, T.; Momose, T.; Tamura, T.; Matsumoto, T.; Chang, F.C.; Goto, J.; Nambara, T. Potential bile acid metabolites. 13. Improved routes to 3,6 and 3,6-dihydroxy-5-cholanoic acids. J. Lipid. Res. 1988, 29, 165–171. [Google Scholar]
- Ma, X.; Shah, Y.M.; Guo, G.L.; Wang, T.; Krausz, K.W.; Idle, J.R.; Gonzalez, F.J. Rifaximin is a gut-specific human pregnane X receptor activator. J. Pharmacol. Exp. Ther. 2007, 322, 391–398. [Google Scholar] [CrossRef]
- Watkins, R.E.; Davis-Searles, P.R.; Lambert, M.H.; Redinbo, M.R. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J. Mol. Biol. 2003, 331, 815–828. [Google Scholar] [CrossRef]
- Chianese, G.; Sepe, V.; Limongelli, V.; Renga, B.; D’Amore, C.; Zampella, A.; Taglialatela-Scafati, O.; Fiorucci, S. Incisterols, highly degraded marine sterols, are a new chemotype of PXR agonists. Steroids 2014, 83, 80–85. [Google Scholar] [CrossRef]
- Sepe, V.; D’Amore, C.; Ummarino, R.; Renga, B.; D’Auria, M.V.; Novellino, E.; Sinisi, A.; Taglialatela-Scafati, O.; Nakao, Y.; Limongelli, V.; et al. Insights on pregnane-X-receptor modulation. Natural and semisynthetic steroids from Theonella marine sponges. Eur. J. Med. Chem. 2014, 12, 126–134. [Google Scholar]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Di Leva, F.S.; Festa, C.; D’Amore, C.; de Marino, S.; Renga, B.; D’Auria, M.V.; Novellino, E.; Limongelli, V.; Zampella, A.; Fiorucci, S. Binding mechanism of the farnesoid X receptor marine antagonist suvanine reveals a strategy to forestall drug modulation on nuclear receptors. Design, synthesis, and biological evaluation of novel ligands. J. Med. Chem. 2013, 56, 4701–4717. [Google Scholar] [CrossRef]
- D’Amore, C.; di Leva, F.S.; Sepe, V.; Renga, B.; del Gaudio, C.; D’Auria, M.V.; Zampella, A.; Fiorucci, S.; Limongelli, V. Design, synthesis, and biological evaluation of potent dual agonists of nuclear and membrane bile acid receptors. J. Med. Chem. 2014, 57, 937–954. [Google Scholar] [CrossRef]
- Ekins, S.; Chang, C.; Mani, S.; Krasowski, M.D.; Reschly, E.J.; Iyer, M.; Kholodovych, V.; Ai, N.; Welsh, W.J.; Sinz, M.; et al. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 2007, 72, 592–603. [Google Scholar] [CrossRef]
- Wagner, B.L.; Pollio, G.; Giangrande, P.; Webster, J.C.; Breslin, M.; Mais, D.E.; Cook, C.E.; Vedeckis, W.V.; Cidlowski, J.A.; McDonnell, D.P. The novel progesterone receptor antagonists RTI 3021–012 and RTI 3021–022 exhibit complex glucocorticoid receptor antagonist activities: Implications for the development of dissociated antiprogestins. Endocrinology 1999, 140, 1449–1458. [Google Scholar]
- Link, J.T.; Sorensen, B.; Patel, J.; Grynfarb, M.; Goos-Nilsson, A.; Wang, J.; Fung, S.; Wilcox, D.; Zinker, B.; Nguyen, P.; et al. Antidiabetic activity of passive nonsteroidal glucocorticoid receptor modulators. J. Med. Chem. 2005, 48, 5295–5304. [Google Scholar] [CrossRef]
- Primer3 (v. 0.4.0). Available online: http://frodo.wi.mit.edu/primer3/ (accessed on 16 November 2011).
- Maestro; Version 9.3; Schrodinger, LLC: New York, NY, USA, 2012.
- MacroModel; Version 9.9; Schrodinger, LLC: New York, NY, USA, 2012.
- Glide; Version 5.8; Schrodinger, LLC: New York, NY, USA, 2011.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System. Version 1.5.0.4. Schrödinger, LLC. Available online: http://www.pymol.org (accessed on 14 September 2010).
- Weihrauch, J.L.; Gardner, J.M. Sterol content of foods of plant origin. J. Am. Diet. Assoc. 1978, 73, 39–47. [Google Scholar]
- Plat, J.; Kerckhoffs, D.A.J.M.; Mensink, R.P. Therapeutic potential of plant sterols and stanols. Curr. Opin. Lipidol. 2000, 11, 571–576. [Google Scholar] [CrossRef]
- Chan, Y.M.; Varady, K.A.; Lin, Y.; Trautwein, E.; Mesnsink, R.P.; Plat, J.; Jones, P.J. Plasma concentrations of plant sterols: Physiology and relationship with coronary heart disease. Nutr. Rev. 2006, 64, 385–402. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sepe, V.; Di Leva, F.S.; D'Amore, C.; Festa, C.; De Marino, S.; Renga, B.; D'Auria, M.V.; Novellino, E.; Limongelli, V.; D'Souza, L.; et al. Marine and Semi-Synthetic Hydroxysteroids as New Scaffolds for Pregnane X Receptor Modulation. Mar. Drugs 2014, 12, 3091-3115. https://doi.org/10.3390/md12063091
Sepe V, Di Leva FS, D'Amore C, Festa C, De Marino S, Renga B, D'Auria MV, Novellino E, Limongelli V, D'Souza L, et al. Marine and Semi-Synthetic Hydroxysteroids as New Scaffolds for Pregnane X Receptor Modulation. Marine Drugs. 2014; 12(6):3091-3115. https://doi.org/10.3390/md12063091
Chicago/Turabian StyleSepe, Valentina, Francesco Saverio Di Leva, Claudio D'Amore, Carmen Festa, Simona De Marino, Barbara Renga, Maria Valeria D'Auria, Ettore Novellino, Vittorio Limongelli, Lisette D'Souza, and et al. 2014. "Marine and Semi-Synthetic Hydroxysteroids as New Scaffolds for Pregnane X Receptor Modulation" Marine Drugs 12, no. 6: 3091-3115. https://doi.org/10.3390/md12063091
APA StyleSepe, V., Di Leva, F. S., D'Amore, C., Festa, C., De Marino, S., Renga, B., D'Auria, M. V., Novellino, E., Limongelli, V., D'Souza, L., Majik, M., Zampella, A., & Fiorucci, S. (2014). Marine and Semi-Synthetic Hydroxysteroids as New Scaffolds for Pregnane X Receptor Modulation. Marine Drugs, 12(6), 3091-3115. https://doi.org/10.3390/md12063091