Targeting Nuclear Receptors with Marine Natural Products

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
| NR | Related Diseases | Drug Development |
|---|---|---|
| CAR | cholestatic liver disease [9] | Phenobarbital [12] |
| type 2 diabetes [10] | ||
| hematopoietic malignancies [11] | ||
| ER(α, β) | breast cancer [13] ovarian cancer, colon cancer [14] prostate cancer [15] | Bazedoxifene [16] |
| Tamoxifen [17] | ||
| Raloxifene [18] | ||
| Lasofoxifene [19] | ||
| FXR | biliary cirrhosis, non-alcoholic fatty liver disease [9] | Fexaramine GW4064 [20] |
| INT-747 [21] | ||
| GR | allergic, inflammatory, haematological disorders [22] | Dexamethasone [23] |
| RU486 [24] | ||
| HNF4α | maturity onset diabetes of the young [25] | MEDICA 16 [26] |
| LXR(α, β) | non-alcoholic fatty liver disease [27] | GW3965 [31] N-Acylthiadiazolines [32] T00901317 [33] |
| Alzheimer’s disease [28] | ||
| breast cancer [29] | ||
| atherosclerosis [30] | ||
| PPAR(α, β, γ) | dyslipidemia [34] diabetes [35] | Fibrates [36] |
| GW9662, GW501516 [37] | ||
| Rosiglitazone [38] | ||
| Thiazolidinediones [39] | ||
| PXR | endothelial detoxification [40] | Rifampicin [43] |
| liver injury [41] | ||
| cholestatic liver disease [9] | ||
| cancers [42] | ||
| RXR | metabolic diseases [44] | Bexarotene [46] |
| cancers [45] | ||
| TR(α, β) | thyroid hormone resistance syndrome [47] | Levothyroxine [49] |
| thyroid cancer [48] | Liothyronine | |
| VDR | diabetic nephropathy, hypertension, atherosclerosis [50,51,52] | Doxercalciferol [53] |
| MR | cardiovascular disease [54] | |
| chronic kidney disease [55,56] | ||
| vascular Disease [57] | ||
| PR | breast cancer [58,59] | RU-486 [24] |
| endometriosis [60] | ||
| AR | androgen insensitivity syndrome [61] | |
| prostate cancer [62] | ||
| osteoporosis [63] | ||
| RAR(α, β, γ) | acute promyelocytic leukemia [64] | |
| kidney disease [65] | ||
| Alzheimer’s Disease [66] | ||
| skin diseases [67] | ||
| cancer [44] |
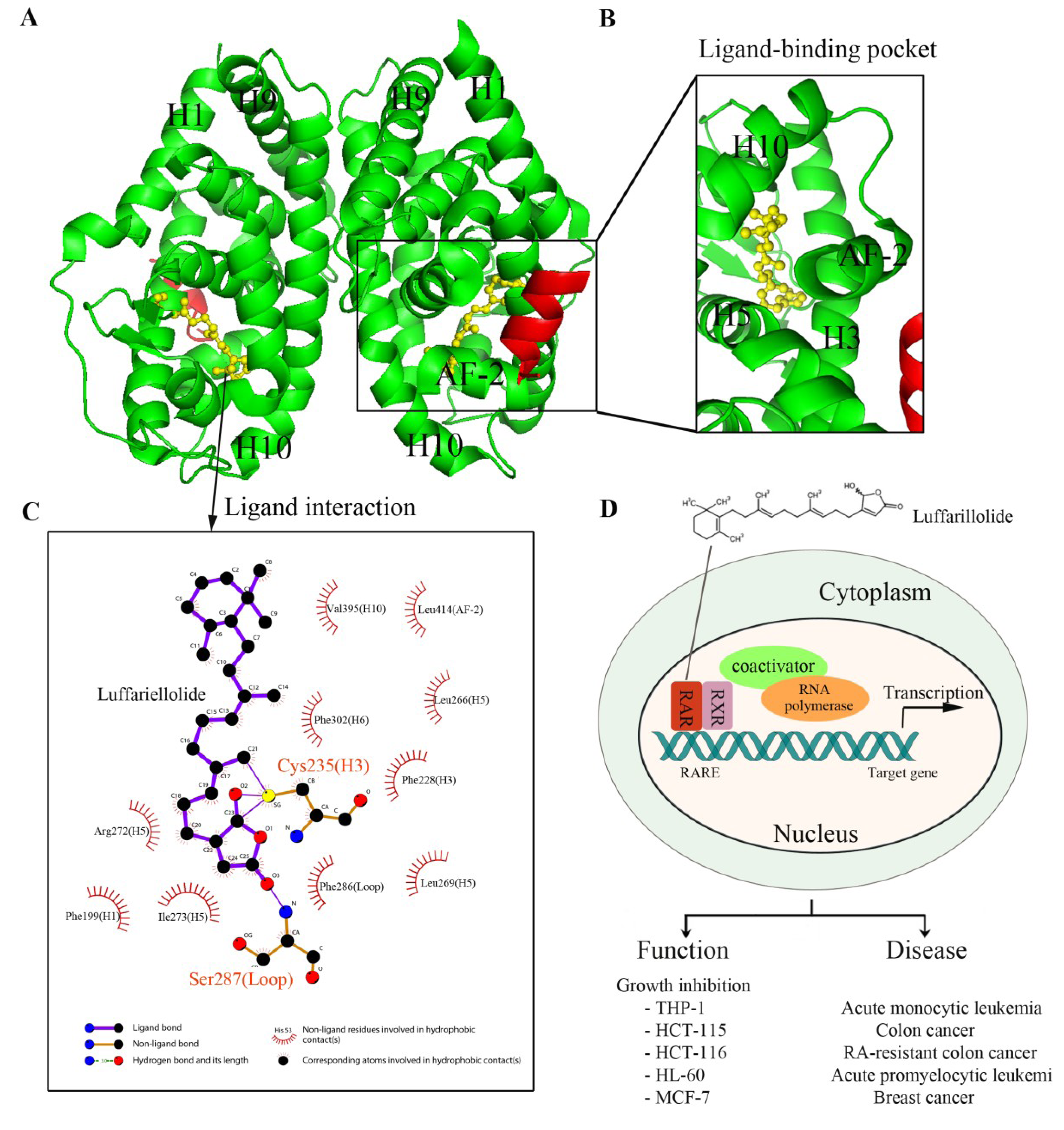
2. Nuclear Receptors: Structure and Function
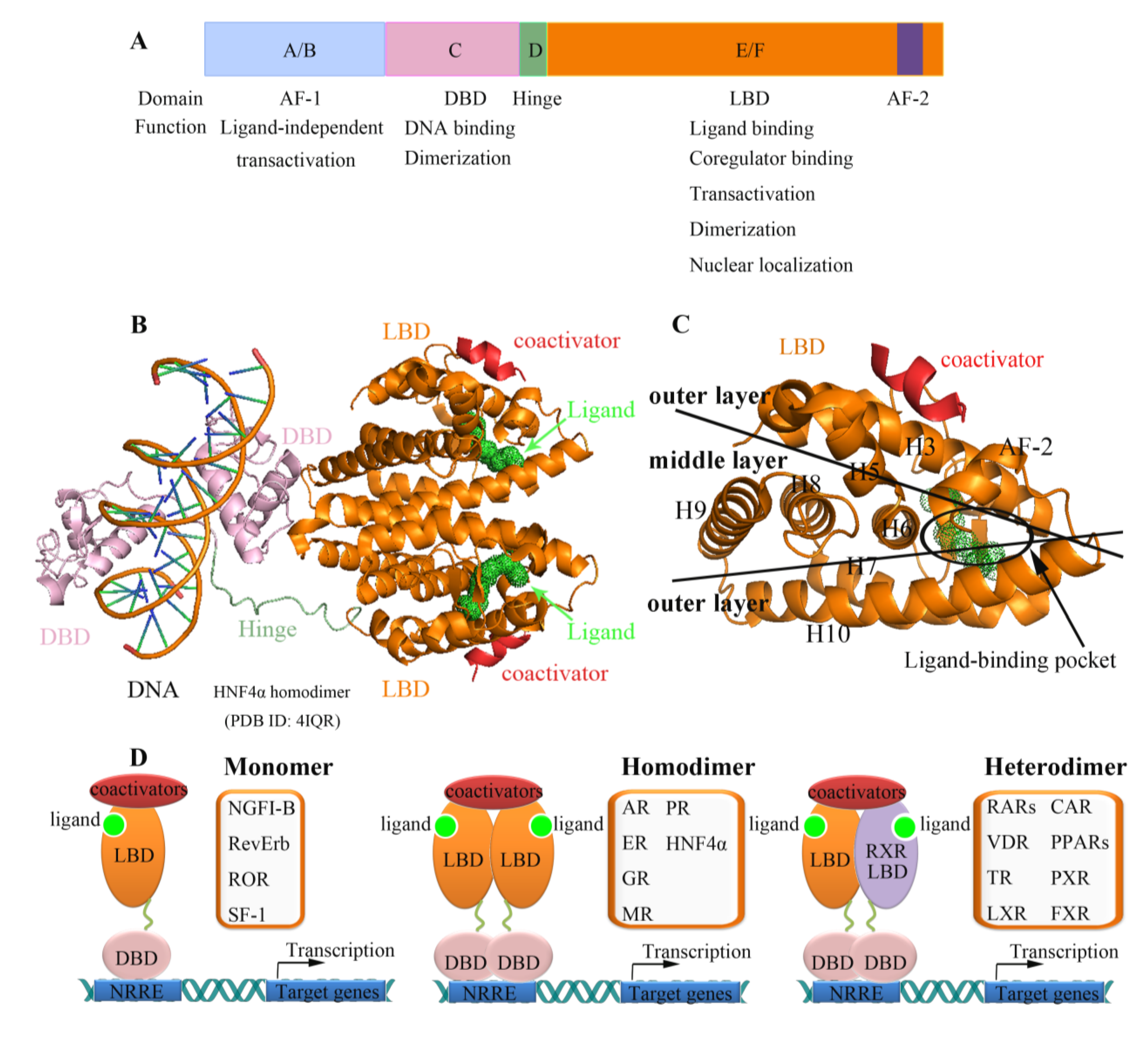
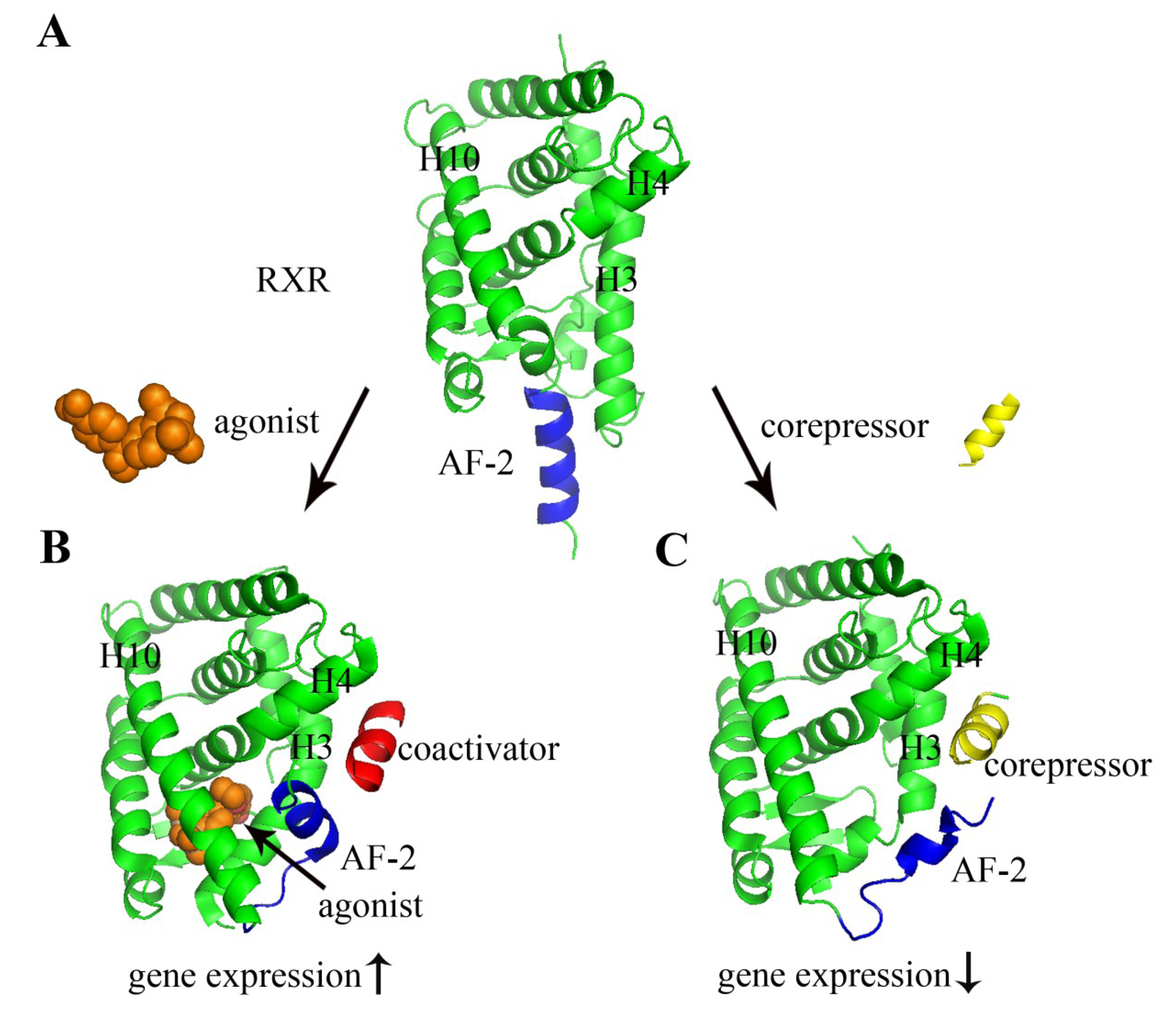
3. Nuclear Receptors as Drug Targets in Related Disease Signaling
3.1. Peroxisome Proliferator-Activated Receptor (PPAR)
3.2. Farnesoid X Receptor (FXR)
3.3. Retinoic Acid Receptor (RAR)
3.4. Pregnane X Receptor (PXR)
4. Strategies for the Discovery of Novel Ligands for Nuclear Receptors
4.1. Fluorescence Polarization (FP) and Fluorescence Resonance Energy Transfer (FRET)
4.2. AlphaScreen (Cofactor Binding Assays)
4.3. Transactivation Reporter Gene Assays (Transient Transfection Assays)
5. Search for Marine Natural Products Targeting Nuclear Receptors
| Compounds | Origin | Target(s) | Comments/References | Method |
|---|---|---|---|---|
| luffariellolide | Marine sponges Luffariella sp. and Fascaplysinopsis | RAR | agonist of RAR with inhibitory effects on cancer cells [196] | AlphaScreen |
| 7-hydroxy retinoic acid | cyanobacteria Microcystis aeruginosa and Spirulina sp. | RAR | agonist of RAR [214] | yeast two hybrid |
| SQA | Brown alga Sargassum yezoense | PPARα/γ | PPARα/γ dual agonists [207] | transfection assay |
| SHQA | Brown alga Sargassum yezoense | PPARα/γ | PPARα/γ dual agonists [207] | transfection assay |
| Ionomycin | Streptomyces conglobatus | PPARγ | partial agonist of PPARγ [69] | AlphaScreen |
| Tuberatolide A | Korean marine tunicate Botryllus tuberatus | FXR | antagonized the (CDCA)-activated FXR [206] | transfection assay |
| Meroterpenoids tuberatolide B | Korean marine tunicate Botryllus tuberatus | FXR | antagonized the (CDCA)-activated FXR [206] | transfection assay |
| 2′-epi-tuberatolide B | Korean marine tunicate Botryllus tuberatus | FXR | antagonized the (CDCA)-activated FXR [206] | transfection assay |
| yezoquinolide | Korean marine tunicate Botryllus tuberatus | FXR | antagonized the (CDCA)-activated FXR [206] | transfection assay |
| (R)-sargachromenol | Korean marine tunicate Botryllus tuberatus | FXR | antagonized the (CDCA)-activated FXR [206] | transfection assay |
| (S)-sargachromenol | Korean marine tunicate Botryllus tuberatus | FXR | antagonized the (CDCA)-activated FXR [206] | transfection assay |
| Compounds 1–5 | marine sponge Spongia sp. | FXR | FXR antagonistic activity [215] | transfection assay |
| 4-methylenesterols | marine sponge Theonellaswinhoei | FXR, PXR | potent agonists of PXR and antagonists of FXR [213,216] | transfection assay |
| Conicasterol E | marine sponge Theonella swinhoei | FXR, PXR | dual FXR and PXR agonist [217] | transfection assay |
| Malaitasterol A | marine sponge Theonella swinhoei | PXR | potent agonists of PXR [218] | transfection assay |
| suvanine | marine sponge | FXR | antagonist of FXR [200] | transfection assay |
| sulfated sterol (compound 8) | marine invertebrates | FXR | antagonist of FXR [219] | transfection assay |
| solomonsterols A and B | marine sponge Theonellaswinhoei | PXR | agonist of PXR [220] | transfection assay |
| okadaic acid | microalgae | CiVDR/PXRa, hPXR | activation at nanomolar concentration [211] | transfection assay |
| pectenotoxin-2 | microalgae | CiVDR/PXRa | activation at nanomolar concentration [211] | transfection assay |
| Phosphoiodyns A | Korean marine sponge Placospongia sp. | PPARδ | highly potent hPPARδ activity (EC50 = 23.7 nm) [208] | NMR spectrum |
| Herdmanine I and K | marine ascidian Herdmania momus | PPARγ | similar PPARγ agonistic activities to rosiglitazone [221] | transfection assay |
| gracilioether B and plakilactone C | marine sponge Plakinastrella mamillaris | PPARγ | selective PPARγ ligands [222] | transfection assay |
| Niphatenones | Marine sponge Niphates digitalis | AR | block androgen receptor transcriptional activity in prostate cancer cells [209] | transfection assay |
| Psammaplin A | marine sponge Pseudoceratina rhax | PPARγ | activates PPARγ in a MCF-7 cell-based reporter assay [223] | transfection assay |
| chlorinated peptides sintokamides A to E | sponge Dysidea sp. | AR | inhibitor of N-terminus transactivation of the androgen receptor in prostate cancer cells [224] | transfection assay |
| theonellasterol | marine sponge Theonella swinhoei | FXR | FXR antagonist [225] | transfection assay |
| steroids 3-oxocholest-1,22-dien-12beta-ol and 3-oxocholest-1,4-dien-20beta-ol | soft coral Dendronephthya gigantea | FXR | inhibitory activity against FXR with IC(50)’s 14 and 15 µM [226] | transfection assay |
| Bendigoles D | marine sponge derived bacterium Actinomadura sp. SBMs009 | GR | inhibitor of GR [210] | transfection assay |
| (3R)-cyclocymopol monomethyl ether | marine alga Cymopolia barbata | PR | PR antagonist [212] | transfection assay |
| (3S)-cyclocymopol monomethyl ether | marine alga Cymopolia barbata | PR | PR agonist [212] | transfection assay |
5.1. Luffariellolide and RARs
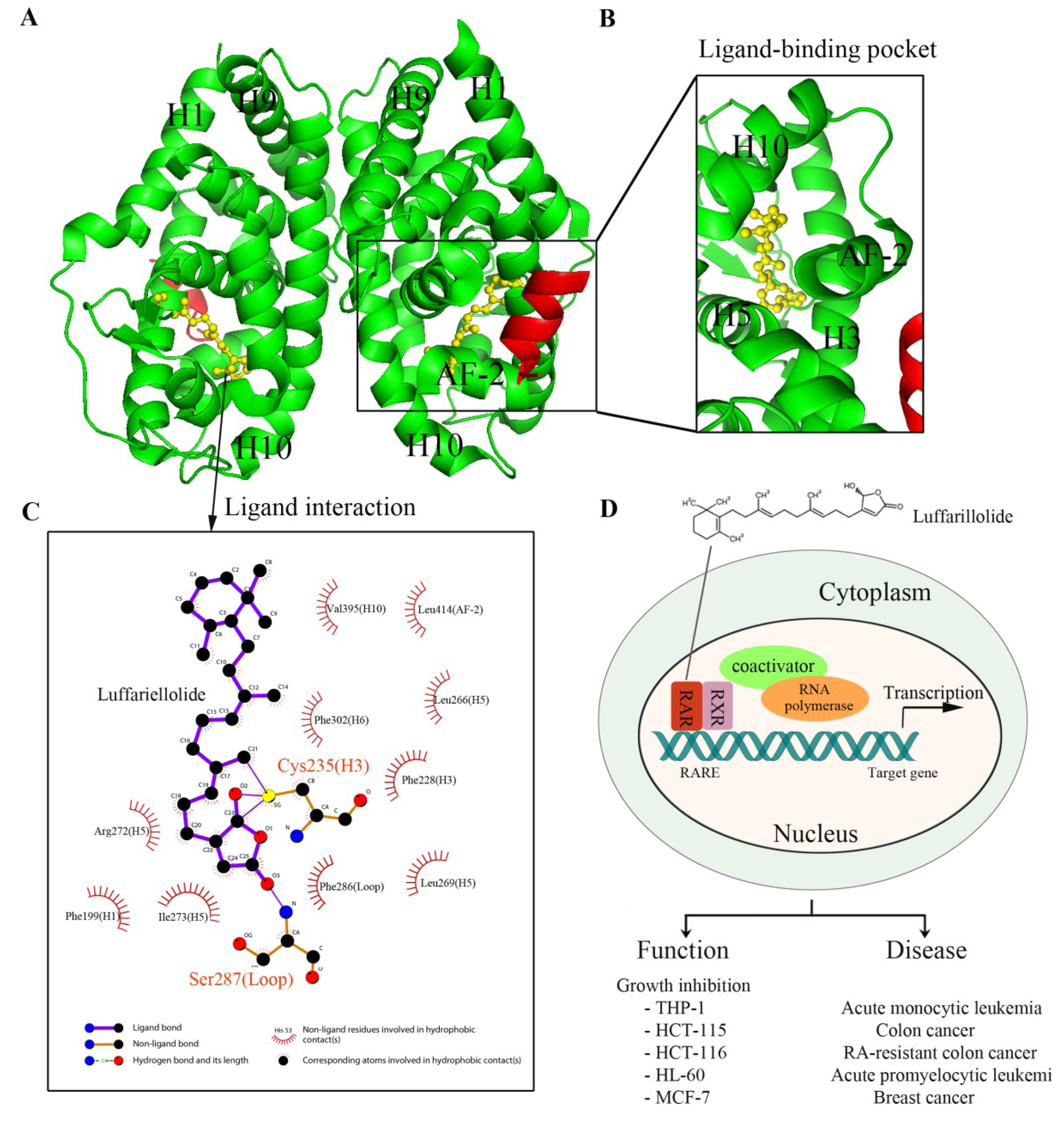
5.2. Marine Products Targeting FXR
5.3. Marine Products Targeting PPARs and AR
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Adis International Limited. Trabectedin: Ecteinascidin 743, Ecteinascidin-743, ET 743, ET-743, NSC 684766. Drugs R D 2006, 7, 317–328. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Li, Y.; Lambert, M.H.; Xu, H.E. Activation of nuclear receptors: A perspective from structural genomics. Structure 2003, 11, 741–746. [Google Scholar]
- Jin, L.; Li, Y. Structural and functional insights into nuclear receptor signaling. Adv. Drug Deliv. Rev. 2010, 62, 1218–1226. [Google Scholar] [CrossRef]
- Halilbasic, E.; Baghdasaryan, A.; Trauner, M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin. Liver Dis. 2013, 17, 161–189. [Google Scholar] [CrossRef]
- Jiang, M.; Xie, W. Role of the constitutive androstane receptor in obesity and type 2 diabetes: A case study of the endobiotic function of a xenobiotic receptor. Drug Metab. Rev. 2013, 45, 156–163. [Google Scholar] [CrossRef]
- Wang, D.; Li, L.; Yang, H.; Ferguson, S.S.; Baer, M.R.; Gartenhaus, R.B.; Wang, H. The constitutive androstane receptor is a novel therapeutic target facilitating cyclophosphamide-based treatment of hematopoietic malignancies. Blood 2013, 121, 329–338. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Phenobarbital for the treatment of epilepsy in the 21st century: A critical review. Epilepsia 2004, 45, 1141–1149. [Google Scholar] [CrossRef]
- Dunnwald, L.K.; Rossing, M.A.; Li, C.I. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007, 9, R6. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.; Lynch, I.J.; Bhardwaj, B. Expression of estrogen receptor (ER) subtypes and ERbeta isoforms in colon cancer. Cancer Res. 2001, 61, 632–640. [Google Scholar]
- Bonkhoff, H.; Fixemer, T.; Hunsicker, I.; Remberger, K. Estrogen receptor expression in prostate cancer and premalignant prostatic lesions. Am. J. Pathol. 1999, 155, 641–647. [Google Scholar] [CrossRef]
- Biskobing, D.M. Update on bazedoxifene: A novel selective estrogen receptor modulator. Clin. Interv. Aging 2007, 2, 299–303. [Google Scholar]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar]
- Cummings, S.R.; Eckert, S.; Krueger, K.A.; Grady, D.; Powles, T.J.; Cauley, J.A.; Norton, L.; Nickelsen, T.; Bjarnason, N.H.; Morrow, M.; et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: Results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 1999, 281, 2189–2197. [Google Scholar] [CrossRef]
- Cummings, S.R.; Ensrud, K.; Delmas, P.D.; LaCroix, A.Z.; Vukicevic, S.; Reid, D.M.; Goldstein, S.; Sriram, U.; Lee, A.; Thompson, J.; et al. Lasofoxifene in postmenopausal women with osteoporosis. N. Engl. J. Med. 2010, 362, 686–696. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, Y.; Yan, L.; Gao, M.; Liu, D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm. Res. 2013, 30, 1447–1457. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef]
- Quax, R.A.; Manenschijn, L.; Koper, J.W.; Hazes, J.M.; Lamberts, S.W.; van Rossum, E.F.; Feelders, R.A. Glucocorticoid sensitivity in health and disease. Nat. Rev. Endocrinol. 2013, 9, 670–686. [Google Scholar]
- Yuan, F.; Nelson, R.K.; Tabor, D.E.; Zhang, Y.; Akhter, M.P.; Gould, K.A.; Wang, D. Dexamethasone prodrug treatment prevents nephritis in lupus-prone (NZB × NZW)F1 mice without causing systemic side effects. Arthritis Rheum. 2012, 64, 4029–4039. [Google Scholar] [CrossRef]
- Gallagher, P.; Young, A.H. Mifepristone (RU-486) treatment for depression and psychosis: A review of the therapeutic implications. Neuropsychiatr. Dis. Treat. 2006, 2, 33–42. [Google Scholar]
- Arya, V.B.; Rahman, S.; Senniappan, S.; Flanagan, S.E.; Ellard, S.; Hussain, K. HNF4A mutation: Switch from hyperinsulinaemic hypoglycaemia to maturity-onset diabetes of the young, and incretin response. Diabet. Med. 2013. [Google Scholar] [CrossRef]
- Kalderon, B.; Mayorek, N.; Ben-Yaacov, L.; Bar-Tana, J. Adipose tissue sensitization to insulin induced by troglitazone and MEDICA 16 in obese Zucker rats in vivo. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E795–E803. [Google Scholar]
- Ducheix, S.; Montagner, A.; Theodorou, V.; Ferrier, L.; Guillou, H. The liver X receptor: A master regulator of the gut-liver axis and a target for non alcoholic fatty liver disease. Biochem. Pharmacol. 2013, 86, 96–105. [Google Scholar] [CrossRef]
- Sodhi, R.K.; Singh, N. Liver X receptors: Emerging therapeutic targets for Alzheimer’s disease. Pharmacol. Res. 2013, 72, 45–51. [Google Scholar] [CrossRef]
- El Roz, A.; Bard, J.M.; Huvelin, J.M.; Nazih, H. LXR agonists and ABCG1-dependent cholesterol efflux in MCF-7 breast cancer cells: Relation to proliferation and apoptosis. Anticancer Res. 2012, 32, 3007–3013. [Google Scholar]
- Kappus, M.S.; Murphy, A.J.; Abramowicz, S.; Ntonga, V.; Welch, C.L.; Tall, A.R.; Westerterp, M. Activation of liver X receptor decreases atherosclerosis in Ldlr−/− mice in the absence of ATP-binding cassette transporters A1 and G1 in myeloid cells. Arterioscler. Thromb. Vasc. Biol. 2013. [Google Scholar] [CrossRef]
- Miao, B.; Zondlo, S.; Gibbs, S.; Cromley, D.; Hosagrahara, V.P.; Kirchgessner, T.G.; Billheimer, J.; Mukherjee, R. Raising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulator. J. Lipid Res. 2004, 45, 1410–1417. [Google Scholar] [CrossRef]
- Molteni, V.; Li, X.; Nabakka, J.; Liang, F.; Wityak, J.; Koder, A.; Vargas, L.; Romeo, R.; Mitro, N.; Mak, P.A.; et al. N-Acylthiadiazolines, a new class of liver X receptor agonists with selectivity for LXRbeta. J. Med. Chem. 2007, 50, 4255–4259. [Google Scholar] [CrossRef]
- Grefhorst, A.; Elzinga, B.M.; Voshol, P.J.; Plosch, T.; Kok, T.; Bloks, V.W.; van der Sluijs, F.H.; Havekes, L.M.; Romijn, J.A.; Verkade, H.J.; et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J. Biol. Chem. 2002, 277, 34182–34190. [Google Scholar] [CrossRef]
- Keller, H.; Dreyer, C.; Medin, J.; Mahfoudi, A.; Ozato, K.; Wahli, W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. USA 1993, 90, 2160–2164. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Andersson, C.X.; Rotter Sopasakis, V.; Smith, U. The effect of PPARgamma ligands on the adipose tissue in insulin resistance. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 65–75. [Google Scholar] [CrossRef]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar]
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E. The peroxisome proliferator-activated receptor beta/delta agonist, GW501516, regulates the expression of genes involved in lipid catabolism and energy uncoupling in skeletal muscle cells. Mol. Endocrinol. 2003, 17, 2477–2493. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- Krentz, A.J.; Friedmann, P.S. Type 2 diabetes, psoriasis and thiazolidinediones. Int. J. Clin. Pract. 2006, 60, 362–363. [Google Scholar] [CrossRef]
- Wang, X.; Fang, X.; Zhou, J.; Chen, Z.; Zhao, B.; Xiao, L.; Liu, A.; Li, Y.S.; Shyy, J.Y.; Guan, Y.; et al. Shear stress activation of nuclear receptor PXR in endothelial detoxification. Proc. Natl. Acad. Sci. USA 2013, 110, 13174–13179. [Google Scholar] [CrossRef]
- Li, T.; Yu, R.T.; Atkins, A.R.; Downes, M.; Tukey, R.H.; Evans, R.M. Targeting the pregnane X receptor in liver injury. Expert Opin. Ther. Targets 2012, 16, 1075–1083. [Google Scholar] [CrossRef]
- Pondugula, S.R.; Mani, S. Pregnane xenobiotic receptor in cancer pathogenesis and therapeutic response. Cancer Lett. 2013, 328, 1–9. [Google Scholar] [CrossRef]
- Gregorio, G.V.; Ball, C.S.; Mowat, A.P.; Mieli-Vergani, G. Effect of rifampicin in the treatment of pruritus in hepatic cholestasis. Arch. Dis. Child. 1993, 69, 141–143. [Google Scholar] [CrossRef]
- Altucci, L.; Leibowitz, M.D.; Ogilvie, K.M.; de Lera, A.R.; Gronemeyer, H. RAR and RXR modulation in cancer and metabolic disease. Nat. Rev. Drug Discov. 2007, 6, 793–810. [Google Scholar] [CrossRef]
- Tanaka, T.; de Luca, L.M. Therapeutic potential of “rexinoids” in cancer prevention and treatment. Cancer Res. 2009, 69, 4945–4947. [Google Scholar] [CrossRef]
- Straus, D.J.; Duvic, M.; Horwitz, S.M.; Hymes, K.; Goy, A.; Hernandez-Ilizaliturri, F.J.; Feldman, T.; Wegner, B.; Myskowski, P.L. Final results of phase II trial of doxorubicin HCl liposome injection followed by bexarotene in advanced cutaneous T-cell lymphoma. Ann. Oncol. 2013. [Google Scholar] [CrossRef]
- Monzani, A.; Moia, S.; Prodam, F.; Walker, G.E.; Bellone, S.; Petri, A.; Bona, G. A novel familial variation of the thyroid hormone receptor beta gene (I276N) associated with resistance to thyroid hormone. Thyroid 2012, 22, 440–441. [Google Scholar] [CrossRef]
- Rosen, M.D.; Privalsky, M.L. Thyroid hormone receptor mutations in cancer and resistance to thyroid hormone: Perspective and prognosis. J. Thyroid Res. 2011, 2011, 361304:1–361304:20. [Google Scholar]
- Negro, R.; Formoso, G.; Mangieri, T.; Pezzarossa, A.; Dazzi, D.; Hassan, H. Levothyroxine treatment in euthyroid pregnant women with autoimmune thyroid disease: Effects on obstetrical complications. J. Clin. Endocrinol. Metab. 2006, 91, 2587–2591. [Google Scholar] [CrossRef]
- Yang, L.; Ma, J.; Zhang, X.; Fan, Y.; Wang, L. Protective role of the vitamin D receptor. Cell. Immunol. 2012, 279, 160–166. [Google Scholar]
- hakhtoura, M.; Azar, S.T. The role of vitamin d deficiency in the incidence, progression, and complications of type 1 diabetes mellitus. Int. J. Endocrinol. 2013, 2013. [Google Scholar] [CrossRef]
- Gardner, D.G.; Chen, S.; Glenn, D.J. Vitamin D and the heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R969–R977. [Google Scholar] [CrossRef]
- Sprague, S.M.; Ho, L.T. Oral doxercalciferol therapy for secondary hyperparathyroidism in a peritoneal dialysis patient. Clin. Nephrol. 2002, 58, 155–160. [Google Scholar] [CrossRef]
- Young, M.J. Targeting the mineralocorticoid receptor in cardiovascular disease. Expert Opin. Ther. Targets 2013, 17, 321–331. [Google Scholar] [CrossRef]
- Volk, M.J.; Bomback, A.S.; Klemmer, P.J. Mineralocorticoid receptor blockade in chronic kidney disease. Curr. Hypertens. Rep. 2011, 13, 282–288. [Google Scholar] [CrossRef]
- Bertocchio, J.P.; Warnock, D.G.; Jaisser, F. Mineralocorticoid receptor activation and blockade: An emerging paradigm in chronic kidney disease. Kidney Int. 2011, 79, 1051–1060. [Google Scholar] [CrossRef]
- McGraw, A.P.; McCurley, A.; Preston, I.R.; Jaffe, I.Z. Mineralocorticoid receptors in vascular disease: Connecting molecular pathways to clinical implications. Curr. Atheroscler. Rep. 2013, 15, 340. [Google Scholar] [CrossRef]
- Gao, X.; Nawaz, Z. Progesterone receptors—Animal models and cell signaling in breast cancer: Role of steroid receptor coactivators and corepressors of progesterone receptors in breast cancer. Breast Cancer Res. 2002, 4, 182–186. [Google Scholar] [CrossRef]
- Fuqua, S.A.; Cui, Y.; Lee, A.V.; Osborne, C.K.; Horwitz, K.B. Insights into the role of progesterone receptors in breast cancer. J. Clin. Oncol. 2005, 23, 931–932. [Google Scholar] [CrossRef]
- Kim, J.J.; Kurita, T.; Bulun, S.E. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr. Rev. 2013, 34, 130–162. [Google Scholar] [CrossRef]
- Mukherjee, S.; Cruz-Rodriguez, O.; Bolton, E.; Iniguez-Lluhi, J.A. The in vivo role of androgen receptor SUMOylation as revealed by androgen insensitivity syndrome and prostate cancer mutations targeting the proline/glycine residues of synergy control motifs. J. Biol. Chem. 2012, 287, 31195–31206. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sakari, M.; Okada, M.; Yokoyama, A.; Takahashi, S.; Kouzmenko, A.; Kato, S. The androgen receptor in health and disease. Annu. Rev. Physiol. 2013, 75, 201–224. [Google Scholar] [CrossRef]
- Manolagas, S.C.; O’Brien, C.A.; Almeida, M. The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 2013, 9, 699–712. [Google Scholar] [CrossRef]
- Licht, J.D. Reconstructing a disease: What essential features of the retinoic acid receptor fusion oncoproteins generate acute promyelocytic leukemia? Cancer Cell 2006, 9, 73–74. [Google Scholar] [CrossRef]
- Zhong, Y.; Wu, Y.; Liu, R.; Li, Z.; Chen, Y.; Evans, T.; Chuang, P.; Das, B.; He, J.C. Novel retinoic acid receptor alpha agonists for treatment of kidney disease. PLoS One 2011, 6, e27945. [Google Scholar]
- Lee, H.P.; Casadesus, G.; Zhu, X.; Lee, H.G.; Perry, G.; Smith, M.A.; Gustaw-Rothenberg, K.; Lerner, A. All-trans retinoic acid as a novel therapeutic strategy for Alzheimer’s disease. Expert Rev. Neurother. 2009, 9, 1615–1621. [Google Scholar] [CrossRef]
- Fisher, G.J.; Voorhees, J.J. Molecular mechanisms of retinoid actions in skin. FASEB J. 1996, 10, 1002–1013. [Google Scholar]
- Jin, L.; Feng, X.; Rong, H.; Pan, Z.; Inaba, Y.; Qiu, L.; Zheng, W.; Lin, S.; Wang, R.; Wang, Z.; et al. The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Zheng, W.; Feng, X.; Qiu, L.; Pan, Z.; Wang, R.; Lin, S.; Hou, D.; Jin, L.; Li, Y. Identification of the antibiotic ionomycin as an unexpected peroxisome proliferator-activated receptor gamma (PPARgamma) ligand with a unique binding mode and effective glucose-lowering activity in a mouse model of diabetes. Diabetologia 2013, 56, 401–411. [Google Scholar] [CrossRef]
- Fujita-Sato, S.; Ito, S.; Isobe, T.; Ohyama, T.; Wakabayashi, K.; Morishita, K.; Ando, O.; Isono, F. Structural basis of digoxin that antagonizes RORgamma t receptor activity and suppresses Th17 cell differentiation and interleukin (IL)-17 production. J. Biol. Chem. 2011, 286, 31409–31417. [Google Scholar]
- Li, Y.; Suino, K.; Daugherty, J.; Xu, H.E. Structural and biochemical mechanisms for the specificity of hormone binding and coactivator assembly by mineralocorticoid receptor. Mol. Cell 2005, 19, 367–380. [Google Scholar] [CrossRef]
- Bourguet, W.; Ruff, M.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature 1995, 375, 377–382. [Google Scholar] [CrossRef]
- Lippert, W.P.; Burschka, C.; Gotz, K.; Kaupp, M.; Ivanova, D.; Gaudon, C.; Sato, Y.; Antony, P.; Rochel, N.; Moras, D.; et al. Silicon analogues of the RXR-selective retinoid agonist SR11237 (BMS649): Chemistry and biology. ChemMedChem 2009, 4, 1143–1152. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L.; Chen, J.; Jiang, H.; Shen, X. Structural basis for retinoic X receptor repression on the tetramer. J. Biol. Chem. 2011, 286, 24593–24598. [Google Scholar] [CrossRef]
- Gampe, R.T., Jr.; Montana, V.G.; Lambert, M.H.; Miller, A.B.; Bledsoe, R.K.; Milburn, M.V.; Kliewer, S.A.; Willson, T.M.; Xu, H.E. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol. Cell 2000, 5, 545–555. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Xu, H.E.; Stanley, T.B.; Montana, V.G.; Lambert, M.H.; Shearer, B.G.; Cobb, J.E.; McKee, D.D.; Galardi, C.M.; Plunket, K.D.; Nolte, R.T.; et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 2002, 415, 813–817. [Google Scholar] [CrossRef]
- Phelan, C.A.; Gampe, R.T., Jr.; Lambert, M.H.; Parks, D.J.; Montana, V.; Bynum, J.; Broderick, T.M.; Hu, X.; Williams, S.P.; Nolte, R.T.; et al. Structure of Rev-erbalpha bound to N-CoR reveals a unique mechanism of nuclear receptor-co-repressor interaction. Nat. Struct. Mol. Biol. 2010, 17, 808–814. [Google Scholar] [CrossRef]
- Kallen, J.; Schlaeppi, J.M.; Bitsch, F.; Filipuzzi, I.; Schilb, A.; Riou, V.; Graham, A.; Strauss, A.; Geiser, M.; Fournier, B. Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERRalpha): Crystal structure of ERRalpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1alpha. J. Biol. Chem. 2004, 279, 49330–49337. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Orans, J.; Moore, L.B.; Xue, Y.; Peng, L.; Collins, J.L.; Wisely, G.B.; Lambert, M.H.; Kliewer, S.A.; Redinbo, M.R. Structural disorder in the complex of human pregnane X receptor and the macrolide antibiotic rifampicin. Mol. Endocrinol. 2005, 19, 1125–1134. [Google Scholar] [CrossRef]
- Kruse, S.W.; Suino-Powell, K.; Zhou, X.E.; Kretschman, J.E.; Reynolds, R.; Vonrhein, C.; Xu, Y.; Wang, L.; Tsai, S.Y.; Tsai, M.J.; et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 2008, 6, e227. [Google Scholar] [CrossRef]
- Krasowski, M.D.; Ni, A.; Hagey, L.R.; Ekins, S. Evolution of promiscuous nuclear hormone receptors: LXR, FXR, VDR, PXR, and CAR. Mol. Cell. Endocrinol. 2011, 334, 39–48. [Google Scholar] [CrossRef]
- Giguere, V.; McBroom, L.D.; Flock, G. Determinants of target gene specificity for ROR alpha 1: Monomeric DNA binding by an orphan nuclear receptor. Mol. Cell. Biol. 1995, 15, 2517–2526. [Google Scholar]
- Wilson, T.E.; Fahrner, T.J.; Milbrandt, J. The orphan receptors NGFI-B and steroidogenic factor 1 establish monomer binding as a third paradigm of nuclear receptor-DNA interaction. Mol. Cell. Biol. 1993, 13, 5794–5804. [Google Scholar]
- Harding, H.P.; Lazar, M.A. The monomer-binding orphan receptor Rev-Erb represses transcription as a dimer on a novel direct repeat. Mol. Cell. Biol. 1995, 15, 4791–4802. [Google Scholar]
- Li, Y.; Choi, M.; Cavey, G.; Daugherty, J.; Suino, K.; Kovach, A.; Bingham, N.C.; Kliewer, S.A.; Xu, H.E. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol. Cell 2005, 17, 491–502. [Google Scholar] [CrossRef]
- Luisi, B.F.; Xu, W.X.; Otwinowski, Z.; Freedman, L.P.; Yamamoto, K.R.; Sigler, P.B. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991, 352, 497–505. [Google Scholar] [CrossRef]
- Schwabe, J.W.; Chapman, L.; Finch, J.T.; Rhodes, D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: How receptors discriminate between their response elements. Cell 1993, 75, 567–578. [Google Scholar] [CrossRef]
- Zhao, Q.; Khorasanizadeh, S.; Miyoshi, Y.; Lazar, M.A.; Rastinejad, F. Structural elements of an orphan nuclear receptor-DNA complex. Mol. Cell 1998, 1, 849–861. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Greschik, H.; Wurtz, J.M.; Sanglier, S.; Bourguet, W.; van Dorsselaer, A.; Moras, D.; Renaud, J.P. Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol. Cell 2002, 9, 303–313. [Google Scholar] [CrossRef]
- Chandra, V.; Huang, P.; Potluri, N.; Wu, D.; Kim, Y.; Rastinejad, F. Multidomain integration in the structure of the HNF-4alpha nuclear receptor complex. Nature 2013, 495, 394–398. [Google Scholar] [CrossRef]
- Bledsoe, R.K.; Montana, V.G.; Stanley, T.B.; Delves, C.J.; Apolito, C.J.; McKee, D.D.; Consler, T.G.; Parks, D.J.; Stewart, E.L.; Willson, T.M.; et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 2002, 110, 93–105. [Google Scholar] [CrossRef]
- Bourguet, W.; Vivat, V.; Wurtz, J.M.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol. Cell 2000, 5, 289–298. [Google Scholar] [CrossRef]
- Pogenberg, V.; Guichou, J.F.; Vivat-Hannah, V.; Kammerer, S.; Perez, E.; Germain, P.; de Lera, A.R.; Gronemeyer, H.; Royer, C.A.; Bourguet, W. Characterization of the interaction between retinoic acid receptor/retinoid X receptor (RAR/RXR) heterodimers and transcriptional coactivators through structural and fluorescence anisotropy studies. J. Biol. Chem. 2005, 280, 1625–1633. [Google Scholar]
- Putcha, B.D.; Wright, E.; Brunzelle, J.S.; Fernandez, E.J. Structural basis for negative cooperativity within agonist-bound TR:RXR heterodimers. Proc. Natl. Acad. Sci. USA 2012, 109, 6084–6087. [Google Scholar] [CrossRef]
- Svensson, S.; Ostberg, T.; Jacobsson, M.; Norstrom, C.; Stefansson, K.; Hallen, D.; Johansson, I.C.; Zachrisson, K.; Ogg, D.; Jendeberg, L. Crystal structure of the heterodimeric complex of LXRalpha and RXRbeta ligand-binding domains in a fully agonistic conformation. EMBO J. 2003, 22, 4625–4633. [Google Scholar] [CrossRef]
- Suino, K.; Peng, L.; Reynolds, R.; Li, Y.; Cha, J.Y.; Repa, J.J.; Kliewer, S.A.; Xu, H.E. The nuclear xenobiotic receptor CAR: Structural determinants of constitutive activation and heterodimerization. Mol. Cell 2004, 16, 893–905. [Google Scholar]
- Xu, R.X.; Lambert, M.H.; Wisely, B.B.; Warren, E.N.; Weinert, E.E.; Waitt, G.M.; Williams, J.D.; Collins, J.L.; Moore, L.B.; Willson, T.M.; et al. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol. Cell 2004, 16, 919–928. [Google Scholar] [CrossRef]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar]
- Khorasanizadeh, S.; Rastinejad, F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem. Sci. 2001, 26, 384–390. [Google Scholar] [CrossRef]
- Williams, S.P.; Sigler, P.B. Atomic structure of progesterone complexed with its receptor. Nature 1998, 393, 392–396. [Google Scholar] [CrossRef]
- Sack, J.S.; Kish, K.F.; Wang, C.; Attar, R.M.; Kiefer, S.E.; An, Y.; Wu, G.Y.; Scheffler, J.E.; Salvati, M.E.; Krystek, S.R., Jr.; et al. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl. Acad. Sci. USA 2001, 98, 4904–4909. [Google Scholar] [CrossRef]
- Zhang, J.; Chalmers, M.J.; Stayrook, K.R.; Burris, L.L.; Wang, Y.; Busby, S.A.; Pascal, B.D.; Garcia-Ordonez, R.D.; Bruning, J.B.; Istrate, M.A.; et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat. Struct. Mol. Biol. 2011, 18, 556–563. [Google Scholar] [CrossRef]
- Orlov, I.; Rochel, N.; Moras, D.; Klaholz, B.P. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. EMBO J. 2012, 31, 291–300. [Google Scholar] [CrossRef]
- Kersten, S.; Dong, D.; Lee, W.; Reczek, P.R.; Noy, N. Auto-silencing by the retinoid X receptor. J. Mol. Biol. 1998, 284, 21–32. [Google Scholar] [CrossRef]
- Kersten, S.; Kelleher, D.; Chambon, P.; Gronemeyer, H.; Noy, N. Retinoid X receptor alpha forms tetramers in solution. Proc. Natl. Acad. Sci. USA 1995, 92, 8645–8649. [Google Scholar] [CrossRef]
- Jouravel, N.; Sablin, E.; Togashi, M.; Baxter, J.D.; Webb, P.; Fletterick, R.J. Molecular basis for dimer formation of TRbeta variant D355R. Proteins 2009, 75, 111–117. [Google Scholar]
- Fradera, X.; Vu, D.; Nimz, O.; Skene, R.; Hosfield, D.; Wynands, R.; Cooke, A.J.; Haunso, A.; King, A.; Bennett, D.J.; et al. X-ray structures of the LXRalpha LBD in its homodimeric form and implications for heterodimer signaling. J. Mol. Biol. 2010, 399, 120–132. [Google Scholar] [CrossRef]
- Cowley, S.M.; Hoare, S.; Mosselman, S.; Parker, M.G. Estrogen receptors alpha and beta form heterodimers on DNA. J. Biol. Chem. 1997, 272, 19858–19862. [Google Scholar] [CrossRef]
- Kumar, V.; Chambon, P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 1988, 55, 145–156. [Google Scholar] [CrossRef]
- Williams, C.; Lin, C.Y. Oestrogen receptors in breast cancer: Basic mechanisms and clinical implications. Ecancermedicalscience 2013, 7, 370. [Google Scholar]
- Aranda, A.; Alonso-Merino, E.; Zambrano, A. Receptors of thyroid hormones. Pediatr. Endocrinol. Rev. 2013, 11, 2–13. [Google Scholar]
- Shah, P.D.; Gucalp, A.; Traina, T.A. The role of the androgen receptor in triple-negative breast cancer. Womens Health 2013, 9, 351–360. [Google Scholar]
- Nakatsuka, A.; Wada, J.; Makino, H. Cell cycle abnormality in metabolic syndrome and nuclear receptors as an emerging therapeutic target. Acta Med. Okayama 2013, 67, 129–134. [Google Scholar]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar]
- Cermenati, G.; Brioschi, E.; Abbiati, F.; Melcangi, R.C.; Caruso, D.; Mitro, N. Liver X receptors, nervous system, and lipid metabolism. J. Endocrinol. Investig. 2013, 36, 435–443. [Google Scholar]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef]
- Ribon, V.; Johnson, J.H.; Camp, H.S.; Saltiel, A.R. Thiazolidinediones and insulin resistance: Peroxisome proliferatoractivated receptor gamma activation stimulates expression of the CAP gene. Proc. Natl. Acad. Sci. USA 1998, 95, 14751–14756. [Google Scholar] [CrossRef]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef]
- Mochizuki, K.; Suruga, K.; Fukami, H.; Kiso, Y.; Takase, S.; Goda, T. Selectivity of fatty acid ligands for PPARalpha which correlates both with binding to cis-element and DNA binding-independent transactivity in Caco-2 cells. Life Sci. 2006, 80, 140–145. [Google Scholar] [CrossRef]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Nolan, J.J.; Ludvik, B.; Beerdsen, P.; Joyce, M.; Olefsky, J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N. Engl. J. Med. 1994, 331, 1188–1193. [Google Scholar] [CrossRef]
- Berger, J.; Bailey, P.; Biswas, C.; Cullinan, C.A.; Doebber, T.W.; Hayes, N.S.; Saperstein, R.; Smith, R.G.; Leibowitz, M.D. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-gamma: Binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology 1996, 137, 4189–4195. [Google Scholar]
- Kobayashi, J.; Nagashima, I.; Taira, K.; Hikita, M.; Tamura, K.; Bujo, H.; Morisaki, N.; Saito, Y. A novel frameshift mutation in exon 6 (the site of Asn 291) of the lipoprotein lipase gene in type I hyperlipidemia. Clin. Chim. Acta Int. J. Clin. Chem. 1999, 285, 173–182. [Google Scholar] [CrossRef]
- Okuno, A.; Tamemoto, H.; Tobe, K.; Ueki, K.; Mori, Y.; Iwamoto, K.; Umesono, K.; Akanuma, Y.; Fujiwara, T.; Horikoshi, H.; et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J. Clin. Investig. 1998, 101, 1354–1361. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef]
- Zanchi, A.; Maillard, M.; Jornayvaz, F.R.; Vinciguerra, M.; Deleaval, P.; Nussberger, J.; Burnier, M.; Pechere-Bertschi, A. Effects of the peroxisome proliferator-activated receptor (PPAR)-gamma agonist pioglitazone on renal and hormonal responses to salt in diabetic and hypertensive individuals. Diabetologia 2010, 53, 1568–1575. [Google Scholar] [CrossRef]
- Allen, T.; Zhang, F.; Moodie, S.A.; Clemens, L.E.; Smith, A.; Gregoire, F.; Bell, A.; Muscat, G.E.; Gustafson, T.A. Halofenate is a selective peroxisome proliferator-activated receptor gamma modulator with antidiabetic activity. Diabetes 2006, 55, 2523–2533. [Google Scholar] [CrossRef]
- Zhang, F.; Lavan, B.E.; Gregoire, F.M. Selective modulators of PPAR-gamma activity: Molecular Aspects related to obesity and side-effects. PPAR Res. 2007, 2007, 32696. [Google Scholar]
- Higgins, L.S.; Depaoli, A.M. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am. J. Clin. Nutr. 2010, 91, 267S–272S. [Google Scholar] [CrossRef]
- Lin, S.; Han, Y.; Shi, Y.; Rong, H.; Zheng, S.; Jin, S.; Lin, S.Y.; Lin, S.C.; Li, Y. Revealing a steroid receptor ligand as a unique PPARgamma agonist. Cell Res. 2012, 22, 746–756. [Google Scholar] [CrossRef]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Moore, D.D.; Huang, W. FXR: A metabolic regulator and cell protector. Cell Res. 2008, 18, 1087–1095. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Jonker, J.W.; Liddle, C.; Downes, M. FXR and PXR: Potential therapeutic targets in cholestasis. J. Steroid Biochem. Mol. Biol. 2012, 130, 147–158. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar]
- Han, S.; Li, T.; Ellis, E.; Strom, S.; Chiang, J.Y. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol. Endocrinol. 2010, 24, 1151–1164. [Google Scholar] [CrossRef]
- Wang, Q.; Blackford, J.A., Jr.; Song, L.N.; Huang, Y.; Cho, S.; Simons, S.S., Jr. Equilibrium interactions of corepressors and coactivators with agonist and antagonist complexes of glucocorticoid receptors. Mol. Endocrinol. 2004, 18, 1376–1395. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef]
- Zhang, Y.; Castellani, L.W.; Sinal, C.J.; Gonzalez, F.J.; Edwards, P.A. Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev. 2004, 18, 157–169. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar]
- Pellicciari, R.; Gioiello, A.; Costantino, G.; Sadeghpour, B.M.; Rizzo, G.; Meyer, U.; Parks, D.J.; Entrena-Guadix, A.; Fiorucci, S. Back door modulation of the farnesoid X receptor: Design, synthesis, and biological evaluation of a series of side chain modified chenodeoxycholic acid derivatives. J. Med. Chem. 2006, 49, 4208–4215. [Google Scholar] [CrossRef]
- Rochette-Egly, C.; Germain, P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e005. [Google Scholar]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef]
- Soprano, D.R.; Qin, P.; Soprano, K.J. Retinoic acid receptors and cancers. Annu. Rev. Nutr. 2004, 24, 201–221. [Google Scholar] [CrossRef]
- Hansen, L.A.; Sigman, C.C.; Andreola, F.; Ross, S.A.; Kelloff, G.J.; de Luca, L.M. Retinoids in chemoprevention and differentiation therapy. Carcinogenesis 2000, 21, 1271–1279. [Google Scholar] [CrossRef]
- Okuno, M.; Kojima, S.; Matsushima-Nishiwaki, R.; Tsurumi, H.; Muto, Y.; Friedman, S.L.; Moriwaki, H. Retinoids in cancer chemoprevention. Curr. Cancer Drug Targets 2004, 4, 285–298. [Google Scholar] [CrossRef]
- Moise, A.R.; Noy, N.; Palczewski, K.; Blaner, W.S. Delivery of retinoid-based therapies to target tissues. Biochemistry 2007, 46, 4449–4458. [Google Scholar] [CrossRef]
- Germain, P.; Chambon, P.; Eichele, G.; Evans, R.M.; Lazar, M.A.; Leid, M.; de Lera, A.R.; Lotan, R.; Mangelsdorf, D.J.; Gronemeyer, H. International Union of Pharmacology. LX. Retinoic acid receptors. Pharmacol. Rev. 2006, 58, 712–725. [Google Scholar] [CrossRef]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar]
- Samarut, E.; Rochette-Egly, C. Nuclear retinoic acid receptors: Conductors of the retinoic acid symphony during development. Mol. Cell. Endocrinol. 2012, 348, 348–360. [Google Scholar] [CrossRef]
- Gillespie, R.F.; Gudas, L.J. Retinoic acid receptor isotype specificity in F9 teratocarcinoma stem cells results from the differential recruitment of coregulators to retinoic response elements. J. Biol. Chem. 2007, 282, 33421–33434. [Google Scholar] [CrossRef]
- Mongan, N.P.; Gudas, L.J. Diverse actions of retinoid receptors in cancer prevention and treatment. Differ. Res. Biol. Divers. 2007, 75, 853–870. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Kliewer, S.A.; Kakizuka, A.; Umesono, K.; Evans, R.M. Retinoid receptors. Recent Prog. Horm. Res. 1993, 48, 99–121. [Google Scholar]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef]
- Moreau, A.; Vilarem, M.J.; Maurel, P.; Pascussi, J.M. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol. Pharm. 2008, 5, 35–41. [Google Scholar] [CrossRef]
- Mencarelli, A.; Migliorati, M.; Barbanti, M.; Cipriani, S.; Palladino, G.; Distrutti, E.; Renga, B.; Fiorucci, S. Pregnane-X-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem. Pharmacol. 2010, 80, 1700–1707. [Google Scholar] [CrossRef]
- Kakizaki, S.; Yamazaki, Y.; Takizawa, D.; Negishi, M. New insights on the xenobiotic-sensing nuclear receptors in liver diseases—CAR and PXR. Curr. Drug Metab. 2008, 9, 614–621. [Google Scholar] [CrossRef]
- Cheng, J.; Shah, Y.M.; Ma, X.; Pang, X.; Tanaka, T.; Kodama, T.; Krausz, K.W.; Gonzalez, F.J. Therapeutic role of rifaximin in inflammatory bowel disease: Clinical implication of human pregnane X receptor activation. J. Pharmacol. Exp. Ther. 2010, 335, 32–41. [Google Scholar] [CrossRef]
- Bertilsson, G.; Heidrich, J.; Svensson, K.; Asman, M.; Jendeberg, L.; Sydow-Backman, M.; Ohlsson, R.; Postlind, H.; Blomquist, P.; Berkenstam, A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc. Natl. Acad. Sci. USA 1998, 95, 12208–12213. [Google Scholar] [CrossRef]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef]
- Falkner, K.C.; Pinaire, J.A.; Xiao, G.H.; Geoghegan, T.E.; Prough, R.A. Regulation of the rat glutathione S-transferase A2 gene by glucocorticoids: Involvement of both the glucocorticoid and pregnane X receptors. Mol. Pharmacol. 2001, 60, 611–619. [Google Scholar]
- Rae, J.M.; Johnson, M.D.; Lippman, M.E.; Flockhart, D.A. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: Studies with cDNA and oligonucleotide expression arrays. J. Pharmacol. Exp. Ther. 2001, 299, 849–857. [Google Scholar]
- Sonoda, J.; Xie, W.; Rosenfeld, J.M.; Barwick, J.L.; Guzelian, P.S.; Evans, R.M. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR). Proc. Natl. Acad. Sci. USA 2002, 99, 13801–13806. [Google Scholar] [CrossRef]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; LaTour, A.; Liu, Y.; Klaassen, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef]
- Synold, T.W.; Dussault, I.; Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 2001, 7, 584–590. [Google Scholar] [CrossRef]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef]
- Watkins, R.E.; Wisely, G.B.; Moore, L.B.; Collins, J.L.; Lambert, M.H.; Williams, S.P.; Willson, T.M.; Kliewer, S.A.; Redinbo, M.R. The human nuclear xenobiotic receptor PXR: Structural determinants of directed promiscuity. Science 2001, 292, 2329–2333. [Google Scholar] [CrossRef]
- Xue, Y.; Moore, L.B.; Orans, J.; Peng, L.; Bencharit, S.; Kliewer, S.A.; Redinbo, M.R. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol. Endocrinol. 2007, 21, 1028–1038. [Google Scholar] [CrossRef]
- Ekins, S.; Chang, C.; Mani, S.; Krasowski, M.D.; Reschly, E.J.; Iyer, M.; Kholodovych, V.; Ai, N.; Welsh, W.J.; Sinz, M.; et al. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 2007, 72, 592–603. [Google Scholar] [CrossRef]
- Zhou, C.; Poulton, E.J.; Grun, F.; Bammler, T.K.; Blumberg, B.; Thummel, K.E.; Eaton, D.L. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol. Pharmacol. 2007, 71, 220–229. [Google Scholar]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, W.; Yuan, L.; Ma, W.; Li, X.; Lu, W.; Zhao, Y.; Chen, G. One-pot synthesis of glycopolymer-porphyrin conjugate as photosensitizer for targeted cancer imaging and photodynamic therapy. Macromol. Biosci. 2013. [Google Scholar] [CrossRef]
- Meanwell, N.A. Improving drug candidates by design: A focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef]
- Burke, T.J.; Loniello, K.R.; Beebe, J.A.; Ervin, K.M. Development and application of fluorescence polarization assays in drug discovery. Comb. Chem. High Throughput Screen. 2003, 6, 183–194. [Google Scholar] [CrossRef]
- Chen, T.; Xie, W.; Agler, M.; Banks, M. Coactivators in assay design for nuclear hormone receptor drug discovery. Assay Drug Dev. Technol. 2003, 1, 835–842. [Google Scholar] [CrossRef]
- Shukla, S.J.; Nguyen, D.T.; Macarthur, R.; Simeonov, A.; Frazee, W.J.; Hallis, T.M.; Marks, B.D.; Singh, U.; Eliason, H.C.; Printen, J.; et al. Identification of pregnane X receptor ligands using time-resolved fluorescence resonance energy transfer and quantitative high-throughput screening. Assay Drug Dev. Technol. 2009, 7, 143–169. [Google Scholar] [CrossRef]
- Cali, J.J.; Niles, A.; Valley, M.P.; O’Brien, M.A.; Riss, T.L.; Shultz, J. Bioluminescent assays for ADMET. Expert Opin. Drug Metab. Toxicol. 2008, 4, 103–120. [Google Scholar] [CrossRef]
- Ullman, E.F.; Kirakossian, H.; Switchenko, A.C.; Ishkanian, J.; Ericson, M.; Wartchow, C.A.; Pirio, M.; Pease, J.; Irvin, B.R.; Singh, S.; et al. Luminescent oxygen channeling assay (LOCI): Sensitive, broadly applicable homogeneous immunoassay method. Clin. Chem. 1996, 42, 1518–1526. [Google Scholar]
- Ullman, E.F.; Kirakossian, H.; Singh, S.; Wu, Z.P.; Irvin, B.R.; Pease, J.S.; Switchenko, A.C.; Irvine, J.D.; Dafforn, A.; Skold, C.N.; et al. Luminescent oxygen channeling immunoassay: Measurement of particle binding kinetics by chemiluminescence. Proc. Natl. Acad. Sci. USA 1994, 91, 5426–5430. [Google Scholar] [CrossRef]
- Dafforn, A.; Kirakossian, H.; Lao, K. Miniaturization of the luminescent oxygen channeling immunoassay (LOCI(TM)) for use in multiplex array formats and other biochips. Clin. Chem. 2000, 46, 1495–1497. [Google Scholar]
- Liu, Y.P.; de Keczer, S.; Alexander, S.; Pirio, M.; Davalian, D.; Kurn, N.; Ullman, E.F. Homogeneous, rapid luminescent oxygen channeling immunoassay (LOCI(TM)) for homocysteine. Clin. Chem. 2000, 46, 1506–1507. [Google Scholar]
- Mathis, G.; Socquet, F.; Viguier, M.; Darbouret, B. Homogeneous immunoassays using rare earth cryptates and time resolved fluorescence: Principles and specific advantages for tumor markers. Anticancer Res. 1997, 17, 3011–3014. [Google Scholar]
- Marchese, A.; George, S.R.; Kolakowski, L.F., Jr.; Lynch, K.R.; O’Dowd, B.F. Novel GPCRs and their endogenous ligands: Expanding the boundaries of physiology and pharmacology. Trends Pharmacol. Sci. 1999, 20, 370–375. [Google Scholar] [CrossRef]
- Gray, A.; Olsson, H.; Batty, I.H.; Priganica, L.; Peter Downes, C. Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 2003, 313, 234–245. [Google Scholar] [CrossRef]
- Guenat, S.; Rouleau, N.; Bielmann, C.; Bedard, J.; Maurer, F.; Allaman-Pillet, N.; Nicod, P.; Bielefeld-Sevigny, M.; Beckmann, J.S.; Bonny, C.; et al. Homogeneous and nonradioactive high-throughput screening platform for the characterization of kinase inhibitors in cell lysates. J. Biomol. Screen. 2006, 11, 1015–1026. [Google Scholar] [CrossRef]
- Li, Y.; Choi, M.; Suino, K.; Kovach, A.; Daugherty, J.; Kliewer, S.A.; Xu, H.E. Structural and biochemical basis for selective repression of the orphan nuclear receptor liver receptor homolog 1 by small heterodimer partner. Proc. Natl. Acad. Sci. USA 2005, 102, 9505–9510. [Google Scholar] [CrossRef]
- Glickman, J.F.; Wu, X.; Mercuri, R.; Illy, C.; Bowen, B.R.; He, Y.; Sills, M. A comparison of ALPHAScreen, TR-FRET, and TRF as assay methods for FXR nuclear receptors. J. Biomol. Screen. 2002, 7, 3–10. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Z.; Lin, S.; Zheng, W.; Wang, R.; Jin, S.; Chen, J.; Jin, L.; Li, Y. Revealing a natural marine product as a novel agonist for retinoic acid receptors with a unique binding mode and inhibitory effects on cancer cells. Biochem. J. 2012, 446, 79–87. [Google Scholar] [CrossRef]
- Jin, L.; Martynowski, D.; Zheng, S.; Wada, T.; Xie, W.; Li, Y. Structural basis for hydroxycholesterols as natural ligands of orphan nuclear receptor RORgamma. Mol. Endocrinol. 2010, 24, 923–929. [Google Scholar] [CrossRef]
- Jin, L.; Lin, S.; Rong, H.; Zheng, S.; Jin, S.; Wang, R.; Li, Y. Structural basis for iloprost as a dual peroxisome proliferator-activated receptor alpha/delta agonist. J. Biol. Chem. 2011, 286, 31473–31479. [Google Scholar] [CrossRef]
- Mani, S.; Huang, H.; Sundarababu, S.; Liu, W.; Kalpana, G.; Smith, A.B.; Horwitz, S.B. Activation of the steroid and xenobiotic receptor (human pregnane X receptor) by nontaxane microtubule-stabilizing agents. Clin. Cancer Res. 2005, 11, 6359–6369. [Google Scholar] [CrossRef]
- Di Leva, F.S.; Festa, C.; D’Amore, C.; de Marino, S.; Renga, B.; D’Auria, M.V.; Novellino, E.; Limongelli, V.; Zampella, A.; Fiorucci, S. Binding mechanism of the farnesoid X receptor marine antagonist suvanine reveals a strategy to forestall drug modulation on nuclear receptors. Design, synthesis, and biological evaluation of novel ligands. J. Med. Chem. 2013, 56, 4701–4717. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, H.; Lu, Y.; Liu, S.; Zhang, Q.; Huang, J.; Zhu, R.; Yang, J.; Zhang, R.; Zhang, D.; et al. Identification of chemerin as a novel FXR target gene down-regulated in the progression of nonalcoholic steatohepatitis. Endocrinology 2013, 154, 1794–1801. [Google Scholar] [CrossRef]
- Chu, V.; Einolf, H.J.; Evers, R.; Kumar, G.; Moore, D.; Ripp, S.; Silva, J.; Sinha, V.; Sinz, M.; Skerjanec, A. In vitro and in vivo induction of cytochrome p450: A survey of the current practices and recommendations: A pharmaceutical research and manufacturers of america perspective. Drug Metab. Dispos. 2009, 37, 1339–1354. [Google Scholar] [CrossRef]
- Zwart, W.; Theodorou, V.; Kok, M.; Canisius, S.; Linn, S.; Carroll, J.S. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. EMBO J. 2011, 30, 4764–4776. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; D’Amore, C.; Cipriani, S.; D’Auria, M.V.; Sepe, V.; Chini, M.G.; Monti, M.C.; Bifulco, G.; Zampella, A.; et al. Discovery that theonellasterol a marine sponge sterol is a highly selective FXR antagonist that protects against liver injury in cholestasis. PLoS One 2012, 7, e30443. [Google Scholar] [CrossRef]
- McEwan, I.J. What lies beneath: Natural products from marine organisms as nuclear receptor modulators. Biochem. J. 2012, 446, e1–e3. [Google Scholar] [CrossRef]
- Choi, H.; Hwang, H.; Chin, J.; Kim, E.; Lee, J.; Nam, S.J.; Lee, B.C.; Rho, B.J.; Kang, H. Tuberatolides, potent FXR antagonists from the Korean marine tunicate Botryllus tuberatus. J. Nat. Prod. 2011, 74, 90–94. [Google Scholar]
- Kim, S.N.; Choi, H.Y.; Lee, W.; Park, G.M.; Shin, W.S.; Kim, Y.K. Sargaquinoic acid and sargahydroquinoic acid from Sargassum yezoense stimulate adipocyte differentiation through PPARalpha/gamma activation in 3T3-L1 cells. FEBS Lett. 2008, 582, 3465–3472. [Google Scholar] [CrossRef]
- Kim, H.; Chin, J.; Choi, H.; Baek, K.; Lee, T.G.; Park, S.E.; Wang, W.; Hahn, D.; Yang, I.; Lee, J.; et al. Phosphoiodyns A and B, unique phosphorus-containing iodinated polyacetylenes from a Korean sponge Placospongia sp. Org. Lett. 2013, 15, 100–103. [Google Scholar] [CrossRef]
- Meimetis, L.G.; Williams, D.E.; Mawji, N.R.; Banuelos, C.A.; Lal, A.A.; Park, J.J.; Tien, A.H.; Fernandez, J.G.; de Voogd, N.J.; Sadar, M.D.; et al. Niphatenones, glycerol ethers from the sponge Niphates digitalis block androgen receptor transcriptional activity in prostate cancer cells: Structure elucidation, synthesis, and biological activity. J. Med. Chem. 2012, 55, 503–514. [Google Scholar] [CrossRef]
- Simmons, L.; Kaufmann, K.; Garcia, R.; Schwar, G.; Huch, V.; Muller, R. Bendigoles D–F, bioactive sterols from the marine sponge-derived Actinomadura sp. SBMs009. Bioorg. Med. Chem. 2011, 19, 6570–6575. [Google Scholar] [CrossRef]
- Fidler, A.E.; Holland, P.T.; Reschly, E.J.; Ekins, S.; Krasowski, M.D. Activation of a tunicate (Ciona intestinalis) xenobiotic receptor orthologue by both natural toxins and synthetic toxicants. Toxicon 2012, 59, 365–372. [Google Scholar]
- Pathirana, C.; Stein, R.B.; Berger, T.S.; Fenical, W.; Ianiro, T.; Mais, D.E.; Torres, A.; Goldman, M.E. Nonsteroidal human progesterone receptor modulators from the marine alga Cymopolia barbata. Mol. Pharmacol. 1995, 47, 630–635. [Google Scholar]
- De Marino, S.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; D’Amore, C.; Renga, B.; Mencarelli, A.; Petek, S.; Fiorucci, S.; et al. 4-Methylenesterols from Theonella swinhoei sponge are natural pregnane-X-receptor agonists and farnesoid-X-receptor antagonists that modulate innate immunity. Steroids 2012, 77, 484–495. [Google Scholar] [CrossRef]
- Kaya, K.; Shiraishi, F.; Uchida, H.; Sano, T. A novel retinoic acid analogue, 7-hydroxy retinoic acid, isolated from cyanobacteria. Biochim. Biophys. Acta 2011, 1810, 414–419. [Google Scholar]
- Nam, S.J.; Ko, H.; Shin, M.; Ham, J.; Chin, J.; Kim, Y.; Kim, H.; Shin, K.; Choi, H.; Kang, H. Farnesoid X-activated receptor antagonists from a marine sponge Spongia sp. Bioorg. Med. Chem. Lett. 2006, 16, 5398–5402. [Google Scholar]
- De Marino, S.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; Renga, B.; D’Amore, C.; Fiorucci, S.; Debitus, C.; Zampella, A. Theonellasterols and conicasterols from Theonella swinhoei. Novel marine natural ligands for human nuclear receptors. J. Med. Chem. 2011, 54, 3065–3075. [Google Scholar] [CrossRef]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Chini, M.G.; Bifulco, G.; Renga, B.; D’Amore, C.; Debitus, C.; Fiorucci, S.; Zampella, A. Conicasterol E, a small heterodimer partner sparing farnesoid X receptor modulator endowed with a pregnane X receptor agonistic activity, from the marine sponge Theonella swinhoei. J. Med. Chem. 2012, 55, 84–93. [Google Scholar] [CrossRef]
- De Marino, S.; Sepe, V.; D’Auria, M.V.; Bifulco, G.; Renga, B.; Petek, S.; Fiorucci, S.; Zampella, A. Towards new ligands of nuclear receptors. Discovery of malaitasterol A, an unique bis-secosterol from marine sponge Theonella swinhoei. Org. Biomol. Chem. 2011, 9, 4856–4862. [Google Scholar] [CrossRef]
- Sepe, V.; Bifulco, G.; Renga, B.; D’Amore, C.; Fiorucci, S.; Zampella, A. Discovery of sulfated sterols from marine invertebrates as a new class of marine natural antagonists of farnesoid-X-receptor. J. Med. Chem. 2011, 54, 1314–1320. [Google Scholar] [CrossRef]
- Festa, C.; de Marino, S.; D’Auria, M.V.; Bifulco, G.; Renga, B.; Fiorucci, S.; Petek, S.; Zampella, A. Solomonsterols A and B from Theonella swinhoei. The first example of C-24 and C-23 sulfated sterols from a marine source endowed with a PXR agonistic activity. J. Med. Chem. 2011, 54, 401–405. [Google Scholar] [CrossRef]
- Li, J.L.; Xiao, B.; Park, M.; Yoo, E.S.; Shin, S.; Hong, J.; Chung, H.Y.; Kim, H.S.; Jung, J.H. PPAR-gamma agonistic metabolites from the ascidian Herdmania momus. J. Nat. Prod. 2012, 75, 2082–2087. [Google Scholar] [CrossRef]
- Festa, C.; Lauro, G.; de Marino, S.; D’Auria, M.V.; Monti, M.C.; Casapullo, A.; D’Amore, C.; Renga, B.; Mencarelli, A.; Petek, S.; et al. Plakilactones from the marine sponge Plakinastrella mamillaris. Discovery of a new class of marine ligands of peroxisome proliferator-activated receptor gamma. J. Med. Chem. 2012, 55, 8303–8317. [Google Scholar] [CrossRef]
- Mora, F.D.; Jones, D.K.; Desai, P.V.; Patny, A.; Avery, M.A.; Feller, D.R.; Smillie, T.; Zhou, Y.D.; Nagle, D.G. Bioassay for the identification of natural product-based activators of peroxisome proliferator-activated receptor-gamma (PPARgamma): The marine sponge metabolite psammaplin A activates PPARgamma and induces apoptosis in human breast tumor cells. J. Nat. Prod. 2006, 69, 547–552. [Google Scholar] [CrossRef]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Soest, R.V.; Andersen, R.J. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org. Lett. 2008, 10, 4947–4950. [Google Scholar] [CrossRef]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Taglialatela-Scafati, O.; Marino, S.D.; D’Amore, C.; Renga, B.; Chini, M.G.; Bifulco, G.; Nakao, Y.; et al. Preliminary structure-activity relationship on theonellasterol, a new chemotype of FXR antagonist, from the marine sponge Theonella swinhoei. Mar. Drugs 2012, 10, 2448–2466. [Google Scholar] [CrossRef]
- Shin, K.; Chin, J.; Hahn, D.; Lee, J.; Hwang, H.; Won, D.H.; Ham, J.; Choi, H.; Kang, E.; Kim, H.; et al. Sterols from a soft coral, Dendronephthya gigantea as farnesoid X-activated receptor antagonists. Steroids 2012, 77, 355–359. [Google Scholar] [CrossRef]
- Suzuki, T.; Nishimaki-Mogami, T.; Kawai, H.; Kobayashi, T.; Shinozaki, Y.; Sato, Y.; Hashimoto, T.; Asakawa, Y.; Inoue, K.; Ohno, Y.; et al. Screening of novel nuclear receptor agonists by a convenient reporter gene assay system using green fluorescent protein derivatives. Phytomedicine 2006, 13, 401–411. [Google Scholar] [CrossRef]
- D’Auria, M.V.; Sepe, V.; Zampella, A. Natural ligands for nuclear receptors: Biology and potential therapeutic applications. Curr. Top. Med. Chem. 2012, 12, 637–669. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Festa, C.; de Marino, S.; Sepe, V.; D’Auria, M.V.; Bifulco, G.; Debitus, C.; Bucci, M.; Vellecco, V.; Zampella, A. Solomonamides A and B, new anti-inflammatory peptides from Theonella swinhoei. Org. Lett. 2011, 13, 1532–1535. [Google Scholar] [CrossRef]
- De Marino, S.; Festa, C.; D’Auria, M.V.; Cresteil, T.; Debitus, C.; Zampella, A. Swinholide J, a potent cytotoxin from the marine sponge Theonella swinhoei. Mar. Drugs 2011, 9, 1133–1141. [Google Scholar] [CrossRef]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Mencarelli, A.; D’Amore, C.; Renga, B.; Zampella, A.; Fiorucci, S. Total synthesis and pharmacological characterization of solomonsterol A, a potent marine pregnane-X-receptor agonist endowed with anti-inflammatory activity. J. Med. Chem. 2011, 54, 4590–4599. [Google Scholar] [CrossRef]
- Renga, B.; Migliorati, M.; Mencarelli, A.; Cipriani, S.; D’Amore, C.; Distrutti, E.; Fiorucci, S. Farnesoid X receptor suppresses constitutive androstane receptor activity at the multidrug resistance protein-4 promoter. Biochim. Biophys. Acta 2011, 1809, 157–165. [Google Scholar] [CrossRef]
- Urizar, N.L.; Liverman, A.B.; Dodds, D.T.; Silva, F.V.; Ordentlich, P.; Yan, Y.; Gonzalez, F.J.; Heyman, R.A.; Mangelsdorf, D.J.; Moore, D.D. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science 2002, 296, 1703–1706. [Google Scholar] [CrossRef]
- Burris, T.P.; Montrose, C.; Houck, K.A.; Osborne, H.E.; Bocchinfuso, W.P.; Yaden, B.C.; Cheng, C.C.; Zink, R.W.; Barr, R.J.; Hepler, C.D.; et al. The hypolipidemic natural product guggulsterone is a promiscuous steroid receptor ligand. Mol. Pharmacol. 2005, 67, 948–954. [Google Scholar]
- Younk, L.M.; Uhl, L.; Davis, S.N. Pharmacokinetics, efficacy and safety of aleglitazar for the treatment of type 2 diabetes with high cardiovascular risk. Expert Opin. Drug Metab. Toxicol. 2011, 7, 753–763. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, C.; Li, Q.; Li, Y. Targeting Nuclear Receptors with Marine Natural Products. Mar. Drugs 2014, 12, 601-635. https://doi.org/10.3390/md12020601
Yang C, Li Q, Li Y. Targeting Nuclear Receptors with Marine Natural Products. Marine Drugs. 2014; 12(2):601-635. https://doi.org/10.3390/md12020601
Chicago/Turabian StyleYang, Chunyan, Qianrong Li, and Yong Li. 2014. "Targeting Nuclear Receptors with Marine Natural Products" Marine Drugs 12, no. 2: 601-635. https://doi.org/10.3390/md12020601
APA StyleYang, C., Li, Q., & Li, Y. (2014). Targeting Nuclear Receptors with Marine Natural Products. Marine Drugs, 12(2), 601-635. https://doi.org/10.3390/md12020601