Continuous Drug Release by Sea Anemone Nematostella vectensis Stinging Microcapsules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
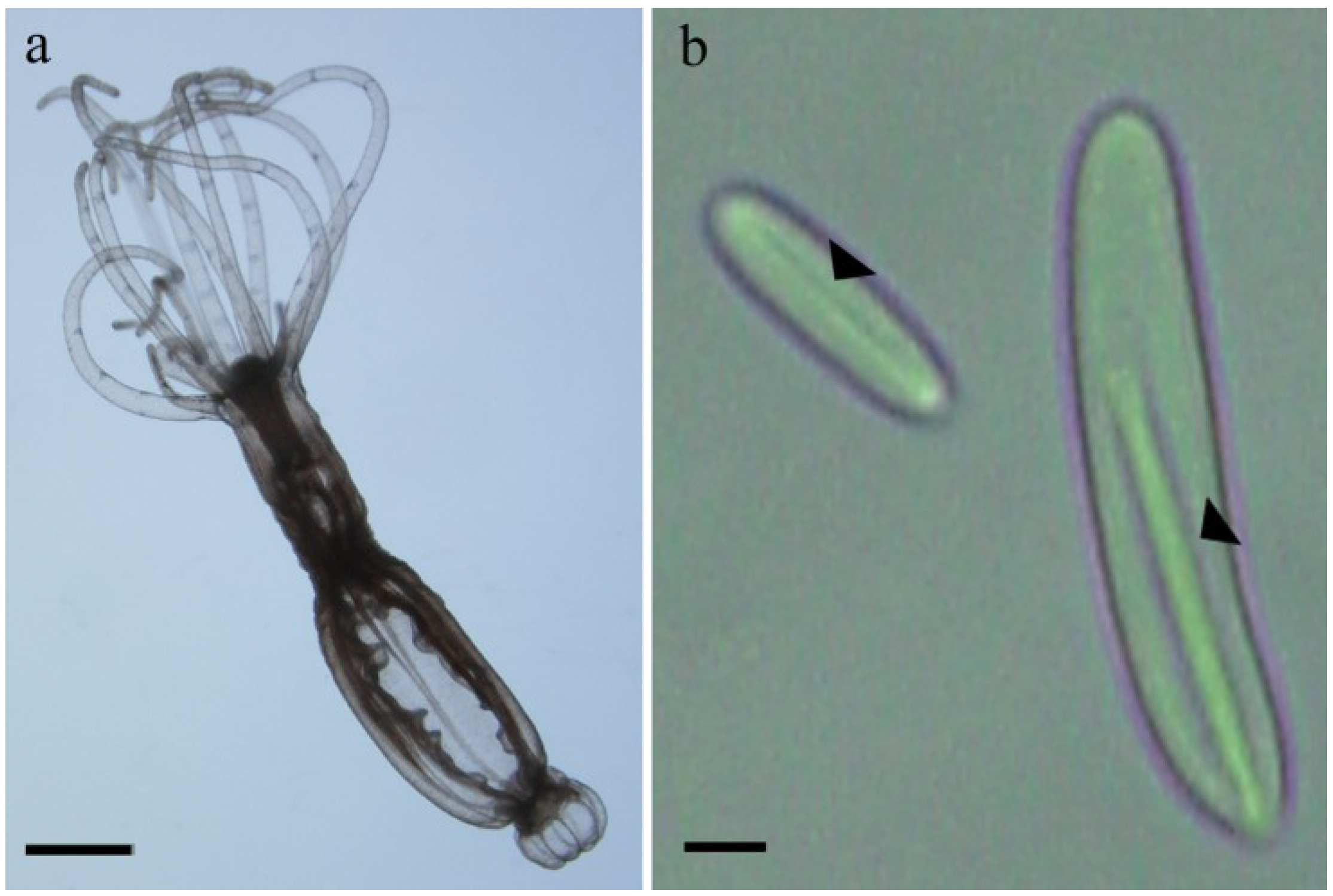
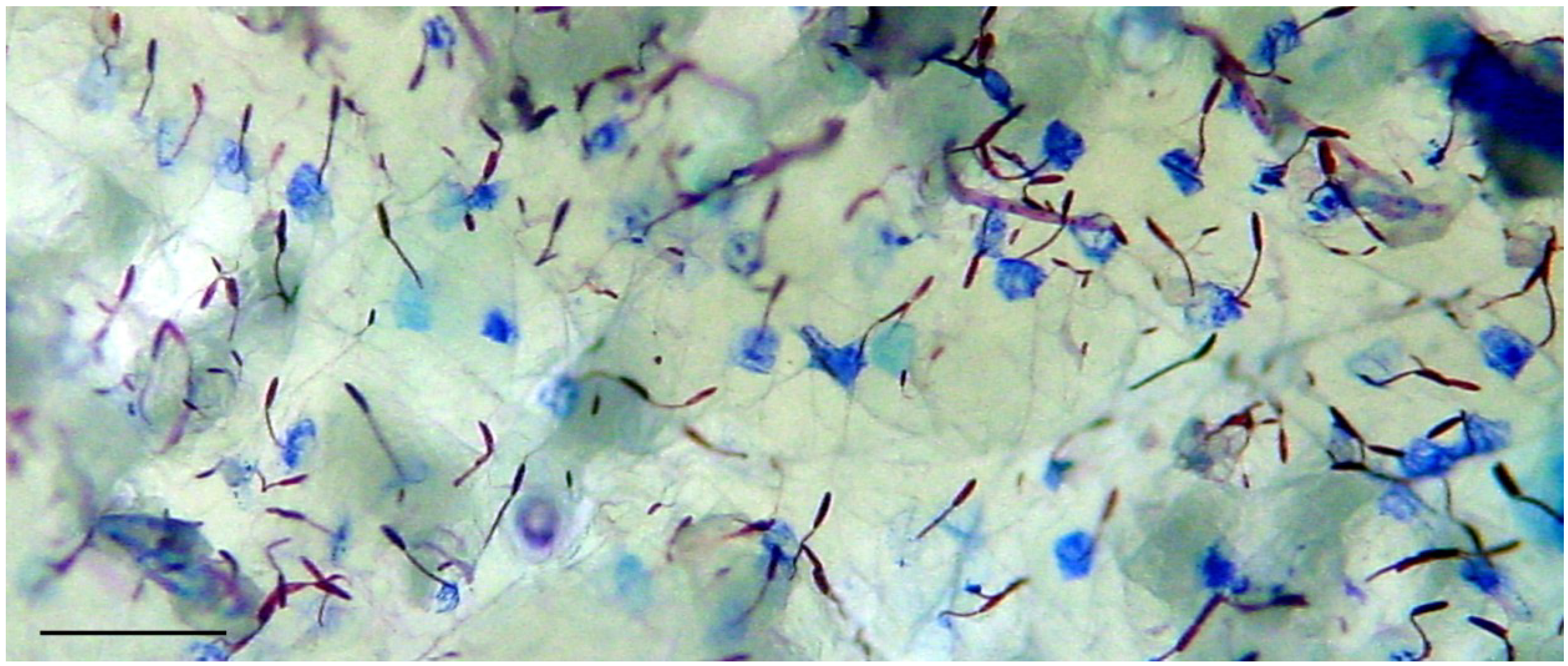
2.1. Preparation of Nematocysts for Skin Penetration
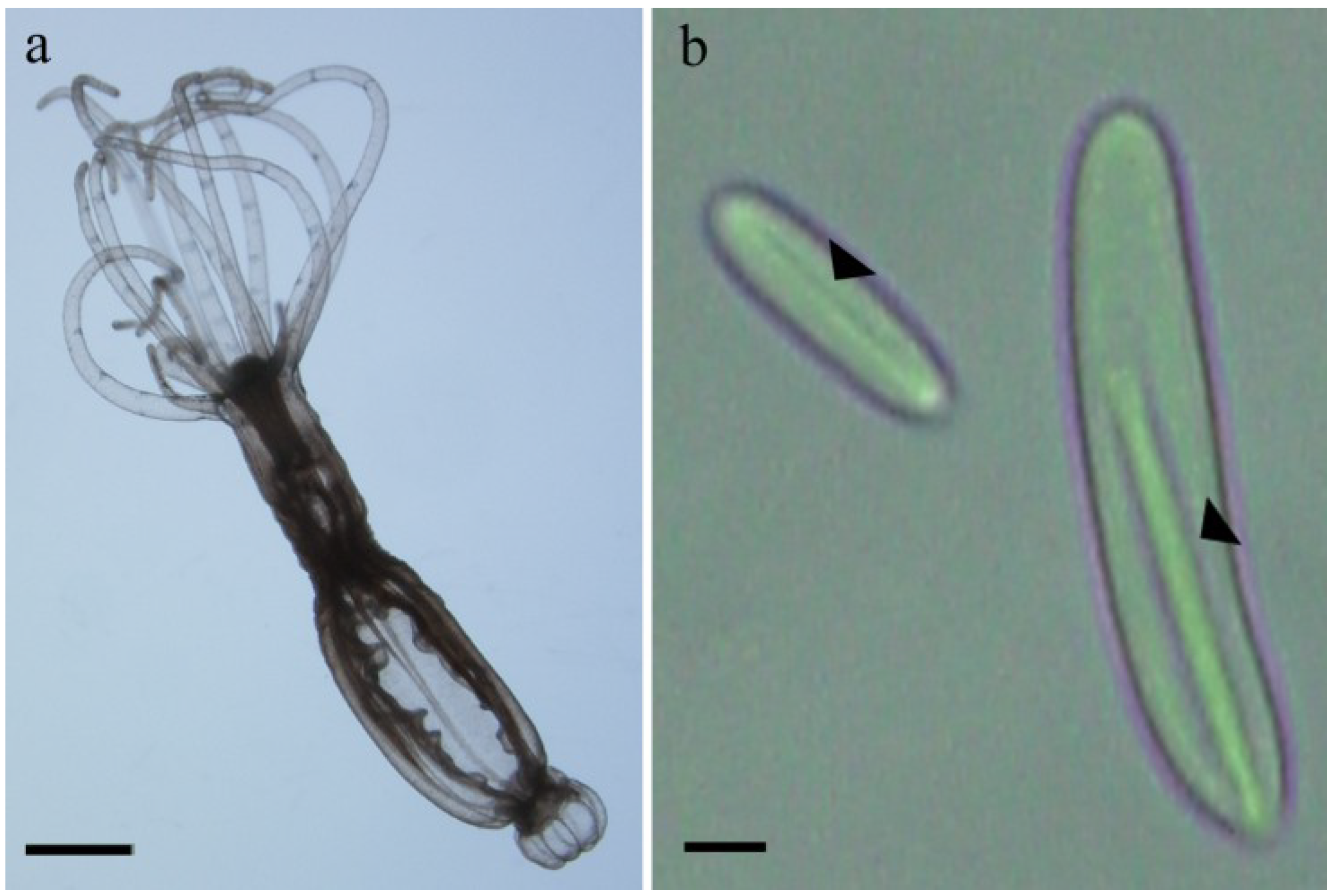
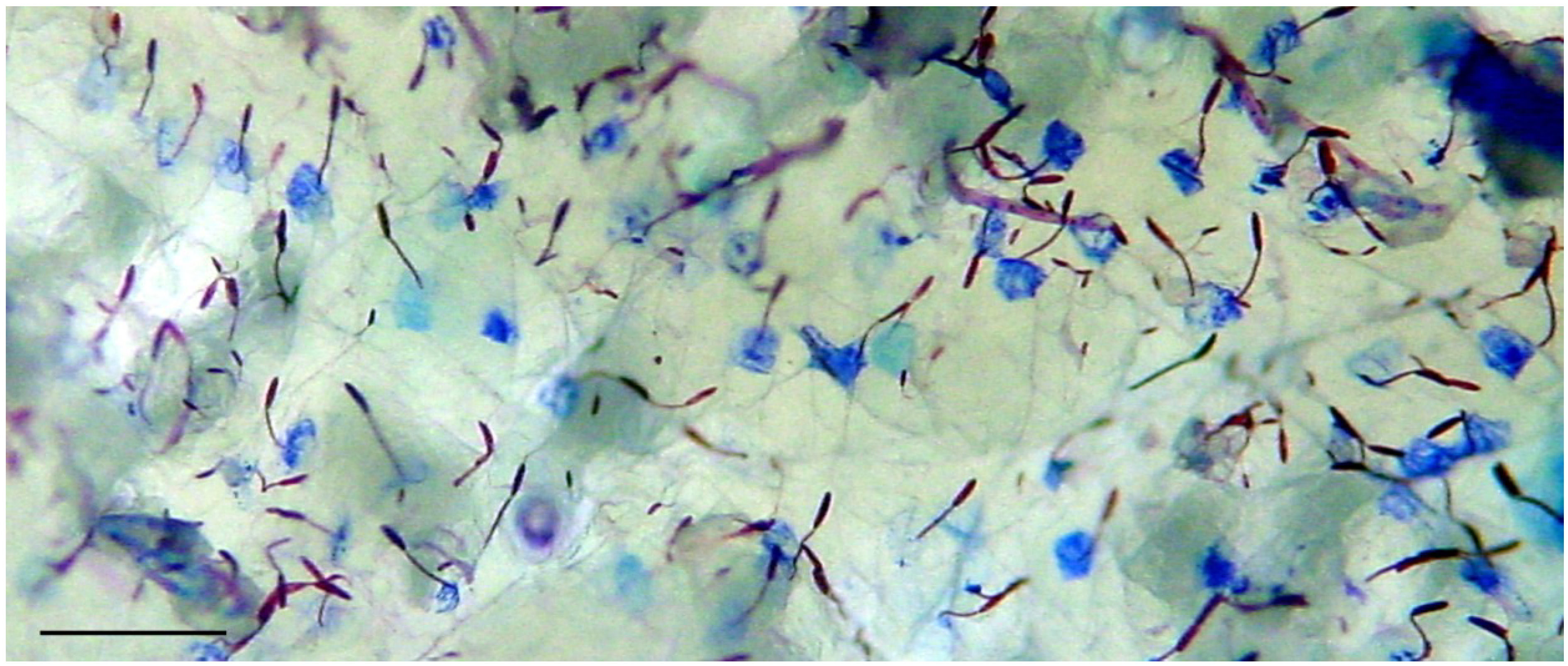
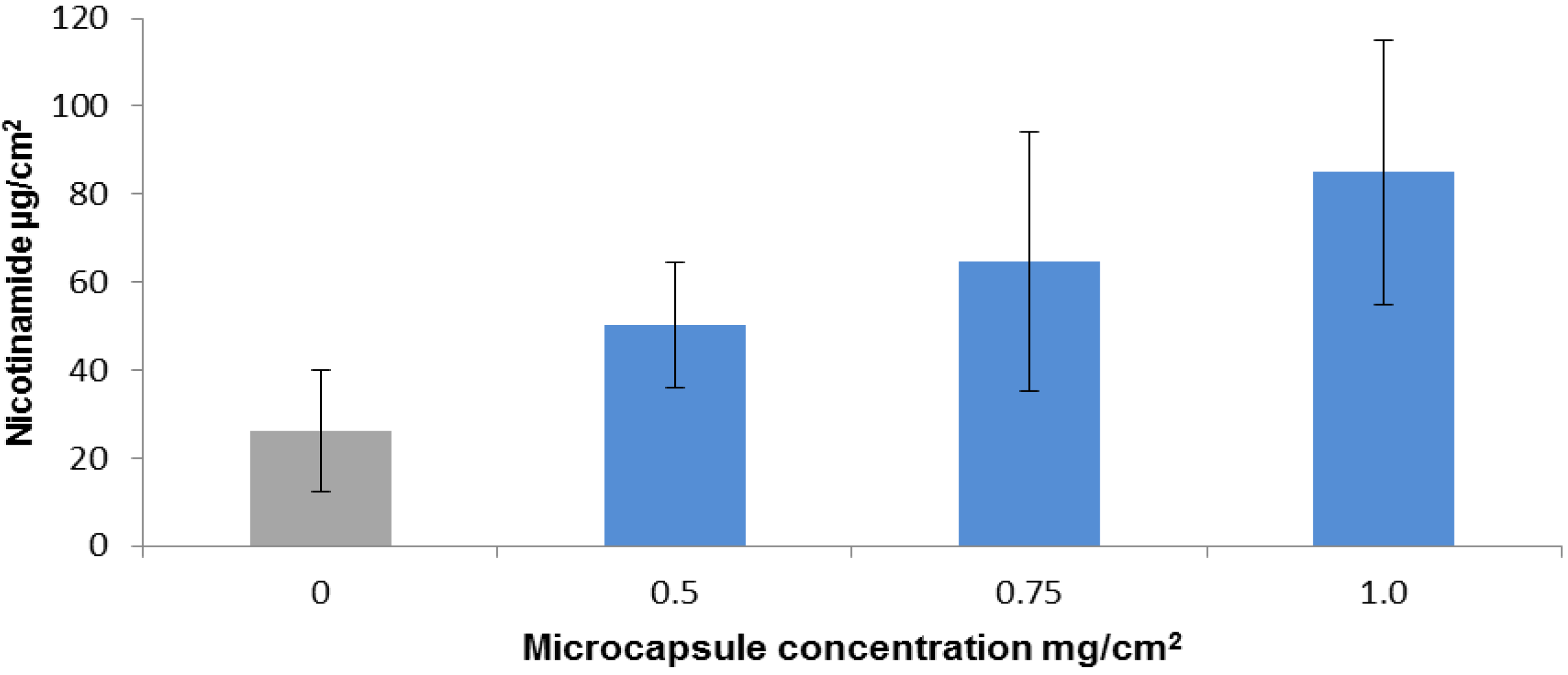
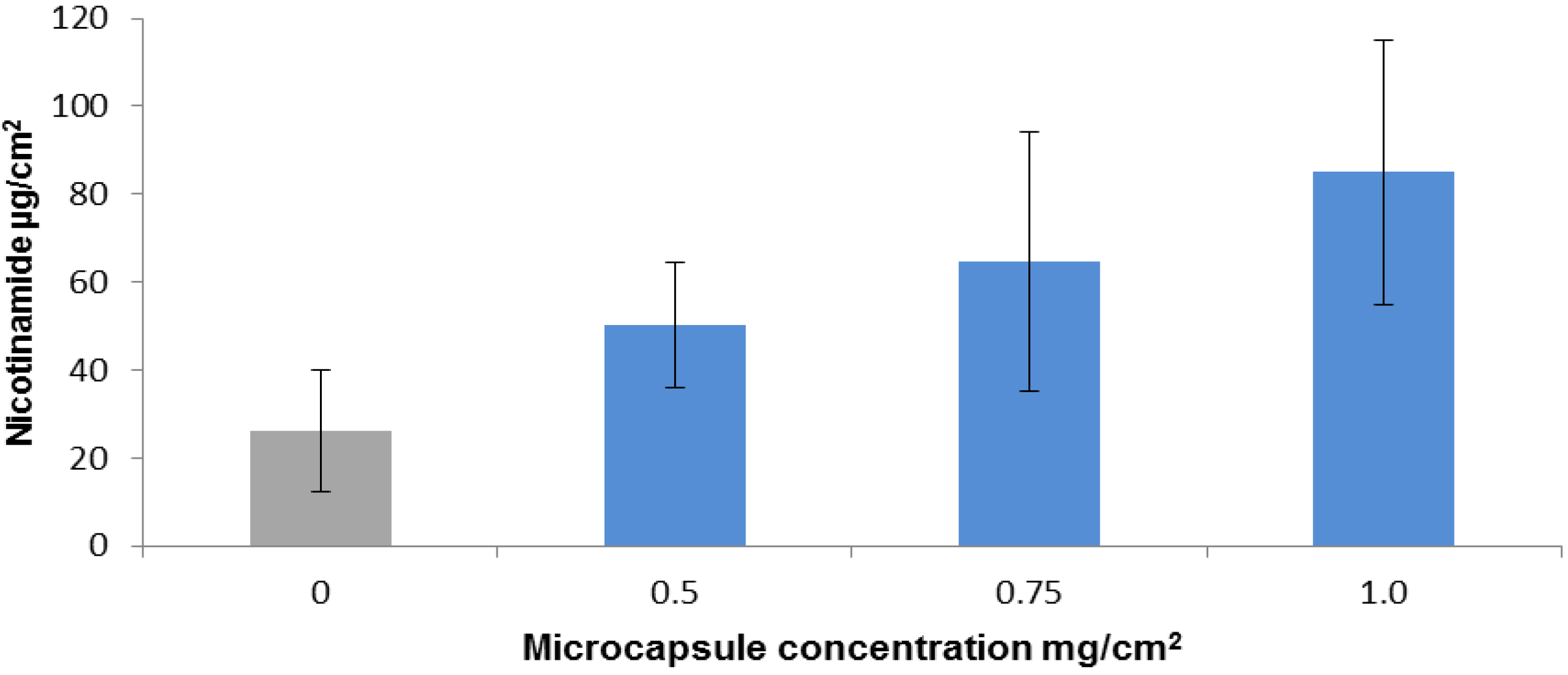
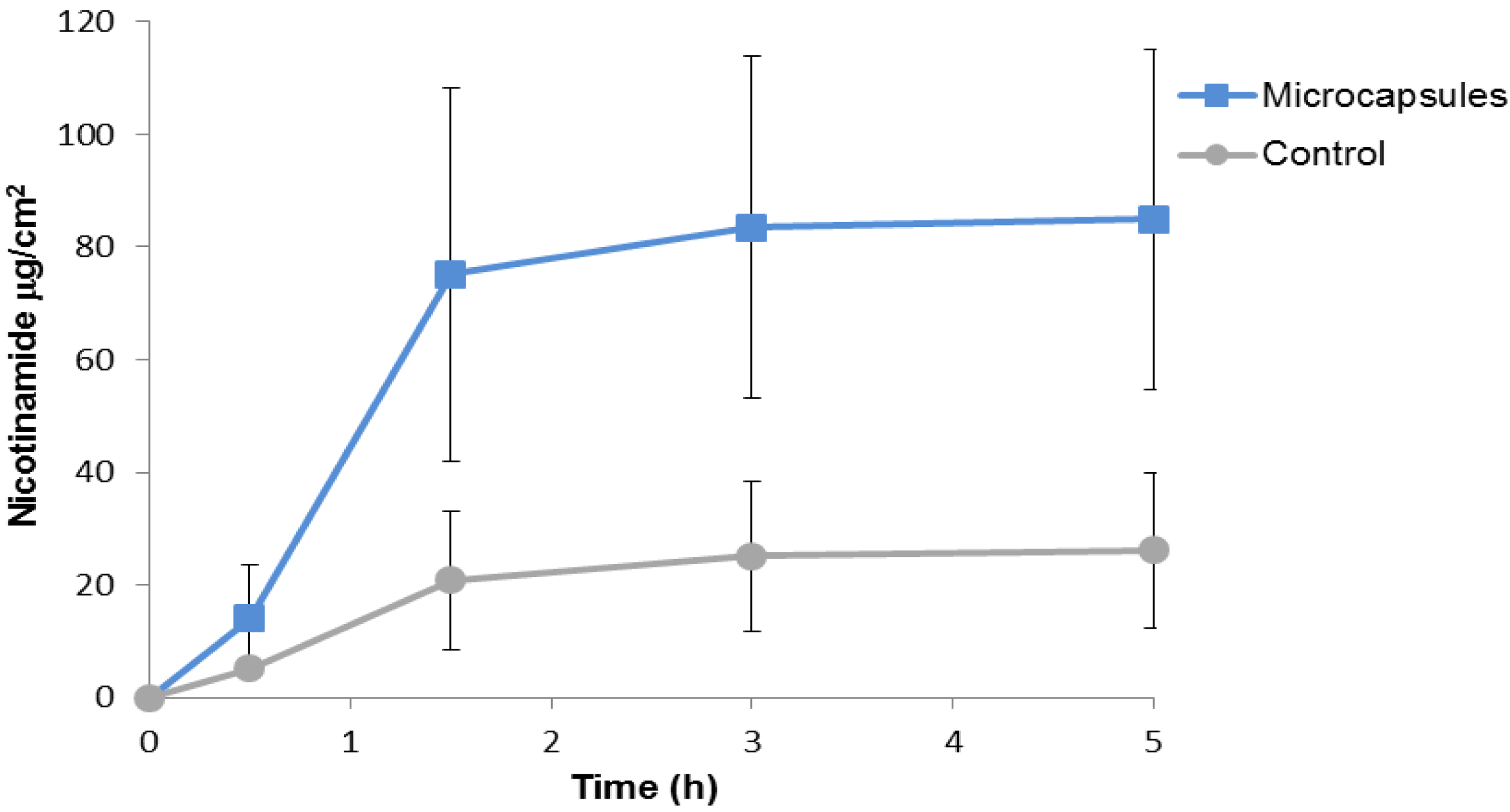
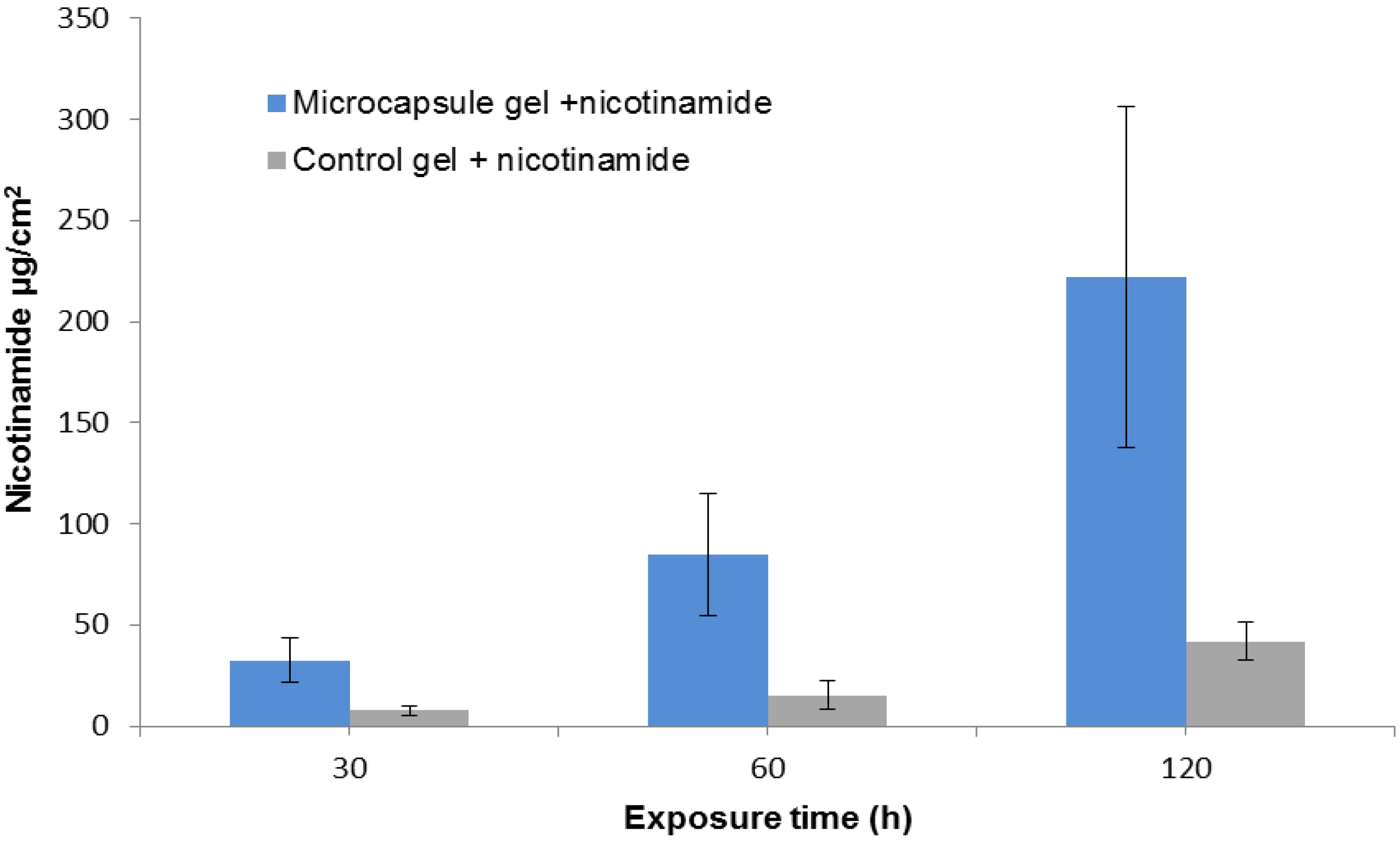
2.2. Nicotinamide Permeation across Nude Mouse Skin in Vitro
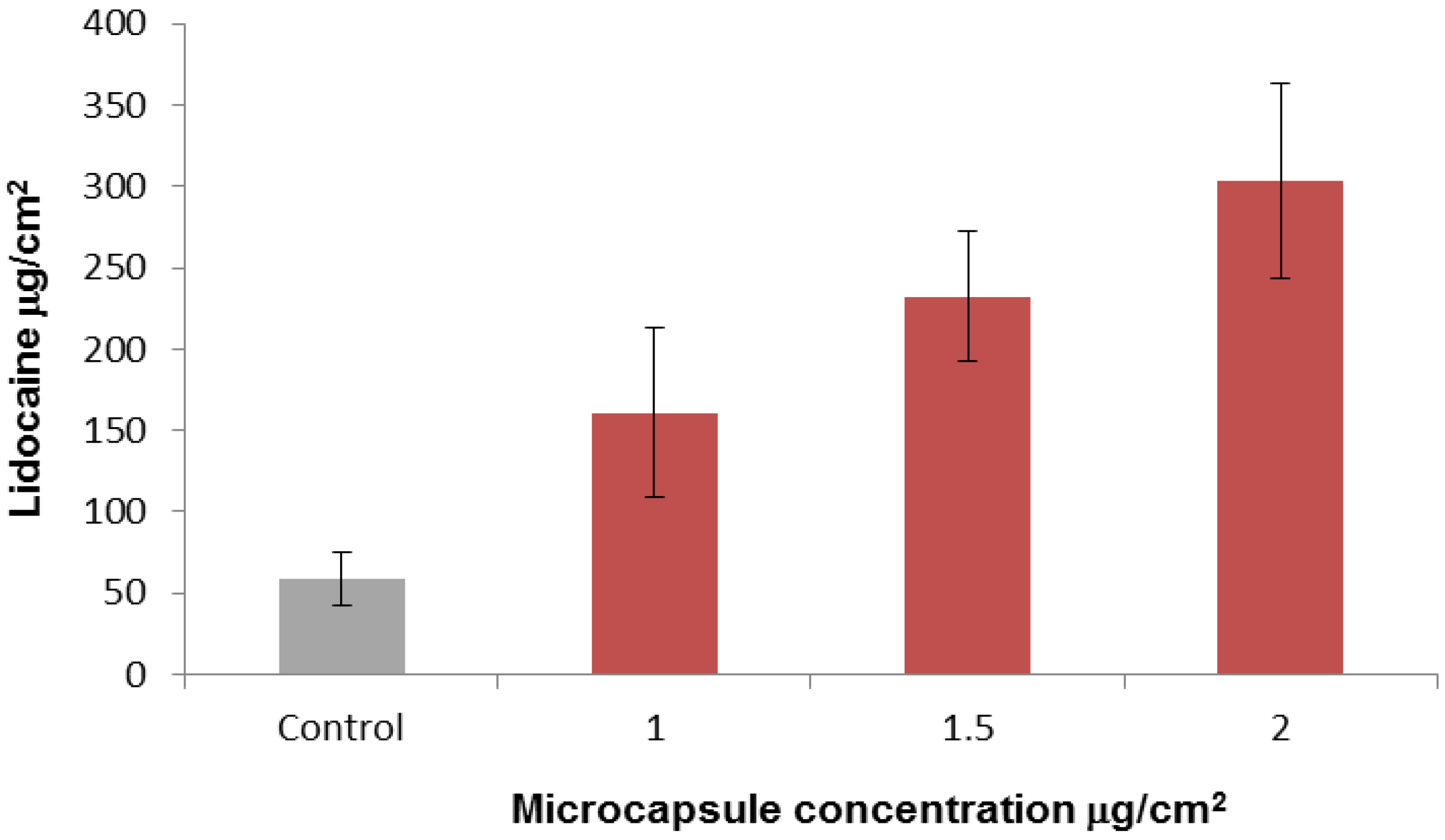
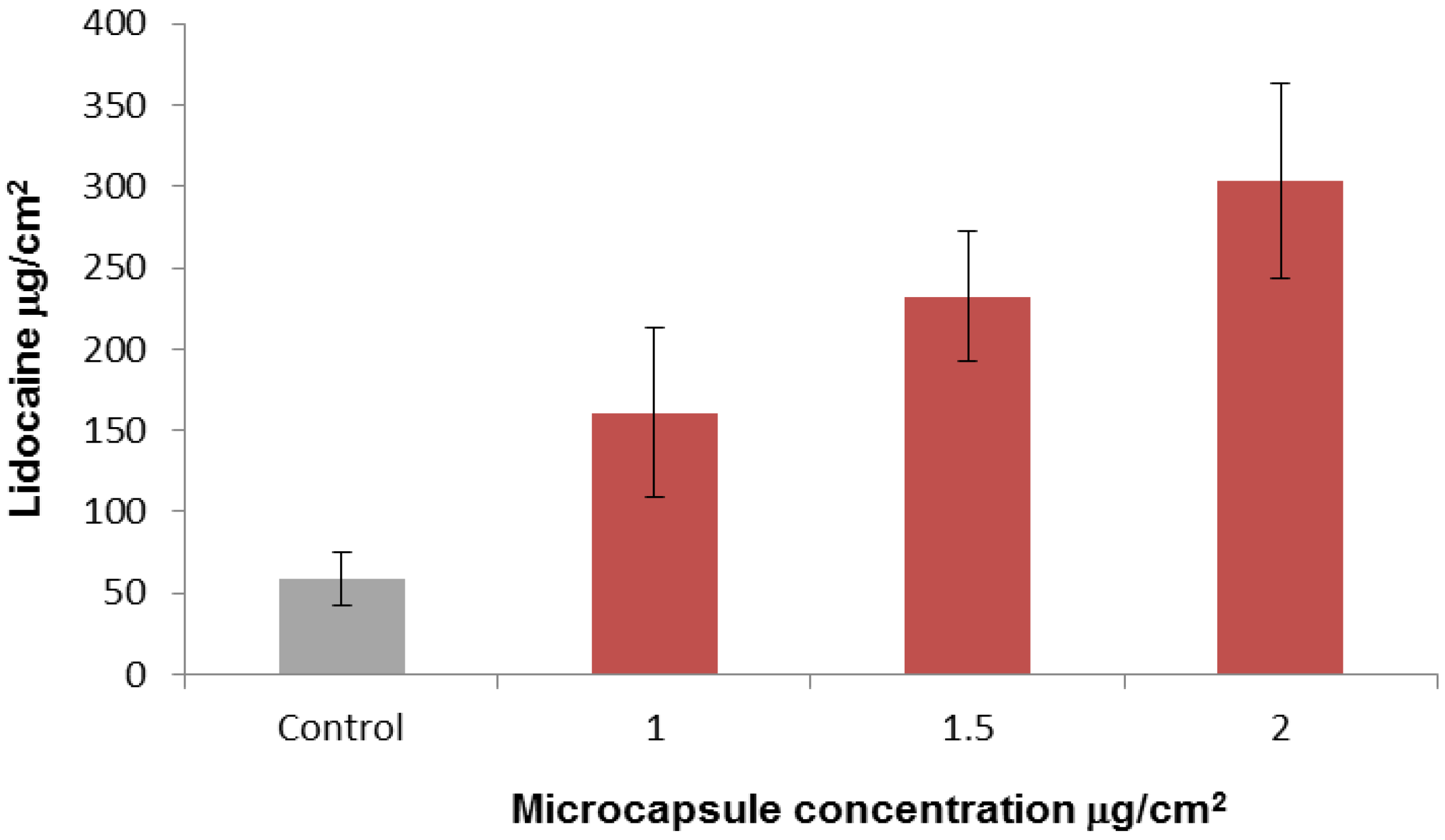
2.3. In-Vitro Delivery of Lidocaine Hydrochloride Using Applied Microcapsules as a Penetration Enhancer
2.4. In-Vitro Skin Irritation Test
3. Experimental Section
3.1. Isolation of Microcapsules
3.2. Chemicals and Formulations
3.3. Diffusion Cell Method
3.4. HPLC Analysis
3.5. Data Analysis
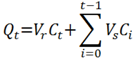
3.6. In-Vitro Skin Irritation Test
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Park, E.; Hwang, D.-S.; Lee, J.-S.; Song, J.-I.; Seo, T.-K.; Won, Y.-J. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Mol. Phylogenet. Evol. 2012, 62, 329–345. [Google Scholar] [CrossRef]
- David, C.N.; Ozbek, S.; Adamczyk, P.; Meier, S.; Pauly, B.; Chapman, J.; Hwang, J.S.; Gojobori, T.; Holstein, T.W. Evolution of complex structures: Minicollagens shape the cnidarian nematocyst. Trends Genet. 2008, 24, 431–438. [Google Scholar] [CrossRef]
- Nüchter, T.; Benoit, M.; Engel, U.; Ozbek, S.; Holstein, T.W. Nanosecond-scale kinetics of nematocyst discharge. Curr. Biol. 2006, 16, R316–R318. [Google Scholar] [CrossRef]
- Szczepanek, S.; Cikala, M.; David, C.N. Poly-γ-glutamate synthesis during formation of nematocyst capsules in Hydra. J. Cell Sci. 2002, 115, 745–751. [Google Scholar]
- Weber, J. Poly(gamma-glutamic acid)s are the major constituents of nematocysts in Hydra (Hydrozoa, Cnidaria). J. Biol. Chem. 1990, 265, 9664–9669. [Google Scholar]
- Tardent, P. The cnidarian cnidocyte, a hightech cellular weaponry. Bioessays 1995, 17, 351–362. [Google Scholar] [CrossRef]
- Lotan, T. Immediate Topical Drug Delivery Using Natural Nano-Injectors. In Modified-Release Drug Delivery Technology, 2nd ed.; Rathbone, M.J., Hadgraft, J., Roberts, M.S., Lane, M.E., Eds.; Informa Healthcare: New York, NY, USA, 2008; Volume 2, pp. 395–404. [Google Scholar]
- Ayalon, A.; Shichor, I.; Tal, Y.; Lotan, T. Immediate topical drug delivery by natural submicron injectors. Int. J. Pharm. 2011, 419, 147–153. [Google Scholar] [CrossRef]
- Shaoul, E.; Ayalon, A.; Tal, Y.; Lotan, T. Transdermal delivery of scopolamine by natural submicron injectors: In-vivo study in pig. PLoS One 2012, 7, e31922. [Google Scholar] [CrossRef]
- Prausnitz, M.R.; Langer, R. Transdermal drug delivery. Nat. Biotechnol. 2008, 26, 1261–1268. [Google Scholar] [CrossRef]
- Subedi, R.; Oh, S.; Chun, M.-K.; Choi, H.-K. Recent advances in transdermal drug delivery. Arch. Pharm. Res. 2010, 33, 339–351. [Google Scholar] [CrossRef]
- Cevc, G.; Vierl, U. Nanotechnology and the transdermal route: A state of the art review and critical appraisal. J. Control. Release 2010, 141, 277–299. [Google Scholar] [CrossRef]
- Barry, B.W. Breaching the skin’s barrier to drugs. Nat. Biotechnol. 2004, 22, 165–167. [Google Scholar] [CrossRef]
- Darling, J.A.; Reitzel, A.R.; Burton, P.M.; Mazza, M.E.; Ryan, J.F.; Sullivan, J.C.; Finnerty, J.R. Rising starlet: The starlet sea anemone, Nematostella vectensis. Bioessays 2005, 27, 211–221. [Google Scholar] [CrossRef]
- Technau, U.; Steele, R.E. Evolutionary crossroads in developmental biology: Cnidaria. Development 2011, 138, 1447–1458. [Google Scholar] [CrossRef]
- Putnam, N.; Srivastava, M.; Hellsten, U.; Dirks, B.; Chapman, J.; Salamov, A.; Terry, A.; Shapiro, H.; Lindquist, E.; Kapitonov, V.; et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 2007, 317, 86–94. [Google Scholar] [CrossRef]
- Nakanishi, N.; Renfer, E.; Technau, U.; Rentzsch, F. Nervous systems of the sea anemone Nematostella vectensis are generated by ectoderm and endoderm and shaped by distinct mechanisms. Development 2012, 139, 347–357. [Google Scholar] [CrossRef]
- Renfer, E.; Amon-Hassenzahl, A.; Steinmetz, P.R.H.; Technau, U. A muscle-specific transgenic reporter line of the sea anemone, Nematostella vectensis. Proc. Natl. Acad. Sci. USA 2010, 107, 104–108. [Google Scholar] [CrossRef]
- Zenkert, C.; Takahashi, T.; Diesner, M.-O.; Özbek, S. Morphological and molecular analysis of the nematostella vectensis cnidom. PLoS One 2011, 6, e22725. [Google Scholar]
- Williams, R.B. A redescription of the brackish-water sea anemone Nematostella vectensis Stephenson, with an appraisal of congeneric species. J. Nat. Hist. 1975, 9, 51–64. [Google Scholar] [CrossRef]
- Moran, Y.; Praher, D.; Schlesinger, A.; Ayalon, A.; Tal, Y.; Technau, U. Analysis of soluble protein contents from the nematocysts of a model sea anemone sheds light on venom evolution. Mar. Biotechnol. 2013, 15, 329–339. [Google Scholar] [CrossRef]
- Hand, C.; Uhlinger, K.R. The culture, sexual and asexual reproduction, and growth of the Sea Anemone Nematostella vectensis. Biol. Bull. 1992, 182, 169–176. [Google Scholar] [CrossRef]
- Stefanik, D.J.; Friedman, L.E.; Finnerty, J.R. Collecting, rearing, spawning and inducing regeneration of the starlet sea anemone, Nematostella vectensis. Nat. Protoc. 2013, 8, 916–923. [Google Scholar] [CrossRef]
- Weber, J. Nematocysts (stinging capsules of Cnidaria) as Donnan-potential-dominated osmotic systems. Eur. J. Biochem. 1989, 184, 465–476. [Google Scholar] [CrossRef]
- Simon, G.; Maibach, H. Relevance of hairless mouse as an experimental model of percutaneous penetration in man. Skin Pharmacol. Appl. Skin Physiol. 1998, 11, 80–86. [Google Scholar] [CrossRef]
- Otte, N.; Borelli, C.; Korting, H.C. Nicotinamide–biologic actions of an emerging cosmetic ingredient. Int. J. Cosmet. Sci. 2005, 27, 255–261. [Google Scholar] [CrossRef]
- Damian, D.L.; Patterson, C.R.S.; Stapelberg, M.; Park, J.; Barnetson, R.S.C.; Halliday, G.M. UV radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J. Invest. Dermatol. 2007, 128, 447–454. [Google Scholar]
- Moser, K.; Kriwet, K.; Naik, A.; Kalia, Y.N.; Guy, R.H. Passive skin penetration enhancement and its quantification in vitro. Eur. J. Pharm. Biopharm. 2001, 52, 103–112. [Google Scholar] [CrossRef]
- Williams, A.C.; Barry, B.W. Penetration enhancers. Adv. Drug Del. Rev. 2012, 64, 128–137. [Google Scholar] [CrossRef]
- Prausnitz, M.R. Microneedles for transdermal drug delivery. Adv. Drug Deliver. Rev. 2004, 56, 581–587. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Singh, T.R.R.; Woolfson, A.D. Microneedle-based drug delivery systems: Microfabrication, drug delivery, and safety. Drug Deliv. 2010, 17, 187–207. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tal, Y.; Ayalon, A.; Sharaev, A.; Kazir, Z.; Brekhman, V.; Lotan, T. Continuous Drug Release by Sea Anemone Nematostella vectensis Stinging Microcapsules. Mar. Drugs 2014, 12, 734-745. https://doi.org/10.3390/md12020734
Tal Y, Ayalon A, Sharaev A, Kazir Z, Brekhman V, Lotan T. Continuous Drug Release by Sea Anemone Nematostella vectensis Stinging Microcapsules. Marine Drugs. 2014; 12(2):734-745. https://doi.org/10.3390/md12020734
Chicago/Turabian StyleTal, Yossi, Ari Ayalon, Agnesa Sharaev, Zoya Kazir, Vera Brekhman, and Tamar Lotan. 2014. "Continuous Drug Release by Sea Anemone Nematostella vectensis Stinging Microcapsules" Marine Drugs 12, no. 2: 734-745. https://doi.org/10.3390/md12020734
APA StyleTal, Y., Ayalon, A., Sharaev, A., Kazir, Z., Brekhman, V., & Lotan, T. (2014). Continuous Drug Release by Sea Anemone Nematostella vectensis Stinging Microcapsules. Marine Drugs, 12(2), 734-745. https://doi.org/10.3390/md12020734