
Pulmonary Artery Pulsatility Index in Acute and Chronic Pulmonary Embolism

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Data
2.2. Ethics
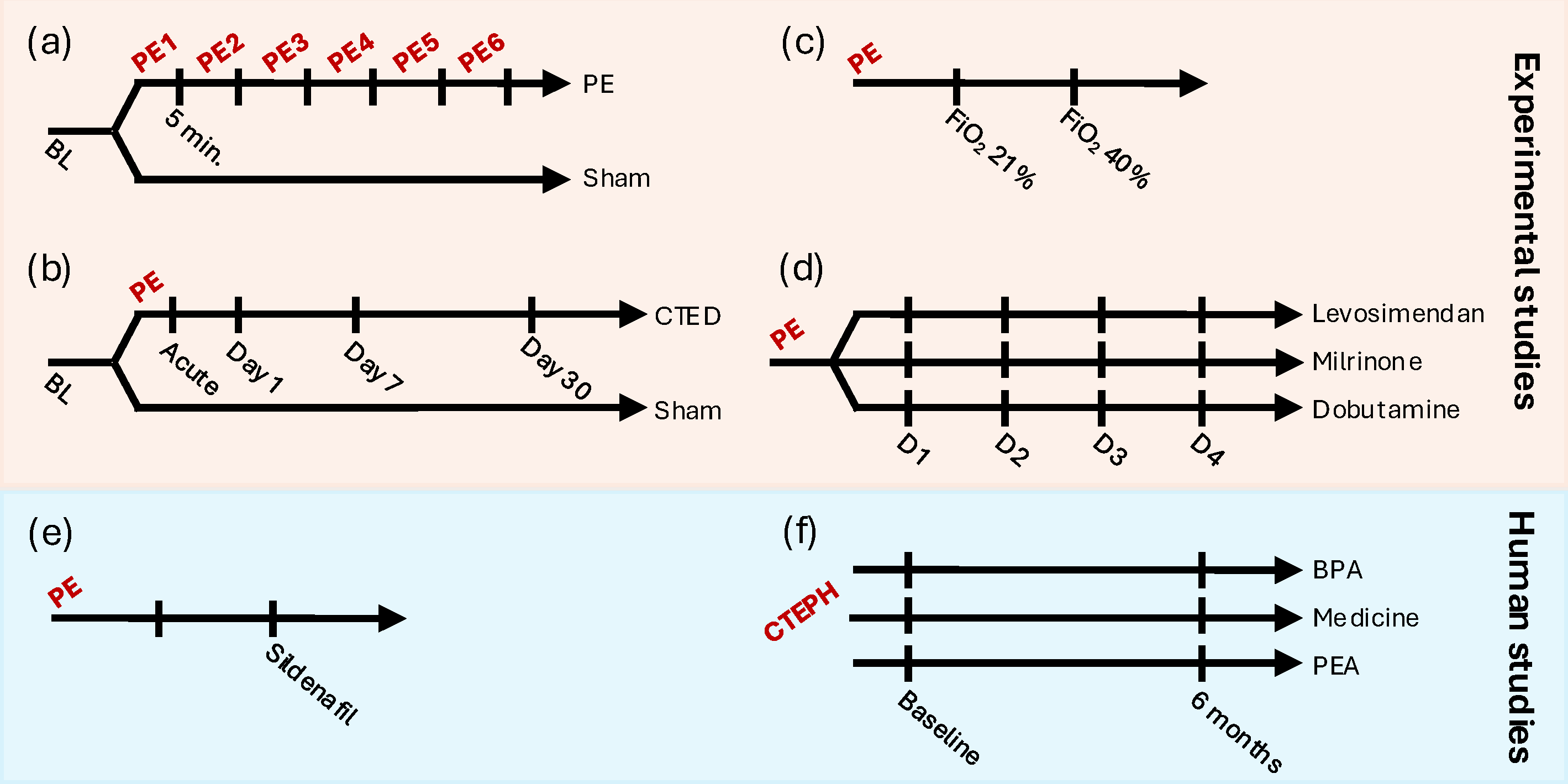
2.3. Experimental Studies
2.4. Human Studies
2.5. Statistical Analysis
3. Results
3.1. Experimental Results
3.2. Human Results
4. Discussion
4.1. Acute Pulmonary Embolism
4.2. Chronic Thromboembolic Pulmonary Hypertension

{kind=link}
{kind=link}
{kind=link}
| Strengths | Limitations |
|---|---|
|
|
|
|
|
|
| Results from the present study | |
|
|
|
|
| |
4.3. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Konstantinides, S.V.; Meyer, G.; Becattini, C.; Bueno, H.; Geersing, G.J.; Harjola, V.-P.; Huisman, M.V.; Humbert, M.; Jennings, C.S.; Jiménez, D.; et al. 2019 ESC Guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS). Eur. Heart J. 2020, 41, 543–603. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2018, 137, e578–e622. [Google Scholar] [CrossRef]
- Sanz, J.; Sánchez-Quintana, D.; Bossone, E.; Bogaard, H.J.; Naeije, R. Anatomy, Function, and Dysfunction of the Right Ventricle. J. Am. Coll. Cardiol. 2019, 73, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Manek, G.; Gupta, M.; Chhabria, M.; Bajaj, D.; Agrawal, A.; Tonelli, A.R. Hemodynamic indices in pulmonary hypertension: A narrative review. Cardiovasc. Diagn. Ther. 2022, 12, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Gustafsson, F. Pulmonary artery pulsatility index: Physiological basis and clinical application. Eur. J. Heart Fail. 2020, 22, 32–38. [Google Scholar] [CrossRef]
- Zochios, V.; Yusuff, H.; Schmidt, M. Acute Right Ventricular Injury Phenotyping in ARDS. Intensive Care Med. 2023, 49, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Dugar, S.; Cheungpasitporn, W.; Schleicher, M.; Collier, P.; Vallabhajosyula, S.; Duggal, A. The Impact of Right Ventricular Injury on the Mortality in Patients with Acute Respiratory Distress Syndrome: A Systematic Review and Meta-Analysis. Crit. Care 2021, 25, 172. [Google Scholar] [CrossRef]
- Akamkam, A.; Galand, V.; Jungling, M.; Delmas, C.; Dambrin, C.; Pernot, M.; Kindo, M.; Gaudard, P.; Rouviere, P.; Senage, T.; et al. Association between pulmonary artery pulsatility and mortality after implantation of left ventricular assist device. ESC Heart Fail. 2024, 11, 2100–2112. [Google Scholar] [CrossRef] [PubMed]
- Kochav, S.M.; Flores, R.J.; Truby, L.K.; Topkara, V.K. Prognostic Impact of Pulmonary Artery Pulsatility Index (PAPi) in Patients With Advanced Heart Failure: Insights From the ESCAPE Trial. J. Card. Fail. 2018, 24, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Mazimba, S.; Welch, T.S.; Mwansa, H.; Breathett, K.K.; Kennedy, J.L.; Mihalek, A.D.; Harding, W.C.; Mysore, M.M.; Zhuo, D.X.; Bilchick, K.C. Haemodynamically Derived Pulmonary Artery Pulsatility Index Predicts Mortality in Pulmonary Arterial Hypertension. Heart Lung Circ. 2019, 28, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Martin-Suarez, S.; Gliozzi, G.; Cavalli, G.G.; Orioli, V.; Loforte, A.; Pastore, S.; Rossi, B.; Zardin, D.; Galiè, N.; Palazzini, M.; et al. Is Pulmonary Artery Pulsatility Index (PAPi) a Predictor of Outcome after Pulmonary Endarterectomy? J. Clin. Med. 2022, 11, 4353. [Google Scholar] [CrossRef]
- Dragsbaek, S.J.; Lyhne, M.D.; Hansen, J.V.; Pedersen, C.C.; Jujo-Sanada, T.; Karout, L.; Kalra, M.K.; Nielsen-Kudsk, J.E.; Andersen, A. A porcine model of human-like chronic thromboembolic pulmonary disease. Thromb. Res. 2023, 231, 25–28. [Google Scholar] [CrossRef]
- Lyhne, M.D.; Schultz, J.G.; Mortensen, C.S.; Kramer, A.; Nielsen-Kudsk, J.E.; Andersen, A. Immediate cardiopulmonary responses to consecutive pulmonary embolism: A randomized, controlled, experimental study. BMC Pulm. Med. 2024, 24, 233. [Google Scholar] [CrossRef] [PubMed]
- Lyhne, M.D.; Hansen, J.V.; Dragsbæk, S.J.; Mortensen, C.S.; Nielsen-Kudsk, J.E.; Andersen, A. Oxygen Therapy Lowers Right Ventricular Afterload in Experimental Acute Pulmonary Embolism. Crit. Care Med. 2021, 49, e891–e901. [Google Scholar] [CrossRef] [PubMed]
- Lyhne, M.D.; Dragsbaek, S.J.; Hansen, J.V.; Schultz, J.G.; Andersen, A.; Nielsen-Kudsk, J.E. Levosimendan, milrinone, and dobutamine in experimental acute pulmonary embolism. Pulm. Circ. 2021, 11, 20458940211022977. [Google Scholar] [CrossRef]
- Andersen, A.; Waziri, F.; Schultz, J.G.; Holmboe, S.; Becker, S.W.; Jensen, T.; Søndergaard, H.M.; Dodt, K.K.; May, O.; Mortensen, U.M.; et al. Pulmonary vasodilation by sildenafil in acute intermediate-high risk pulmonary embolism: A randomized explorative trial. BMC Pulm. Med. 2021, 21, 72. [Google Scholar] [CrossRef] [PubMed]
- Lyhne, M.D.; Hansen, J.V.; Andersen, S.; Schultz, J.G.; Sørensen, S.G.; Kirk, M.E.; Merit, V.T.; Andersen, M.J.; Mellemkjær, S.; Ilkjær, L.B.; et al. Right ventricular to pulmonary artery coupling in chronic thromboembolic pulmonary hypertension. Int. J. Cardiol. 2025, 418, 132639. [Google Scholar] [CrossRef] [PubMed]
- Brener, M.I.; Kanwar, M.K.; Lander, M.M.; Hamid, N.B.; Raina, A.; Sethi, S.S.; Finn, M.T.; Fried, J.A.; Raikhelkar, J.; Masoumi, A.; et al. Impact of Interventricular Interaction on Ventricular Function Insights From Right Ventricular Pressure-Volume Analysis. JACC Heart Fail. 2024, 12, 1179–1192. [Google Scholar] [CrossRef]
- Tello, K.; Wan, J.; Dalmer, A.; Vanderpool, R.; Ghofrani, H.A.; Naeije, R.; Roller, F.; Mohajerani, E.; Seeger, W.; Herberg, U.; et al. Validation of the Tricuspid Annular Plane Systolic Excursion/Systolic Pulmonary Artery Pressure Ratio for the Assessment of Right Ventricular-Arterial Coupling in Severe Pulmonary Hypertension. Circ. Cardiovasc. Imaging 2019, 12, e009047. [Google Scholar] [CrossRef] [PubMed]
- Du Sert, N.P.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J. Physiol. 2020, 598, 3793–3801. [Google Scholar] [CrossRef]
- Rudski, L.G.; Lai, W.W.; Afilalo, J.; Hua, L.; Handschumacher, M.D.; Chandrasekaran, K.; Solomon, S.D.; Louie, E.K.; Schiller, N.B. Guidelines for the Echocardiographic Assessment of the Right Heart in Adults: A Report from the American Society of Echocardiography Endorsed by the European Association of Echocardiography, a Registered Branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J. Am. Soc. Echocardiogr. 2010, 23, 685–713. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for Cardiac Chamber Quantification by Echocardiography in Adults: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39e14. [Google Scholar] [CrossRef]
- Zern, E.K.; Wang, D.; Rambarat, P.; Bernard, S.; Paniagua, S.M.; Liu, E.E.; McNeill, J.; Wang, J.K.; Andrews, C.T.; Pomerantsev, E.V.; et al. Association of Pulmonary Artery Pulsatility Index With Adverse Cardiovascular Events Across a Hospital-Based Sample. Circ. Heart Fail. 2022, 15, e009085. [Google Scholar] [CrossRef]
- Omar, H.R.; Barlow, M.; Guglin, M. Discharge pulmonary artery pulsatility index predicts morbidity and mortality after acute heart failure: From the ESCAPE trial. Am. Heart J. Plus 2021, 1, 100003. [Google Scholar] [CrossRef] [PubMed]
- Smulders, Y.M. Contribution of pulmonary vasoconstriction to haemodynamic instability after acute pulmonary embolism. Implications for treatment? Neth. J. Med. 2001, 58, 241–247. [Google Scholar] [CrossRef]
- Sławek-Szmyt, S.; Araszkiewicz, A.; Jankiewicz, S.; Grygier, M.; Mularek-Kubzdela, T.; Lesiak, M. Prognostic Value of Pulmonary Artery Pulsatility Index in Right Ventricle Failure-Related Mortality in Inoperable Chronic Thromboembolic Pulmonary Hypertension. J. Clin. Med. 2022, 11, 2735. [Google Scholar] [CrossRef] [PubMed]
- Waziri, F.; Mellemkjær, S.; Clemmensen, T.S.; Hjortdal, V.E.; Ilkjær, L.B.; Nielsen, S.L.; Poulsen, S.H. Long-Term Changes of Exercise Hemodynamics and Physical Capacity in Chronic Thromboembolic Pulmonary Hypertension after Pulmonary Thromboendarterectomy. Int. J. Cardiol. 2020, 317, 181–187. [Google Scholar] [CrossRef]
- Korshin, A.; Grønlykke, L.; Nilsson, J.C.; Møller-Sørensen, H.; Ihlemann, N.; Kjøller, S.M.; Damgaard, S.; Lehnert, P.; Hassager, C.; Kjaergaard, J.; et al. Tricuspid Annular Plane Systolic Excursion Is Significantly Reduced during Uncomplicated Coronary Artery Bypass Surgery: A Prospective Observational Study. J. Thorac. Cardiovasc. Surg. 2019, 158, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Cao, L.; Movahed, A. Altered Right Ventricular Contractile Pattern after Cardiac Surgery: Monitoring of Septal Function Is Essential. Echocardiography 2014, 31, 1159–1165. [Google Scholar] [CrossRef]
- Lim, Y.; Low, T.; Chan, S.P.; Lin, W.; Teo, T.W.; Jang, J.J.; Kuntjoro, I.; Tay, E.L.; Yip, J.W. Does pulmonary artery pulsatility index predict mortality in pulmonary arterial hypertension? ESC Heart Fail. 2021, 8, 3835–3844. [Google Scholar] [CrossRef]
- Witkin, A.S.; Channick, R.N. Chronic Thromboembolic Pulmonary Hypertension: The End Result of Pulmonary Embolism. Curr. Cardiol. Rep. 2015, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Golbin, J.M.; Shukla, N.; Nero, N.; Hockstein, M.A.; Tonelli, A.R.; Siuba, M.T. Non-invasive Surrogates for Right Ventricular-Pulmonary Arterial Coupling: A Systematic Review and Meta-Analysis. Pulm. Circ. 2024, 14, e70004. [Google Scholar] [CrossRef] [PubMed]


| Acute PE (n = 10) [16] | CTEPH (n = 130) [17] | |
|---|---|---|
| Age, years | 63 ± 9 | 70 [59–75] |
| Sex, male | 7 (70%) | 72 (52%) |
| BMI, kg/m2 | 30 ± 4 | 27 [24–31] |
| COPD | 2 (20%) | 37 (27%) |
| Smoking | 10 (100%) | 83 (60%) |
| Previous VTE | 1 (10%) | 129 (93%) |
| Heart rate, bpm | 85 ± 12 | 78 [70–87] |
| Systolic arterial pressure, mmHg | 137 ± 24 | 131 [119–148] |
| Right heart catheterization | ||
| RAP, mmHg | 7 ± 2 | 8 [5–11] |
| mPAP, mmHg | 27 ± 4 | 40 ± 10 |
| PASP, mmHg | 47 ± 8 | 77 ± 19 |
| PADP, mmHg | 17 ± 3 | 27 ± 8 |
| PVR, WU | 2.3 ± 1.0 | 8.4 [5.6–11.8] |
| Transthoracic echocardiography | ||
| TAPSE, mm | 17 ± 4 | 18 ± 5 |
| TRG, m/s | 2.9 ± 1.4 | 4.1 ± 0.7 |
| RV/LV | 1.1 ± 0.1 | 1.2 ± 0.3 |
| RV FAC, % | 34 ± 15 | 25 ± 12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyhne, M.D.; Yuriditsky, E.; Zochios, V.; Dragsbaek, S.J.; Hansen, J.V.; Andersen, M.J.; Mellemkjær, S.; Kabrhel, C.; Andersen, A. Pulmonary Artery Pulsatility Index in Acute and Chronic Pulmonary Embolism. Medicina 2025, 61, 363. https://doi.org/10.3390/medicina61020363
Lyhne MD, Yuriditsky E, Zochios V, Dragsbaek SJ, Hansen JV, Andersen MJ, Mellemkjær S, Kabrhel C, Andersen A. Pulmonary Artery Pulsatility Index in Acute and Chronic Pulmonary Embolism. Medicina. 2025; 61(2):363. https://doi.org/10.3390/medicina61020363
Chicago/Turabian StyleLyhne, Mads Dam, Eugene Yuriditsky, Vasileios Zochios, Simone Juel Dragsbaek, Jacob Valentin Hansen, Mads Jønsson Andersen, Søren Mellemkjær, Christopher Kabrhel, and Asger Andersen. 2025. "Pulmonary Artery Pulsatility Index in Acute and Chronic Pulmonary Embolism" Medicina 61, no. 2: 363. https://doi.org/10.3390/medicina61020363
APA StyleLyhne, M. D., Yuriditsky, E., Zochios, V., Dragsbaek, S. J., Hansen, J. V., Andersen, M. J., Mellemkjær, S., Kabrhel, C., & Andersen, A. (2025). Pulmonary Artery Pulsatility Index in Acute and Chronic Pulmonary Embolism. Medicina, 61(2), 363. https://doi.org/10.3390/medicina61020363