
Whole Exome Sequencing in 26 Saudi Patients Expands the Mutational and Clinical Spectrum of Diabetic Nephropathy


 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Sample Collection and DNA Extraction
2.3. Whole Exome Sequencing (WES) and Library Preparation
2.4. Quality Control and Read Alignment
2.5. Variant Calling and Filtration
- Genome Analysis Toolkit (GATK) Pipeline:
- ○
- Base Quality Score Recalibration (BQSR): adjusts for systematic errors in base quality scores.
- ○
- HaplotypeCaller: simultaneously identifies SNPs and small INDELs.
- ○
- Genotype Refinement: generates a preliminary VCF file.
- SAMtools:
- ○
- mpileup: builds read alignments across the exome.
- ○
- bcftools call: discerns SNPs and small INDELs, creating a separate VCF file.
2.6. Functional Annotation and Impact Prediction
2.7. Prioritization of Genes of Interest
- INSR, ABCC8, KCNJ11, MAFA, ACE, IKBKB, TNF, MAPK1, MAPK8, CACNA1C, CACNA1D, PRKCD, PRKCZ, IRS2, SOCS1, and PIK3R3.
2.8. Pathway Analysis and Gene Ontology (GO) Enrichment
- KEGG Mapper was used to map gene IDs carrying high- or moderate-impact variants onto known canonical pathways, notably the T2D pathway.
- Significant Pathways: genes related to insulin signaling (INSR, IRS2, PIK3R3), β-cell K_ATP channels (ABCC8, KCNJ11), and inflammation (TNF, IKBKB) were frequently enriched (q-value < 0.0001).
- ✓
- BPs: glucose homeostasis, lipid metabolism, and cytokine-mediated inflammatory responses.
- ✓
- MFs: ATP binding, kinase activity, receptor ligand binding.
- ✓
- CCs: plasma membrane receptors, intracellular signaling complexes.
2.9. Statistical Analyses
2.10. Identification of PolyPhen-2 and SIFT Scores
- PolyPhen-2
- ○
- Input: for each missense SNP, the reference and alternate amino acids and their positions in the canonical protein sequence were submitted along with additional protein structure/function metadata.
- ○
- Algorithm: PolyPhen-2 integrates multiple features—sequence conservation, presence/absence of structural domains, known functional residues, and evolutionary relationships among homologous proteins.
- ○
- Result Interpretation:
- ▪
- Benign: low likelihood of damaging protein structure/function.
- ▪
- Possibly Damaging: intermediate level of confidence requires further validation.
- ▪
- Probably Damaging: high confidence that the variant impairs normal protein function.
- SIFT
- ○
- Input: the same missense variant data, but with particular emphasis on alignments against sequences from multiple species to gauge evolutionary conservation.
- ○
- Algorithm: SIFT calculates a normalized probability for each possible amino acid substitution at a given position. The more conserved the position, the higher the likelihood that a substitution will be deleterious.
- ○
- Result Interpretation:
- ▪
- Tolerated (score > 0.05): the substitution is less likely to impact protein function.
- ▪
- Deleterious (score ≤ 0.05): the substitution is likely to disrupt the protein’s normal activity.
- Intersection of Predictions
- ○
- Stringent Criteria: variants that both PolyPhen-2 labeled as “probably damaging” and SIFT labeled as “deleterious” were prioritized in subsequent analyses of T2DM predisposition and renal complications.
- ○
- Biological Relevance: positions flagged by both algorithms often represent evolutionarily conserved and structurally or functionally crucial residues.
3. Result
3.1. Overview of Variant Identification and Classification
3.2. Chromosomal Distribution of Variants
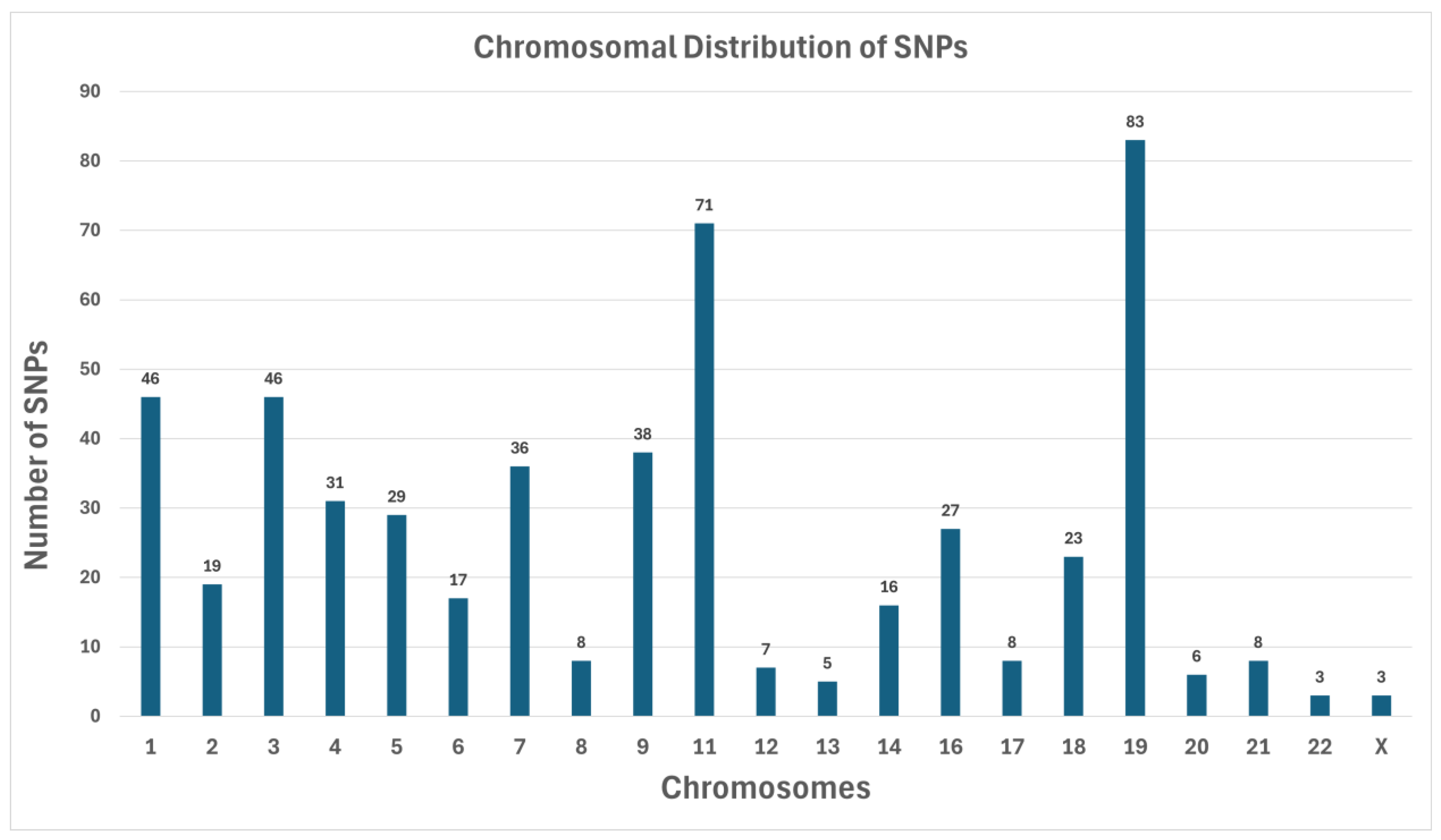
SNP Counts by Chromosome
- Chromosome 19 has the highest number of SNPs (83) (Figure 1). This observation is consistent with longstanding findings that Chromosome 19 is relatively gene-dense and carries many loci implicated in metabolic regulation.
- Chromosome 1 had 46 SNPs, along with Chromosome 3, which also had 46. Historically, Chromosome 1 is known to contain multiple diabetes-associated loci (e.g., regions near INSR).
- Chromosome 11 contained 71 SNPs, notable because it houses genes like KCNJ11 and ABCC8, both of which are critical for insulin secretion.
- A small number of chromosomes (e.g., Chromosomes 20, 21, 22, and X) showed minimal SNP counts, each having fewer than 10 identified variants in our filtered list. The lowest counts were Chromosome 22 (3 SNPs) and Chromosome X (3 SNPs), possibly reflecting either a lack of highly damaging variants in the coding regions for these chromosomes in our cohort or limited representation of X-linked metabolic disruptions in this sample set.
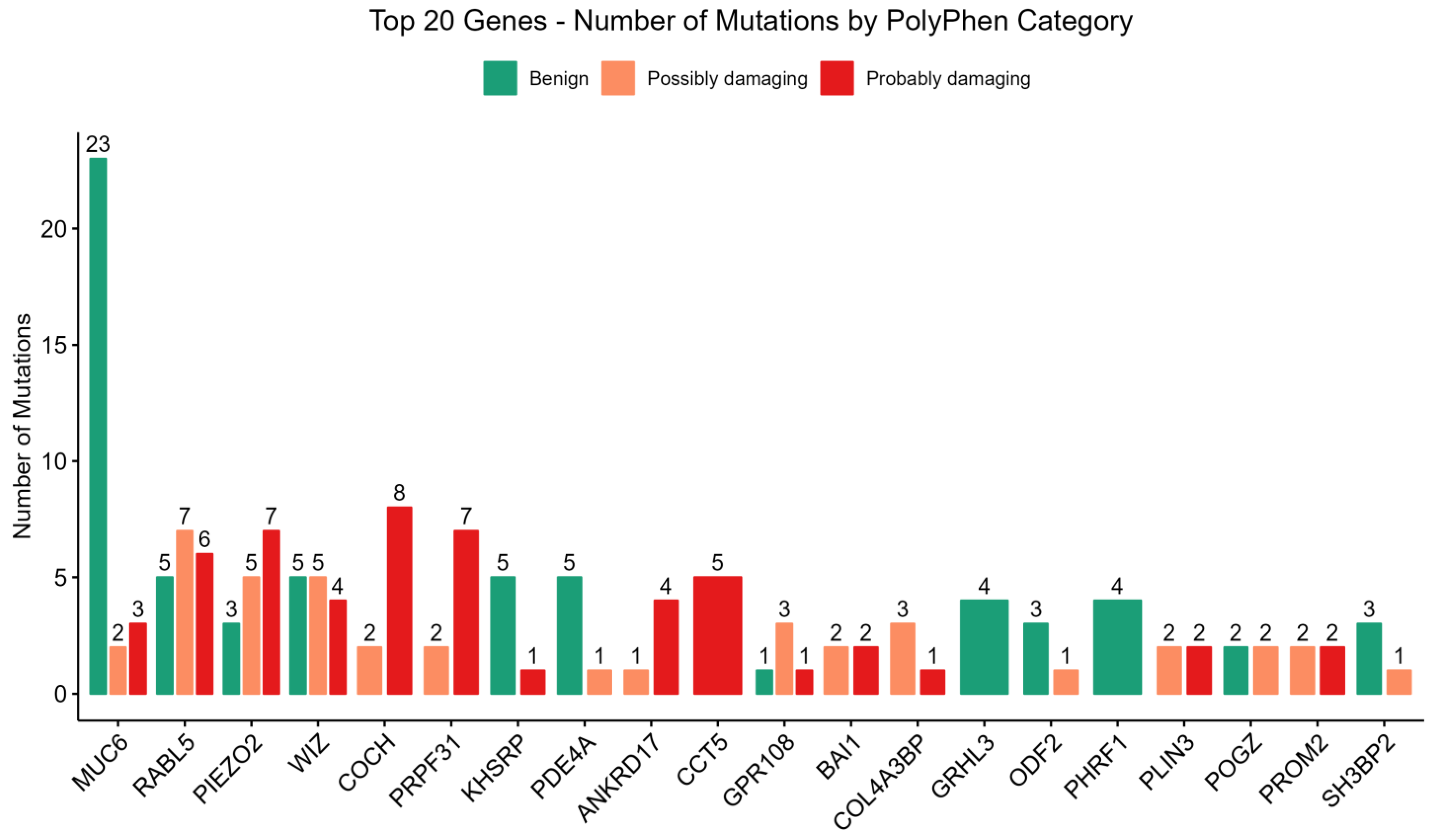
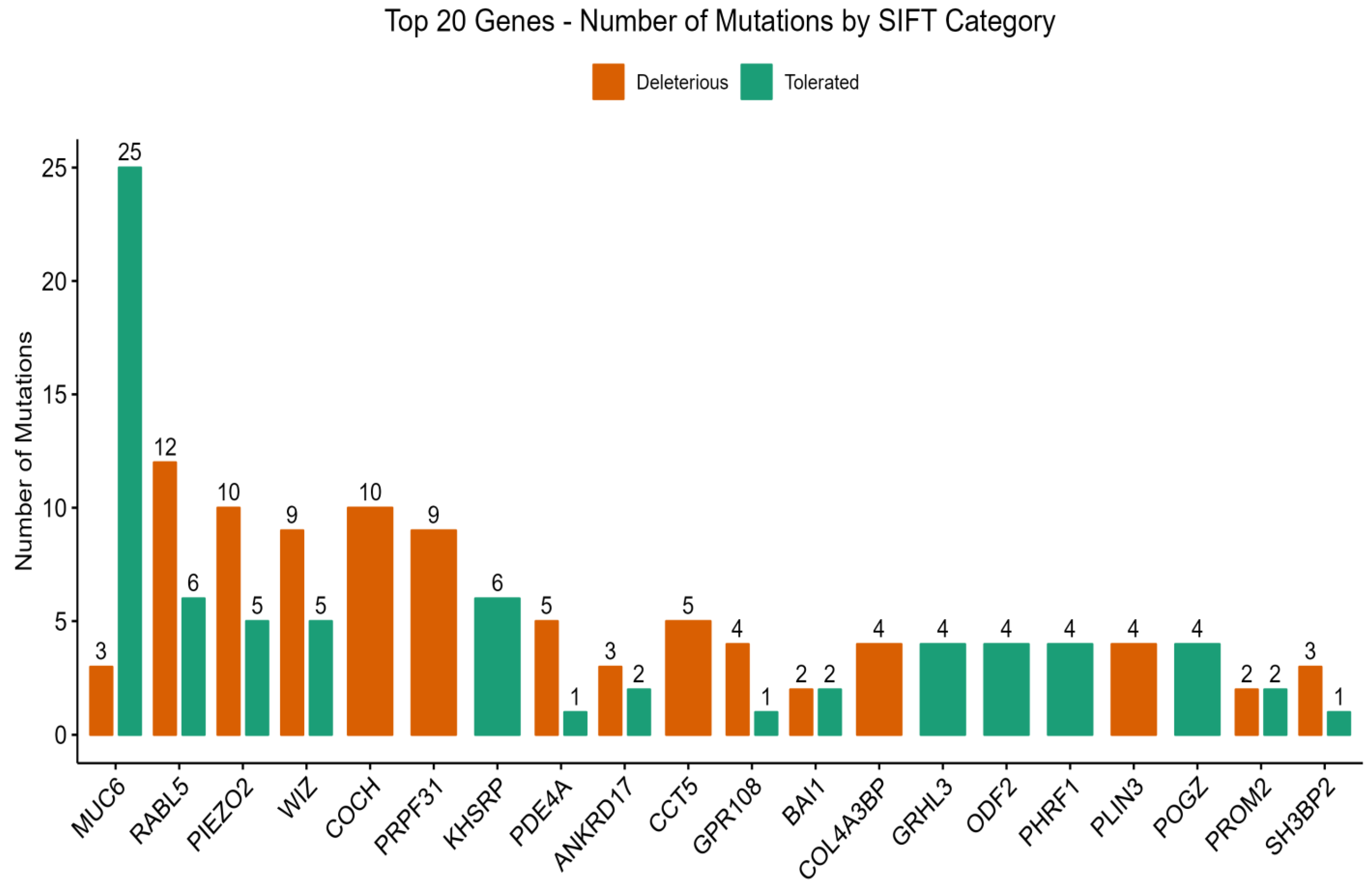
3.3. PolyPhen-2 and SIFT Score Distributions
3.4. Key Genes Linked to Type 2 Diabetes Mellitus (T2DM) and Diabetic Nephropathy (DN)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Location | Allele | Variant Type |
|---|---|---|---|
| ABCC8 | 11:17493912-17493912 | TGTT | intron_variant |
| 12:2550682-2550682 | C | intron_variant | |
| CACNA1D | 3:53848646-53848646 | T | downstream_gene_variant |
| IKBKB | 8:42156045-42156045 | ATG | intron_variant |
| INSR | 19:7293887-7293887 | C | upstream_gene_variant |
| IRS2 | 13:110424850-110424850 | A | intron_variant |
| KCNJ11 | 11:17415389-17415389 | G | upstream_gene_variant |
| MAFA | 8:144512253-144512253 | G | synonymous_variant |
| 8:144517130-144517130 | G | upstream_gene_variant | |
| MAPK1 | 22:22162633-22162633 | C | intron_variant |
| MAPK8 | 10:49515638-49515638 | A | intron_variant |
| 10:49515970-49515970 | T | intron_variant | |
| 10:49610716-49610716 | T | intron_variant | |
| 10:49612299-49612299 | A | intron_variant | |
| 10:49614180-49614180 | G | intron_variant | |
| 10:49620460-49620460 | A | downstream_gene_variant | |
| 10:49632740-49632740 | C | upstream_gene_variant | |
| 10:49648606-49648606 | A | downstream_gene_variant | |
| PIK3R3 | 1:46547692-46547692 | C | intron_variant |
| PRKCD | 3:53189911-53189911 | G | upstream_gene_variant |
| PRKCZ | 1:2074301-2074301 | C | upstream_gene_variant |
| SOCS1 | 16:11350612-11350612 | T | upstream_gene_variant |
| TNF | 6:31538847-31538847 | C | upstream_gene_variant |
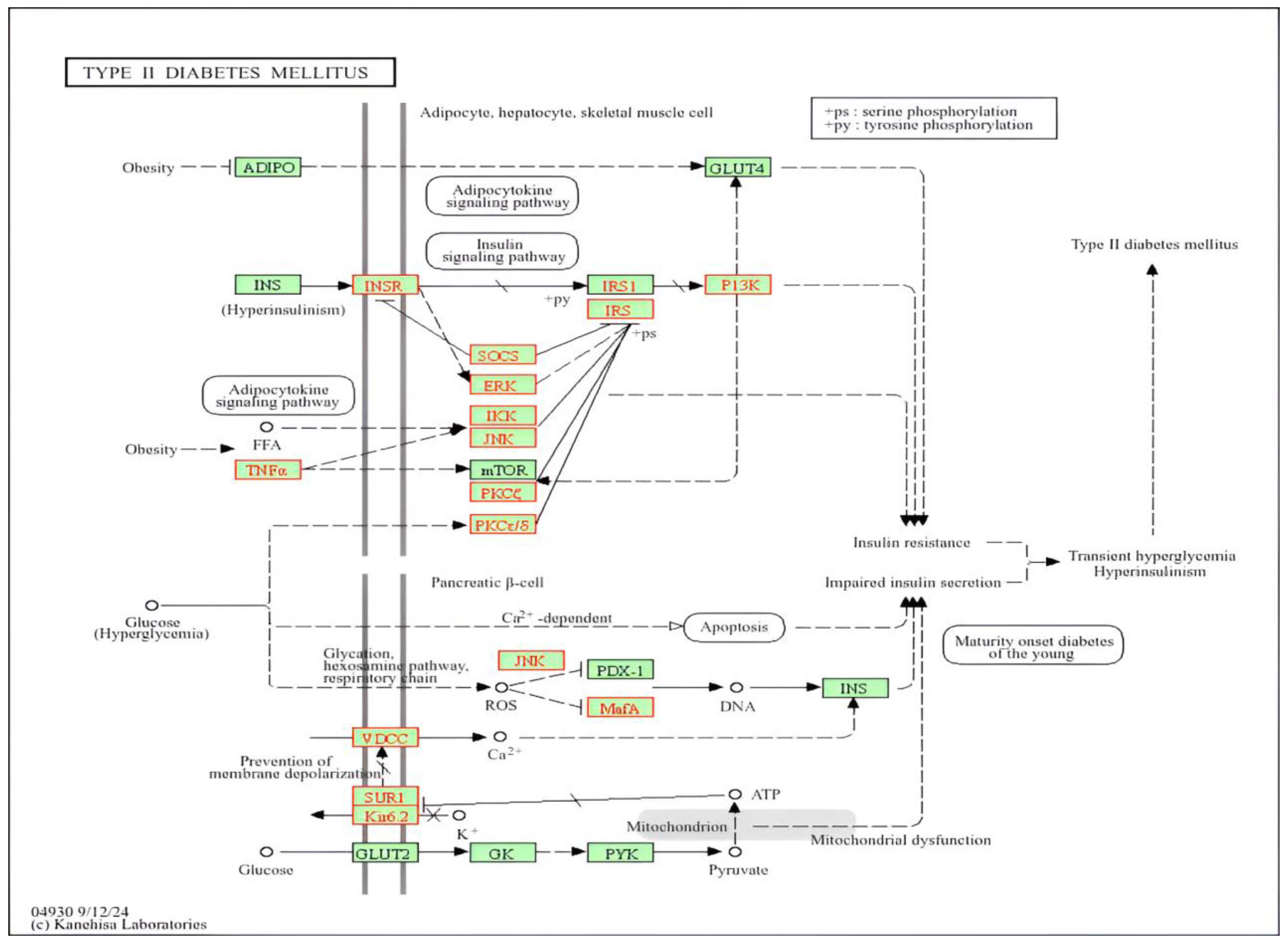
3.5. Biological Pathway Involvement
- a.
- Insulin Signaling: Insulin signaling includes genes INSR, IRS2, PIK3R3, PRKCZ, and various MAPK family members. Disruption in this axis will result in decreased glucose uptake, hyperglycemia, and increased lipolysis.
- b.
- β-Cell Function and Insulin Secretion: The important gene is CACNA1C. Changes in IKBKB, SOCS1, and MAPK8 aggravate pro-inflammatory networks, thereby fueling systemic insulin resistance and provoking local renal inflammation. Such persistent low-grade inflammation disturbs homeostasis in the kidney and may speed the progression toward end-stage renal disease (ESRD).
- c.
- Inflammatory Pathways: IKBKB, and some MAPK family members, highlight how immune regulators are intimately involved in the development of insulin resistance and the vascular inflammation that are the hallmark of diabetic complications. SOCS1 and MAPK8 aggravate pro-inflammatory networks.
3.6. Biological Processes Potentially Affected
3.7. Linking Genetic Findings to T2D Severity and Progression
- Polygenic Risk Load: a patient with damaging variants in both INSR and KCNJ11, for example, might experience concurring β-cell failure and insulin resistance, which will exacerbate hyperglycemia.
- Inflammatory Burden: other variants in TNF or IKBKB could fuel an inflammatory cascade that may, in turn, damage micro-vessels.
- Renal-Specific Dysregulation: opportunistically damaging variants at PRKCD, or certain MAPK genes, would additionally confer a renal stress element in an environment that dwells upon sugars, hence progressing kidney injury.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collaboration NCDRF. Worldwide trends in diabetes prevalence and treatment from 1990 to 2022: A pooled analysis of 1108 population-representative studies with 141 million participants. Lancet 2024, 404, 2077–2093. [Google Scholar] [CrossRef] [PubMed]
- Naaman, S.C.; Bakris, G.L. Diabetic Nephropathy: Update on Pillars of Therapy Slowing Progression. Diabetes Care 2023, 46, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Aljulifi, M.Z. Prevalence and reasons of increased type 2 diabetes in Gulf Cooperation Council Countries. Saudi Med. J. 2021, 42, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Pokharel, P.; Piganelli, J.D. Decoding the immune dance: Unraveling the interplay between beta cells and type 1 diabetes. Mol. Metab. 2024, 88, 101998. [Google Scholar] [CrossRef]
- Mlynarska, E.; Czarnik, W.; Dzieza, N.; Jedraszak, W.; Majchrowicz, G.; Prusinowski, F.; Stabrawa, M.; Rysz, J.; Franczyk, B. Type 2 Diabetes Mellitus: New Pathogenetic Mechanisms, Treatment and the Most Important Complications. Int. J. Mol. Sci. 2025, 26, 1094. [Google Scholar] [CrossRef]
- Choudhury, A.A.; Devi Rajeswari, V. Gestational diabetes mellitus—A metabolic and reproductive disorder. Biomed. Pharmacother. 2021, 143, 112183. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, W.; Liu, J.; Xie, M.; Liu, Q.; Li, S. Vascular complications of diabetes: A narrative review. Medicine 2023, 102, e35285. [Google Scholar] [CrossRef]
- Elfaki, I.; Mir, R.; Elnageeb, M.E.; Hamadi, A.; Alharbi, Z.M.; Bedaiwi, R.I.; Javid, J.; Alrasheed, T.; Alatawi, D.; Alrohaf, B.M.; et al. Identification of Interactive Genetic Loci Linked to Insulin Resistance in Metabolic Syndrome—An Update. Medicina 2025, 61, 83. [Google Scholar] [CrossRef]
- Natesan, V.; Kim, S.J. Diabetic Nephropathy—A Review of Risk Factors, Progression, Mechanism, and Dietary Management. Biomol. Ther. 2021, 29, 365–372. [Google Scholar] [CrossRef]
- Poulsen, C.G.; Jesse, K.; Carstensen, B.; Frimodt-Moller, M.; Hansen, T.W.; Persson, F.; Vistisen, D.; Rossing, P. Prognosis for Type 1 Diabetes with Diabetic Nephropathy between 2000 and 2020—Changes in Kidney Function Decline Over Time and Development of Cardiovascular Disease, Kidney Failure, and Mortality. Kidney Int. Rep. 2024, 9, 3403–3413. [Google Scholar] [CrossRef]
- Ma, X.; Liu, R.; Xi, X.; Zhuo, H.; Gu, Y. Global burden of chronic kidney disease due to diabetes mellitus, 1990-2021, and projections to 2050. Front. Endocrinol. 2025, 16, 1513008. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, I.T.; Stangou, M.; Sarafidis, P.; Gouliovaki, A.; Giamalis, P.; Tsouchnikas, I.; Didangelos, T.; Papagianni, A. TNF-alpha pathway and T-cell immunity are activated early during the development of diabetic nephropathy in Type II Diabetes Mellitus. Clin. Immunol. 2020, 215, 108423. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Matoba, K.; Utsunomiya, K. Signaling pathways in diabetic nephropathy. Histol. Histopathol. 2016, 31, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, R.; Wu, X.; Chen, Y.; Ji, W.; Wang, J.; Zhang, Y.; Xia, Y.; Tang, Y.; Yuan, J. The Wnt Signaling Pathway in Diabetic Nephropathy. Front. Cell Dev. Biol. 2021, 9, 701547. [Google Scholar] [CrossRef]
- John, S. Complication in diabetic nephropathy. Diabetes Metab. Syndr. 2016, 10, 247–249. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Fang, Y.; Zhou, S.; Liu, X.; Li, Z. Estimating the global prevalence of secondary hyperparathyroidism in patients with chronic kidney disease. Front. Endocrinol. 2024, 15, 1400891. [Google Scholar] [CrossRef]
- Tsai, S.F.; Tarng, D.C. Anemia in patients of diabetic kidney disease. Crit. Care Med. 2019, 82, 752–755. [Google Scholar] [CrossRef]
- Singh, S.; Kriti, M.; K S, A.; Sarma, D.K.; Verma, V.; Nagpal, R.; Mohania, D.; Tiwari, R.; Kumar, M. Deciphering the complex interplay of risk factors in type 2 diabetes mellitus: A comprehensive review. Metabol. Open 2024, 22, 100287. [Google Scholar] [CrossRef]
- McGrath, K.; Edi, R. Diabetic Kidney Disease: Diagnosis, Treatment, and Prevention. Am. Fam. Physician 2019, 99, 751–759. [Google Scholar]
- Simo, R.; Hernandez, C. What else can we do to prevent diabetic retinopathy? Diabetologia 2023, 66, 1614–1621. [Google Scholar] [CrossRef]
- Kornilov, S.A.; Rakhlin, N.; Koposov, R.; Lee, M.; Yrigollen, C.; Caglayan, A.O.; Magnuson, J.S.; Mane, S.; Chang, J.T.; Grigorenko, E.L. Genome-Wide Association and Exome Sequencing Study of Language Disorder in an Isolated Population. Pediatrics 2016, 137, e20152469. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.; Altemani, F.H.; Algehainy, N.A.; Alanazi, M.A.; Elfaki, I.; Alsayed, B.A.; Mir, M.M.; Mustafa, S.K.; Moawadh, M.S.; Tayeb, F.J.; et al. Identification of Novel Genomic Variants in COVID-19 Patients Using Whole-Exome Sequencing: Exploring the Plausible Targets of Functional Genomics. Biochem. Genet. 2024, 62, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.; Elfaki, I.; Alanazi, M.A.; Algehainy, N.A.; Altemani, F.H.; Alsayed, B.A.; Mohamed, E.I.; Mustafa, S.K.; Moawadh, M.S.; Tayeb, F.J.; et al. Whole-Exome Sequencing Detecting a Recurrent Pathogenic Mutation, HFE p.His63Asp (H63D) in COVID-19 Patients and Its Effect on Mortality. Discov. Med. 2024, 36, 1513–1526. [Google Scholar] [CrossRef]
- Elliott, A.M.; Adam, S.; du Souich, C.; Lehman, A.; Nelson, T.N.; van Karnebeek, C.; Alderman, E.; Armstrong, L.; Aubertin, G.; Blood, K.; et al. Genome-wide sequencing and the clinical diagnosis of genetic disease: The CAUSES study. HGG Adv. 2022, 3, 100108. [Google Scholar] [CrossRef] [PubMed]
- Javid, J.; Mir, R.; Elfaki, I.; Almotairi, R.; Barnawi, J.; Algehainy, N.A.; Jalal, M.M.; Altayar, M.A.; Alanazi, M.A.; Albalawi, S.O.; et al. Dysregulated Vitamin D, CYP2R1, TCF7L2, and CCR5 Delta32 Gene Variations are Associated with Coronary Artery Disease. Discov. Med. 2024, 36, 2287–2299. [Google Scholar] [CrossRef]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef]
- Sato, G.; Shirai, Y.; Namba, S.; Edahiro, R.; Sonehara, K.; Hata, T.; Uemura, M.; Biobank Japan, P.; Matsuda, K.; Doki, Y.; et al. Pan-cancer and cross-population genome-wide association studies dissect shared genetic backgrounds underlying carcinogenesis. Nat. Commun. 2023, 14, 3671. [Google Scholar] [CrossRef]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes-2023. Diabetes Care 2023, 46 (Suppl. S1), S19–S40. [Google Scholar] [CrossRef]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]
- Sandholm, N.; Van Zuydam, N.; Ahlqvist, E.; Juliusdottir, T.; Deshmukh, H.A.; Rayner, N.W.; Di Camillo, B.; Forsblom, C.; Fadista, J.; Ziemek, D.; et al. The Genetic Landscape of Renal Complications in Type 1 Diabetes. J. Am. Soc. Nephrol. 2017, 28, 557–574. [Google Scholar] [CrossRef]
- Rotroff, D.M.; Yee, S.W.; Zhou, K.; Marvel, S.W.; Shah, H.S.; Jack, J.R.; Havener, T.M.; Hedderson, M.M.; Kubo, M.; Herman, M.A.; et al. Genetic Variants in CPA6 and PRPF31 Are Associated with Variation in Response to Metformin in Individuals with Type 2 Diabetes. Diabetes 2018, 67, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Oba, R.; Ueno, H.; Oishi, A.; Nagahama, K.; Kanzaki, G.; Tsuboi, N.; Yokoo, T.; Nagase, M. Upregulation of Piezo2 and increased extracellular matrix protein in diabetic kidney disease mice. Hypertens. Res. 2025, 48, 1514–1528. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mesa, Y.; Cabo, R.; Gonzalez-Gay, M.; Garcia-Piqueras, J.; Vina, E.; Martinez, I.; Cobo, T.; Garcia-Suarez, O. Relationship of PIEZO1 and PIEZO2 vascular expression with diabetic neuropathy. Front. Physiol. 2023, 14, 1243966. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Shen, Y. Rab-like small GTPases in the regulation of ciliary Bardet-Biedl syndrome (BBS) complex transport. FEBS J. 2022, 289, 7359–7367. [Google Scholar] [CrossRef]
- Tian, X.; Zhao, H.; Zhou, J. Organization, functions, and mechanisms of the BBSome in development, ciliopathies, and beyond. eLife 2023, 12, e87623. [Google Scholar] [CrossRef]
- Okui, N.; Hachiya, T.; Horie, S. Pilot study using a discrete mathematical approach for topological analysis and ssGSEA of gene expression in autosomal recessive polycystic kidney disease. Sci. Rep. 2025, 15, 15559. [Google Scholar] [CrossRef]
- Xue, B.; Liu, Y.X.; Dong, B.; Wingfield, J.L.; Wu, M.; Sun, J.; Lechtreck, K.F.; Fan, Z.C. Intraflagellar transport protein RABL5/IFT22 recruits the BBSome to the basal body through the GTPase ARL6/BBS3. Proc. Natl. Acad. Sci. USA 2020, 117, 2496–2505. [Google Scholar] [CrossRef]
- Antona, V.; Scalia, F.; Giorgio, E.; Radio, F.C.; Brusco, A.; Oliveri, M.; Corsello, G.; Lo Celso, F.; Vadala, M.; Conway de Macario, E.; et al. A Novel CCT5 Missense Variant Associated with Early Onset Motor Neuropathy. Int. J. Mol. Sci. 2020, 21, 7631. [Google Scholar] [CrossRef]
- Min, W.; Angileri, F.; Luo, H.; Lauria, A.; Shanmugasundaram, M.; Almerico, A.M.; Cappello, F.; de Macario, E.C.; Lednev, I.K.; Macario, A.J.; et al. A human CCT5 gene mutation causing distal neuropathy impairs hexadecamer assembly in an archaeal model. Sci. Rep. 2014, 4, 6688. [Google Scholar] [CrossRef]
- Chopra, M.; McEntagart, M.; Clayton-Smith, J.; Platzer, K.; Shukla, A.; Girisha, K.M.; Kaur, A.; Kaur, P.; Pfundt, R.; Veenstra-Knol, H.; et al. Heterozygous ANKRD17 loss-of-function variants cause a syndrome with intellectual disability, speech delay, and dysmorphism. Am. J. Hum. Genet. 2021, 108, 1138–1150. [Google Scholar] [CrossRef]
- Bukova, I.; Szczerkowska, K.I.; Prochazkova, M.; Beck, I.M.; Prochazka, J.; Sedlacek, R. Loss of Wiz Function Affects Methylation Pattern in Palate Development and Leads to Cleft Palate. Front. Cell Dev. Biol. 2021, 9, 620692. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Ni, A.; Sun, L.; Li, S.; Li, G. Analysis of the Upregulated Expression Mechanism of Apoptotic Chromatin Condensation Inducer 1 in Hepatocellular Carcinoma Based on Bioinformatics. Turk. J. Gastroenterol. 2024, 35, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Mousa, M.; Albarguthi, S.; Albreiki, M.; Farooq, Z.; Sajid, S.; El Hajj Chehadeh, S.; ElBait, G.D.; Tay, G.; Deeb, A.A.; Alsafar, H. Whole-Exome Sequencing in Family Trios Reveals De Novo Mutations Associated with Type 1 Diabetes Mellitus. Biology 2023, 12, 413. [Google Scholar] [CrossRef] [PubMed]
- Pujar, M.; Vastrad, B.; Kavatagimath, S.; Vastrad, C.; Kotturshetti, S. Identification of candidate biomarkers and pathways associated with type 1 diabetes mellitus using bioinformatics analysis. Sci. Rep. 2022, 12, 9157. [Google Scholar] [CrossRef]
- Zou, L.; Li, W.; Han, J.; Yang, Y.; Jin, J.; Xiao, F.; Xu, X.; Zhai, Z. Identification of a low frequency missense mutation in MUC6 contributing to pulmonary artery hypertension by whole-exome sequencing. Pulm. Circ. 2018, 8, 2045894018794374. [Google Scholar] [CrossRef]
- Shiu, F.H.; Wong, J.C.; Yamamoto, T.; Lala, T.; Purcell, R.H.; Owino, S.; Zhu, D.; Van Meir, E.G.; Hall, R.A.; Escayg, A. Mice lacking full length Adgrb1 (Bai1) exhibit social deficits, increased seizure susceptibility, and altered brain development. Exp. Neurol. 2022, 351, 113994. [Google Scholar] [CrossRef]
- Alghamdi, A.; Alhotti, D.Z.; Sabico, S.; Al-Attas, O.S.; Al-Daghri, N.M. Associations of Perilipin 3 with Insulin Resistance in Arab Adults with Type 2 Diabetes. Dis. Markers. 2021, 2021, 4791915. [Google Scholar] [CrossRef]
- Saha, S.K.; Islam, S.M.R.; Kwak, K.S.; Rahman, M.S.; Cho, S.G. PROM1 and PROM2 expression differentially modulates clinical prognosis of cancer: A multiomics analysis. Cancer Gene Ther. 2020, 27, 147–167. [Google Scholar] [CrossRef]
- Jaszai, J.; Farkas, L.M.; Fargeas, C.A.; Janich, P.; Haase, M.; Huttner, W.B.; Corbeil, D. Prominin-2 is a novel marker of distal tubules and collecting ducts of the human and murine kidney. Histochem. Cell Biol. 2010, 133, 527–539. [Google Scholar] [CrossRef]
- Briata, P.; Bordo, D.; Puppo, M.; Gorlero, F.; Rossi, M.; Perrone-Bizzozero, N.; Gherzi, R. Diverse roles of the nucleic acid-binding protein KHSRP in cell differentiation and disease. Wiley Interdiscip. Rev. RNA 2016, 7, 227–240. [Google Scholar] [CrossRef]
- Barssotti, L.; Soares, G.M.; Marconato-Junior, E.; Lourenconi Alves, B.; Oliveira, K.M.; Carneiro, E.M.; Boschero, A.C.; Barbosa, H.C.L. KSRP improves pancreatic beta cell function and survival. Sci. Rep. 2024, 14, 6136. [Google Scholar] [CrossRef] [PubMed]
- Beysel, S.; Eyerci, N.; Pinarli, F.A.; Kizilgul, M.; Ozcelik, O.; Caliskan, M.; Cakal, E. HNF1A gene p.I27L is associated with early-onset, maturity-onset diabetes of the young-like diabetes in Turkey. BMC Endocr. Disord. 2019, 19, 51. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Ahmed, U.; Sim, M.F.M.; Bejar, A.; Zhang, X.; Talukder, M.M.U.; Rice, R.; Flannick, J.; Podgornaia, A.I.; Reilly, D.F.; et al. Discovering metabolic disease gene interactions by correlated effects on cellular morphology. Mol. Metab. 2019, 24, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Zhou, H.; Na, S.Y.; Niedra, R.; Peng, Y.; Wang, H.; Seed, B.; Zhou, G.L. GPR108, an NF-kappaB activator suppressed by TIRAP, negatively regulates TLR-triggered immune responses. PLoS ONE 2018, 13, e0205303. [Google Scholar] [CrossRef]
- Foresto-Neto, O.; Albino, A.H.; Arias, S.C.A.; Faustino, V.D.; Zambom, F.F.F.; Cenedeze, M.A.; Elias, R.M.; Malheiros, D.; Camara, N.O.S.; Fujihara, C.K.; et al. NF-kappaB System Is Chronically Activated and Promotes Glomerular Injury in Experimental Type 1 Diabetic Kidney Disease. Front. Physiol. 2020, 11, 84. [Google Scholar] [CrossRef]
- Yehualashet, A.S. Toll-like Receptors as a Potential Drug Target for Diabetes Mellitus and Diabetes-associated Complications. Diabetes Metab. Syndr. Obes. 2020, 13, 4763–4777. [Google Scholar] [CrossRef]
- Aly, R.H.; Ahmed, A.E.; Hozayen, W.G.; Rabea, A.M.; Ali, T.M.; El Askary, A.; Ahmed, O.M. Patterns of Toll-Like Receptor Expressions and Inflammatory Cytokine Levels and Their Implications in the Progress of Insulin Resistance and Diabetic Nephropathy in Type 2 Diabetic Patients. Front. Physiol. 2020, 11, 609223. [Google Scholar] [CrossRef]
- Tengholm, A. Cyclic AMP dynamics in the pancreatic beta-cell. Ups. J. Med. Sci. 2012, 117, 355–369. [Google Scholar] [CrossRef]
- Ookawara, M.; Nio, Y. Phosphodiesterase 4 inhibitors in diabetic nephropathy. Cell Signal 2022, 90, 110185. [Google Scholar] [CrossRef]
- Xu, R.; Fu, J.; Hu, Y.; Yang, X.; Tao, X.; Chen, L.; Huang, K.; Fu, Q. Roflumilast-Mediated Phosphodiesterase 4D Inhibition Reverses Diabetes-Associated Cardiac Dysfunction and Remodeling: Effects Beyond Glucose Lowering. Diabetes 2022, 71, 1660–1678. [Google Scholar] [CrossRef]
- Srivastava, T.; Sharma, M. Emerging Role of SH3BP2 as Regulator of Immune and Nonimmune Cells in Nephrotic Syndrome. Glomerular Dis. 2025, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Vairo, F.; Johnson, M.B.; Caswell, R.; Laver, T.W.; Lango Allen, H.; Hussain, K.; Ellard, S. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr. Diabetes 2017, 18, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Reinbothe, T.M.; Alkayyali, S.; Ahlqvist, E.; Tuomi, T.; Isomaa, B.; Lyssenko, V.; Renstrom, E. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia 2013, 56, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Mercader, J.M.; Puiggros, M.; Segre, A.V.; Planet, E.; Sorianello, E.; Sebastian, D.; Rodriguez-Cuenca, S.; Ribas, V.; Bonas-Guarch, S.; Draghici, S.; et al. Identification of novel type 2 diabetes candidate genes involved in the crosstalk between the mitochondrial and the insulin signaling systems. PLoS Genet. 2012, 8, e1003046. [Google Scholar] [CrossRef]
- Meyerovich, K.; Ortis, F.; Cardozo, A.K. The non-canonical NF-kappaB pathway and its contribution to beta-cell failure in diabetes. J. Mol. Endocrinol. 2018, 61, F1–F6. [Google Scholar] [CrossRef]
- Kubota, N.; Kubota, T.; Kajiwara, E.; Iwamura, T.; Kumagai, H.; Watanabe, T.; Inoue, M.; Takamoto, I.; Sasako, T.; Kumagai, K.; et al. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat. Commun. 2016, 7, 12977. [Google Scholar] [CrossRef]
- Hirose, M.; Inoue, K.; Matoba, S.; Tatebe, T.; Tokita, S.; Dodo, Y.; Tomishima, T.; Hasegawa, A.; Honda, A.; Ozaki, M.; et al. Disruption of insulin receptor substrate 2 (IRS2) causes non-obese type 2 diabetes with beta-cell dysfunction in the golden (Syrian) hamster. Sci. Rep. 2024, 14, 17450. [Google Scholar] [CrossRef]
- Cataldo, L.R.; Singh, T.; Achanta, K.; Bsharat, S.; Prasad, R.B.; Luan, C.; Renstrom, E.; Eliasson, L.; Artner, I. MAFA and MAFB regulate exocytosis-related genes in human beta-cells. Acta Physiol 2022, 234, e13761. [Google Scholar] [CrossRef]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific beta cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Flanagan, S.E.; Walker, E.; Quezado, R.; de Sousa Barros, F.A.; Caswell, R.; Johnson, M.B.; Wakeling, M.; Brandle, M.; Guo, M.; et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc. Natl. Acad. Sci. USA 2018, 115, 1027–1032. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Tsuchiya, K.; Yasuda, K.; Fujita, M.; Takinishi, A.; Furukawa, M.; Nitta, K.; Maeda, A. MafA is a Key Molecule in Glucose and Energy Balance in the Central Nervous System and Peripheral Organs. Int. J. Biomed. Sci. 2011, 7, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Zheng, Z.; Niu, B.; Su, J.; Su, H. Different MAPK signal transduction pathways play different roles in the impairment of glucose-stimulated insulin secretion in response to IL-1beta. Mol. Med. Rep. 2020, 22, 2973–2980. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Yu, H.; Choi, E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp. Mol. Med. 2020, 52, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Han, S.; Wang, C.; Chen, H.; Xu, Q.; Feng, S.; Wang, Y.; Yao, J.; Zhou, Q.; Tang, X.; et al. MAPK1 Mediates MAM Disruption and Mitochondrial Dysfunction in Diabetic Kidney Disease via the PACS-2-Dependent Mechanism. Int. J. Biol. Sci. 2024, 20, 569–584. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Sun, Z.; Wang, H.; Ning, R.; Xu, L.; Zhao, Y.; Yang, K.; Xi, X.; Tian, J. MAPK8 and CAPN1 as potential biomarkers of intervertebral disc degeneration overlapping immune infiltration, autophagy, and ceRNA. Front. Immunol. 2023, 14, 1188774. [Google Scholar] [CrossRef]
- Cassidy, H.; Radford, R.; Slyne, J.; O’Connell, S.; Slattery, C.; Ryan, M.P.; McMorrow, T. The role of MAPK in drug-induced kidney injury. J. Signal Transduct. 2012, 2012, 463617. [Google Scholar] [CrossRef]
- Yang, X.; Fu, Y.; Hu, F.; Luo, X.; Hu, J.; Wang, G. PIK3R3 regulates PPARalpha expression to stimulate fatty acid beta-oxidation and decrease hepatosteatosis. Exp. Mol. Med. 2018, 50, e431. [Google Scholar] [CrossRef]
- Gemeda, D.; Abebe, E.; Duguma, A. Metabolic Syndrome and Its Associated Factors among Type 2 Diabetic Patients in Southwest Ethiopia, 2021/2022. J. Diabetes Res. 2022, 2022, 8162342. [Google Scholar] [CrossRef]
- Fleming, A.K.; Storz, P. Protein kinase C isoforms in the normal pancreas and in pancreatic disease. Cell Signal 2017, 40, 1–9. [Google Scholar] [CrossRef]
- Langham, R.G.; Kelly, D.J.; Gow, R.M.; Zhang, Y.; Cox, A.J.; Qi, W.; Thai, K.; Pollock, C.A.; Christensen, P.K.; Parving, H.H.; et al. Increased renal gene transcription of protein kinase C-beta in human diabetic nephropathy: Relationship to long-term glycaemic control. Diabetologia 2008, 51, 668–674. [Google Scholar] [CrossRef]
- Bezy, O.; Tran, T.T.; Pihlajamaki, J.; Suzuki, R.; Emanuelli, B.; Winnay, J.; Mori, M.A.; Haas, J.; Biddinger, S.B.; Leitges, M.; et al. PKCdelta regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J. Clin. Investig. 2011, 121, 2504–2517. [Google Scholar] [CrossRef] [PubMed]
- Talior, I.; Tennenbaum, T.; Kuroki, T.; Eldar-Finkelman, H. PKC-delta-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice: Role for NADPH oxidase. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E405–E411. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Cheung, S.C.; Lan, L.L.; Ho, S.K.; Chan, J.C.; Tong, P.C. The pivotal role of protein kinase C zeta (PKCzeta) in insulin- and AMP-activated protein kinase (AMPK)-mediated glucose uptake in muscle cells. Cell Signal 2010, 22, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; He, A.B.; Liu, X.J.; Li, Y.; Chang, Y.S.; Fang, F.D. Protein kinase Czeta and glucose uptake. Biochemistry 2006, 71, 701–706. [Google Scholar] [CrossRef]
- Pan, D.; Xu, L.; Guo, M. The role of protein kinase C in diabetic microvascular complications. Front. Endocrinol. 2022, 13, 973058. [Google Scholar] [CrossRef]
- Galic, S.; Sachithanandan, N.; Kay, T.W.; Steinberg, G.R. Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem. J. 2014, 461, 177–188. [Google Scholar] [CrossRef]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol. Cell Biol. 2004, 24, 5434–5446. [Google Scholar] [CrossRef]
- Suchy, D.; Labuzek, K.; Machnik, G.; Kozlowski, M.; Okopien, B. SOCS and diabetes--ups and downs of a turbulent relationship. Cell Biochem. Funct. 2013, 31, 181–195. [Google Scholar] [CrossRef]
- Elfaki, I.; Bayer, P.; Mueller, J.W. A potential transcriptional regulator is out-of-frame translated from the metallothionein 2A messenger RNA. Anal. Biochem. 2011, 409, 159–161. [Google Scholar] [CrossRef]
- Elfaki, I.; Knitsch, A.; Matena, A.; Bayer, P. Identification and characterization of peptides that bind the PPIase domain of Parvulin17. J. Pept. Sci. 2013, 19, 362–369. [Google Scholar] [CrossRef]
- Sotomayor-Vivas, C.; Hernandez-Lemus, E.; Dorantes-Gilardi, R. Linking protein structural and functional change to mutation using amino acid networks. PLoS ONE 2022, 17, e0261829. [Google Scholar] [CrossRef]
| Characteristic | N = 26 1 |
|---|---|
| Age (Years) | 63 (59, 66) |
| Age Groups | |
| >60 Years | 16 (61.53%) |
| 40–60 Years | 10 (38.46%) |
| Gender | |
| Female | 8 (30.76%) |
| Male | 18 (69.23%) |
| Duration of T2D (Years) | 9.00 (7.00, 10.00) |
| Blood Sugar (Fasting) | 156 (145, 160) |
| HBA1c (%) | 7.40 (7.05, 7.50) |
| HBA1c Groups | |
| Diabetic | 24 (90.30%) |
| Control diabetic | 2 (7.69%) |
| BMI (kg/m2) | 27.68 (22.24, 30.05) |
| BMI Groups | |
| Normal | 10 (38.46%) |
| Obese | 6 (23.07%) |
| Overweight | 10 (38.46%) |
| Triglyceride (mg/dL) | 210 (189, 242) |
| Triglyceride Levels | |
| <150 mg/dL | 2 (7.70%) |
| ≥150 mg/dL | 24 (92.30%) |
| Total cholesterol (mg/dL) | |
| <200 mg/dL | 12(46.15%) |
| >200 mg/dL | 14(53.84%) |
| HDL (mg/dL) | 34 (29, 48) |
| <40 mg/dL | 15 (57.69%) |
| ≥40 mg/dL | 11 (42.30%) |
| LDL (mg/dL) | |
| >190 mg/dl | 13 (50%) |
| 160–190 mg/dL | 10 (38.46%) |
| 100–160 mg/dL | 03 (11.53%) |
| VLDL (mg/dL) | |
| >40 mg/dL | 10 (38.46%) |
| 5–40 mg/dL | 16 (61.53%) |
| Creatinine (mg/dL) | 2.50 (1.90, 2.60) |
| BILIRUBIN (mg/dL) | 1 (0, 2) |
| AST (U/L) | 30 (20, 40) |
| ALT (U/L) | 42 (37, 48) |
| ALP (U/L) | 97 (73, 110) |
| Estimated glomerular filtration rate; | |
| eGFR, mL/min per 1.73 m2 | 41.0 (14.0–59.0) |
| Urinary albumin excretion (UAE) | |
| UAE, mg/g | 90.5 (35.8–388.7) |
| 30 mg/g | 0 |
| >30 to 300 mg/g | 26 |
| Arterial hypertension, % | 90.3 |
| Blood pressure | |
| SBP [mmHg] | 150.0 [140.3–162.0] |
| DBP [mmHg] | 87.0 [78.0–92.0] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elfaki, I.; Mir, R.; Almowallad, S.; Almassabi, R.F.; Albalawi, W.; Albalawi, A.D.; Bhat, A.A.; Barnawi, J.; Tayeb, F.J.; Jalal, M.M.; et al. Whole Exome Sequencing in 26 Saudi Patients Expands the Mutational and Clinical Spectrum of Diabetic Nephropathy. Medicina 2025, 61, 1017. https://doi.org/10.3390/medicina61061017
Elfaki I, Mir R, Almowallad S, Almassabi RF, Albalawi W, Albalawi AD, Bhat AA, Barnawi J, Tayeb FJ, Jalal MM, et al. Whole Exome Sequencing in 26 Saudi Patients Expands the Mutational and Clinical Spectrum of Diabetic Nephropathy. Medicina. 2025; 61(6):1017. https://doi.org/10.3390/medicina61061017
Chicago/Turabian StyleElfaki, Imadeldin, Rashid Mir, Sanaa Almowallad, Rehab F. Almassabi, Wed Albalawi, Aziz Dhaher Albalawi, Ajaz A. Bhat, Jameel Barnawi, Faris J. Tayeb, Mohammed M. Jalal, and et al. 2025. "Whole Exome Sequencing in 26 Saudi Patients Expands the Mutational and Clinical Spectrum of Diabetic Nephropathy" Medicina 61, no. 6: 1017. https://doi.org/10.3390/medicina61061017
APA StyleElfaki, I., Mir, R., Almowallad, S., Almassabi, R. F., Albalawi, W., Albalawi, A. D., Bhat, A. A., Barnawi, J., Tayeb, F. J., Jalal, M. M., Altayar, M. A., & Altemani, F. H. (2025). Whole Exome Sequencing in 26 Saudi Patients Expands the Mutational and Clinical Spectrum of Diabetic Nephropathy. Medicina, 61(6), 1017. https://doi.org/10.3390/medicina61061017