Tetrodotoxin (TTX) as a Therapeutic Agent for Pain

 ,
,
Abstract
:
1. Introduction
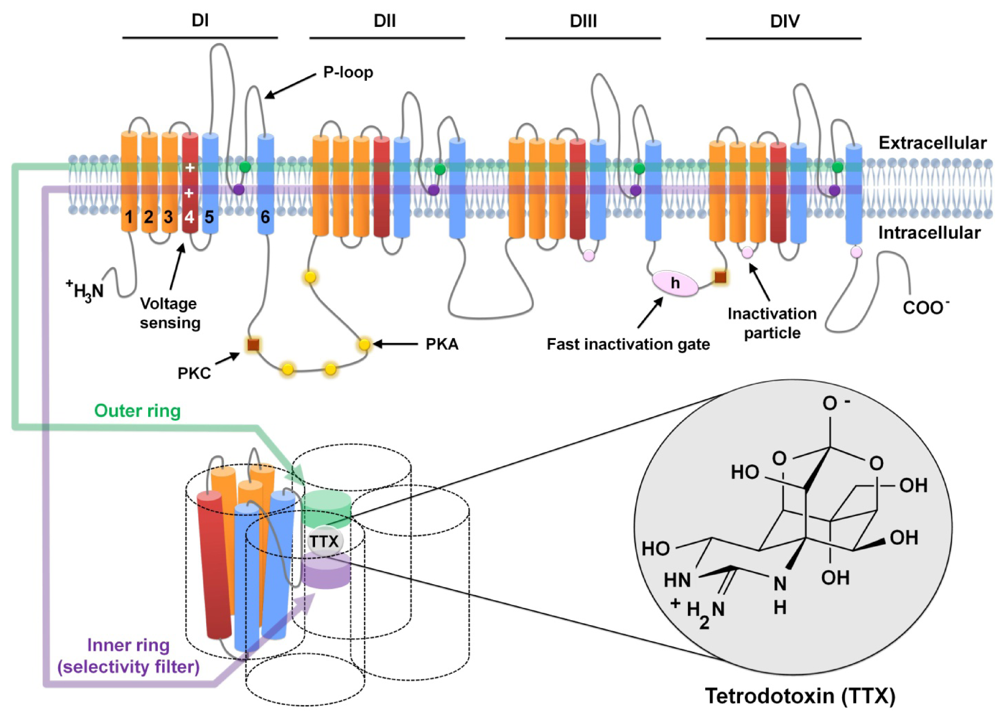
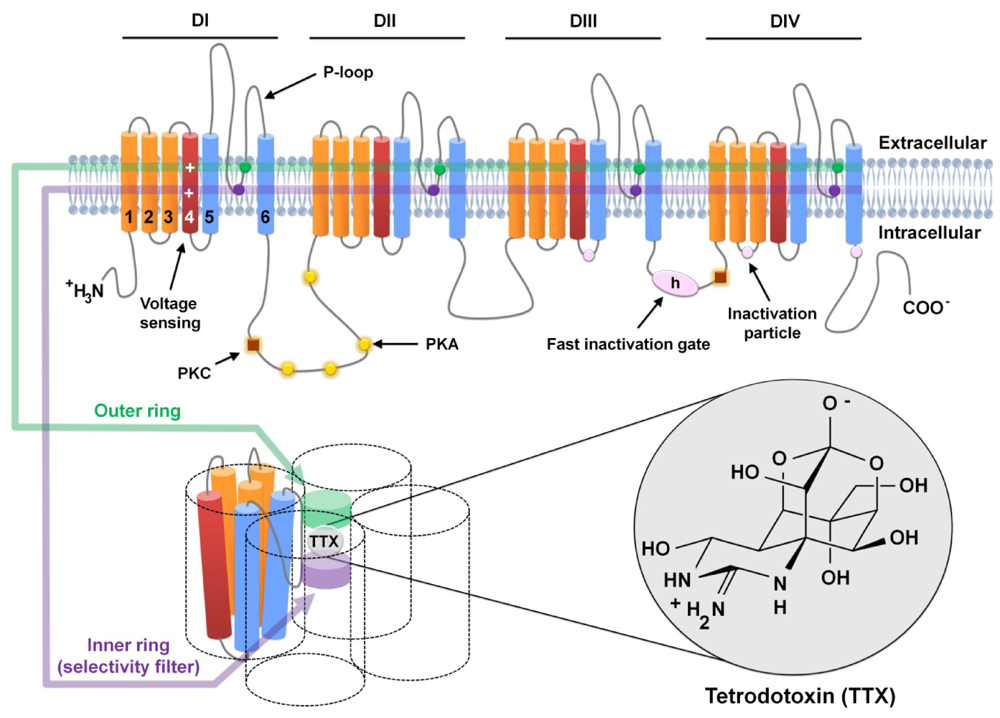
2. Voltaged-Gated Sodium Channels and TTX
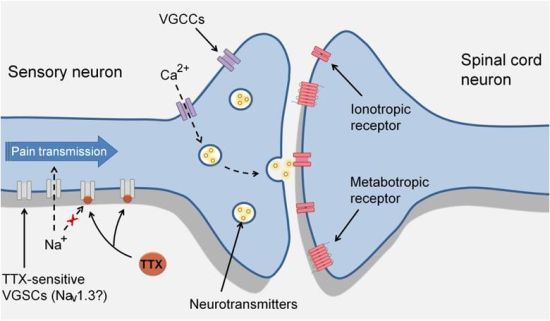
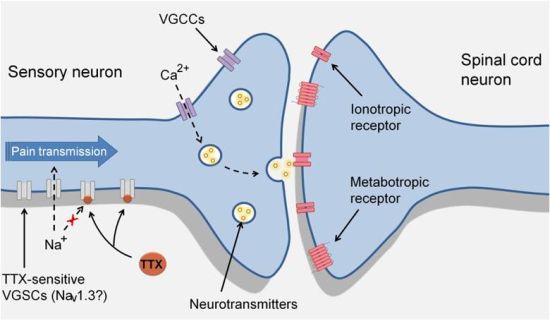
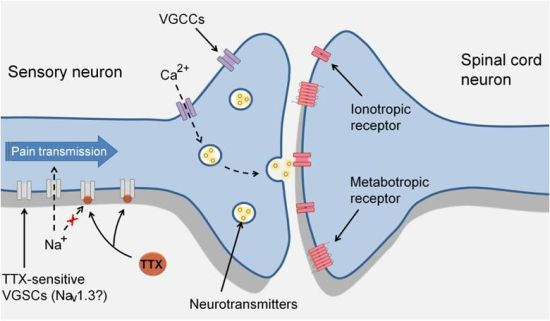
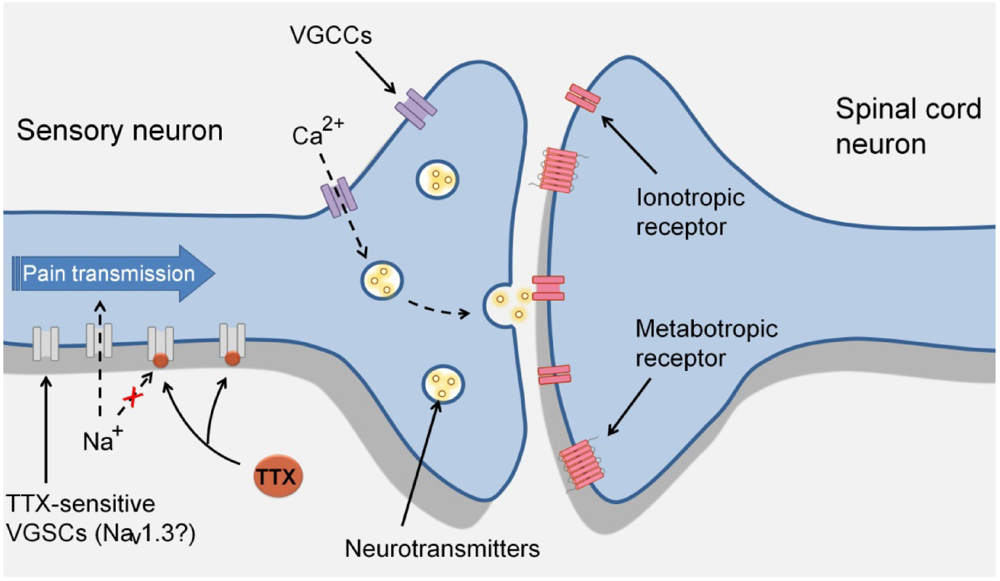
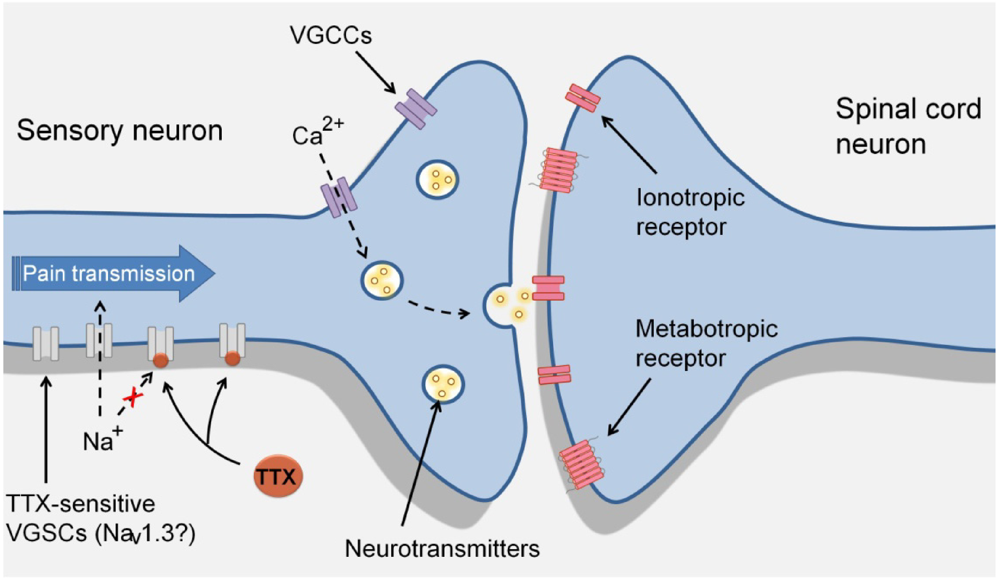
3. TTX-Sensitive Voltage-Gated Sodium Channels and Pain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Channel | Normal localization | Changes of expression in pain states | Knockdown/Knockout | Mutations related to pain states |
|---|---|---|---|---|
| Nav1.1 | -CNS, PNS (in DRG mainly in A-fiber neurons) -Microglia | Animal -Unclear after PNI in NP | ― | -Inherited hemiplegic migraine |
| Nav1.2 | -Mainly CNS, very low expression in PNS -In SC in lamina I/II | Animal -Unclear after PNI in NP | ― | ― |
| Nav1.3 | -Embryonic sodium channel -In adult neurons, in lamina I/II of SC, negligible in DRG | Human -↑ in human neuromas -↑ in human nerves after PNI -↑ in human trigeminal neuralgia Animal -↑ in DRG in inflammatory pain -↑ in DRG after PNI in NP -↑ in trigeminal ganglion after PNI in NP -↑ in SC dorsal horn after PNI in NP -↑ in rat neuromas -↓ in ferret trigeminal neuralgia | -Contradictory data with i.t. antisense ODNs -Knockout mice developed normally acute, inflammatory and neuropathic pain | ― |
| Nav1.4 | - In skeletal muscle | ― | ― | ― |
| Nav1.6 | -Mainly in Nodes of Ranvier -In SC and PNS (in DRG mainly in A-fiber neurons) -In epidermal free nerve terminals and keratinocytes -Main sodium channel in microglia | Human -↑ in skin of patients with complex regional pain syndrome and post-herpetic neuralgia Animal -Unclear in diabetic neuropathy -↑ in nerve after infraorbital nerve injury -↓ in DRG after PNI in NP | ― | ― |
| Nav1.7 | -Mainly in PNS in all types of DRG neurons -In SC and PNS (in DRG, mainly in A-fiber neurons) -In epidermal free nerve terminals | Human -↑ in human neuromas -↑ in skin of patients with complex regional pain syndrome and post-herpetic neuralgia -↑ painful human dental pulp -↑ in idiopathic rectal hypersensitivity and fecal urgency -↓ in human DRG after PNI -↓ in human trigeminal neuralgia Animal -↑ in DRG in inflammatory pain -↑ in rat neuromas -Unclear in diabetic neuropathy -↓ in DRG after PNI in NP -↓ in sciatic nerve after PNI in NP -↓ in ferret trigeminal neuralgia | -Knockdown of Nav1.7 ↓ inflammatory pain and Nav1.7 expression in primary afferents in mice -Knockout mice showed ↑ mechanical and thermal pain thresholds and ↓ inflammatory pain - Knockout mice developed normally neuropathic pain | -Inherited erythermalgia -Paroxysmal extreme pain disorder -Congenital insensitivity to pain |
3.1. Nav1.1
3.2. Nav1.2
3.3. Nav1.3
3.4. Nav1.6
3.5. Nav1.7
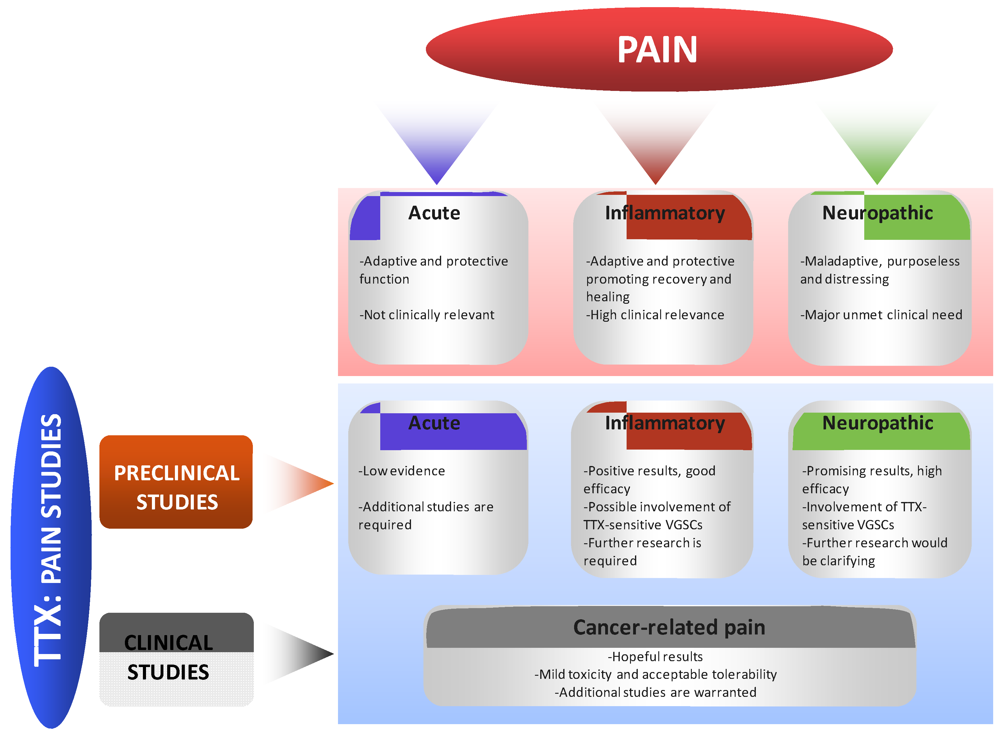
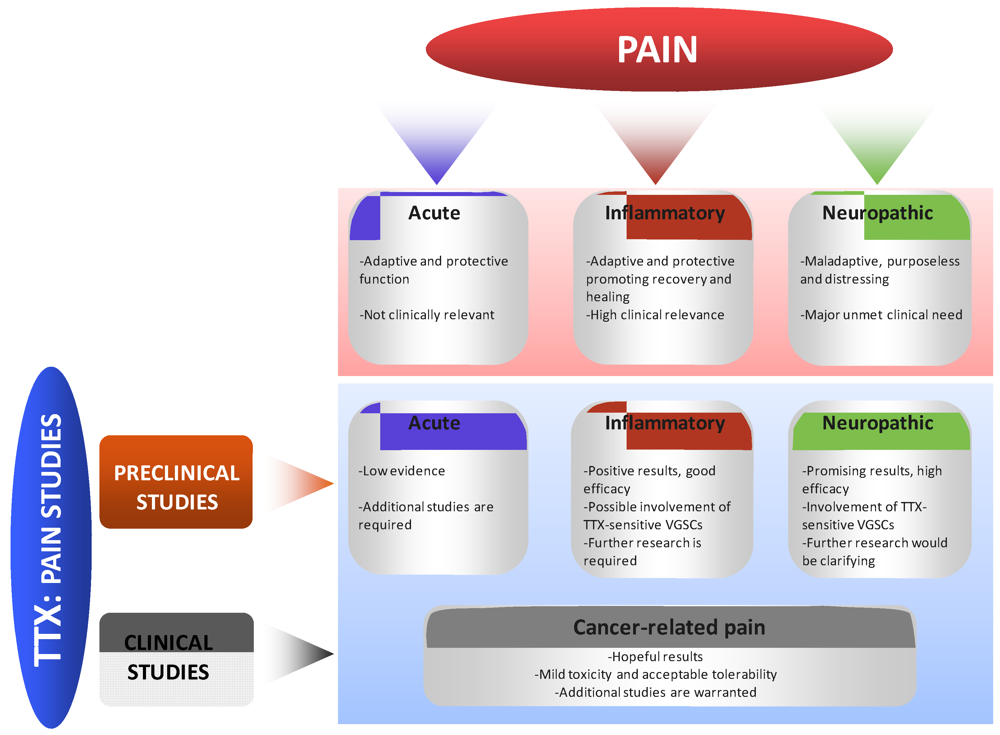
4. Effects of TTX in Pain States
4.1. Preclinical Studies
4.1.1. Effects of TTX in Acute Pain
4.1.2. Effects of TTX in Inflammatory Pain
4.1.3. Effects of TTX in Neuropathic Pain
4.1.4. Effects of TTX in the Electrophysiological Abnormalities Associated with Neuropathic Pain
| Type of pain | Administration of TTX | TTX doses | Effect (+, +/- or -) | Test | Reference |
|---|---|---|---|---|---|
| Acute pain | Sciatic nerve blockage | TTX osmotic pump | + | Thermal and mechanical sensitivity | [104] |
| Intraneural (10 mM/4 µL) | +/- | Pain induced by ET-1 | [105] | ||
| Intrathecal | 0.2–6 pM/20 µL | + (2–6 pM) | Thermal hypersensitivity | [111] | |
| Systemic | 0.3–6 µg s.c. | - | 1º phase of formalin test | [107] | |
| 0.3–6 µg s.c. | + (3–6 µg) | Writhing test | [107] | ||
| 1–6 µg s.c. | - | Mechanical, cold and heat sensitivity | [108] | ||
| 6 µg s.c. | - | Mechanical nociceptive pain | [109] | ||
| Acute and subchronic TTX (1–6 µg s.c.) | - | Thermal and mechanical sensitivity | [110] | ||
| Inflammatory pain | Sciatic nerve blockage | 50 µM/0.2 mL | + | Carrageenan-induced paw inflammatory edema and mechanical and thermal hyperalgesia. | [113] |
| Intrathecal | 0.2–6 pmM/20 µL | + (0.2–6 pM) | Thermal hypersensitivity induced by CFA | [111] | |
| Systemic | 0.3–6 µg s.c. | + (6 µg) | 2° phase of formalin test | [107] | |
| 50 µM/0.2 mL s.c. | - | Carrageenan-induced paw inflammatory edema and mechanical and thermal hyperalgesia. | [113] | ||
| 2.5 µg s.c. | + (2.5 µg) | Carrageenan-induced mechanical hyperalgesia | [114] | ||
| Neuropathic pain | Sciatic nerve blockage | TTX osmotic pump | + | Thermal and mechanical hypersensitivity and spontaneous activity induced by SNI and CCI | [104] |
| Topical DRG | 12.5–50 nM/5 µL | + (12.5–50 µg) | Mechanical allodynia induced by SNL | [116] | |
| Epidural | 25 nM/5 µL | + (25 µg) | Mechanical allodynia induced by SNL | [116] | |
| Topical median nerve | Gel pads with TTX | + | Mechanical hypersensitivity and the increment of astrocyte activation in the cuneate nucleus after CCI of median nerve | [117] | |
| Systemic | 25 nM/5 µL i.p. | - | Mechanical allodynia induced by SNL | [116] | |
| 0.3–6 µg s.c. | + (1–6 µg) | Mechanical allodynia and thermal hyperalgesia induced by SNL | [107] | ||
| Acute and subchronic TTX (1–6 µg s.c.) | + | Thermal and mechanical hypersensitivity and c-fos expression induced by CCI of sciatic nerve | [110] | ||
| Acute and subchronic TTX (1–6 µg s.c.) | +/- | Thermal and mechanical hypersensitivity induced by CCI of infraorbital nerve | [110] | ||
| 8 µg i.p | - | Mechanical allodynia induced by vincristine | [118] | ||
| 1–6 µg s.c. | + | Mechanical, cold and heat hypersensitivity induced by paclitaxel | [108] | ||
| 6 µg s.c. | + | Mechanical hypersensitivity induced by intraplantar capsaicin | [109] |
4.2. Clinical Studies
5. Concluding Remarks
Acknowledgments
References
- Woolf, C.J. What is this thing called pain? J. Clin. Invest. 2010, 120, 3742–3744. [Google Scholar] [CrossRef]
- Goldberg, D.S.; McGee, S.J. Pain as a global public health priority. BMC Public Health 2011, 11. [Google Scholar] [CrossRef]
- Narahashi, T. Tetrodotoxin: A brief history. Proc. Jpn. Acad. Ser. B 2008, 84, 147–154. [Google Scholar]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar]
- Yu, F.H.; Catterall, W.A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003, 4. [Google Scholar] [CrossRef]
- Chahine, M.; O’Leary, M.E. Regulatory role of voltage-gated Na channel β Subunits in sensory neurons. Front. Pharmacol. 2011, 2. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Ulbricht, W. Sodium channel inactivation: Molecular determinants and modulation. Physiol. Rev. 2005, 85, 1271–1301. [Google Scholar]
- Ragsdale, D.S., McPhee; Scheuer, T.; Catterall, W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA 1996, 93, 9270–9275. [Google Scholar]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar]
- Fozzard, H.A.; Lipkind, G.M. The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Mar. Drugs 2010, 8, 219–234. [Google Scholar]
- Lee, C.H.; Ruben, P.C. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2008, 2, 407–412. [Google Scholar]
- Geffeney, S.L.; Ruben, P.C. The structural basis and functional consequences of interactions between tetrodotoxin and voltage-gated sodium channels. Mar. Drugs 2006, 4, 143–156. [Google Scholar]
- Black, J.A.; Waxman, S.G. Sodium channel expression: A dynamic process in neurons and non-neuronal cells. Dev. Neurosci. 1996, 18, 139–152. [Google Scholar]
- Black, J.A.; Waxman, S.G. Sodium channels and microglial function. Exp. Neurol. 2011, (in press). [Google Scholar]
- Verkhratsky, A.; Steinhäuser, C. Ion channels in glial cells. Brain Res. Rev. 2000, 32, 380–412. [Google Scholar]
- Sontheimer, H.; Waxman, S.G. Ion channels in spinal cord astrocytes in vitro: II. Biophysical and pharmacological analysis of two Na+ current types. J. Neurophysiol. 1992, 68, 1001–1011. [Google Scholar]
- Sontheimer, H.; Fernandez-Marques, E.; Ullrich, N.; Pappas, C.; Waxman, S.G. Astrocyte Na+ channels are required for maintenance of Na+/K+-ATPase activity. J. Neurosci. 1994, 14, 2464–2475. [Google Scholar]
- Amin, A.S.; Asghari-Roodsari, A.; Tan, H.L. Cardiac sodium channelopathies. Pflugers Arch. 2010, 460, 223–237. [Google Scholar]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar]
- Shi, X.; Yasumoto, S.; Kurahashi, H.; Nakagawa, E.; Fukasawa, T.; Uchiya, S.; Hirose, S. Clinical spectrum of SCN2A mutations. Brain Dev. 2011, in press. [Google Scholar]
- Venance, S.L.; Cannon, S.C.; Fialho, D.; Fontaine, B.; Hanna, M.G.; Ptacek, L.J.; Tristani-Firouzi, M.; Tawil, R.; Griggs, R.C. CINCH investigators. The primary periodic paralyses: Diagnosis, pathogenesis and treatment. Brain 2006, 129, 8–17. [Google Scholar]
- Waxman, S.G. Transcriptional channelopathies: An emerging class of disorders. Nat. Rev. Neurosci. 2006, 2, 652–659. [Google Scholar]
- Liu, M.; Wood, J.N. The roles of sodium channels in nociception: Implications for mechanisms of neuropathic pain. Pain Med. 2011, 12, S93–S99. [Google Scholar]
- Bhattacharya, A.; Wickenden, A.D.; Chaplan, S.R. Sodium channel blockers for the treatment of neuropathic pain. Neurotherapeutics 2009, 6, 663–678. [Google Scholar]
- Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. Voltage-gated sodium channels: Therapeutic targets for pain. Pain Med. 2009, 10, 1260–1269. [Google Scholar]
- Waxman, S.G.; Kocsis, J.D.; Black, J.A. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J. Neurophysiol. 1994, 72, 466–470. [Google Scholar]
- Dib-Hajj, S.D.; Black, J.A.; Felts, P.; Waxman, S.G. Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc. Natl. Acad. Sci. USA 1996, 93, 14950–14954. [Google Scholar]
- Dib-Hajj, S.D.; Tyrrell, L.; Black, J.A.; Waxman, S.G. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. USA 1998, 95, 8963–8968. [Google Scholar]
- Dib-Hajj, S.D.; Fjell, J.; Cummins, T.R.; Zheng, Z.; Fried, K.; LaMotte, R.; Black, J.A.; Waxman, S.G. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 1999, 83, 591–600. [Google Scholar]
- Kim, C.H.; Oh, Y.; Chung, J.M.; Chung, K. The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res. Mol. Brain Res. 2001, 95, 153–161. [Google Scholar]
- Rizzo, M.A.; Kocsis, J.D.; Waxman, S.G. Selective loss of slow and enhancement of fast Na+ currents in cutaneous afferent dorsal root ganglion neurones following axotomy. Neurobiol. Dis. 1995, 2, 87–96. [Google Scholar]
- Cummins, T.R.; Waxman, S.G. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J. Neurosci. 1997, 17, 3503–3514. [Google Scholar]
- Black, J.A.; Cummins, T.R.; Plumpton, C.; Chen, Y.H.; Hormuzdiar, W.; Clare, J.J.; Waxman, S.G. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J. Neurophysiol. 1999, 82, 2776–2785. [Google Scholar]
- Rush, A.M.; Cummins, T.R.; Waxman, S.G. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 2007, 579, 1–14. [Google Scholar]
- Trimmer, J.S.; Cooperman, S.S.; Tomiko, S.A.; Zhou, J.; Crean, S.M.; Boyle, M.B.; Kalen, R.G.; Sheng, Z.; Barchi, R.L.; Sigworth, F.J.; et al. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron 1989, 3, 33–49. [Google Scholar] [CrossRef]
- Black, J.A.; Dib-Hajj, S.; McNabola, K.; Jeste, S.; Rizzo, M.A.; Kocsis, J.D.; Waxman, S.G. Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res. Mol. Brain Res. 1996, 43, 117–131. [Google Scholar]
- Fukuoka, T.; Kobayashi, K.; Yamanaka, H.; Obata, K.; Dai, Y.; Noguchi, K. Comparative study of the distribution of the alpha-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J. Comp. Neurol. 2008, 5, 188–206. [Google Scholar]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef]
- Dichgans, M.; Freilinger, T.; Eckstein, G.; Babini, E.; Lorenz-Depiereux, B.; Biskup, S.; Ferrari, M.D.; Herzog, J.; van den Maagdenberg, A.M.; Pusch, M.; Strom, T.M. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 2005, 366, 371–377. [Google Scholar]
- Black, J.A.; Nikolajsen, L.; Kroner, K.; Jensen, T.S.; Waxman, S.G. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 2008, 64, 644–653. [Google Scholar]
- Black, J.A.; Liu, S.; Tanaka, M.; Cummins, T.R.; Waxman, S.G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 2004, 108, 237–247. [Google Scholar]
- Zhao, P.; Waxman, S.G.; Hains, B.C. Sodium channel expression in the ventral posterolateral nucleus of the thalamus after peripheral nerve injury. Mol. Pain 2006, 2. [Google Scholar] [CrossRef]
- Berta, T.; Poirot, O.; Pertin, M.; Ji, R.R.; Kellenberger, S.; Decosterd, I. Transcriptional and functional profiles of voltage-gated Na(+) channels in injured and non-injured DRG neurons in the SNI model of neuropathic pain. Mol. Cell. Neurosci. 2008, 37, 196–208. [Google Scholar]
- Wang, W.; Atianjoh, F.; Gauda, E.B.; Yaster, M.; Li, Y.; Tao, Y.X. Increased expression of sodium channel subunit Nav1.1 in the injured dorsal root ganglion after peripheral nerve injury. Anat. Rec. (Hoboken) 2011, 294, 1406–1411. [Google Scholar] [CrossRef]
- Vacher, H.; Mohapatra, D.P.; Trimmer, J.S. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 2008, 88, 1407–1447. [Google Scholar]
- Hildebrand, M.E.; Mezeyova, J.; Smith, P.L.; Salter, M.W.; Tringham, E.; Snutch, T.P. Identification of sodium channel isoforms that mediate action potential firing in lamina I/II spinal cord neurons. Mol. Pain 2011, 7. [Google Scholar] [CrossRef]
- Lindia, J.A.; Abbadie, C. Distribution of the voltage gated sodium channel Na(v)1.3-like immunoreactivity in the adult rat central nervous system. Brain Res. 2003, 960, 132–141. [Google Scholar] [CrossRef]
- Fukuoka, T.; Kobayashi, K.; Noguchi, K. Laminae-specific distribution of alpha-subunits of voltage-gated sodium channels in the adult rat spinal cord. Neuroscience 2010, 169, 994–1006. [Google Scholar]
- Coward, K.; Aitken, A.; Powell, A.; Plumpton, C.; Birch, R.; Tate, S.; Bountra, C.; Anand, P. Plasticity of TTX-sensitive sodium channels PN1 and brain III in injured human nerves. Neuroreport 2001, 12, 495–500. [Google Scholar]
- Siqueira, S.R.; Alves, B.; Malpartida, H.M.; Teixeira, M.J.; Siqueira, J.T. Abnormal expression of voltage-gated sodium channels Nav1.7, Nav1.3 and Nav1.8 in trigeminal neuralgia. Neuroscience 2009, 164, 573–577. [Google Scholar] [CrossRef]
- Abe, M.; Kurihara, T.; Han, W.; Shinomiya, K; Tanabe, T. Changes in expression of voltage-dependent ion channel subunits in dorsal root ganglia of rats with radicular injury and pain. Spine 2002, 27, 1517–1524. [Google Scholar] [CrossRef]
- Craner, M.J.; Klein, J.P.; Renganathan, M.; Black, J.A.; Waxman, S.G. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann. Neurol. 2002, 52, 786–792. [Google Scholar]
- Wallace, V.C.; Cottrell, D.F.; Brophy, P.J.; Fleetwood-Walker, S.M. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J. Neurosci. 2003, 23, 3221–3233. [Google Scholar]
- Hong, S.; Morrow, T.J.; Paulson, P.E.; Isom, L.L.; Wiley, J.W. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 2004, 279, 29341–29350. [Google Scholar]
- Shah, B.S.; Rush, A.M.; Liu, S.; Tyrrell, L.; Black, J.A.; Dib-Hajj, S.D.; Waxman, S.G. Contactin associates with sodium channel Nav1.3 in native tissues and increases channel density at the cell surface. J. Neurosci. 2004, 24, 7387–7399. [Google Scholar]
- Garry, E.M.; Delaney, A.; Anderson, H.A.; Sirinathsinghji, E.C.; Clapp, R.H.; Martin, W.J.; Kinchington, P.R.; Krah, D.L.; Abbadie, C.; Fleetwood-Walker, S.M. Varicella zoster virus induces neuropathic changes in rat dorsal root ganglia and behavioral reflex sensitisation that is attenuated by gabapentin or sodium channel blocking drugs. Pain 2005, 118, 97–111. [Google Scholar]
- Lindia, J.A.; Köhler, M.G.; Martin, W.J.; Abbadie, C. Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain 2005, 117, 145–153. [Google Scholar] [CrossRef]
- He, X.H.; Zang, Y.; Chen, X.; Pang, R.P.; Xu, J.T.; Zhou, X.; Wei, X.H.; Li, Y.Y.; Xin, W.J.; Qin, Z.H.; Liu, X.G. TNF-α contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain 2010, 151, 266–279. [Google Scholar] [CrossRef]
- Zhang, Y.; Guzinski, M.; Eger, E.I., 2nd.; Laster, M.J.; Sharma, M.; Harris, R.A.; Hemmings, H.C., Jr. Bidirectional modulation of isoflurane potency by intrathecal tetrodotoxin and veratridine in rats. Br. J. Pharmacol. 2010, 159, 872–878. [Google Scholar] [CrossRef]
- Cheng, K.I.; Lai, C.S.; Wang, F.Y.; Wang, H.C.; Chang, L.L.; Ho, S.T.; Tsai, H.P.; Kwan, A.L. Intrathecal lidocaine pretreatment attenuates immediate neuropathic pain by modulating Nav1.3 expression and decreasing spinal microglial activation. BMC Neurol. 2011, 11. [Google Scholar] [CrossRef]
- Mo, G.; Grant, R.; O’Donnell, D.; Ragsdale, D.S.; Cao, C.Q.; Séguéla, P. Neuropathic Nav1.3-mediated sensitization to P2X activation is regulated by protein kinase C. Mol. Pain 2011, 7. [Google Scholar] [CrossRef]
- Fukuoka, T.; Yamanaka, H.; Kobayashi, K.; Okubo, M.; Miyoshi, K.; Dai, Y.; Noguchi, K. Re-evaluation of the phenotypic changes in L4 dorsal root ganglion neurons after L5 spinal nerve ligation. Pain 2012, 153, 68–79. [Google Scholar]
- Eriksson, J.; Jablonski, A.; Persson, A.K.; Hao, J.X.; Kouya, P.F.; Wiesenfeld-Hallin, Z.; Xu, X.J.; Fried, K. Behavioral changes and trigeminal ganglion sodium channel regulation in an orofacial neuropathicpain model. Pain 2005, 119, 82–94. [Google Scholar]
- Hains, B.C.; Klein, J.P.; Saab, C.Y.; Craner, M.J.; Black, J.A.; Waxman, S.G. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 2003, 23, 8881–8892. [Google Scholar]
- Hains, B.C.; Saab, C.Y.; Klein, J.P.; Craner, M.J.; Waxman, S.G. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J. Neurosci. 2004, 24, 4832–4839. [Google Scholar]
- Hains, B.C.; Saab, C.Y.; Waxman, S.G. Changes in electrophysiological properties and sodium channel Nav1.3 expression in thalamic neurons after spinal cord injury. Brain 2005, 128, 2359–2371. [Google Scholar] [CrossRef]
- Xiao, H.S.; Huang, Q.H.; Zhang, F.X.; Bao, L.; Lu, Y.J.; Guo, C.; Yang, L.; Huang, W.J.; Fu, G.; Xu, S.H.; et al. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc. Natl. Acad. Sci. USA 2002, 99, 8360–8365. [Google Scholar]
- Cummins, T.R.; Aglieco, F.; Renganathan, M.; Herzog, R.I.; Dib-Hajj, S.D.; Waxman, S.G. Nav1.3 sodium channels: Rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 2001, 21, 5952–5961. [Google Scholar]
- Lampert, A.; Hains, B.C.; Waxman, S.G. Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp. Brain Res. 2006, 174, 660–666. [Google Scholar]
- Huang, H.L.; Cendan, C.M.; Roza, C.; Okuse, K.; Cramer, R.; Timms, J.F.; Wood, J.N. Proteomic profiling of neuromas reveals alterations in protein composition and local protein synthesis in hyper-excitable nerves. Mol. Pain 2008, 4. [Google Scholar] [CrossRef]
- Thakor, D.K.; Lin, A.; Matsuka, Y.; Meyer, E.M.; Ruangsri, S.; Nishimura, I.; Spigelman, I. Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painfulneuropathy. Mol. Pain 2009, 5. [Google Scholar] [CrossRef]
- Ohno, K.; Yokota, A.; Hirofuji, S.; Kanbara, K.; Ohtsuka, H.; Kinoshita, M. Altered expression of sodium channel distribution in the dorsal root ganglion after gradualelongation of rat sciatic nerves. J. Orthop. Res. 2010, 28, 481–486. [Google Scholar]
- Davies, S.L.; Loescher, A.R.; Clayton, N.M.; Bountra, C.; Robinson, P.P.; Boissonade, F.M. Changes in sodium channel expression following trigeminal nerve injury. Exp. Neurol. 2006, 202, 207–216. [Google Scholar]
- Nassar, M.A.; Baker, M.D.; Levato, A.; Ingram, R.; Mallucci, G.; McMahon, S.B.; Wood, J.N. Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol. Pain 2006, 2. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Schaller, K.L.; Lasher, R.S.; Peles, E.; Levinson, S.R. Sodium channel Nav1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 5616–5620. [Google Scholar]
- Tzoumaka, E.; Tischler, A.C.; Sangameswaran, L.; Eglen, R.M.; Hunter, J.C.; Novakovic, S.D. Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. J. Neurosci. Res. 2000, 60, 37–44. [Google Scholar] [CrossRef]
- Black, J.A.; Renganathan, M.; Waxman, S.G. Sodium channel Nav1.6 is expressed along nonmyelinated axons and it contributes to conduction. Brain Res. Mol. Brain Res. 2002, 105, 19–28. [Google Scholar]
- Persson, A.K.; Black, J.A.; Gasser, A.; Cheng, X.; Fischer, T.Z.; Waxman, S.G. Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol. Pain 2010, 6. [Google Scholar] [CrossRef]
- Zhao, P.; Barr, T.P.; Hou, Q.; Dib-Hajj, S.D.; Black, J.A.; Albrecht, P.J.; Petersen, K.; Eisenberg, E.; Wymer, J.P.; Rice, F.L.; Waxman, S.G. Voltage-gated sodium channel expression in rat and human epidermal keratinocytes: Evidence for a role in pain. Pain 2008, 139, 90–105. [Google Scholar]
- Kim, C.H.; Oh, Y.; Chung, J.M.; Chung, K. Changes in three subtypes of tetrodotoxin sensitive sodium channel expression in the axotomized dorsal root ganglion in the rat. Neurosci. Lett. 2002, 323, 125–128. [Google Scholar]
- Raymond, C.K.; Castle, J.; Garrett-Engele, P.; Armour, C.D.; Kan, Z.; Tsinoremas, N.; Johnson, J.M. Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia. J. Biol. Chem. 2004, 279, 46234–46241. [Google Scholar]
- Persson, A.K.; Thun, J.; Xu, X.J.; Wiesenfeld-Hallin, Z.; Ström, M.; Devor, M.; Lidman, O.; Fried, K. Autotomy behavior correlates with the DRG and spinal expression of sodium channels in inbredmouse strains. Brain Res. 2009, 1285, 1–13. [Google Scholar]
- Henry, M.A.; Freking, A.R.; Johnson, L.R.; Levinson, S.R. Sodium channel Nav1.6 accumulates at the site of infraorbital nerve injury. BMC Neurosci. 2007, 8. [Google Scholar] [CrossRef]
- Sangameswaran, L.; Fish, L.M.; Koch, B.D.; Rabert, D.K.; Delgado, S.G.; Ilnicka, M.; Jakeman, L.B.; Novakovic, S.; Wong, K.; Sze, P.; et al. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 1997, 272, 14805–14809. [Google Scholar]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. From genes to pain: Nav 1.7 and human pain disorders. Trends Neurosci. 2007, 30, 555–563. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128, 1847–1854. [Google Scholar] [CrossRef]
- Lee, M.J.; Yu, H.S.; Hsieh, S.T.; Stephenson, D.A.; Lu, C.J.; Yang, C.C. Characterization of a familial case with primary erythromelalgia from Taiwan. J. Neurol. 2007, 254, 210–214. [Google Scholar]
- Nilsen, K.B.; Nicholas, A.K.; Woods, C.G.; Mellgren, S.I.; Nebuchennykh, M.; Aasly, J. Two novel SCN9A mutations causing insensitivity to pain. Pain 2009, 143, 155–158. [Google Scholar]
- Gould, H.J., 3rd.; England, J.D.; Soignier, R.D.; Nolan, P.; Minor, L.D.; Liu, Z.P.; Levinson, S.R.; Paul, D. Ibuprofen blocks changes in Na v 1.7 and 1.8 sodium channels associated with complete Freund's adjuvant-induced inflammation in rat. J. Pain 2004, 5, 270–280. [Google Scholar] [CrossRef]
- Strickland, I.T.; Martindale, J.C.; Woodhams, P.L.; Reeve, A.J.; Chessell, I.P.; McQueen, D.S. Changes in the expression of Nav1.7, Nav1.8 and Nav1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur. J. Pain 2008, 12, 564–572. [Google Scholar] [CrossRef]
- Gould, H.J., 3rd.; Gould, T.N.; England, J.D.; Paul, D.; Liu, Z.P.; Levinson, S.R. A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. 2000, 854, 19–29. [Google Scholar]
- Yeomans, D.C.; Levinson, S.R.; Peters, M.C.; Koszowski, A.G.; Tzabazis, A.Z.; Gilly, W.F.; Wilson, S.P. Decrease in inflammatory hyperalgesia by herpes vector-mediated knockdown of Nav1.7 sodium channels in primary afferents. Hum. Gene Ther. 2005, 16, 271–277. [Google Scholar] [CrossRef]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatorypain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar]
- Nassar, M.A.; Levato, A.; Stirling, L.C.; Wood, J.N. Neuropathic pain develops normally in mice lacking both Nav1.7 and Nav1.8. Mol. Pain 2005, 1. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Mata, M.; Fink, D.J. Continuous delta-opioid receptor activation reduces neuronal voltage-gated sodium channel (NaV1.7) levels through activation of protein kinase C in painful diabetic neuropathy. J. Neurosci. 2008, 28, 6652–6658. [Google Scholar] [CrossRef]
- Persson, A.K.; Gasser, A.; Black, J.A.; Waxman, S.G. Nav1.7 accumulates and co-localizes with phosphorylated ERK1/2 within transected axons in earlyexperimental neuromas. Exp. Neurol. 2011, 230, 273–279. [Google Scholar] [CrossRef]
- Kretschmer, T.; Happel, L.T.; England, J.D.; Nguyen, D.H.; Tiel, R.L.; Beuerman, R.W.; Kline, D.G. Accumulation of PN1 and PN3 sodium channels in painful human neuroma-evidence from immunocytochemistry. Acta Neurochir. 2002, 144, 803–810. [Google Scholar]
- Luo, S.; Perry, G.M.; Levinson, S.R.; Henry, M.A. Nav1.7 expression is increased in painful human dental pulp. Mol. Pain 2008, 4. [Google Scholar] [CrossRef]
- Yiangou, Y.; Facer, P.; Chessell, I.P.; Bountra, C.; Chan, C.; Fertleman, C.; Smith, V.; Anand, P. Voltage-gated ion channel Nav1.7 innervation in patients with idiopathic rectal hypersensitivity and paroxysmal extreme pain disorder (familial rectal pain). Neurosci. Lett. 2007, 427, 77–82. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Duncan, K.G.; Fields, H.L.; Jones, M.R. Tetrodotoxin: Anesthetic activity in the de-epithelialized cornea. Graefes Arch. Clin. Exp. Ophthalmol. 1998, 236, 790–794. [Google Scholar]
- Schwartz, D.M.; Fields, H.L.; Duncan, K.G.; Duncan, J.L.; Jones, M.R. Experimental study of tetrodotoxin, a long-acting topical anesthetic. Am. J. Ophthalmol. 1998, 125, 481–487. [Google Scholar]
- Xie, W.; Strong, J.A.; Meij, J.T.; Zhang, J.M.; Yu, L. Neuropathic pain: Early spontaneous afferent activity is the trigger. Pain 2005, 116, 243–256. [Google Scholar]
- Houck, C.S.; Khodorova, A.; Reale, A.M.; Strichartz, G.R.; Davar, G. Sensory fibers resistant to the actions of tetrodotoxin mediate nocifensive responses to local administration of endothelin-1 in rats. Pain 2004, 110, 719–726. [Google Scholar]
- Barnet, C.S.; Tse, J.Y.; Kohane, D.S. Site 1 sodium channel blockers prolong the duration of sciatic nerve blockade from tricyclic antidepressants. Pain 2004, 110, 432–438. [Google Scholar]
- Marcil, J.; Walczak, J.S.; Guindon, J.; Ngoc, A.H.; Lu, S.; Beaulieu, P. Antinociceptive effects of tetrodotoxin (TTX) in rodents. Br. J. Anaesth. 2006, 96, 761–768. [Google Scholar]
- Nieto, F.R.; Entrena, J.M.; Cendán, C.M.; Pozo, E.D.; Vela, J.M.; Baeyens, J.M. Tetrodotoxin inhibits the development and expression of neuropathic pain induced by paclitaxel in mice. Pain 2008, 137, 520–531. [Google Scholar]
- Entrena, J.M.; Cobos, E.J.; Nieto, F.R.; Cendán, C.M.; Gris, G.; Del Pozo, E.; Zamanillo, D.; Baeyens, J.M. Sigma-1 receptors are essential for capsaicin-induced mechanical hypersensitivity: Studies with selective sigma-1 ligands and sigma-1 knockout mice. Pain 2009, 143, 252–261. [Google Scholar]
- Kayser, V.; Viguier, F.; Ioannidi, M.; Bernard, J.F.; Latrémolière, A.; Michot, B.; Vela, J.M.; Buschmann, H.; Hamon, M.; Bourgoin, S. Differential anti-neuropathic pain effects of tetrodotoxin in sciatic nerve- versus infraorbital nerve-ligated rats-behavioral, pharmacological and immunohistochemical investigations. Neuropharmacology 2010, 58, 474–487. [Google Scholar]
- Iwamoto, T.; Takasugi, Y.; Higashino, H.; Ito, H.; Koga, Y.; Nakao, S. Antinociceptive action of carbamazepine on thermal hypersensitive pain at spinal level in a rat model of adjuvant-induced chronic inflammation. J. Anesth. 2011, 25, 78–86. [Google Scholar]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Beloeil, H.; Ababneh, Z.; Chung, R.; Zurakowski, D.; Mulkern, R.V.; Berde, C.B. Effects of bupivacaine and tetrodotoxin on carrageenan-induced hind paw inflammation in rats (Part 1): Hyperalgesia, edema, and systemic cytokines. Anesthesiology 2006, 105, 128–138. [Google Scholar]
- Alguacil, L.F.; Pérez-García, C.; Salas, E.; González-Martín, C.; Castillo, C.; Polanco, M.J.; Herradón, G.; Morales, L. Subcutaneous tetrodotoxin and inflammatory pain. Br. J. Anaesth. 2008, 100, 729–730. [Google Scholar]
- Craner, M.J.; Damarjian, T.G.; Liu, S; Hains, B.C.; Lo, A.C.; Black, J.A.; Newcombe, J.; Cuzner, M.L.; Waxman, S.G. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 2005, 49, 220–229. [Google Scholar] [CrossRef]
- Lyu, Y.S.; Park, S.K.; Chung, K.; Chung, J.M. Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res. 2000, 871, 98–103. [Google Scholar]
- Chen, J.J.; Lue, J.H.; Lin, L.H.; Huang, C.T.; Chiang, R.P.; Chen, C.L.; Tsai, Y.J. Effects of pre-emptive drug treatment on astrocyte activation in the cuneate nucleus following rat median nerve injury. Pain 2010, 148, 158–166. [Google Scholar]
- Nozaki-Taguchi, N.; Chaplan, S.R.; Higuera, E.S.; Ajakwe, R.C.; Yaksh, T.L. Vincristine-induced allodynia in the rat. Pain 2001, 93, 69–76. [Google Scholar]
- Cavaletti, G.; Marmiroli, P. Chemotherapy-induced peripheral neurotoxicity. Nat. Rev. Neurol. 2010, 6, 657–666. [Google Scholar]
- Klein, T.; Magerl, W.; Rolke, R.; Treede, R.D. Human surrogate models of neuropathic pain. Pain 2005, 115, 227–233. [Google Scholar]
- Xie, W.; Strong, J.A.; Zhang, J.M. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience 2009, 160, 847–857. [Google Scholar]
- Cummins, T.R.; Black, J.A.; Dib-Hajj, S.D.; Waxman, S.G. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J. Neurosci. 2000, 20, 8754–8761. [Google Scholar]
- Matzner, O.; Devor, M. Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J. Neurophysiol. 1994, 72, 349–359. [Google Scholar]
- Omana-Zapata, I.; Khabbaz, M.A.; Hunter, J.C.; Clarke, D.E.; Bley, K.R. Tetrodotoxin inhibits neuropathic ectopic activity in neuromas, dorsal root ganglia and dorsal horn neurons. Pain 1997, 72, 41–49. [Google Scholar]
- Amir, R.; Michaelis, M.; Devor, M. Membrane potential oscillations in dorsal root ganglion neurons: Role in normal electrogenesis and neuropathic pain. J. Neurosci. 1999, 19, 8589–8596. [Google Scholar]
- Liu, X.; Zhou, J.L.; Chung, K.; Chung, J.M. Ion channels associated with the ectopic discharges generated after segmental spinal nerve injury in the rat. Brain Res. 2001, 900, 119–127. [Google Scholar]
- Hagen, N.A.; Fisher, K.M.; Lapointe, B.; du Souich, P.; Chary, S.; Moulin, D.; Sellers, E.; Ngoc, A.H. Canadian Tetrodotoxin Study Group. An open-label, multi-dose efficacy and safety study of intramuscular tetrodotoxin in patients with severe cancer-related pain. J. Pain Symptom Manag. 2007, 34, 171–182. [Google Scholar] [CrossRef]
- Hagen, N.A.; du Souich, P.; Lapointe, B.; Ong-Lam, M.; Dubuc, B.; Walde, D.; Love, R.; Ngoc, A.H. Canadian Tetrodotoxin Study Group. Tetrodotoxin for moderate to severe cancer pain: A randomized, double blind, parallel design multicenter study. J. Pain Symptom Manag. 2008, 35, 420–429. [Google Scholar] [CrossRef]
- Hagen, N.A.; Lapointe, B.; Ong-Lam, M.; Dubuc, B.; Walde, D.; Gagnon, B.; Love, R.; Goel, R.; Hawley, P.; Ngoc, A.H.; du Souich, P. A multicentre open-label safety and efficacy study of tetrodotoxin for cancer pain. Curr. Oncol. 2011, 18, 109–116. [Google Scholar]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nieto, F.R.; Cobos, E.J.; Tejada, M.Á.; Sánchez-Fernández, C.; González-Cano, R.; Cendán, C.M. Tetrodotoxin (TTX) as a Therapeutic Agent for Pain. Mar. Drugs 2012, 10, 281-305. https://doi.org/10.3390/md10020281
Nieto FR, Cobos EJ, Tejada MÁ, Sánchez-Fernández C, González-Cano R, Cendán CM. Tetrodotoxin (TTX) as a Therapeutic Agent for Pain. Marine Drugs. 2012; 10(2):281-305. https://doi.org/10.3390/md10020281
Chicago/Turabian StyleNieto, Francisco Rafael, Enrique José Cobos, Miguel Ángel Tejada, Cristina Sánchez-Fernández, Rafael González-Cano, and Cruz Miguel Cendán. 2012. "Tetrodotoxin (TTX) as a Therapeutic Agent for Pain" Marine Drugs 10, no. 2: 281-305. https://doi.org/10.3390/md10020281
APA StyleNieto, F. R., Cobos, E. J., Tejada, M. Á., Sánchez-Fernández, C., González-Cano, R., & Cendán, C. M. (2012). Tetrodotoxin (TTX) as a Therapeutic Agent for Pain. Marine Drugs, 10(2), 281-305. https://doi.org/10.3390/md10020281