Recruitment of Glycosyl Hydrolase Proteins in a Cone Snail Venomous Arsenal: Further Insights into Biomolecular Features of Conus Venoms

 ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
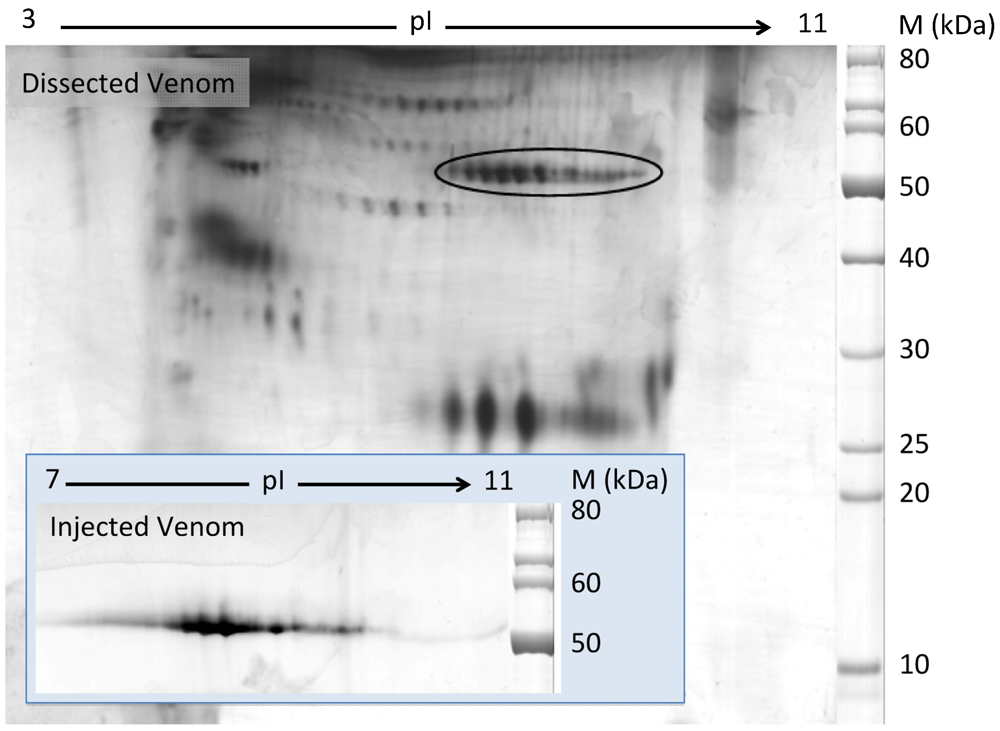
2.1. Discovery of Hyaluronidases in Cone Snail Venom
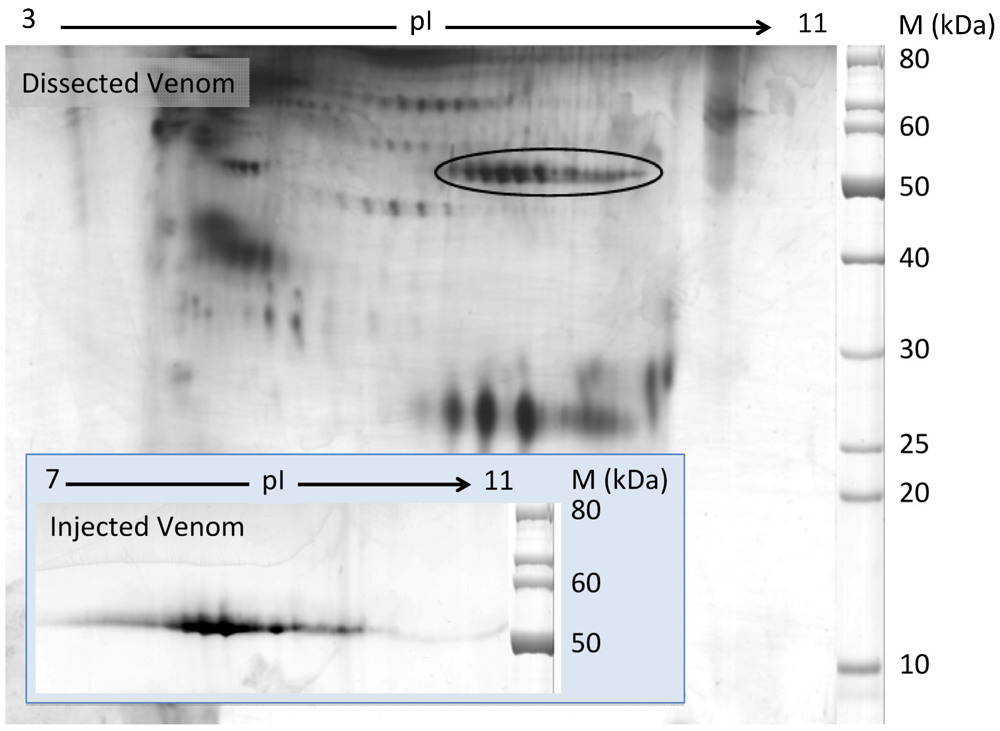
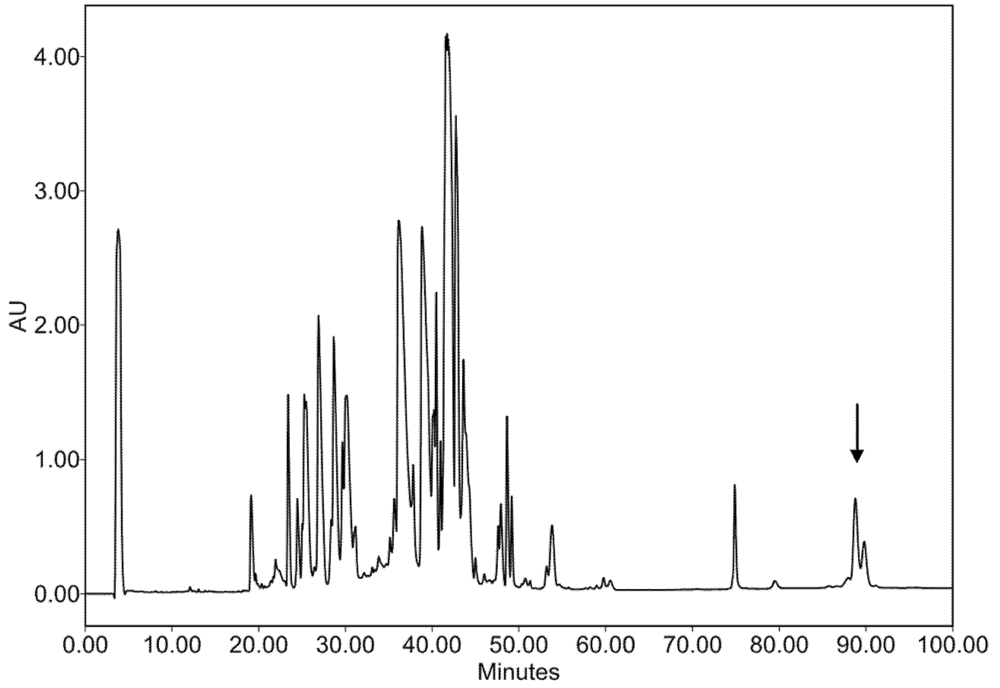
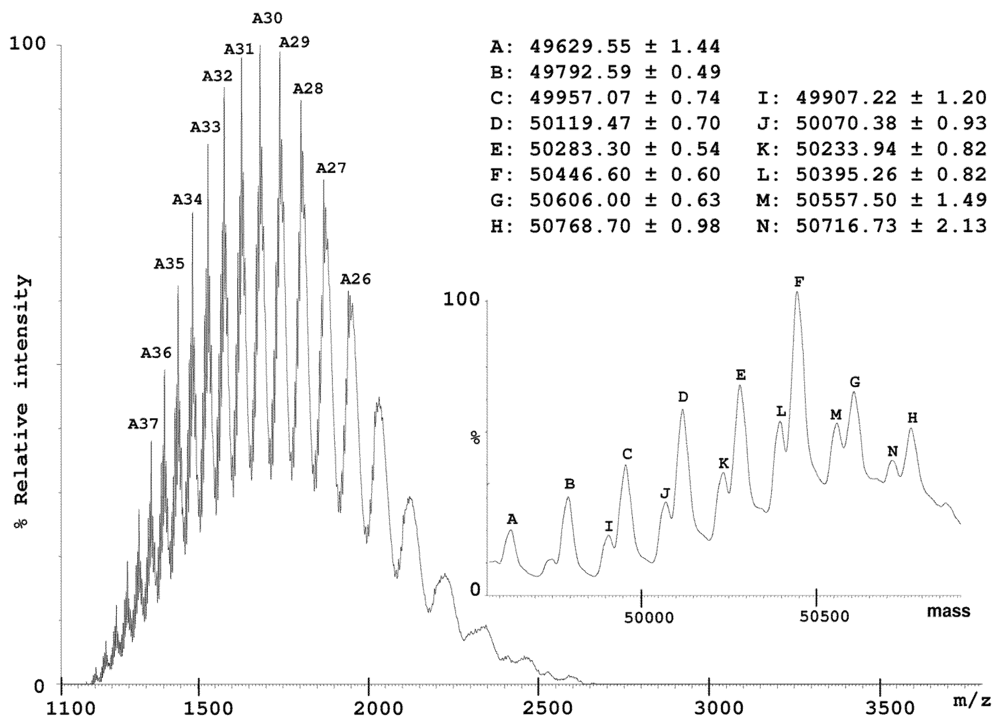
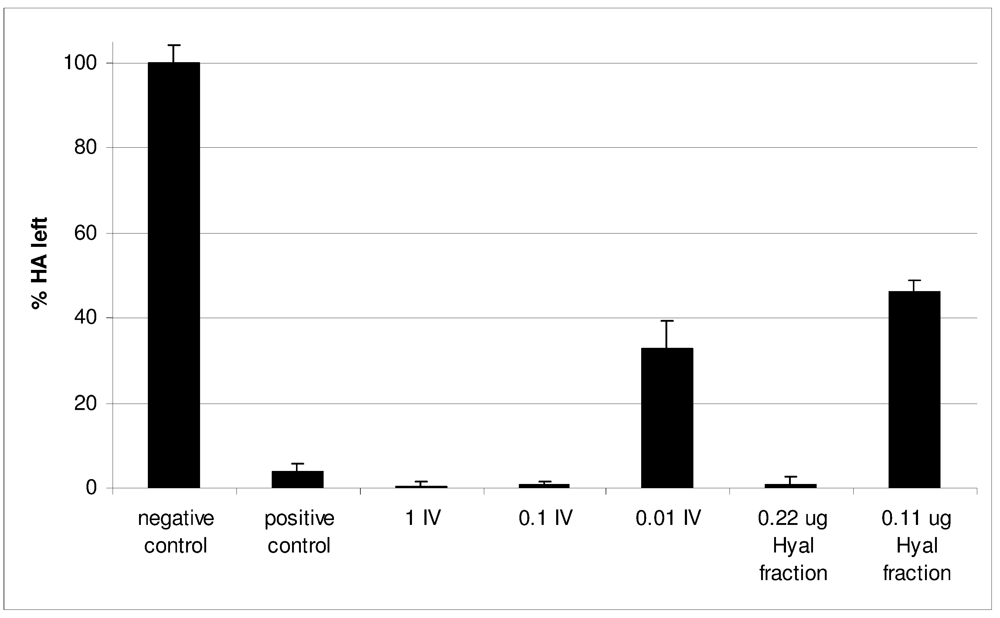
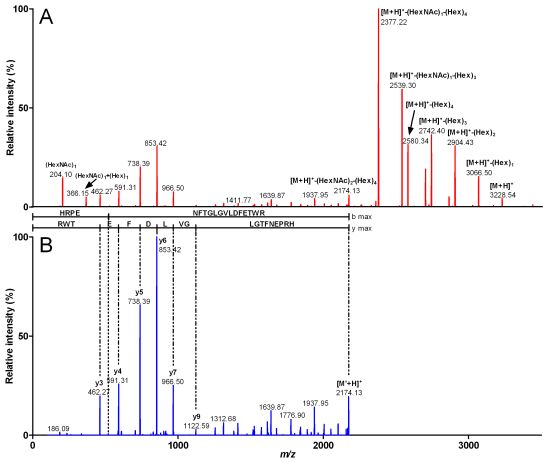
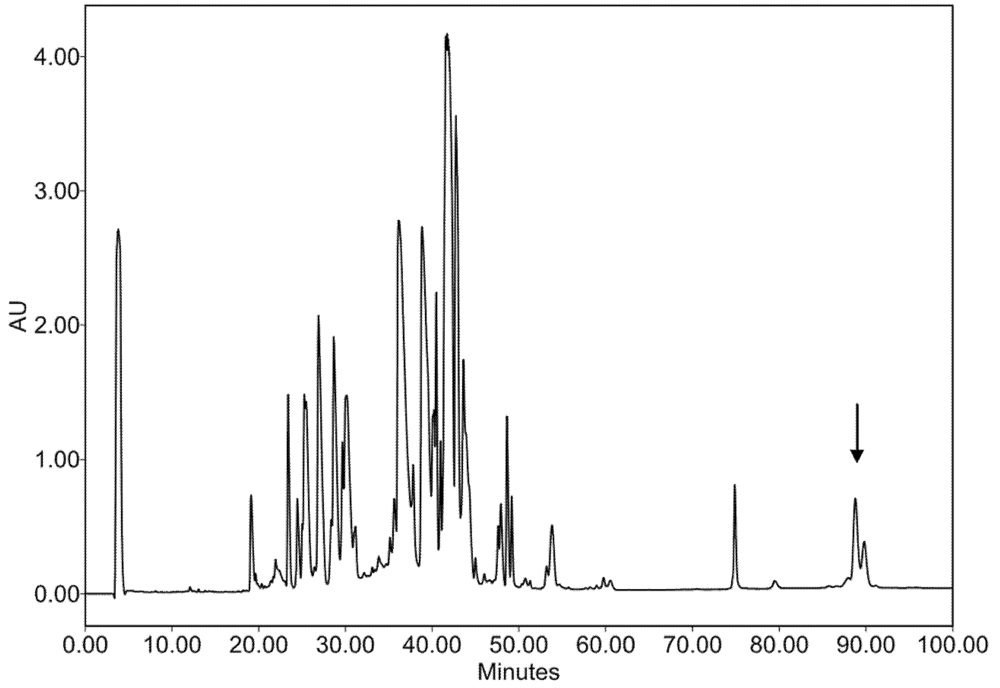
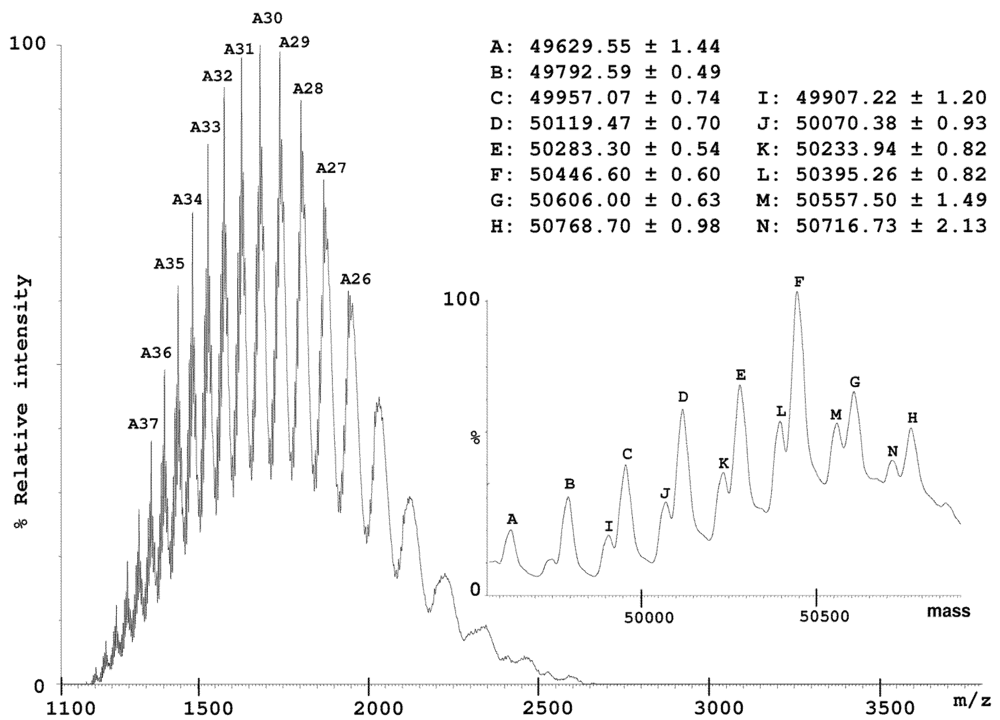
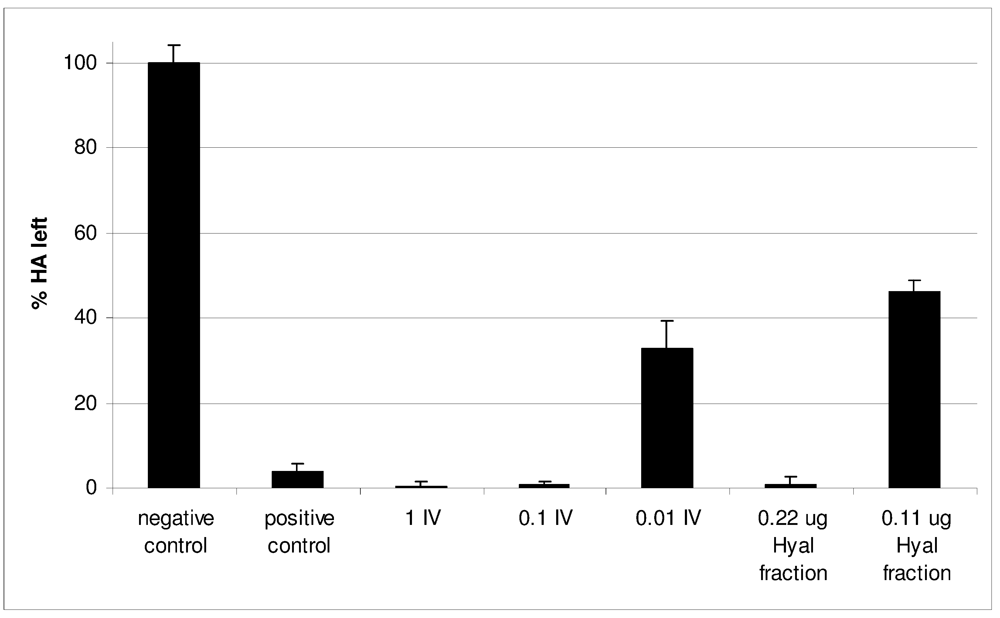
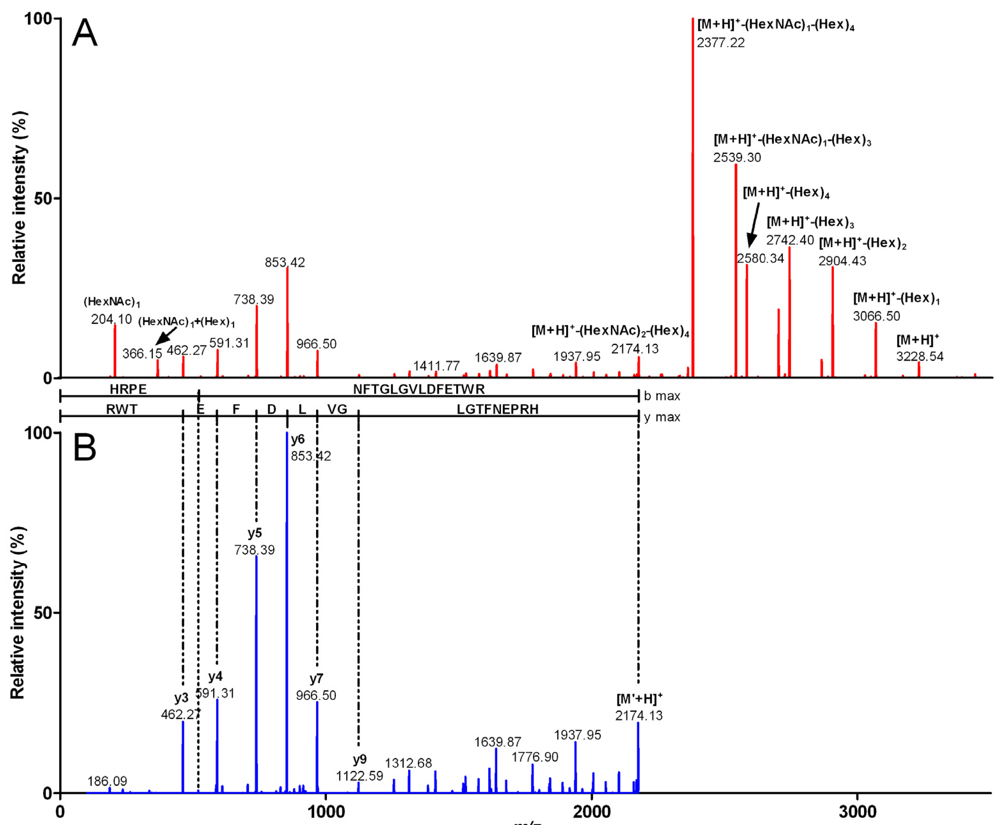
2.2. Characterization of Conohyal-Cn1

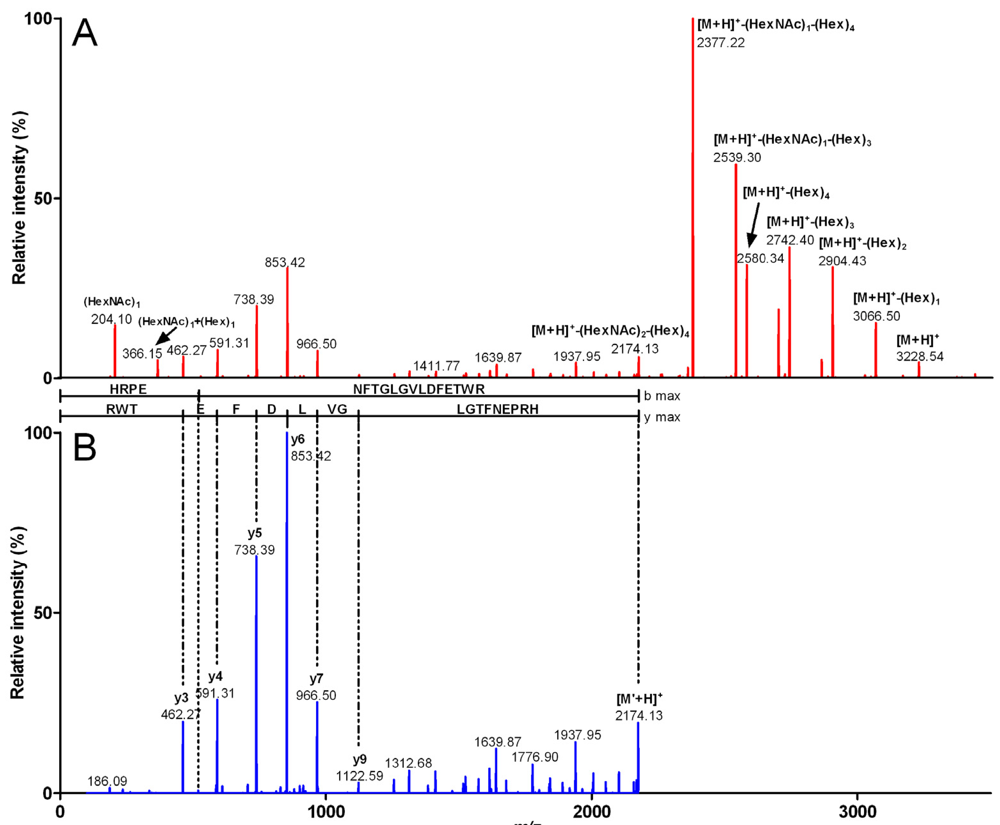

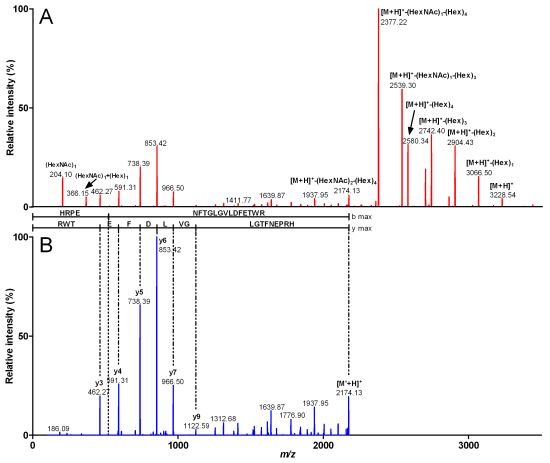
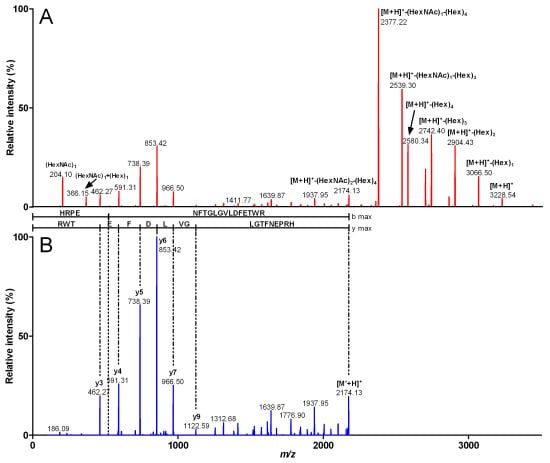{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment mass (Da) | m/z (z) | Peptide sequence | Sequence mass (Da) | Glycosylation | Glycosylation mass (Da) |
|---|---|---|---|---|---|
| 2,557.84 | 853.62 (3) | NVTQMMTDBSR | 1,341.45 | (HexNAc)2 (Hex)5 | 1,216.39 |
| 2,233.74 | 1,117.87 (2) | NVTQMMTDBSR | 1,341.45 | (HexNAc)2 (Hex)3 | 892.29 |
| 2,071.70 | 1,036.85 (2) | NVTQMMTDBSR | 1,341.46 | (HexNAc)2 (Hex)2 | 730.26 |
| 3,540.30 | 1,181.10 (3) | AIYSTNFGPMTIYQNESVK | 2,161.83 | (HexNAc)2 (Hex)6 | 1,378.47 |
| 3,378.12 | 1,127.04 (3) | AIYSTNFGPMTIYQNESVK | 2,161.73 | (HexNAc)2 (Hex)5 | 1,216.39 |
| 3,054.09 | 1,019.03 (3) | AIYSTNFGPMTIYQNESVK | 2,161.8 | (HexNAc)2 (Hex)3 | 892.29 |
| 2,892.14 | 965.05 (3) | AIYSTNFGPMTIYQNESVK | 2,161.88 | (HexNAc)2 (Hex)2 | 730.26 |
| 3,916.49 | 980.12 (4) | HRPENFTGLGVLDFETWR | 2,172.88 | (HexNAc)3 (Hex)7 | 1,743.61 |
| 3,551.27 | 888.81 (4) | HRPENFTGLGVLDFETWR | 2,172.8 | (HexNAc)2 (Hex)6 | 1,378.47 |
| 3,227.22 | 807.81 (4) | HRPENFTGLGVLDFETWR | 2,172.86 | (HexNAc)2 (Hex)4 | 1,054.36 |
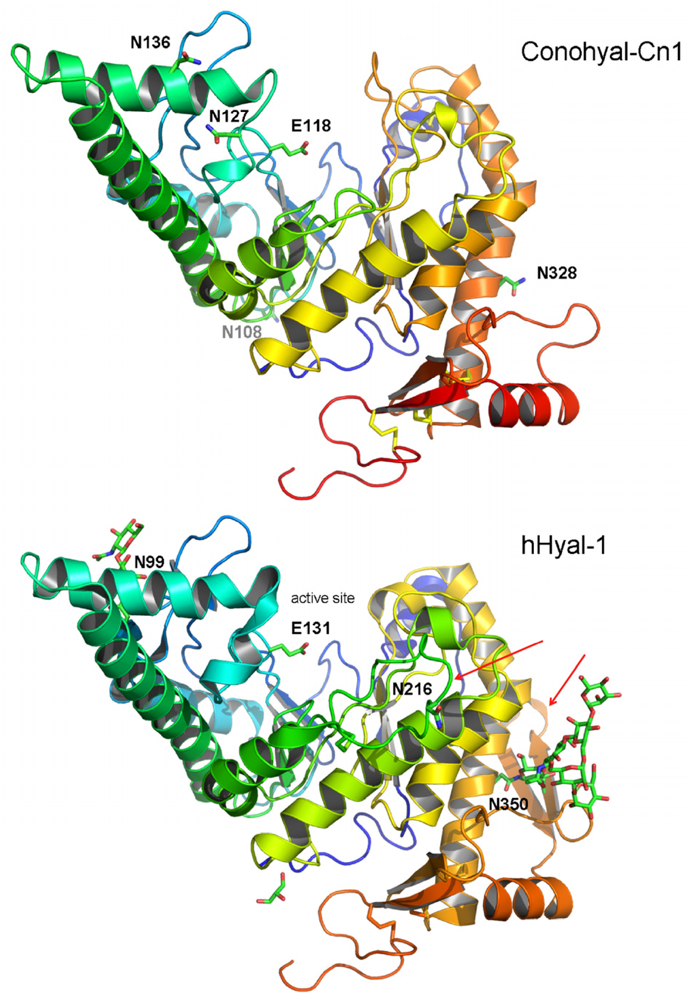
2.3. Structure Modeling
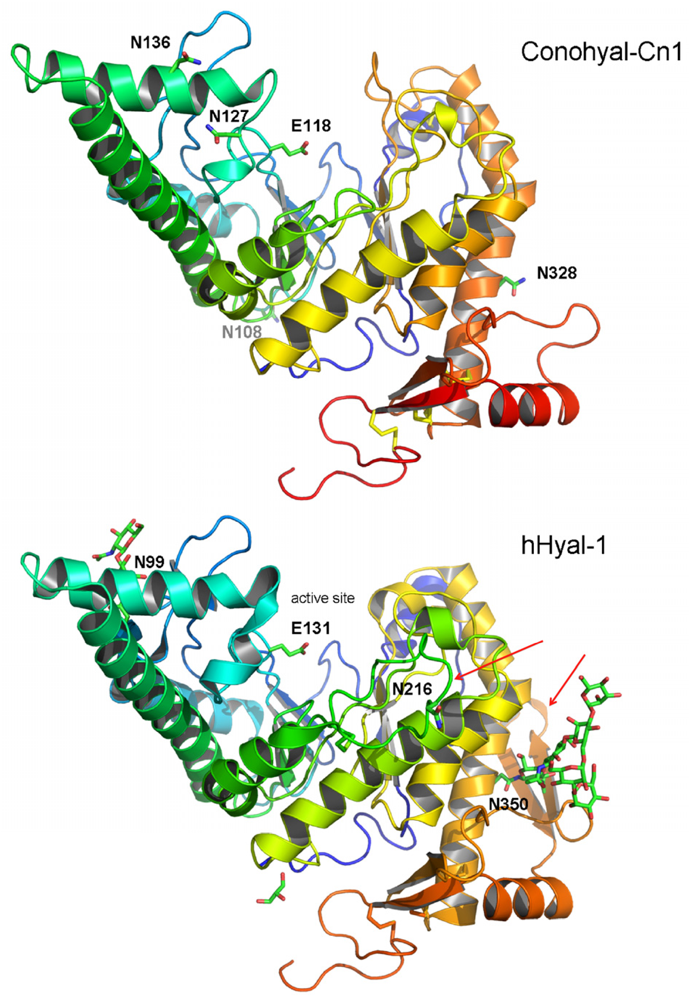
2.4. Phylogenetic Analysis

3. Experimental Section
3.1. Chemicals
3.2. Sample Preparations
3.3. Injectable Venom Fractionation
3.4. cDNA Library Construction, 454 Sequencing and Assembly
3.5. Two-Dimensional Gel Electrophoresis (2-DE)
3.6. Mass Spectrometry (MS and MS/MS) on 2-DE Spots
3.7. Protein Biochemistry and Edman Sequencing
3.8. Mass Spectrometry on HPLC Fractions
3.9. Enzymatic Activity
3.10. Homology Modeling
3.11. Phylogenetic Analysis
4. Conclusions
Acknowledgments
Supplementary Files
References
- Olivera, B.M.; Teichert, R.W. Diversity of the neurotoxic Conus peptides: a model for concerted pharmacological discovery. Mol. Interv. 2007, 7, 251–260. [Google Scholar]
- Favreau, P.; Stöcklin, R. Marine snail venoms: use and trends in receptor and channel neuropharmacology. Curr. Opin. Pharmacol. 2009, 9, 594–601. [Google Scholar]
- Halai, R.; Craik, D.J. Conotoxins: natural product drug leads. Nat. Prod. Rep. 2009, 26, 526–536. [Google Scholar]
- Lewis, R.J. Conotoxins: molecular and therapeutic targets. Prog. Mol. Subcell. Biol 2009, 46, 45–65. [Google Scholar]
- Newcomb, R.; Miljanich, G. Handbook of Neurotoxicology; Massaro, E.J., Ed.; Humana Press Inc: Totowa, NJ, USA, 2002; Volume 1, pp. 617–652, Chapter 28. [Google Scholar]
- Mebs, D. Venomous and Poisonous Animals; Medpharm Scientific Publishers: Stuttgart, Germany, 2002. [Google Scholar]
- Marsh, H. The caseinase activity of some vermivorous cone shell venoms. Toxicon 1971, 9, 63–67. [Google Scholar]
- Balbin, J.A. Acetylcholinesterase from Conus geographus venom: partial purification and characterization. MS Thesis, Dept. of Biochemistry and Molecular Biology, University of the Philippines, Manila, Philippines, 1980. [Google Scholar]
- Miranda, R.R. Partial purification and characterization of Conus textile venom phosphodiesterase I. MS Thesis, Dept. of Biochemistry and Molecular Biology, University of the Philippines, Manila, Philippines, 1982. [Google Scholar]
- Milne, T.J.; Abbenante, G.; Tyndall, J.D.; Halliday, J.; Lewis, R.J. Isolation and characterization of a cone snail protease with homology to CRISP proteins of the pathogenesis-related protein superfamily. J. Biol. Chem. 2003, 278, 31105–31110. [Google Scholar]
- Qian, J.; Guo, Z.Y.; Chi, C.W. Cloning and isolation of a Conus cysteine-rich protein homologous to Tex31 but without proteolytic activity. Acta Biochim. Biophys. Sin. (Shanghai) 2008, 40, 174–181. [Google Scholar] [CrossRef]
- Stanley, T.B.; Stafford, D.W.; Olivera, B.M.; Bandyopadhyay, P.K. Identification of a vitamin K-dependent carboxylase in the venom duct of a Conus snail. FEBS Lett. 1997, 407, 85–88. [Google Scholar]
- Safavi-Hemami, H.; Bulaj, G.; Olivera, B.M.; Williamson, N.A.; Purcell, A.W. Identification of Conus peptidylprolyl cis-trans isomerases (PPIases) and assessment of their role in the oxidative folding of conotoxins. J. Biol. Chem. 2010, 285, 12735–12746. [Google Scholar]
- Buczek, O.; Olivera, B.M.; Bulaj, G. Propeptide does not act as an intramolecular chaperone but facilitates protein disulfide isomerase-assisted folding of a conotoxin precursor. Biochemistry 2004, 43, 1093–1101. [Google Scholar]
- Gowd, K.H.; Krishnan, K.S.; Balaram, P. Identification of Conus amadis disulfide isomerase: minimum sequence length of peptide fragments necessary for protein annotation. Mol. Biosyst. 2007, 3, 554–566. [Google Scholar]
- Price-Carter, M.; Gray, W.R.; Goldenberg, D.P. Folding of omega-conotoxins. 2. Influence of precursor sequences and protein disulfide isomerase. Biochemistry 1996, 35, 15547–15557. [Google Scholar]
- Wang, Z.Q.; Han, Y.H.; Shao, X.X.; Chi, C.W.; Guo, Z.Y. Molecular cloning, expression and characterization of protein disulfide isomerase from Conus marmoreus. FEBS J. 2007, 274, 4778–4787. [Google Scholar]
- Moller, C.; Mari, F. 9.3 KDa components of the injected venom of Conus purpurascens define a new five-disulfide conotoxin framework. Biopolymers 2011, 96, 158–165. [Google Scholar] [CrossRef]
- McIntosh, J.M.; Ghomashchi, F.; Gelb, M.H.; Dooley, D.J.; Stoehr, S.J.; Giordani, A.B.; Naisbitt, S.R.; Olivera, B.M. Conodipine-M, a novel phospholipase A2 isolated from the venom of the marine snail Conus magus. J. Biol. Chem. 1995, 270, 3518–3526. [Google Scholar]
- Kini, R.M. Venom Phospholipase A2 Enzymes, Structure, Function and Mechanism; Kini, R.M., Ed.; John Wiley & Sons: Chichester, UK, 1997. [Google Scholar]
- Stern, R.; Jedrzejas, M.J. Hyaluronidases: their genomics, structures, and mechanisms of action. Chem. Rev. 2006, 106, 818–839. [Google Scholar]
- Terrat, Y.; Biass, D.; Dutertre, S.; Favreau, P.; Remm, M.; Stöcklin, R.; Piquemal, D.; Ducancel, F. High-resolution picture of a venom gland transcriptome: Case study with the marine snail Conus consors. Toxicon 2011, 59, 34–46. [Google Scholar]
- Dutertre, S.; Biass, D.; Stöcklin, R.; Favreau, P. Dramatic intraspecimen variations within the injected venom of Conus consors: an unsuspected contribution to venom diversity. Toxicon 2010, 55, 1453–1462. [Google Scholar]
- Sutherland, S.K.; Lane, W.R. Toxins and mode of envenomation of the common ringed or blue-banded octopus. Med. J. Aust. 1969, 1, 893–898. [Google Scholar]
- Fry, B.G.; Scheib, H.; van der Weerd, L.; Young, B.; McNaughtan, J.; Ramjan, S.F.; Vidal, N.; Poelmann, R.E.; Norman, J.A. Evolution of an arsenal: structural and functional diversification of the venom system in the advanced snakes (Caenophidia). Mol. Cell. Proteomics 2008, 7, 215–246. [Google Scholar]
- Harrison, R.A.; Ibison, F.; Wilbraham, D.; Wagstaff, S.C. Identification of cDNAs encoding viper venom hyaluronidases: cross-generic sequence conservation of full-length and unusually short variant transcripts. Gene 2007, 392, 22–33. [Google Scholar]
- Fry, B.G.; Winter, K.; Norman, J.A.; Roelants, K.; Nabuurs, R.J.; van Osch, M.J.; Teeuwisse, W.M.; van der Weerd, L.; McNaughtan, J.E.; Kwok, H.F.; Scheib, H.; Greisman, L.; Kochva, E.; Miller, L.J.; Gao, F.; Karas, J.; Scanlon, D.; Lin, F.; Kuruppu, S.; Shaw, C.; Wong, L.; Hodgson, W.C. Functional and structural diversification of the Anguimorpha lizard venom system. Mol. Cell. Proteomics 2010, 9, 2369–2390. [Google Scholar]
- Feng, L.; Gao, R.; Meng, J.; Gopalakrishnakone, P. Cloning and molecular characterization of BmHYA1, a novel hyaluronidase from the venom of Chinese red scorpion Buthus martensi Karsch. Toxicon 2010, 56, 474–479. [Google Scholar]
- Madokoro, M.; Ueda, A.; Kiriake, A.; Shiomi, K. Properties and cDNA cloning of a hyaluronidase from the stonefish Synanceia verrucosa venom. Toxicon 2011, 58, 285–292. [Google Scholar]
- Magalhaes, M.R.; da Silva, N.J., Jr.; Ulhoa, C.J. A hyaluronidase from Potamotrygon motoro (freshwater stingrays) venom: isolation and characterization. Toxicon 2008, 51, 1060–1067. [Google Scholar]
- Ng, H.C.; Ranganathan, S.; Chua, K.L.; Khoo, H.E. Cloning and molecular characterization of the first aquatic hyaluronidase, SFHYA1, from the venom of stonefish (Synanceja horrida). Gene 2005, 346, 71–81. [Google Scholar]
- Gmachl, M.; Kreil, G. Bee venom hyaluronidase is homologous to a membrane protein of mammalian sperm. Proc. Natl. Acad. Sci. USA 1993, 90, 3569–3573. [Google Scholar]
- Kolarich, D.; Leonard, R.; Hemmer, W.; Altmann, F. The N-glycans of yellow jacket venom hyaluronidases and the protein sequence of its major isoform in Vespula vulgaris. FEBS J. 2005, 272, 5182–5190. [Google Scholar]
- Feng, L.; Gao, R.; Gopalakrishnakone, P. Isolation and characterization of a hyaluronidase from the venom of Chinese red scorpion Buthus martensi. Comp Biochem. Physiol. C. Toxicol. Pharmacol. 2008, 148, 250–257. [Google Scholar]
- Pessini, A.C.; Takao, T.T.; Cavalheiro, E.C.; Vichnewski, W.; Sampaio, S.V.; Giglio, J.R.; Arantes, E.C. A hyaluronidase from Tityus serrulatus scorpion venom: isolation, characterization and inhibition by flavonoids. Toxicon 2001, 39, 1495–1504. [Google Scholar]
- Nagaraju, S.; Devaraja, S.; Kemparaju, K. Purification and properties of hyaluronidase from Hippasa partita (funnel web spider) venom gland extract. Toxicon 2007, 50, 383–393. [Google Scholar]
- Girish, K.S.; Shashidharamurthy, R.; Nagaraju, S.; Gowda, T.V.; Kemparaju, K. Isolation and characterization of hyaluronidase a "spreading factor" from Indian cobra (Naja naja) venom. Biochimie 2004, 86, 193–202. [Google Scholar]
- Muchmore, D.B.; Vaughn, D.E. Review of the mechanism of action and clinical efficacy of recombinant human hyaluronidase coadministration with current prandial insulin formulations. J. Diabetes Sci. Technol. 2010, 4, 419–428. [Google Scholar]
- Frost, G.I. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin. Drug Deliv. 2007, 4, 427–440. [Google Scholar]
- Farr, C.; Menzel, J.; Seeberger, J.; Schweigle, B. Clinical pharmacology and possible applications of hyaluronidase with reference to Hylase "Dessau". Wien. Med. Wochenschr. 1997, 147, 347–355. [Google Scholar]
- Tanaka, T.; Nakatani, T. New Therapeutic Strategies for Castration-Resistant Prostate Cancer. Recent Pat. Anticancer Drug Discov. 2011, 6, 373–383. [Google Scholar]
- Thompson, C.B.; Shepard, H.M.; O'Connor, P.M.; Kadhim, S.; Jiang, P.; Osgood, R.J.; Bookbinder, L.H.; Li, X.; Sugarman, B.J.; Connor, R.J.; Nadjsombati, S.; Frost, G.I. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol. Cancer Ther. 2010, 9, 3052–3064. [Google Scholar]
- Mikesh, L.M.; Ueberheide, B.; Chi, A.; Coon, J.J.; Syka, J.E.; Shabanowitz, J.; Hunt, D.F. The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta 2006, 1764, 1811–1822. [Google Scholar]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar]
- Craig, A.G.; Bandyopadhyay, P.; Olivera, B.M. Post-translationally modified neuropeptides from Conus venoms. Eur. J. Biochem. 1999, 264, 271–275. [Google Scholar]
- Le Gall, F.; Favreau, P.; Benoit, E.; Mattei, C.; Bouet, F.; Menou, J.L.; Menez, A.; Letourneux, Y.; Molgo, J. A new conotoxin isolated from Conus consors venom acting selectively on axons and motor nerve terminals through a Na+-dependent mechanism. Eur. J. Neurosci. 1999, 11, 3134–3142. [Google Scholar]
- Craig, A.G.; Zafaralla, G.; Cruz, L.J.; Santos, A.D.; Hillyard, D.R.; Dykert, J.; Rivier, J.E.; Gray, W.R.; Imperial, J.; DelaCruz, R.G.; Sporning, A.; Terlau, H.; West, P.J.; Yoshikami, D.; Olivera, B.M. An O-glycosylated neuroexcitatory conus peptide. Biochemistry 1998, 37, 16019–16025. [Google Scholar]
- Chao, K.L.; Muthukumar, L.; Herzberg, O. Structure of human hyaluronidase-1, a hyaluronan hydrolyzing enzyme involved in tumor growth and angiogenesis. Biochemistry 2007, 46, 6911–6920. [Google Scholar]
- Yan, B.; Zhang, W.; Ding, J.; Gao, P. Sequence pattern for the occurrence of N-glycosylation in proteins. J. Protein Chem. 1999, 18, 511–521. [Google Scholar]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar]
- Csoka, A.B.; Frost, G.I.; Stern, R. The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol. 2001, 20, 499–508. [Google Scholar]
- Holford, M.; Zhang, M.M.; Gowd, K.H.; Azam, L.; Green, B.R.; Watkins, M.; Ownby, J.P.; Yoshikami, D.; Bulaj, G.; Olivera, B.M. Pruning nature: Biodiversity-derived discovery of novel sodium channel blocking conotoxins from Conus bullatus. Toxicon 2009, 53, 90–98. [Google Scholar]
- Safavi-Hemami, H.; Siero, W.A.; Gorasia, D.G.; Young, N.D.; Macmillan, D.; Williamson, N.A.; Purcell, A.W. Specialisation of the Venom Gland Proteome in Predatory Cone Snails Reveals Functional Diversification of the Conotoxin Biosynthetic Pathway. J. Proteome Res. 2011, 3904–3919. [Google Scholar]
- Hopkins, C.; Grilley, M.; Miller, C.; Shon, K.J.; Cruz, L.J.; Gray, W.R.; Dykert, J.; Rivier, J.; Yoshikami, D.; Olivera, B.M. A new family of Conus peptides targeted to the nicotinic acetylcholine receptor. J. Biol. Chem. 1995, 270, 22361–22367. [Google Scholar]
- Tastet, C.; Lescuyer, P.; Diemer, H.; Luche, S.; van Dorsselaer, A.; Rabilloud, T. A versatile electrophoresis system for the analysis of high- and low-molecular-weight proteins. Electrophoresis 2003, 24, 1787–1794. [Google Scholar]
- Castellanos-Serra, L.; Vallin, A.; Proenza, W.; Le Caer, J.P.; Rossier, J. An optimized procedure for detection of proteins on carrier ampholyte isoelectric focusing and immobilized pH gradient gels with imidazole and zinc salts: its application to the identification of isoelectric focusing separated isoforms by in-gel proteolysis and mass spectrometry analysis. Electrophoresis 2001, 22, 1677–1685. [Google Scholar]
- Di Ferrante, N. Turbidimetric measurement of acid mucopolysaccharides and hyaluronidase activity. J. Biol Chem. 1956, 220, 303–306. [Google Scholar]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar]
- Edgar, R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5, 113. [Google Scholar]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Kreil, G. Hyaluronidases--a group of neglected enzymes. Protein Sci. 1995, 4, 1666–1669. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Violette, A.; Leonardi, A.; Piquemal, D.; Terrat, Y.; Biass, D.; Dutertre, S.; Noguier, F.; Ducancel, F.; Stöcklin, R.; Križaj, I.; et al. Recruitment of Glycosyl Hydrolase Proteins in a Cone Snail Venomous Arsenal: Further Insights into Biomolecular Features of Conus Venoms. Mar. Drugs 2012, 10, 258-280. https://doi.org/10.3390/md10020258
Violette A, Leonardi A, Piquemal D, Terrat Y, Biass D, Dutertre S, Noguier F, Ducancel F, Stöcklin R, Križaj I, et al. Recruitment of Glycosyl Hydrolase Proteins in a Cone Snail Venomous Arsenal: Further Insights into Biomolecular Features of Conus Venoms. Marine Drugs. 2012; 10(2):258-280. https://doi.org/10.3390/md10020258
Chicago/Turabian StyleViolette, Aude, Adrijana Leonardi, David Piquemal, Yves Terrat, Daniel Biass, Sébastien Dutertre, Florian Noguier, Frédéric Ducancel, Reto Stöcklin, Igor Križaj, and et al. 2012. "Recruitment of Glycosyl Hydrolase Proteins in a Cone Snail Venomous Arsenal: Further Insights into Biomolecular Features of Conus Venoms" Marine Drugs 10, no. 2: 258-280. https://doi.org/10.3390/md10020258
APA StyleViolette, A., Leonardi, A., Piquemal, D., Terrat, Y., Biass, D., Dutertre, S., Noguier, F., Ducancel, F., Stöcklin, R., Križaj, I., & Favreau, P. (2012). Recruitment of Glycosyl Hydrolase Proteins in a Cone Snail Venomous Arsenal: Further Insights into Biomolecular Features of Conus Venoms. Marine Drugs, 10(2), 258-280. https://doi.org/10.3390/md10020258