Neritinaceramides A–E, New Ceramides from the Marine Bryozoan Bugula neritina Inhabiting South China Sea and Their Cytotoxicity


Abstract
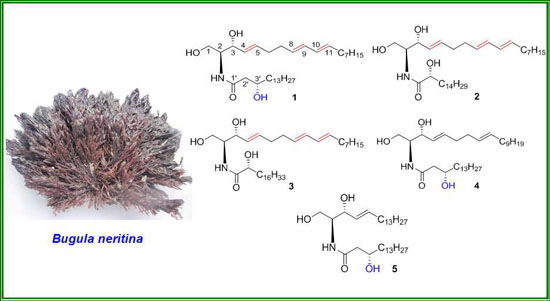
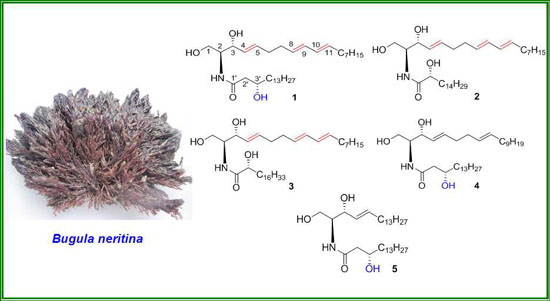
:
1. Introduction
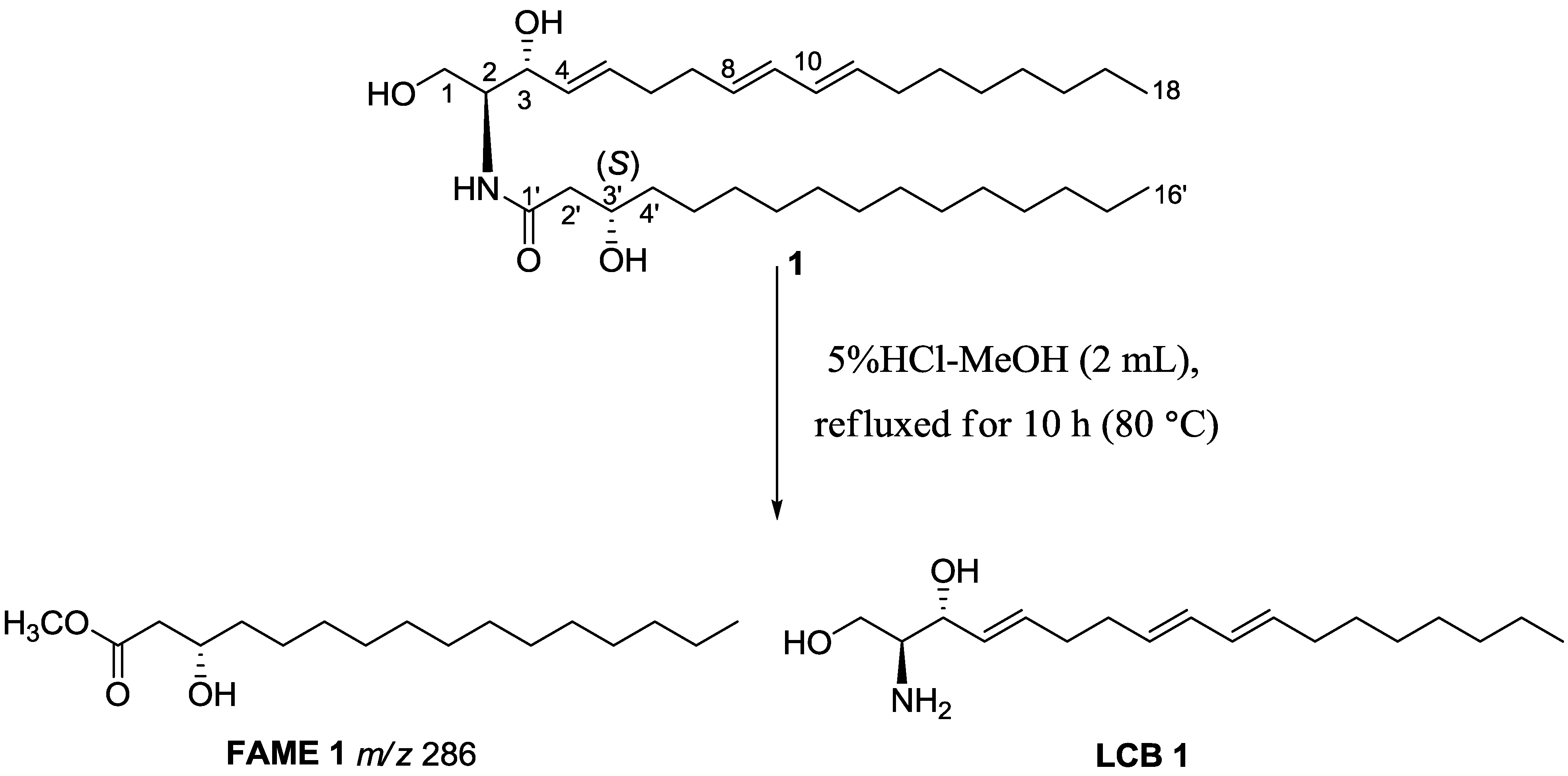
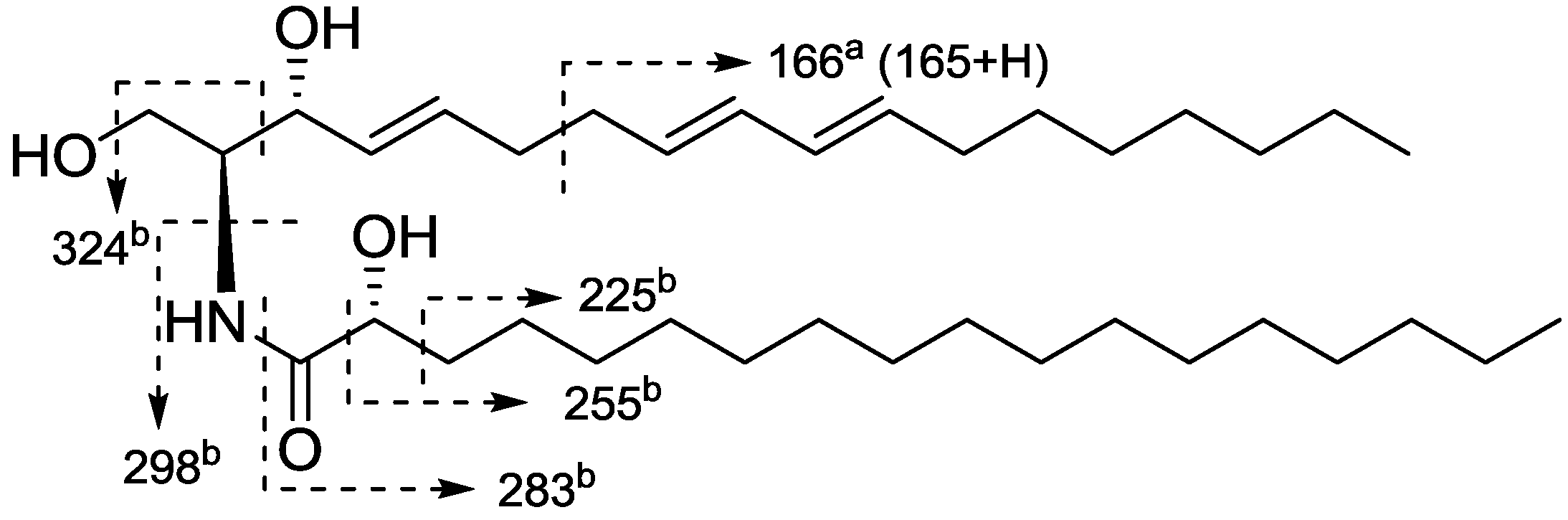
2. Results and Discussion
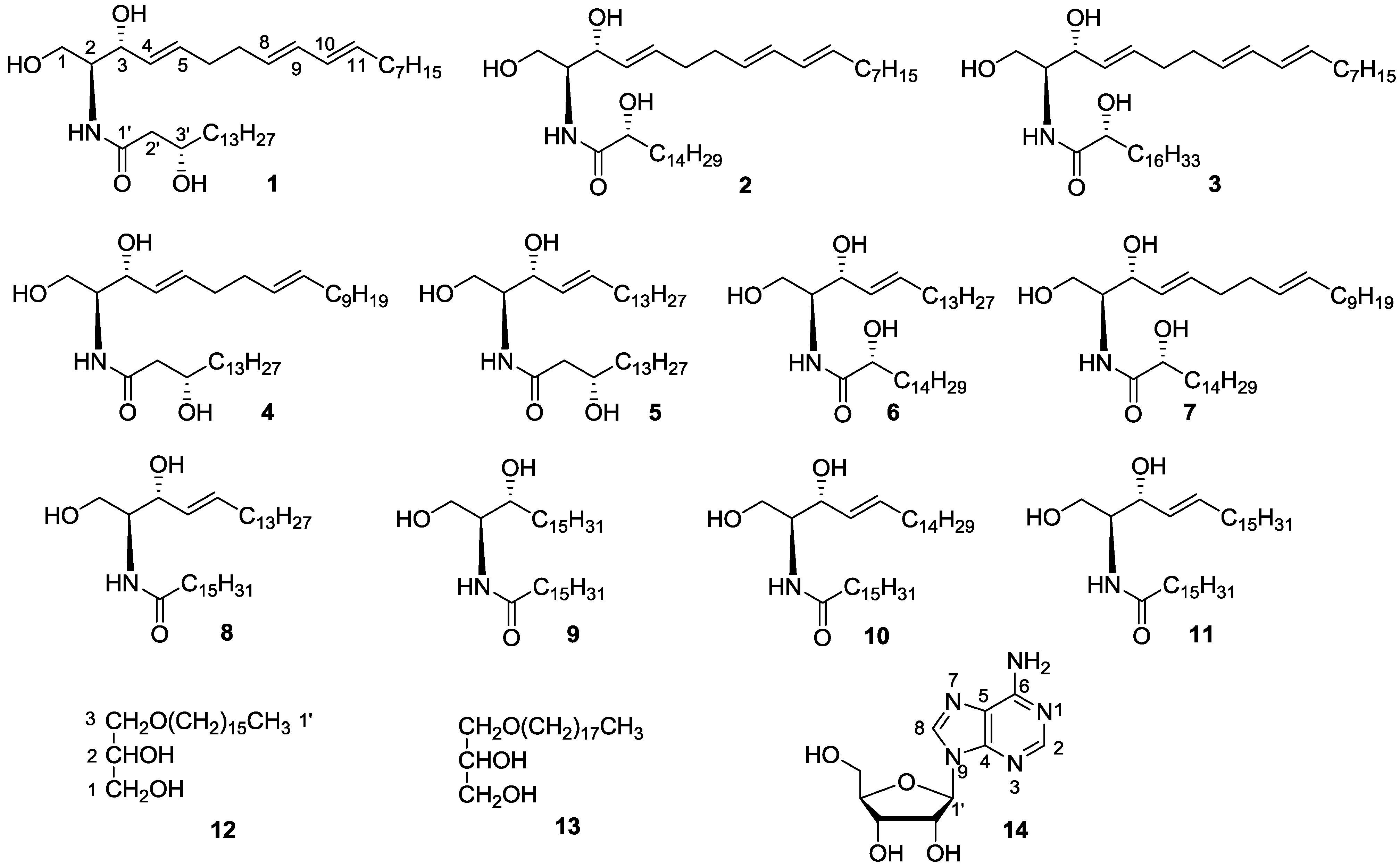

{kind=link}
{kind=link}
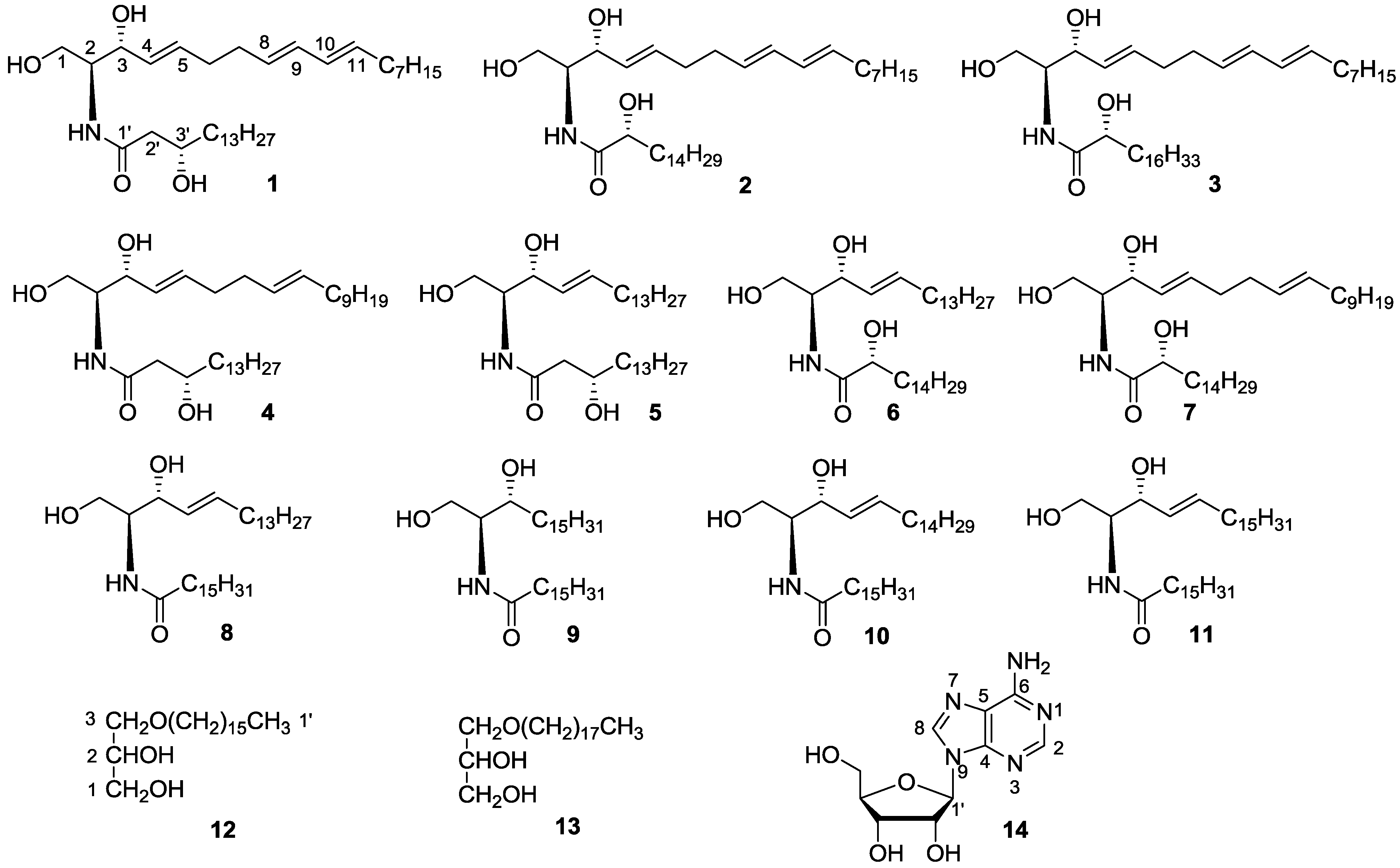{kind=link}
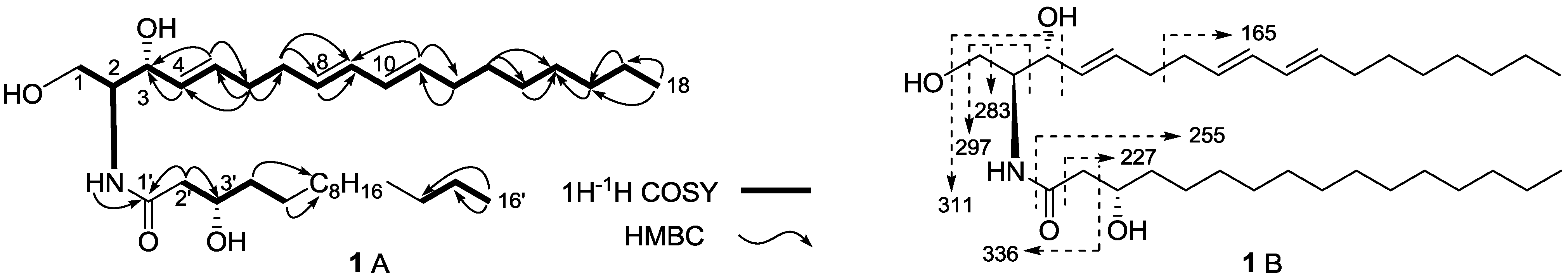{kind=link}
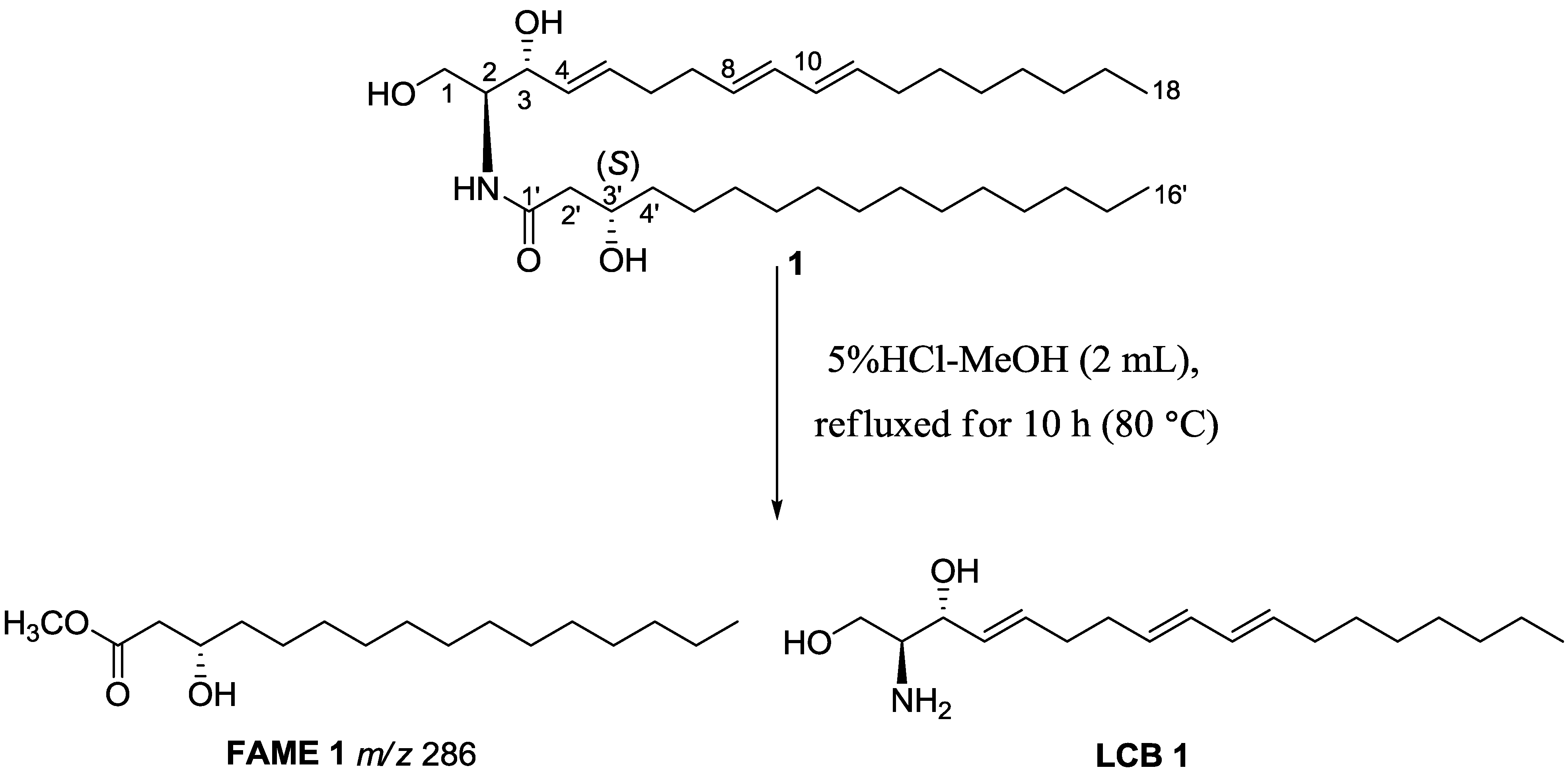{kind=link}
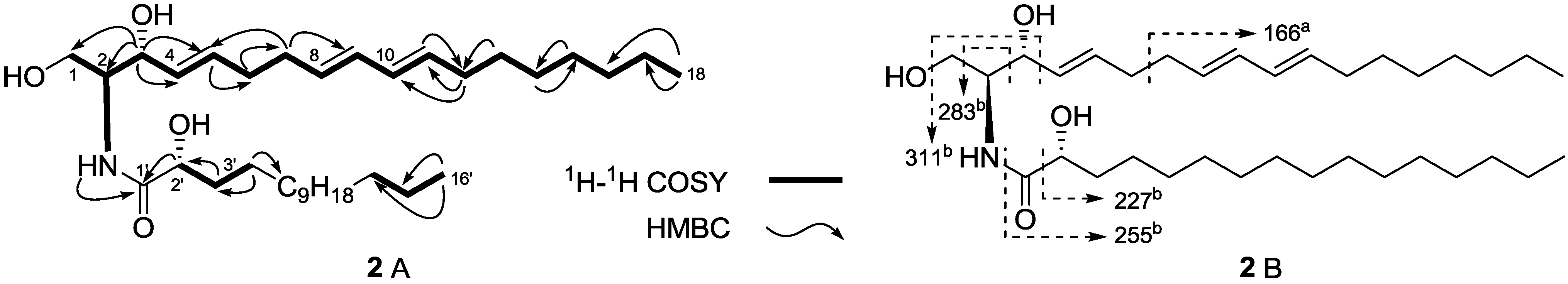{kind=link}
{kind=link}
{kind=link}
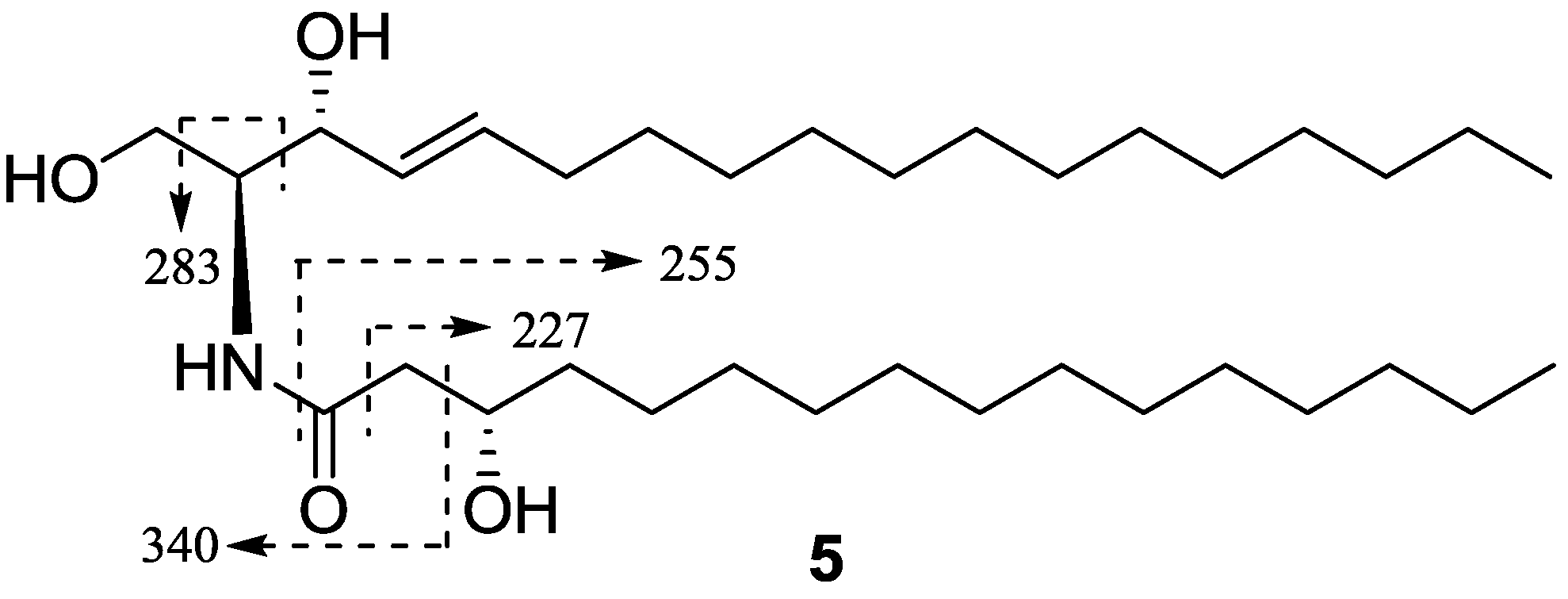{kind=link}
| No. | 1 | 2 | |||
|---|---|---|---|---|---|
| δC, mult. b | δH (int., mult., J in Hz) b | δC, mult. b | δC, mult. c | δH (int., mult., J in Hz) c | |
| 1 | 62.0 (t) | 3.72 (1H, br d, 8.3), 3.90 (1H, br d, 8.3) | 62.0 (t) | 61.9 (t) | 4.22 (1H, dd, 10.8, 4.2), 4.46 (1H, dd, 10.8, 4.5) |
| 2 | 54.4 (d) | 3.92 (1H, m) | 54.4 (d) | 56.1 (d) | 4.67 (1H, m) |
| 3 | 74.2 (d) | 4.31 (1H, br s) | 74.2 (d) | 73.0 (d) | 4.83 (1H, br t, 6.1) |
| 4 | 129.3 (d) | 5.54 (1H, dd, 15.2, 6.2) | 129.1 (d) | 131.4 (d) | 6.13 (1H, m) |
| 5 | 133.2 (d) | 5.77 (1H, dt, 15.2, 6.3) | 133.3 (d) | 132.6 (d) | 6.05 (1H, m) |
| 6 | 31.9 (t) | 2.16 (2H, m) | 31.9 (t) | 32.7 (t) | 2.18 (1H, m) |
| 7 | 32.1 (t) | 2.04 (2H, dt, 14.1, 6.9) | 32.1 (t) | 32.9 (t) | 2.04 (1H, m) |
| 8 | 133.3 (d) | 5.59 (1H, m) | 133.5 (d) | 132.8 (d) | 5.64 (1H, m) |
| 9 | 130.0 (d) | 5.99 (1H, dd, 15.3, 5.0) | 130.0 (d) | 131.0 (d) | 6.11 (1H, m) |
| 10 | 131.2 (d) | 6.00 (1H, dd, 15.3, 5.0) | 131.2 (d) | 131.6 (d) | 5.65 (1H, m) |
| 11 | 130.7 (d) | 5.52 (1H, m) | 130.7 (d) | 131.5 (d) | 5.99 (1H, m) |
| 12 | 32.1 (t) | 2.16 (2H, m) | 32.1 (t) | 32.7 (t) | 2.18 (2H, br s) |
| 13 | 32.6 (t) | 2.04 (2H, dt, 14.1, 6.9) | 32.6 (t) | 32.1 (t) | 1.23 (2H, br s) d |
| 14 | 29.4 (t) | 1.36 (2H, m) d | 29.4 (t) | 29.4 (t) | 1.33 (2H, m) d |
| 15 | 29.2 (t) | 1.36 (2H, m) d | 29.2 (t) | 29.3 (t) | 1.33 (2H, m) d |
| 16 | 31.9 (t) | 1.26 (2H, br s) d | 31.9 (t) | 32.1 (t) | 1.23 (2H, br s) d |
| 17 | 22.7 (t) | 1.26 (2H, br s) d | 22.7 (t) | 22.9 (t) | 1.23 (2H, br s) d |
| 18 | 14.1 (q) | 0.88 (3H, t, 7.1) | 14.1 (q) | 14.2 (q) | 0.84 (3H, t, 7.1) |
| 1′ | 173.0 (s) | - | 175.0 (s) | 175.4 (s) | - |
| 2′ | 43.1 (t) | a 2.30 (1H, m), b 2.43 (1H, d, 2.2) | 72.4 (d) | 72.5 (d) | 4.59 (1H, m) |
| 3′ | 68.9 (d) | 4.00 (1H, m) | 34.8 (t) | 35.8 (t) | 2.22 (1H, m), 2.03 (1H, m) |
| 4′ | 37.1 (t) | a 1.43 (1H, m), b 1.51 (1H, m) | 31.8 (t) | 32.0 (t) | 1.23 (2H, br s) d |
| 5′ | 25.6 (t) | 1.37 (2H, m) | 25.1 (t) | 25.9 (t) | 1.64 (2H, m) |
| 6′~13′ | 29.2–29.7 (t) | 1.26 (16H, br s) d | 29.2–29.7 (t) | 29.3–30.0 (t) | 1.23 (16H, br s) d |
| 14′ | 31.9 (t) | 1.26 (2H, br s) d | 31.9 (t) | 32.1 (t) | 1.23 (2H, br s) d |
| 15′ | 22.7 (t) | 1.26 (2H, br s) d | 22.7 (t) | 22.9 (t) | 1.23 (2H, br s) d |
| 16′ | 14.1 (q) | 0.88 (3H, t, 7.1) | 14.1 (q) | 14.2 (q) | 0.84 (3H, t, 7.1) |
| NH | - | 6.65 (1H, d, 8.0) | - | - | 8.34 (1H, d, 8.7) |
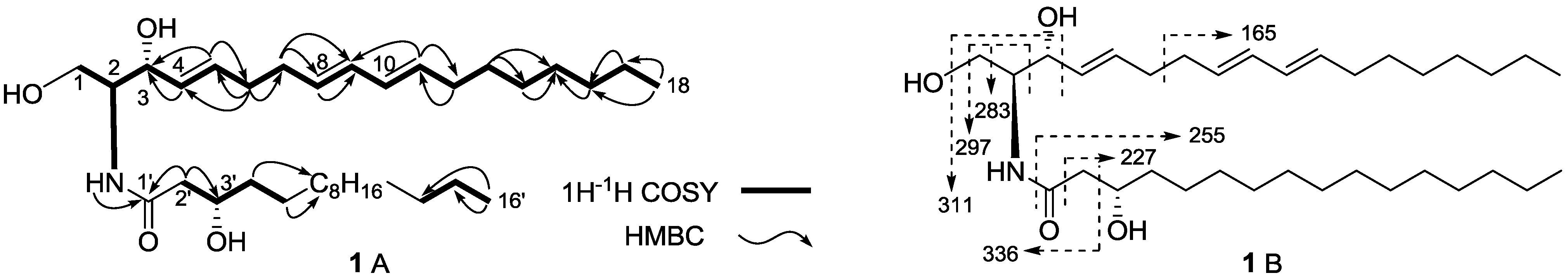
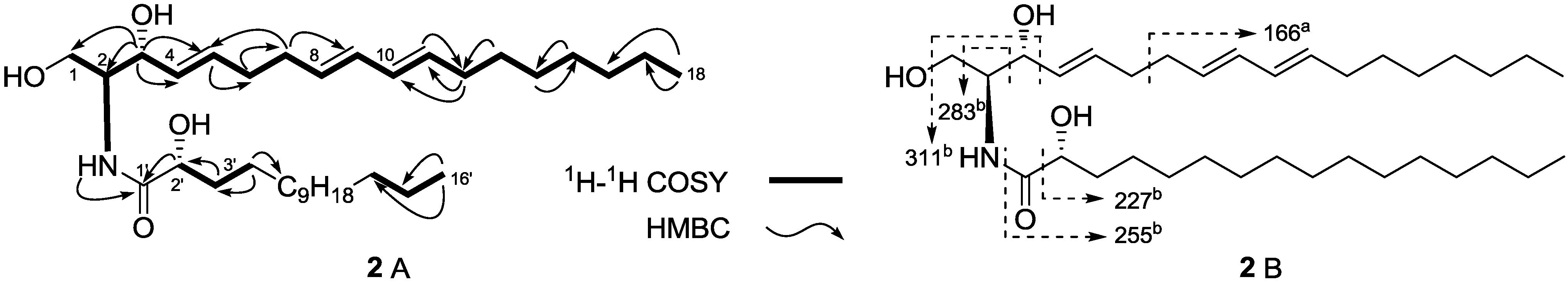
| No. | 3 | 4 | 5 | |||
|---|---|---|---|---|---|---|
| δC, mult. | δH (int., mult., J in Hz) | δC, mult. | δH (int., mult., J in Hz) | δC, mult. | δH (int., mult., J in Hz) | |
| 1 | 62.2 (t) | 3.75 (1H, m), 3.89 (1H, br d, 10.0) | 61.8 (t) | 3.71 (1H, d, 11.0), 3.86 (1H, d, 11.2) | 61.9 (t) | 3.74 (1H, m), 3.87 (1H, m) |
| 2 | 54.4 (d) | 3.91 (1H, br d, 10.0) | 54.5 (d) | 3.92 (1H, m) | 56.1 (d) | 3.93 (1H, m) |
| 3 | 74.2 (d) | 4.28 (1H, m) | 74.1 (d) | 4.28 (1H, br s) | 74.1 (d) | 4.28 (1H, m) |
| 4 | 129.2 (d) | 5.54 (1H, m) | 128.9 (d) | 5.51 (1H, dd, 15.5, 6.0) | 128.6 (d) | 5.53 (1H, br d, 14.0) |
| 5 | 133.3 (d) | 5.77 (1H, m) | 133.5 (d) | 5.77 (1H, m) | 134.3 (d) | 5.65 (1H, dt, 14.3, 6.5) |
| 6 | 31.9 (t) | 2.16 (2H, t, 2.9) | 32.6 (t) | 1.96 (2H, q-like, 6.2) | 32.3 (t) | 2.05 (2H, m) |
| 7 | 32.1 (t) | 2.04 (2H, q, 7.1) | 32.4 (t) | 2.11 (2H, m) | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 8 | 133.5 (d) | 5.59 (1H, dt, 15.0, 6.0) | 131.3 (d) | 5.41 (1H, m) | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 9 | 130.0 (d) | 5.98 (1H, m) | 129.0 (d) | 5.38 (1H, m) | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 10 | 131.2 (d) | 6.00 (1H, m) | 32.2 (t) | 2.07 (2H, m) | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 11 | 130.7 (d) | 5.52 (1H, m) | 29.3–29.7 (t) | 1.26 (2H, br s) b | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 12 | 32.1 (t) | 2.16 (2H, t, 2.9) | 29.3–29.7 (t) | 1.26 (2H, br s) b | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 13 | 32.6 (t) | 2.04 (2H, q, 7.1) | 29.3–29.7 (t) | 1.26 (2H, br s) b | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 14 | 29.4 (t) | 1.36 (2H, m) | 29.3–29.7 (t) | 1.26 (2H, br s) b | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 15 | 29.2 (t) | 1.36 (2H, m) | 29.3–29.7 (t) | 1.26 (2H, br s) b | 29.2–29.7 (t) | 1.26 (2H, br s) b |
| 16 | 31.9 (t) | 1.26 (2H, br s) b | 32.0 (t) | 1.26 (2H, br s) b | 32.0 (t) | 1.26 (2H, br s) b |
| 17 | 22.7 (t) | 1.26 (2H, br s) b | 22.7 (t) | 1.26 (2H, br s) b | 22.7 (t) | 1.26 (2H, br s) b |
| 18 | 14.1 (q) | 0.87 (3H, t, 7.1) | 14.1 (q) | 0.88(3H, t, 7.0) | 14.1 (q) | 0.88(3H, t, 7.1) |
| 1′ | 175.1 (s) | - | 173.0 (s) | - | 173.0 (s) | - |
| 2′ | 72.4 (d) | 4.12 (1H, dd, 7.8, 3.4) | 43.4 (t) | a 2.29 (1H, dd, 14.5, 9.5), | 43.3 (t) | a 2.31 (1H, m), b 2.41 (1H, m) |
| b 2.41 (1H, d, 14.3) | ||||||
| 3′ | 34.8 (t) | 2.16 (2H, t, 2.9) | 68.9 (d) | 4.00 (1H, m) | 68.9 (d) | 4.00 (1H, m) |
| 4′ | 31.8 (t) | 1.26 (2H, br s) b | 37.2 (t) | a 1.43 (1H, m), b 1.51 (1H, m) | 37.1 (t) | a 1.43 (1H, m), b 1.51 (1H, m) |
| 5′ | 25.1 (t) | 1.62 (2H, m) | 25.6 (t) | a 1.26 (1H, br s) b, b 1.43 (1H, m) | 25.6 (t) | a 1.30 (1H, m), b 1.43 (1H, m) |
| 6′~13′ | 29.1–29.7 (t) | 1.26 (16H, br s) b | 29.3–29.7 (t) | 1.26 (16H, br s) b | 29.2–29.7 (t) | 1.26 (16H, br s) b |
| 14′ | 29.1–29.7 (t) | 1.26 (2H, br s) b | 32.0 (t) | 1.26 (2H, br s) b | 32.0 (t) | 1.26 (2H, br s) b |
| 15′ | 29.1–29.7 (t) | 1.26 (2H, br s) b | 22.7 (t) | 1.26 (2H, br s) b | 22.7 (t) | 1.26 (2H, br s) b |
| 16′ | 31.9 (t) | 1.26 (2H, br s) b | 14.1 (q) | 0.88 (3H, t, 7.0) | 14.1 (q) | 0.88 (3H, t, 7.1) |
| 17′ | 22.7 (t) | 1.26 (2H, br s) b | - | - | - | - |
| 18′ | 14.1 (q) | 0.87 (3H, t, 7.1) | - | - | - | - |
| NH | - | 7.20 (1H, d, 7.9) | - | 6.76 (1H, d, 7.8) | - | 6.77 (1H, d, 7.8) |
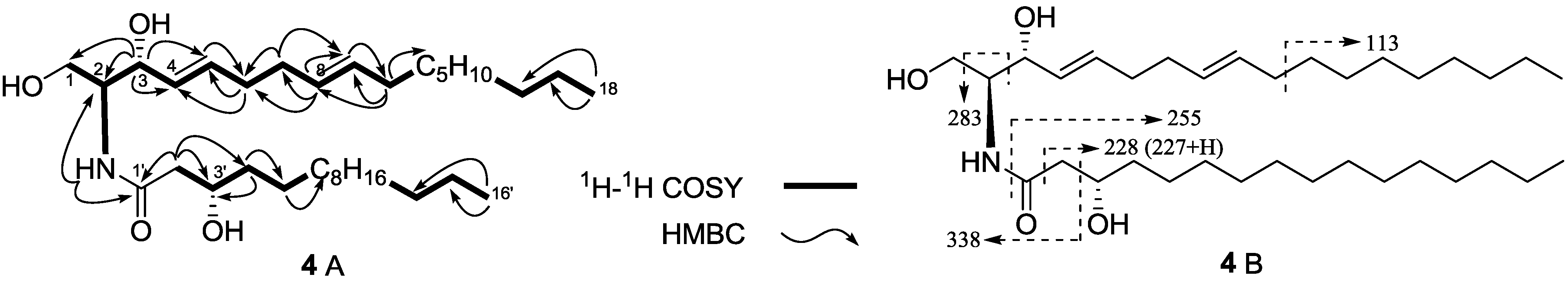
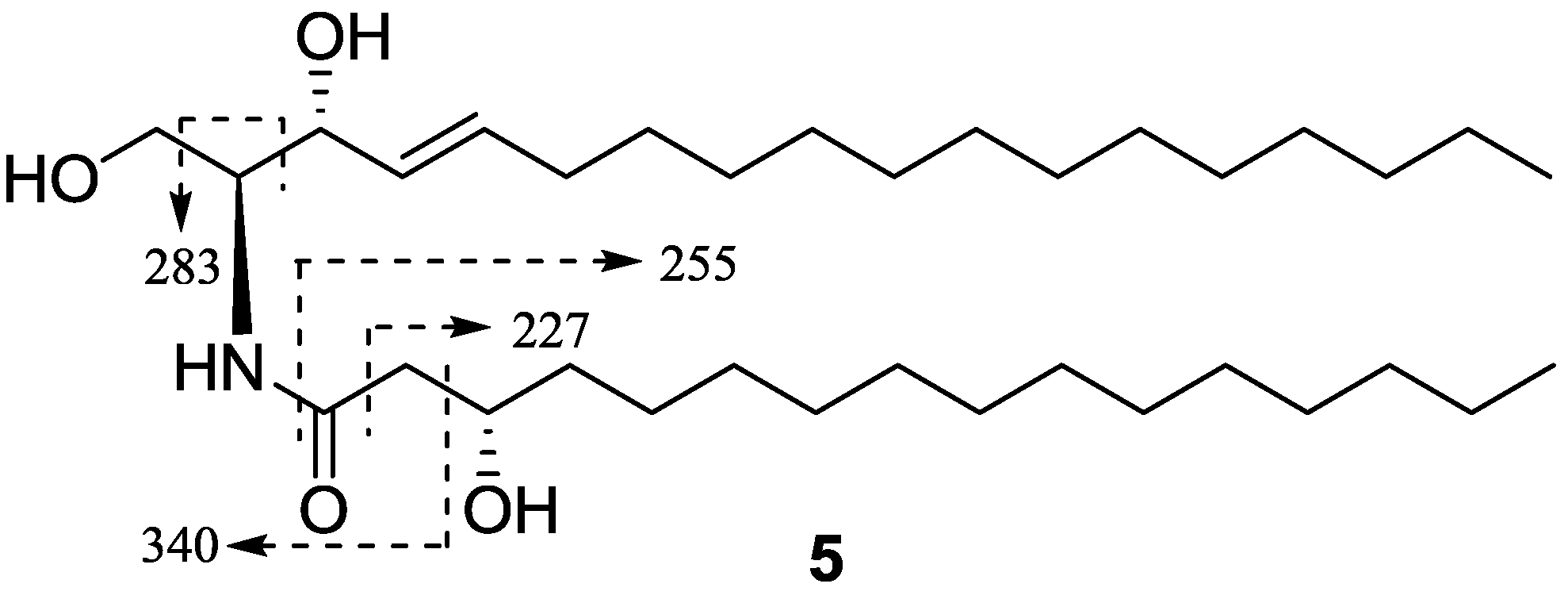
| Compound | HepG2 | NCI-H460 | SGC7901 |
|---|---|---|---|
| 1 | 52.1 | >100 | 57.7 |
| 2 | 47.3 | 84.4 | 52.2 |
| 3 | 47.8 | 85.1 | 52.9 |
| 4 | 53.7 | >100 | 57.9 |
| 5 | 54.6 | >100 | 58.1 |
| Adriamycin b | 0.14 | 0.13 | 0.17 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Extraction and Isolation
3.3.1. Neritinaceramide A (1)
3.3.2. Neritinaceramide B (2)
3.3.3. Neritinaceramide C (3)
3.3.4. Neritinaceramide D (4)
3.3.5. Neritinaceramide E (5)
3.3.6. 1-O-Palmityl Glycerin Ether (12)
3.3.7. 1-O-Octadecyl Glycerin Ether (13)
3.4. Methanolysis of Ceramides 1−5
3.5. MTT Cytotoxicity Assays
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hammad, S.M. Blood sphingolipids in homeostasis and pathobiology. Adv. Exp. Med. Biol. 2011, 721, 57–66. [Google Scholar] [CrossRef]
- Jin, W.; Rinehart, K.L.; Jares-Erijman, E.A. Ophidiacerebrosides: Cytotoxic glycosphingolipids containing a novel sphingosine from a sea star. J. Org. Chem. 1994, 59, 144–147. [Google Scholar] [CrossRef]
- Tan, R.X.; Chen, J.H. The cerebrosides. Nat. Prod. Rep. 2003, 20, 509–534. [Google Scholar] [CrossRef]
- Farokhi, F.; Wielgosz-Collin, G.; Clement, M.; Kornprobst, J.-M.; Barnathan, G. Cytotoxicity on human cancer cells of ophidiacerebrosides isolated from the African starfish Narcissia canariensis. Mar. Drugs 2010, 8, 2988–2998. [Google Scholar] [CrossRef]
- Chebaane, K.; Guyot, M. Occurrence of erythro-docosasphinga-4,8-dienine, as an ester, in Anemonia sulcata. Tetrahedron Lett. 1986, 27, 1495–1496. [Google Scholar]
- Almeida, J.G.L.; Maia, A.I.V.; Wilke, D.V.; Silveira, E.R.; Braz-Filho, R.; La Clair, J.J.; Costa-Lotufo, L.V.; Pesspa, O.D.L. Palyosulfonoceramides A and B: Unique sulfonylated ceramides from the Brazilian zoanthids Palythoa caribaeorum and Protopalyhtoa variabilis. Mar. Drugs 2012, 10, 2846–2860. [Google Scholar] [CrossRef]
- Hattori, T.; Adachi, K.; Shizuri, Y. New ceramides from marine sponge Haliclona koremella and related compounds as antifouling substances against macroalgae. J. Nat. Prod. 1998, 61, 823–826. [Google Scholar]
- Su, J.Y.; Kuang, Y.Y.; Zeng, L.M.; Li, H. New tetracyclic diterpenoid and new ceramides from the soft coral Sinularia conferta. J. Asian Nat. Prod. Res. 2005, 7, 107–113. [Google Scholar] [CrossRef]
- Loukaci, A.; Bultel-Ponce, V.; Longeon, A.; Guyot, M. New lipids from the tunicate Cystodytes cf. dellechiajei, as PLA2 inhibitors. J. Nat. Prod. 2000, 63, 799–802. [Google Scholar] [CrossRef]
- Ojika, M.; Yoshino, G.; Sakagami, Y. Novel ceramide 1-sulfates, potent DNA topoisomerase I inhibitors isolated from the bryozoa Watersipora cucullata. Tetrahddron Lett. 1997, 38, 4235–4238. [Google Scholar] [CrossRef]
- Cateni, F.; Zilic, J.; Zacchigna, M.; Procida, G. Cerebrosides with antiproliferative activity from Euphorbia peplis L. Fitoterapia 2010, 81, 97–103. [Google Scholar] [CrossRef]
- Blackman, A.J.; Walls, J.T. Bryozoan secondary metabolites and their chemical ecology. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 1995; pp. 73–112. [Google Scholar]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Tian, X.R.; Tang, H.F.; Li, Y.S.; Lin, H.W.; Ma, N.; Zhang, W.; Yao, M.N. Ceramides and cerebrosides from the marine bryozoan Bugula neritina inbating South China Sea. J. Asian Nat. Prod. Res. 2009, 11, 1005–1012. [Google Scholar] [CrossRef]
- Tian, X.R.; Tang, H.F.; Li, Y.S.; Lin, H.W.; Ma, N.; Zhang, W. Sterols from marine bryozoan Bugula neritina. Biochem. Syst. Ecol. 2010, 38, 435–437. [Google Scholar] [CrossRef]
- Tian, X.R.; Tang, H.F.; Li, Y.S.; Lin, H.W.; Chen, X.L.; Ma, N.; Yao, M.N.; Zhang, P.H. New cytotoxic oxygenated sterols from the marine bryozoan Cryptosula pallasiana. Mar. Drugs 2011, 9, 162–183. [Google Scholar] [CrossRef]
- Tian, X.R.; Tang, H.F.; Li, Y.S.; Lin, H.W.; Tong, X.Y.; Ma, N. Alkaloids from marine bryozoan Cryptosula pallasiana. Biochem. Syst. Ecol. 2010, 38, 1250–1252. [Google Scholar] [CrossRef]
- De Hann, J.W.; Van de Ven, L.J.M. Configurations and conformations in acyclic unsaturated hydrocarbons. A 13C-NMR study. Org. Magn. Reson. 1973, 5, 147–153. [Google Scholar] [CrossRef]
- Shi, J.; Seo, Y. Isolation of new ceramides from the gorgonian Acabaria undulate. J. Nat. Prod. 1995, 58, 948–953. [Google Scholar] [CrossRef]
- Kim, S.; Lee, S.; Lee, T.; Ko, H.; Kim, D. Efficient synthesis of d-erythro-sphingosine and d-erythro-azidosphingosine from d-ribo-phytosphingosine via a cyclic sulfate intermediate. J. Org. Chem. 2006, 71, 8661–8664. [Google Scholar] [CrossRef]
- Nakagawa, M.; Tsuruoka, A.; Yoshida, J.; Hino, T. Total synthesis of (+)-erythro-N-lauroyldocosasphinga-4,8-dienine from Anemonia sulcata and determination of the absolute configuration. J. Chem. Soc. Chem. Commun. 1990, 603–605. [Google Scholar] [CrossRef]
- Blanc, D.; Madec, J.; Popowyck, F.; Ayad, T.; Phansavath, P.; Ratovelomanana-Vidal, V.; Genet, J.P. General enantioselective synthesis of butyrolactone natural products as ruthenium-SYNPHOS-catalyzed hydrogenation reactions. Adv. Synth. Catal. 2007, 349, 943–950. [Google Scholar] [CrossRef]
- Inagaki, M.; Ikeda, Y.; Kawatake, S.; Nakamura, K.; Tanaka, M.; Misawa, E.; Yamada, M.; Higuchi, R. Isolation and structure of four new ceramides from the starfish Luidia maculate. Chem. Pharm. Bull. 2006, 54, 1647–1649. [Google Scholar] [CrossRef]
- Mori, K.; Funaki, Y. Synthesis of (4E,8E,2S,3R,2′R)-N-2′-hydroxyhexadecanoyl-1-O-β-d-gluco pyranosyl-9-methyl-4,8-sphingadienine, the fruiting inducing cerebroside in a basidiomycete Schizophyllum commune. Tetrahedron 1985, 41, 2379–2386. [Google Scholar] [CrossRef]
- Ishii, T.; Okino, T.; Mino, Y. A ceramide and cerebroside from the starfish Asterias amurensis Lutken and their plant-growth promotion activities. J. Nat. Prod. 2006, 69, 1080–1082. [Google Scholar] [CrossRef]
- Liu, J.K.; Hu, L.; Dong, Z.J. A glucosylceramide with a novel ceramide and three novel ceramides from the basidiomycete Cortinarius umidicola. Lipids 2003, 38, 669–675. [Google Scholar] [CrossRef]
- Dokolina, E.V.; Bushnev, A.S.; Zvonkova, E.N.; Evstigneeva, R.P. Resolution of racemic 2-hydroxypalmitic acid into antipodes. Bioorg. Khim. 1981, 7, 277–279. [Google Scholar]
- Yan, Z.; Lu, J.; Lu, D.; Xue, J.; Chen, S.; Li, P. Chemical constituents from the bile of Anser anser and their anti-MMP activity. Nat. Prod. Res. Dev. 2008, 20, 960–963. [Google Scholar]
- Shibuya, H.; Kawashima, K.; Sakagami, M.; Kawanishi, H.; Shimomura, M.; Ohashi, K.; Kitagawa, I. Sphingolipids and Glycerolipids. 1. Chemical structures and Ionophoretic activites of soyacerebrosides I and II from Soybean. Chem. Pharm. Bull. 1990, 38, 2933–2938. [Google Scholar] [CrossRef]
- Chakrabarty, M.; Batabyal, A.; Barua, A.K.; Patra, A. New ceramides from the hypotensive extract of a sea anemone, Paracondylactis indicus. J. Nat. Prod. 1994, 57, 393–395. [Google Scholar] [CrossRef]
- Zhang, F.; Luo, S.; Gao, B.; Ding, L. Chemical constituents from the sprout of Pteridium aquilinum var. latiusculum. Nat. Prod. Res. Dev. 2004, 16, 121–123. [Google Scholar]
- Wang, G.; Liu, Q.; Zeng, L. The chemical constituents of soft coral Cladiella densa from South China Sea. Acta Sci. Nat. Univ. Sunyatseni 1995, 34, 110–113. [Google Scholar]
- Bayer, E.; Fischer, H.O.L. Studies on acetone-glyceraldehydes, and optically active glycerides. J. Bio. Chem. 1941, 140, 397–410. [Google Scholar]
- Linman, J.M.; Long, M.J.; Korst, D.R.; Bethell, F.H. Studies on the stimulation of hemopoiesis by batyl alcohol. J. Lab. Clin. Med. 1959, 54, 335–343. [Google Scholar]
- Szulc, Z.M.; Bai, A.; Bielawski, J.; Mayroo, N.; Miller, D.E.; Gracz, H.; Hannun, Y.A.; Bielawska, A. Synthesis, NMR characterization and divergent biological actions of 2′-hydroxy-ceramide/dihydroceramide stereoisomers in MCF7 cells. Bioorg. Med. Chem. 2010, 18, 7565–7579. [Google Scholar]
- Pettit, G.R.; Tang, Y.; Knight, J.C. Antineoplastic agents. 545. Isolation and structure of turbostatins 1-4 from the Asian marine mollusk Turbo stenogyrus. J. Nat. Prod. 2005, 68, 974–978. [Google Scholar] [CrossRef]
- Sultan, I.; Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Szulc, Z.; Bielawska, A.; Hannun, Y.A.; Ogretmen, B. Regulation of the sphingosine-recycling pathway for ceramide generation by oxidative stressm and its role in controlling c-Myc/Max function. Biochem. J. 2006, 393, 513–521. [Google Scholar] [CrossRef]
- Ogretmen, B.; Pettus, B.J.; Rossi, M.J.; Wood, R.; Usta, J.; Szulc, Z.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A. Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. J. Biol. Chem. 2002, 277, 12960–12969. [Google Scholar] [CrossRef]
- Zeng, X.; Xiang, L.; Li, C.-Y.; Wang, Y.; Qiu, G.; Zhang, Z.-X.; He, X. Cytotoxic ceramides and glycerides from the roots of Livistona chinensis. Fitoterapia 2012, 83, 609–616. [Google Scholar] [CrossRef]
- Li, H.Y.; Matsunaga, S.; Fusetani, N. Halicylindrosides, antifungaland cytotoxic cerebrosides from the marine sponge Halichondria cylindrata. Tetrahedron 1995, 51, 2273–2280. [Google Scholar] [CrossRef]
- Duran, R.; Zubia, E.; Ortega, M.J.; Naranjo, S.; Salva, J. Phallusides, new glucosphingolipids from the ascidian Phallusia fumigata. Tetrahedron 1998, 54, 14597–14602. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tian, X.-R.; Tang, H.-F.; Feng, J.-T.; Li, Y.-S.; Lin, H.-W.; Fan, X.-P.; Zhang, X. Neritinaceramides A–E, New Ceramides from the Marine Bryozoan Bugula neritina Inhabiting South China Sea and Their Cytotoxicity. Mar. Drugs 2014, 12, 1987-2003. https://doi.org/10.3390/md12041987
Tian X-R, Tang H-F, Feng J-T, Li Y-S, Lin H-W, Fan X-P, Zhang X. Neritinaceramides A–E, New Ceramides from the Marine Bryozoan Bugula neritina Inhabiting South China Sea and Their Cytotoxicity. Marine Drugs. 2014; 12(4):1987-2003. https://doi.org/10.3390/md12041987
Chicago/Turabian StyleTian, Xiang-Rong, Hai-Feng Tang, Jun-Tao Feng, Yu-Shan Li, Hou-Wen Lin, Xiao-Pei Fan, and Xing Zhang. 2014. "Neritinaceramides A–E, New Ceramides from the Marine Bryozoan Bugula neritina Inhabiting South China Sea and Their Cytotoxicity" Marine Drugs 12, no. 4: 1987-2003. https://doi.org/10.3390/md12041987
APA StyleTian, X.-R., Tang, H.-F., Feng, J.-T., Li, Y.-S., Lin, H.-W., Fan, X.-P., & Zhang, X. (2014). Neritinaceramides A–E, New Ceramides from the Marine Bryozoan Bugula neritina Inhabiting South China Sea and Their Cytotoxicity. Marine Drugs, 12(4), 1987-2003. https://doi.org/10.3390/md12041987