Isolation and Structural Determination of Two Novel Phlorotannins from the Brown Alga Ecklonia kurome Okamura, and Their Radical Scavenging Activities

Abstract
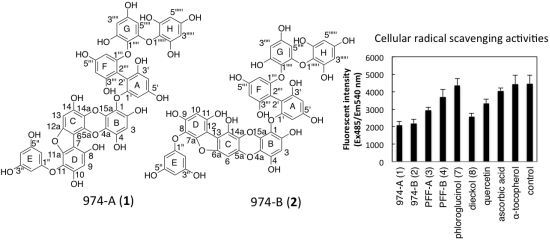
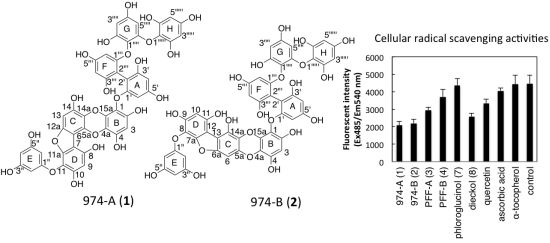
:
1. Introduction
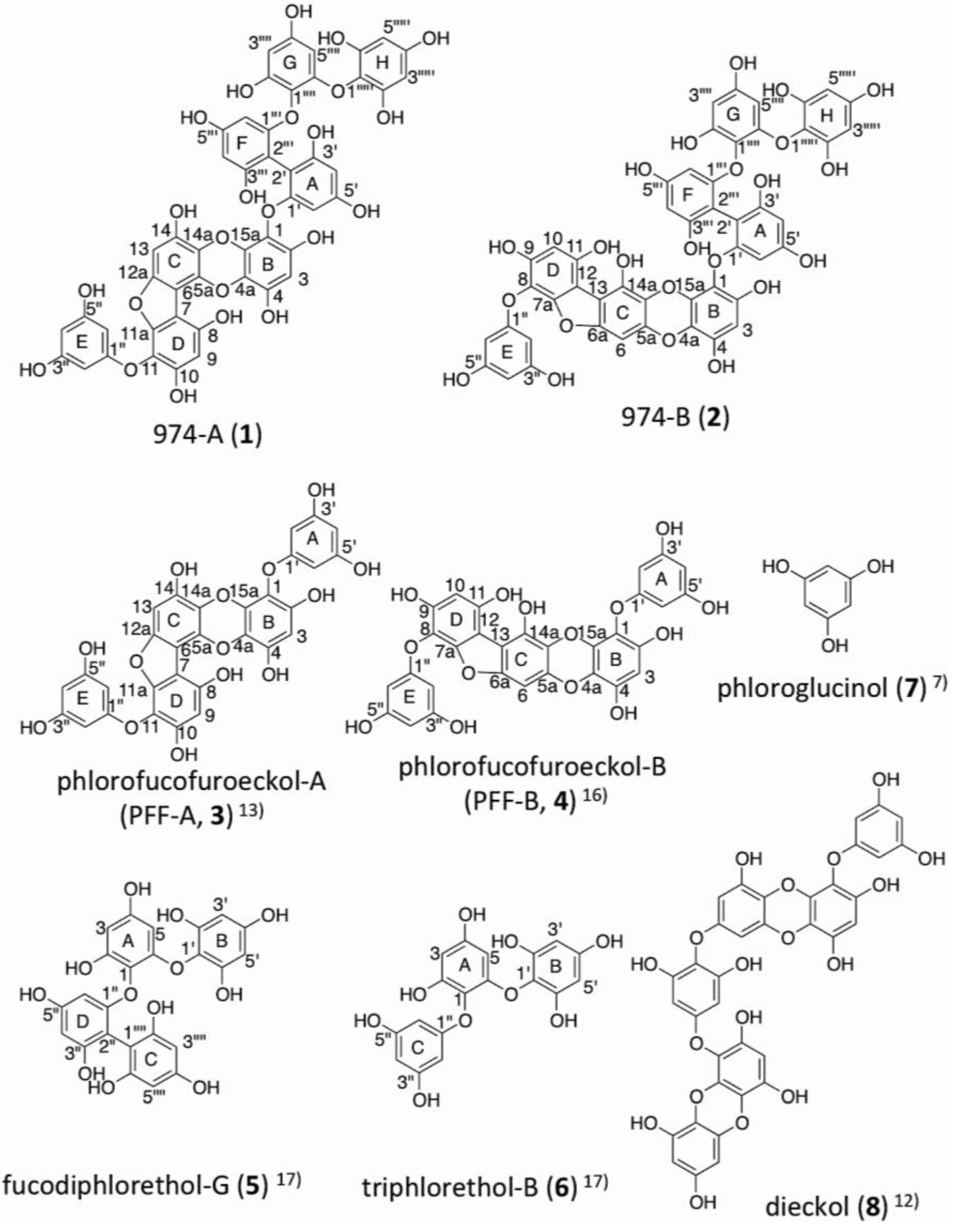
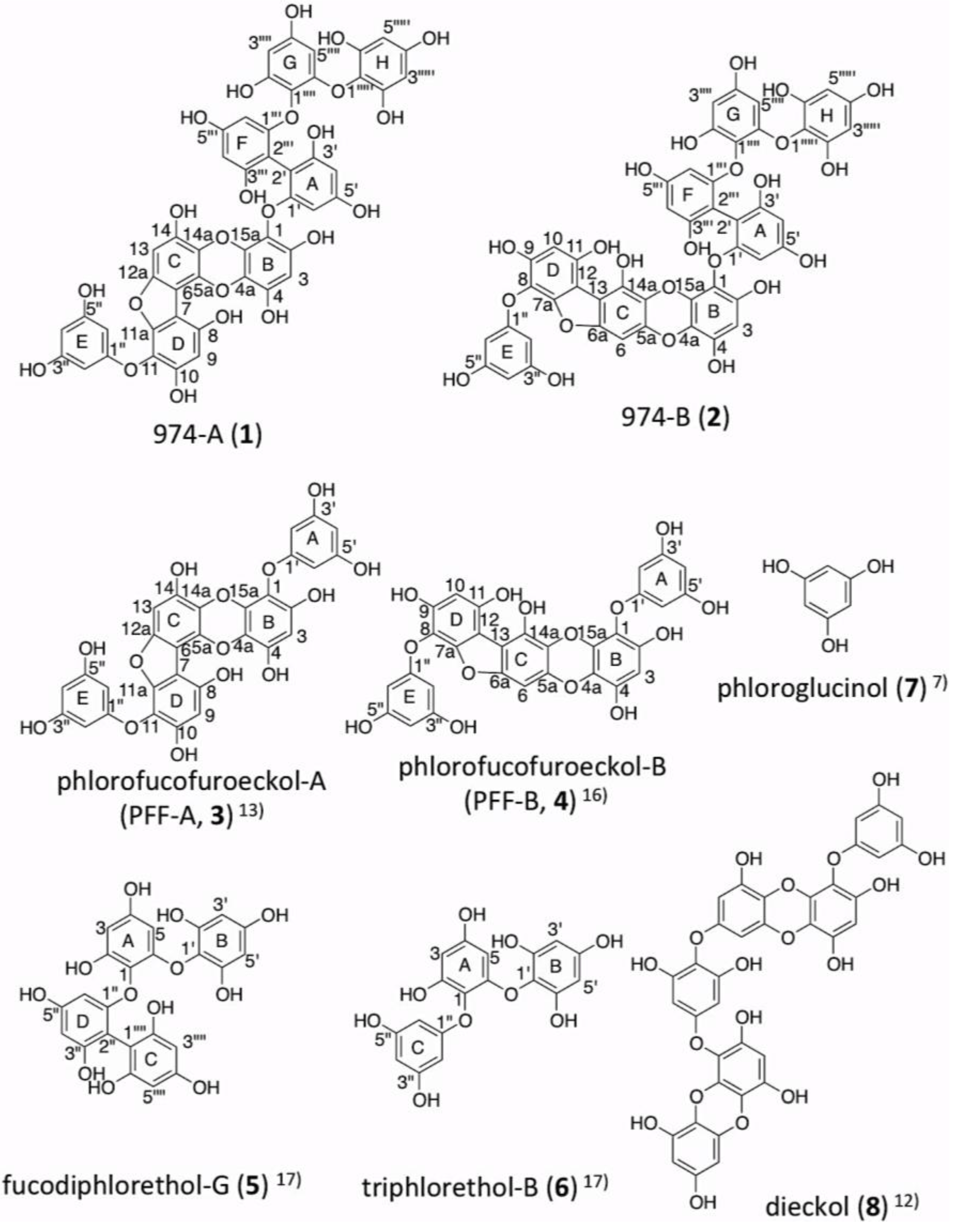
2. Results
2.1. Purification of 974-A (1) and 974-B (2), and Their Molecular Formulas
2.2. Determination of the Numbers of Hydroxyl Groups in 974-A (1) and 974-B (2)
2.3. Determination of the Structure of 974-A (1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 974-A (1) | PFF-A (3) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ring | position | δC | δH (mult, J) | HMBC b | δC | δH (mult, J) | ΔδC (1−3) | ||
| A | 1′ | 159.1 | 161.9 | −2.8 | |||||
| 2′ | 102.4 | 95.4 | 5.96 (d, 2.06) | 7.0 | |||||
| 3′ | 156.6 | 160.2 | −3.6 | ||||||
| 4′ | 98.4 | 6.18 (d, 2.05) | 2′,3′,5′,6′ | 97.6 | 5.91 (t, 2.06) | 0.8 | |||
| 5′ | 159.6 | 160.2 | −0.6 | ||||||
| 6′ | 95.2 | 6.12 (d, 2.05) | 1′,2′,4′,5′ | 95.4 | 5.96 (d, 2.06) | −0.2 | |||
| B | 1 | 124.2 | 124.7 | −0.5 | |||||
| 2 | 147.8 | 148.3 | −0.5 | ||||||
| 3 | 99.3 | 6.25 (s) | 1,2,4,4a,15a | 99.3 | 6.26 (s) | 0.0 | |||
| 4 | 144.3 | 143.9 | 0.4 | ||||||
| 4a | 124.9 | 125.0 | −0.1 | ||||||
| 15a | 138.2 | 138.4 | −0.2 | ||||||
| C | 5a | 135.2 | 135.3 | −0.1 | |||||
| 6 | 105.2 | 105.3 | −0.1 | ||||||
| 12a | 153.4 | 153.2 | 0.2 | ||||||
| 13 | 96.1 | 6.64 (s) | 5a,6,12a,14,14a | 96.2 | 6.63 (s) | −0.1 | |||
| 14 | 145.8 | 146.0 | −0.2 | ||||||
| 14a | 127.6 | 128.1 | −0.5 | ||||||
| D | 7 | 105.2 | 105.3 | −0.1 | |||||
| 8 | 148.2 | 148.2 | 0.0 | ||||||
| 9 | 99.9 | 6.40 (s) | 7,8,10,11 | 99.9 | 6.40 (s) | 0.0 | |||
| 10 | 151.8 | 151.7 | 0.1 | ||||||
| 11 | 122.3 | 122.3 | 0.0 | ||||||
| 11a | 151.2 | 151.2 | 0.0 | ||||||
| E | 1″ | 161.8 | 161.8 | 0.0 | |||||
| 2″,6″ | 95.3 | 5.87 (d, 2.05) | 1″,3″,4″(H2″) | 95.3 | 5.88 (d, 2.06) | 0.0 | |||
| 3″,5″ | 160.2 | 160.2 | 0.0 | ||||||
| 4″ | 97.6 | 5.91 (t, 2.05) | 2″,3″,5″,6″ | 97.7 | 5.94 (t, 2.06) | −0.1 | |||
| fucodiphlorethol G (5, part) [17] | |||||||||
| ring | position | δC | δH (mult, J) | ΔδC (1–5) | |||||
| F | 1′′′ | 159.7 | D | 1″ | 159.4 | 0.3 | |||
| 2′′′ | 102.6 | 2″ | 102.0 | 0.6 | |||||
| 3′′′ | 156.6 | 3″ | 159.2 | −2.6 | |||||
| 4′′′ | 98.7 | 6.20 (d, 2.35) | 2′′′,3′′′,5′′′,6′′′ | 4″ | 97.5 | 6.10 (d, 2.2) | 1.2 | ||
| 5′′′ | 159.7 | 5″ | 159.5 | 0.2 | |||||
| 6′′′ | 94.2 | 5.90 (d, 2.35) | 1′′′,2′′′,4′′′,5′′′ | 6″ | 94.3 | 6.03 (d, 2.2) | −0.1 | ||
| G | 1″″ | 124.9 | A | 1 | 124.9 | 0.0 | |||
| 2″″ | 151.9 | 2 | 152.0 | −0.1 | |||||
| 3″″ | 97.7 | 6.04 (d, 2.64) | 1″″,2″″,4″″,5″″ | 3 | 98.0 | 6.02 c (d, 2.7) | −0.3 | ||
| 4″″ | 156.6 | 4 | 157.5 | −0.9 | |||||
| 5″″ | 94.4 | 5.73 (d, 2.65) | 1″″,3″″,4″″,6″″ | 5 | 94.5 | 5.69 c (d, 2.7) | 0.1 | ||
| 6″″ | 153.9 | 6 | 153.7 | 0.2 | |||||
| H | 1′′′′′ | 124.2 | B | 1′ | 124.3 | −0.1 | |||
| 2′′′′′,6′′′′′ | 152.3 | 2′,6′ | 152.1 | 0.2 | |||||
| 3′′′′′,5′′′′′ | 96.4 | 5.93 (s) | 1′′′′′,2′′′′′,4′′′′′(H3′′′′′) | 3′,5′ | 96.0 | 5.91 (s) | 0.4 | ||
| 4′′′′′ | 156.5 | 4′ | 157.9 | −1.4 | |||||
| 974-B (2) | PFF-B (4) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ring | position | δC | δH (mult, J) | HMBC b | δC | δH (mult, J) | ΔδC (2–4) | ||
| A | 1′ | 159.0 | 161.8 | −2.9 | |||||
| 2′ | 102.5 | 95.1 | 5.97 (d,205) | 7.3 | |||||
| 3′ | 156.9 | 159.9 | −3.1 | ||||||
| 4′ | 98.4 | 6.20 (d,2.05) | 2′,3′,5′,6′ | 97.2 | 5.92 (t,2.05) | 1.1 | |||
| 5′ | 159.6 | 159.9 | −0.4 | ||||||
| 6′ | 95.2 | 6.15 (d,2.05) | 1′,2′,4′,5′ | 95.1 | 5.97 (d 2.05) | 0.0 | |||
| B | 1 | 124.0 | 124.3 | −0.4 | |||||
| 2 | 147.0 | 147.5 | −0.6 | ||||||
| 3 | 99.6 | 6.17 (s) | 1,2,4,4a,15a | 99.2 | 6.16 (s) | 0.3 | |||
| 4 | 143.9 | 143.5 | 0.3 | ||||||
| 4a | 125.3 | 125.2 | 0.0 | ||||||
| 15a | 138.0 | ND c | ND c | ||||||
| C | 5a | 142.9 | 143.0 | −0.2 | |||||
| 6 | 92.7 | 6.69 (s) | 5a,6a,13,14,14a | 92.0 | 6.66 (s) | 0.6 | |||
| 6a | 152.7 | 152.3 | 0.3 | ||||||
| 13 | 109.9 | 109.7 | 0.1 | ||||||
| 14 | 138.3 | 138.2 | 0.0 | ||||||
| 14a | 127.4 | 127.6 | −0.3 | ||||||
| D | 7a | 150.9 | 150.7 | 0.1 | |||||
| 8 | 122.1 | 122.2 | −0.2 | ||||||
| 9 | 151.6 | 151.3 | 0.2 | ||||||
| 10 | 99.5 | 6.38 (s) | 7a,8,9,11,12,13 | 98.9 | 6.37 (s) | 0.5 | |||
| 11 | 148.4 | 147.6 | 0.7 | ||||||
| 12 | 106.4 | 106.7 | −0.4 | ||||||
| E | 1″ | 161.9 | 161.7 | 0.1 | |||||
| 2″,6″ | 95.3 | 5.87 (d,2.06) | 1″,3″,4″(H2″) | 94.9 | 5.87 (d,2.03) | 0.3 | |||
| 3″,5″ | 160.2 | 160.0 | 0.1 | ||||||
| 4″ | 97.6 | 5.91 (t,2.05) | 2″,3″,5″,6″ | 97.2 | 5.94 (t,2.05) | 0.3 | |||
| fucodiphlorethol G (5, part) [17] | |||||||||
| ring | position | δC | δH (mult, J) | ΔδC (2–5) | |||||
| F | 1′′′ | 159.2 | D | 1″ | 159.4 | −0.2 | |||
| 2′′′ | 102.7 | 2″ | 102.0 | 0.7 | |||||
| 3′′′ | 157.5 | 3″ | 159.2 | −1.7 | |||||
| 4′′′ | 98.6 | 6.22 (d, 2.06) | 2′′′,3′′′,5′′′,6′′′ | 4″ | 97.5 | 6.10 (d, 2.2) | 1.1 | ||
| 5′′′ | 159.7 | 5″ | 159.5 | 0.2 | |||||
| 6′′′ | 94.2 | 5.89 (d, 2.05) | 1′′′,2′′′,4′′′,5′′′ | 6″ | 94.3 | 6.03 (d, 2.2) | −0.1 | ||
| G | 1″″ | 124.7 | A | 1 | 124.9 | −0.2 | |||
| 2″″ | 152.2 | 2 | 152.0 | 0.2 | |||||
| 3″″ | 97.6 | 6.05 (d, 2.64) | 1″″,2″″,4″″,5″″ | 3 | 98.0 | 6.02 d (d, 2.7) | −0.4 | ||
| 4″″ | 156.6 | 4 | 157.5 | 0.9 | |||||
| 5″″ | 94.2 | 5.75 (d, 2.64) | 1″″,3″″,4″″,6″″ | 5 | 94.5 | 5.69 d (d, 2.7) | −0.3 | ||
| 6″″ | 153.8 | 6 | 153.7 | 0.1 | |||||
| H | 1′′′′′ | 124.0 | B | 1′ | 124.3 | −0.3 | |||
| 2′′′′′,6′′′′′ | 152.2 | 2′,6′ | 152.1 | 0.1 | |||||
| 3′′′′′,5′′′′′ | 96.5 | 6.00 (s) | 1′′′′′,2′′′′′,4′′′′′(H3′′′′′) | 3′,5′ | 96.0 | 5.91 (s) | 0.5 | ||
| 4′′′′′ | 156.5 | 4′ | 157.9 | −1.4 | |||||
2.4. Determination of the Structure of 974-B (2)
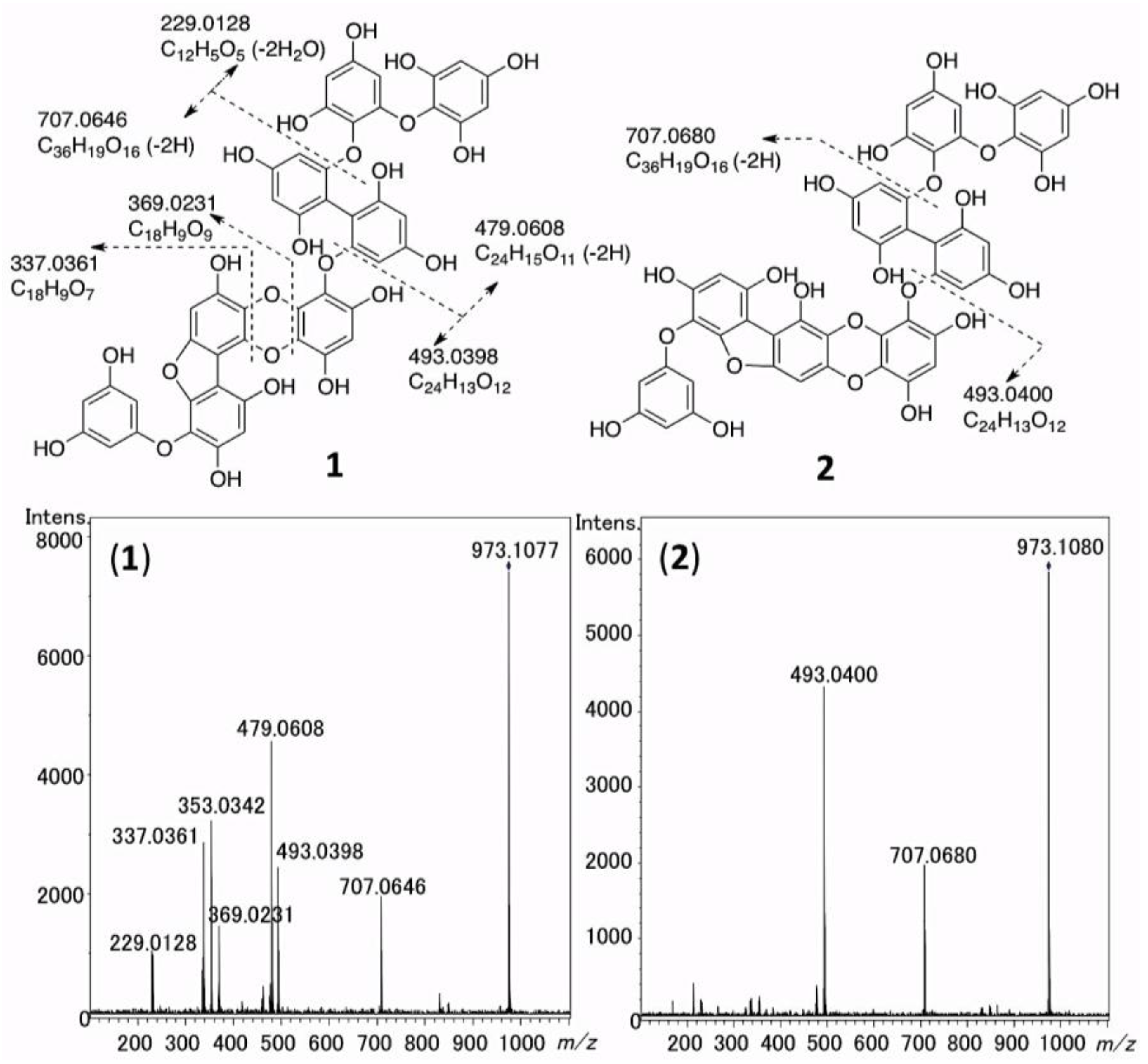
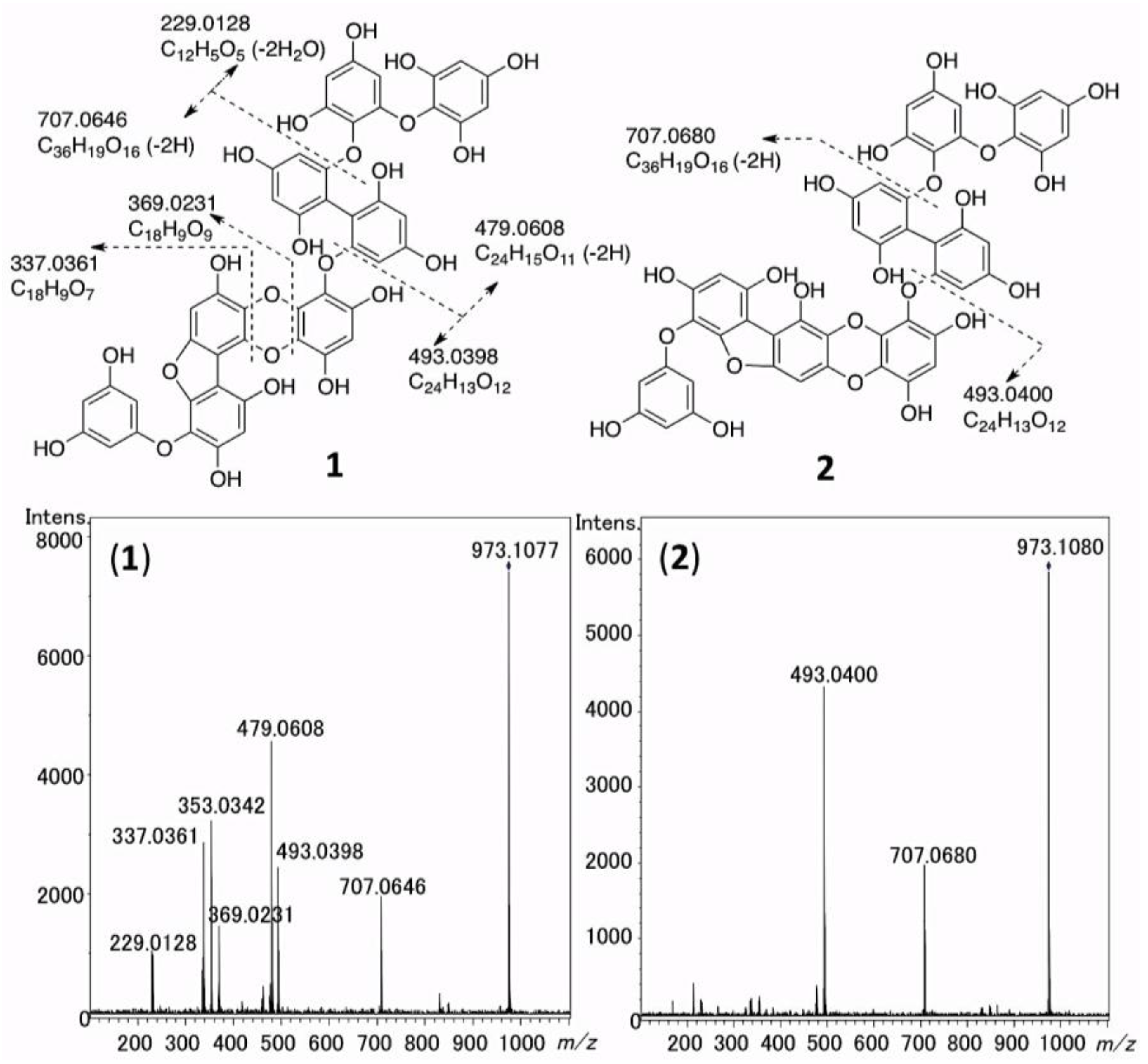
2.5. The MS/MS Fragmentation of 1 and 2
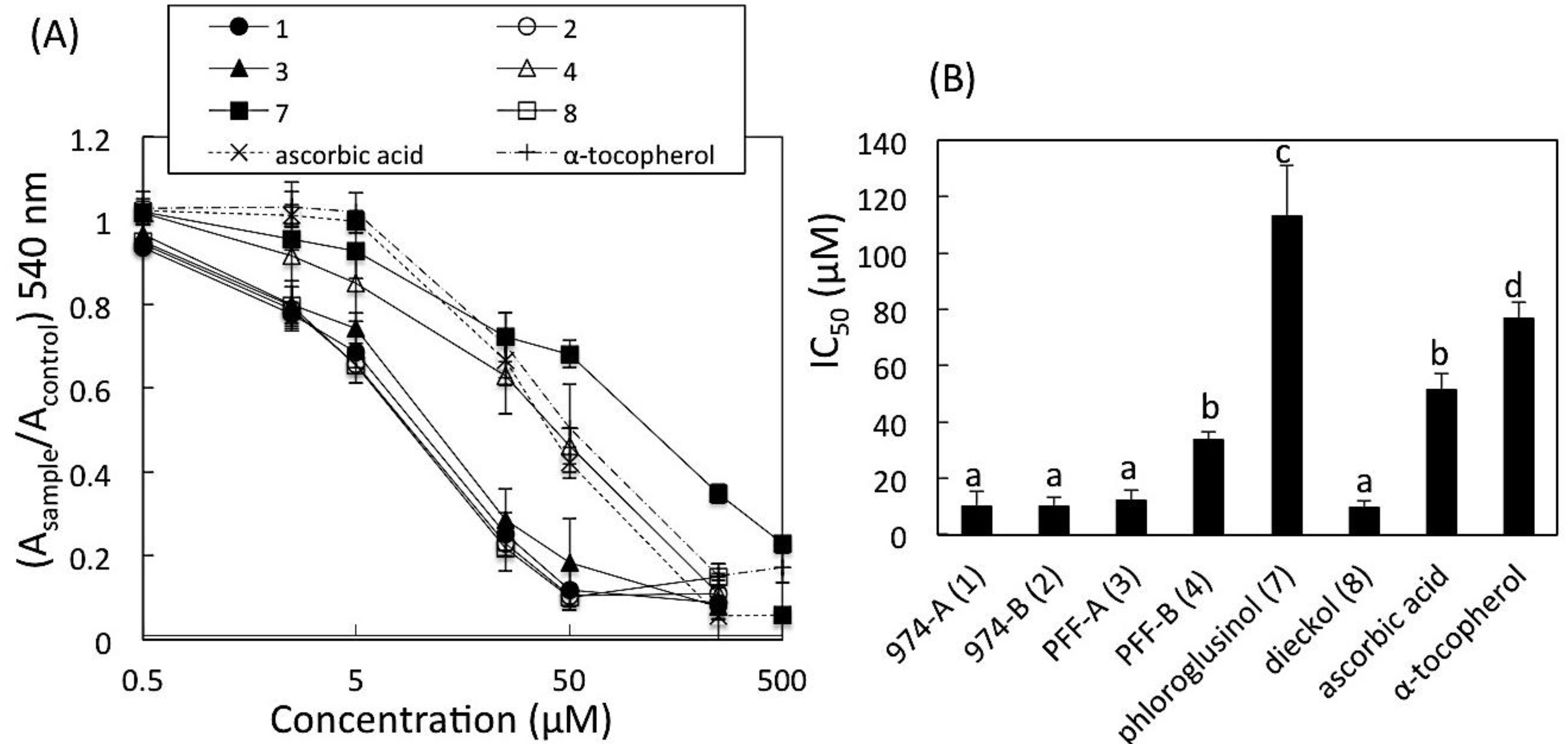
2.6. DPPH Radical Scavenging Activity

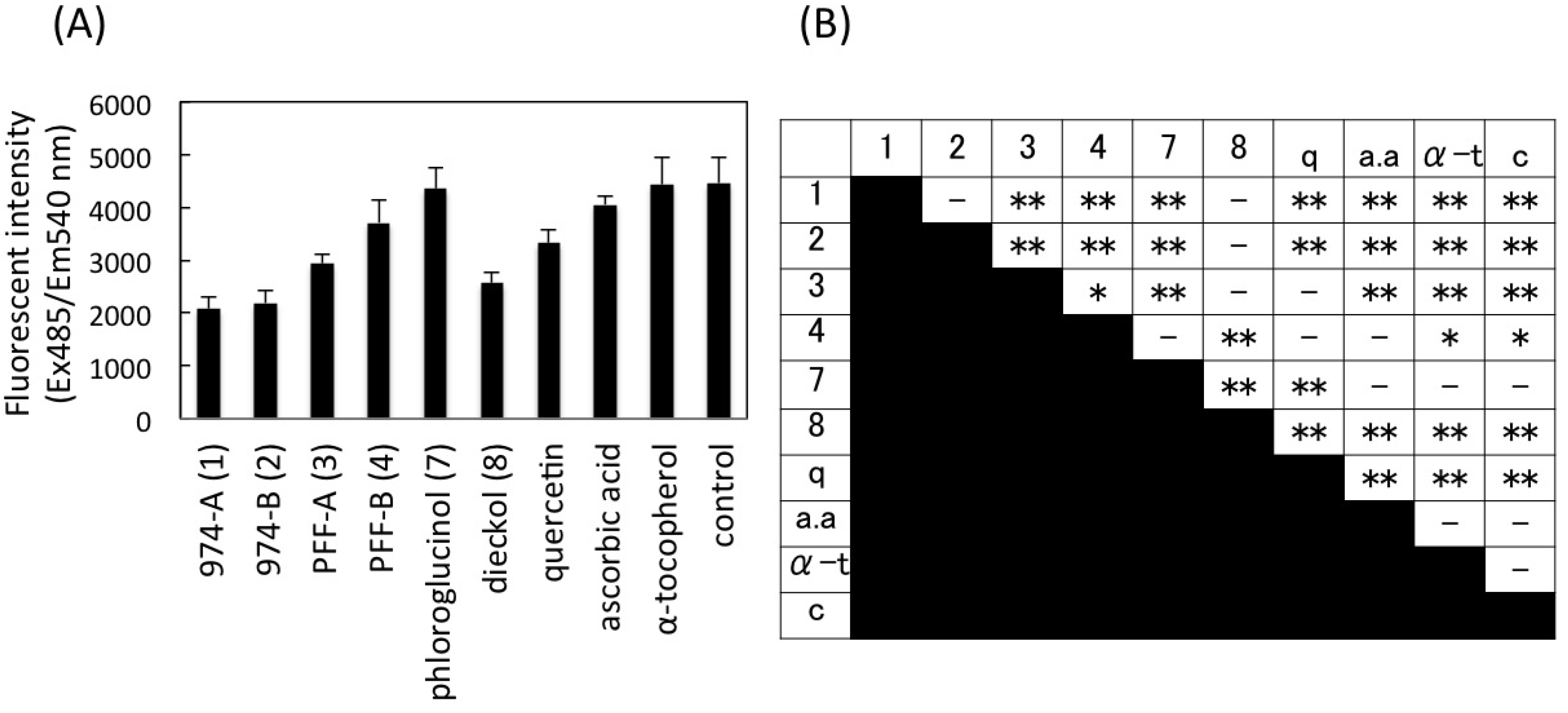
2.7. Intracellular Radical Scavenging Activity Measured Using DCFH-DA

2.8. Cell Viability
3. Discussion
4. Experimental Section
4.1. General Materials
4.2. Isolation of Phlorotannins
4.3. Acetylation of 1 and 2
4.4. HR-ESI-MS/MS Measurement
4.5. DPPH Radical Scavenging Activity
4.6. Cellular Radical Scavenging Activity Measured Using DCFH-DA
4.7. Cell Viability
5. Conclusions
Acknowledgments
Supplementary Files
References
- Singh, I.P.; Bharate, S.B. Phloroglucinol compounds of natural origin. Nat. Prod. Rep. 2006, 23, 558–591. [Google Scholar] [CrossRef]
- Lopes, G.; Sousa, C.; Silva, L.R.; Pinto, E.; Andrade, P.B.; Bernardo, J.; Mouga, T.; Valentao, P. Can phlorotannins purified extracts constitute a novel pharmacological alternative for microbial infections with associated inflammatory conditions? PLoS One 2012, 7, e31145. [Google Scholar]
- Kim, A.R.; Shin, T.S.; Lee, M.S.; Park, J.Y.; Park, K.E.; Yoon, N.Y.; Kim, J.S.; Choi, J.S.; Jang, B.C.; Byun, D.S.; et al. Isolation and identification of phlorotannins from Ecklonia stolonifera with antioxidant and anti-inflammatory properties. J. Agric. Food. Chem. 2009, 57, 3483–3489. [Google Scholar]
- Nagayama, K.; Iwamura, Y.; Shibata, T.; Hirayama, I.; Nakamura, T. Bactericidal activity of phlorotannins from the brown alga Ecklonia kurome. J. Antimicrob. Chemother. 2002, 50, 889–893. [Google Scholar] [CrossRef]
- Li, Y.; Lee, S.H.; Le, Q.T.; Kim, M.M.; Kim, S.K. Anti-allergic effects of phlorotannins on histamine release via binding inhibition between IgE and FcεRI. J. Agric. Food Chem. 2008, 56, 12073–12080. [Google Scholar] [CrossRef]
- Li, Y.; Qian, A.J.; Ryu, B.; Lee, S.H.; Kim, M.M.; Kim, S.K. Chemical components and its antioxidant properties in vitro: An edible marine brown alga, Ecklonia cava. Bioorg. Med. Chem. 2009, 17, 1963–1974. [Google Scholar] [CrossRef]
- Kwon, Y.H.; Jung, S.Y.; Kim, J.W.; Lee, S.H.; Lee, J.H.; Lee, B.Y.; Kwon, S.M. Phloroglucinol inhibits the bioactivities of endothelial progenitor cells and suppresses tumor angiogenesis in LLC-tumor-bearing mice. PLoS One 2012, 7, e33618. [Google Scholar]
- Wijesekara, I.; Kim, S.K. Angiotensin-I-converting enzyme (ACE) inhibitors from marine resources: Prospects in the pharmaceutical industry. Mar. Drugs 2010, 8, 1080–1093. [Google Scholar] [CrossRef]
- Kang, S.M.; Heo, S.J.; Kim, K.N.; Lee, S.H.; Yang, H.M.; Kim, A.D.; Jeon, Y.J. Molecular docking studies of a phlorotannin, dieckol isolated from Ecklonia cava with tyrosinase inhibitory activity. Bioorg. Med. Chem. 2012, 20, 311–316. [Google Scholar] [CrossRef]
- Xu, H.L.; Kitajima, C.; Ito, H.; Miyazaki, T.; Baba, M.; Okuyama, T.; Okada, Y. Antidiabetic effect of polyphenols from brown alga Ecklonia kurome in genetically diabetic KK-A(y) mice. Pharm. Biol. 2012, 50, 393–400. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Miura, I.; Kinzyo, Z.; Mori, H.; Kido, M.; Nakayama, Y.; Takahashi, M.; Ochi, M. Eckols, novel phlorotannins with a dibenzo-p-dioxin skeleton possessing inhibitory effects on α2-macroglobulin from the brown alga Ecklonia kurome Okamura. Chem. Lett. 1985, 14, 739–742. [Google Scholar]
- Fukuyama, Y.; Kodama, M.; Miura, I.; Kinzyo, Z.; Mori, H.; Nakayama, Y.; Takahashi, M. Anti-plasmin inhibitor. V. Structures of novel dimeric eckols isolated from the brown alga Ecklonia kurome Okamura. Chem. Pharm. Bull. 1989, 37, 2438–2440. [Google Scholar] [CrossRef]
- Fukuyama, Y.; Kodama, M.; Miura, I.; Kinzyo, Z.; Mori, H.; Nakayama, Y.; Takahashi, M. Anti-plasmin inhibitor. VI. Structure of phlorofucofuroeckol A, a novel phlorotannin with both dibenzo-1,4-dioxin and dibenzofuran elements, from Ecklonia kurome Okamura. Chem. Pharm. Bull. 1990, 38, 133–135. [Google Scholar] [CrossRef]
- Kang, S.M.; Heo, S.J.; Kim, K.N.; Lee, S.H.; Jeon, Y.J. Isolation and identification of new compound, 2,7″-phloroglucinol-6,6′-bieckol from brown algae, Ecklonia cava and its antioxidant effect. J. Funct. Foods 2012, 4, 158–166. [Google Scholar] [CrossRef]
- Kang, S.M.; Lee, S.H.; Heo, S.J.; Kim, K.N.; Jeon, Y.J. Evaluation of antioxidant properties of a new compound, pyrogallol-phloroglucinol-6,6′-bieckol isolated from brown algae, Ecklonia cav. Nutr. Res. Pract. 2011, 5, 495–502. [Google Scholar] [CrossRef]
- Sugiura, Y.; Matsuda, K.; Yamada, Y.; Nishikawa, M.; Shioya, K.; Katsuzaki, H.; Imai, K.; Amano, H. Isolation of a new anti-allergic phlorotannin, phlorofucofuroeckol-B from an edible brown alga, Eisenia arbore. Biosci. Biotechnol. Boichem. 2006, 70, 2807–2811. [Google Scholar] [CrossRef]
- Ham, Y.M.; Bail, J.S.; Hyun, J.W.; Lee, N.H. Isolation of a new phlorotannin, fucodiphlorethol G, from a brown alga Ecklonia cava. Bull. Korean Chem. Soc. 2007, 28, 1595–1597. [Google Scholar] [CrossRef]
- Bass, D.A.; Parce, J.W.; Dechatelet, L.R.; Szejda, P.; Seeds, M.C.; Thomas, M. Flow cytometric studies of oxidative product formation by neutrophils: A graded response to membrane stimulation. J. Immunol. 1983, 130, 1910–1917. [Google Scholar]
- Estrella, J.L.; Kan-Sutton, C.; Gong, X.; Rajagopalan, M.; Lewis, D.E.; Hunter, R.L.; Eissa, N.T.; Jagannath, C. A novel in vitro human macrophage model to study the persistence of Mycobacterium tuberculosis using vitamin D3 and retinoic acid activated THP-1 macrophages. Front. Microbiol. 2011, 2, 67:1–67:16. [Google Scholar] [CrossRef]
- Girard-Lalancette, K.; Pichette, A.; Legault, J. Sensitive cell-based assay using DCFH oxidation for the determination of pro- and antioxidant properties of compounds and mixtures: Analysis of fruit and vegetable juices. Food Chem. 2009, 115, 720–726. [Google Scholar]
- Ishiyama, M.; Miyazono, Y.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A highly water-soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta 1997, 44, 1299–1305. [Google Scholar] [CrossRef]
- Gülcin, I. Antioxidant activity of food constituents: An overview. Arch. Toxicol. 2012, 86, 345–391. [Google Scholar] [CrossRef]
- Shibata, T.; Ishimaru, K.; Kawaguchi, S.; Yoshikawa, H.; Hama, Y. Antioxidant activities of phlorotannins isolated from Japanese Laminariaceae. J. Appl. Phycol. 2008, 20, 705–711. [Google Scholar] [CrossRef]
- Lee, M.S.; Shin, T.; Utsuki, T.; Choi, J.S.; Byun, D.S.; Kim, H.R. Isolation and identification of phlorotannins from Ecklonia stolonifera with antioxidant and hepatoprotective properties in tacrine-treated HepG2 cells. J. Agric. Food Chem. 2012, 60, 5340–5349. [Google Scholar] [CrossRef]
- Teng, A.; Yuan, C.; Zhang, F.; Huan, M.; Cao, W.; Li, K.; Yang, J.; Cao, D.; Zhou, S.; Mei, Q. Intestinal absorption and first-pass metabolism of polyphenol compounds in rat and their transport dynamics in Caco-2 cells. PLoS One 2012, 7, e29647. [Google Scholar]
- Kang, H.S.; Chung, H.Y.; Jung, J.H.; Son, B.W.; Choi, J.S. A new phlorotannin from the brown alga Ecklonia stolonifera. Chem. Pharm. Bull. 2003, 51, 1012–1014. [Google Scholar] [CrossRef]
- Watanabe, R.; Suzuki, T.; Oshima, Y. Preparation of calibration standards of N1-H paralytic shellfish toxin analogues by large-scale culture of cyanobacterium Anabaena circinalis (TA04). Mar. Drugs 2011, 9, 466–477. [Google Scholar] [CrossRef]
- Nanjo, F.; Goto, K.; Seto, R.; Suzuki, M.; Sakai, M.; Hara, Y. Scavenging effects of tea catechins and their derivatives on 1,1-diphenyl-2-picrylhydrazyl radical. Free Radic. Biol. Med. 1996, 21, 895–902. [Google Scholar] [CrossRef]
- Kimura, T.; Yamagishi, K.; Suzuki, M.; Shinmoto, H. DPPH assay: Relative estimation of the radical scavenging activities of agricultural products (in Japanese). Nippon Shokuhin Kagaku Kogakkaishi 2002, 49, 257–266. [Google Scholar] [CrossRef]
- Zhou, L.; Shen, L.H.; Hu, L.H.; Ge, H.; Pu, J.; Chai, D.J.; Shao, Q.; Wang, L.; Zeng, J.Z.; He, B. Retinoid X receptor agonists inhibit phorbol-12-myristate-13-acetate (PMA)-induced differentiation of monocytic THP-1 cells into macrophages. Mol. Cell. Biochem. 2010, 335, 283–289. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yotsu-Yamashita, M.; Kondo, S.; Segawa, S.; Lin, Y.-C.; Toyohara, H.; Ito, H.; Konoki, K.; Cho, Y.; Uchida, T. Isolation and Structural Determination of Two Novel Phlorotannins from the Brown Alga Ecklonia kurome Okamura, and Their Radical Scavenging Activities. Mar. Drugs 2013, 11, 165-183. https://doi.org/10.3390/md11010165
Yotsu-Yamashita M, Kondo S, Segawa S, Lin Y-C, Toyohara H, Ito H, Konoki K, Cho Y, Uchida T. Isolation and Structural Determination of Two Novel Phlorotannins from the Brown Alga Ecklonia kurome Okamura, and Their Radical Scavenging Activities. Marine Drugs. 2013; 11(1):165-183. https://doi.org/10.3390/md11010165
Chicago/Turabian StyleYotsu-Yamashita, Mari, Sawako Kondo, Shinya Segawa, Yi-Chin Lin, Haruhiko Toyohara, Hisatomi Ito, Keiichi Konoki, Yuko Cho, and Takafumi Uchida. 2013. "Isolation and Structural Determination of Two Novel Phlorotannins from the Brown Alga Ecklonia kurome Okamura, and Their Radical Scavenging Activities" Marine Drugs 11, no. 1: 165-183. https://doi.org/10.3390/md11010165
APA StyleYotsu-Yamashita, M., Kondo, S., Segawa, S., Lin, Y.-C., Toyohara, H., Ito, H., Konoki, K., Cho, Y., & Uchida, T. (2013). Isolation and Structural Determination of Two Novel Phlorotannins from the Brown Alga Ecklonia kurome Okamura, and Their Radical Scavenging Activities. Marine Drugs, 11(1), 165-183. https://doi.org/10.3390/md11010165