

RP1 Dominant p.Ser740* Pathogenic Variant in 20 Knowingly Unrelated Families Affected by Rod–Cone Dystrophy: Potential Founder Effect in Western Sicily

 , , , ,
, , , ,  , ,
, ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical and Instrumental Evaluation
2.2. Transmission Mode Definition
2.3. Genetic Analytic Strategy
2.4. Statistical Evaluation
3. Results
3.1. Molecular Data Analysis
3.2. Phenotype–Genotype Correlation in RP1-Associated ADRP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haim, M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol. Scand. 2002, 233, 1–34. [Google Scholar] [CrossRef]
- Hamel, C. Updated 2014. Available online: https://www.orpha.net (accessed on 20 March 2023).
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 2007, 125, 151–158. [Google Scholar] [CrossRef]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands with Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef]
- Fahim, A.T.; Daiger, S.P.; Weleber, R.G. Nonsyndromic Retinitis Pigmentosa Overview. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- RetNet. Available online: https://sph.uth.edu/retnet (accessed on 20 March 2023).
- OMIM. Available online: https://www.omim.org/ (accessed on 20 March 2023).
- Pierce, E.A.; Quinn, T.; Meehan, T.; McGee, T.L.; Berson, E.L.; Dryja, T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999, 22, 248–254. [Google Scholar] [CrossRef]
- Audo, I.; Mohand-Saïd, S.; Dhaenens, C.M.; Germain, A.; Orhan, E.; Antonio, A.; Hamel, C.; Sahel, J.A.; Bhattacharya, S.S.; Zeitz, C. RP1 and autosomal dominant rod-cone dystrophy: Novel mutations, a review of published variants, and genotype-phenotype correlation. Hum. Mutat. 2012, 33, 73–80. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, J.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; Smith, J.E.; Sullivan, L.S.; Zuo, J.; Milam, A.H.; Pierce, E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Investig. Ophthalmol. Vis. Sci. 2002, 43, 22–32. [Google Scholar]
- Chen, L.J.; Lai, T.Y.; Tam, P.O.; Chiang, S.W.; Zhang, X.; Lam, S.; Lai, R.Y.; Lam, D.S.; Pang, C.P. Compound heterozygosity of two novel truncation mutations in RP1 causing autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2236–2242. [Google Scholar] [CrossRef]
- Khaliq, S.; Abid, A.; Ismail, M.; Hameed, A.; Mohyuddin, A.; Lall, P.; Aziz, A.; Anwar, K.; Mehdi, S.Q. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J. Med. Genet. 2005, 42, 436–438. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, X.; Li, S.; Wang, P.; Sun, W.; Zhang, Q. Dominant RP in the Middle While Recessive in Both the N- and C-Terminals Due to RP1 Truncations: Confirmation, Refinement, and Questions. Front. Cell Dev. Biol. 2021, 9, 634478. [Google Scholar] [CrossRef]
- Payne, A.; Vithana, E.; Khaliq, S.; Hameed, A.; Deller, J.; Abu-Safieh, L.; Kermani, S.; Leroy, B.P.; Mehdi, S.Q.; Moore, A.T.; et al. RP1 protein truncating mutations predominate at the RP1 adRP locus. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4069–4073. [Google Scholar]
- Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V.; Swaroop, A.; Jacobson, S.G.; Stone, E.M. De novo mutation in the RP1 gene (Arg677ter) associated with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3593–3597. [Google Scholar] [CrossRef]
- Kabir, F.; Ullah, I.; Ali, S.; Gottsch, A.D.; Naeem, M.A.; Assir, M.Z.; Khan, S.N.; Akram, J.; Riazuddin, S.; Ayyagari, R.; et al. Loss of function mutations in RP1 are responsible for retinitis pigmentosa in consanguineous familial cases. Mol. Vis. 2016, 22, 610–625. [Google Scholar]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Karali, M.; Testa, F.; Di Iorio, V.; Torella, A.; Zeuli, R.; Scarpato, M.; Romano, F.; Onore, M.E.; Pizzo, M.; Melillo, P.; et al. Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 2022, 12, 20815. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Méjécase, C.; Kozak, I.; Moosajee, M. The genetic landscape of inherited eye disorders in 74 consecutive families from the United Arab Emirates. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 762–772. [Google Scholar] [CrossRef]
- Capelli, C.; Onofri, V.; Brisighelli, F.; Boschi, I.; Scarnicci, F.; Masullo, M.; Ferri, G.; Tofanelli, S.; Tagliabracci, A.; Gusmao, L.; et al. Moors and Saracens in Europe: Estimating the medieval North African male legacy in southern Europe. Eur. J. Hum. Genet. 2009, 17, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Villanueva, A.L.; Soto-Hermida, A.; Duncan, J.L.; Matsui, H.; Borooah, S.; Kurmanov, B.; Richard, G.; Khan, S.Y.; Branham, K.; et al. Deciphering the genetic architecture and ethnographic distribution of IRD in three ethnic populations by whole genome sequence analysis. PLoS Genet. 2021, 17, e1009848. [Google Scholar] [CrossRef] [PubMed]
- Avila-Fernandez, A.; Corton, M.; Nishiguchi, K.M.; Muñoz-Sanz, N.; Benavides-Mori, B.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Garcia-Sandoval, B.; Rivolta, C.; Ayuso, C. Identification of an RP1 prevalent founder mutation and related phenotype in Spanish patients with early-onset autosomal recessive retinitis. Ophthalmology 2012, 119, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Nesta, A.V.; Tafur, D.; Beck, C.R. Hotspots of Human Mutation. Trends Genet. 2021, 37, 717–729. [Google Scholar] [CrossRef]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef]
- Simonelli, F.; Sodi, A.; Falsini, B.; Bacci, G.; Iarossi, G.; Di Iorio, V.; Giorgio, D.; Placidi, G.; Andrao, A.; Reale, L.; et al. Narrative medicine to investigate the quality of life and emotional impact of inherited retinal disorders through the perspectives of patients, caregivers and clinicians: An Italian multicentre project. BMJ Open 2022, 12, e061080. [Google Scholar] [CrossRef]
- Schofield, D.; Kraindler, J.; Tan, O.; Shrestha, R.; Jelovic, D.; West, S.; Ma, A.; Grigg, J.; Jamieson, R.V. Patient-Reported Health-Related Quality of Life in Individuals with Inherited Retinal Diseases. Ophthalmol. Sci. 2021, 2, 100106. [Google Scholar] [CrossRef]
- Hlavatá, L.; Ďuďáková, Ľ.; Trková, M.; Soldátová, I.; Skalická, P.; Kousal, B.; Lišková, P. Preimplantační genetická diagnostika a dědičná onemocnění oka [Preimplantation genetic diagnosis and monogenic inherited eye diseases]. Cesk Slov. Oftalmol. 2016, 72, 167–171. (In Czech) [Google Scholar]
- Marwaha, S.; Knowles, J.W.; Ashley, E.A. A guide for the diagnosis of rare and undiagnosed disease: Beyond the exome. Genome Med. 2022, 14, 23. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef]
- Vössing, C.; Owczarek-Lipska, M.; Nagel-Wolfrum, K.; Reiff, C.; Jüschke, C.; Neidhardt, J. Translational Read-Through Therapy of RPGR Nonsense Mutations. Int. J. Mol. Sci. 2020, 21, 8418. [Google Scholar] [CrossRef]
- Samanta, A.; Stingl, K.; Kohl, S.; Ries, J.; Linnert, J.; Nagel-Wolfrum, K. Ataluren for the Treatment of Usher Syndrome 2A Caused by Nonsense Mutations. Int. J. Mol. Sci. 2019, 20, 6274. [Google Scholar] [CrossRef]
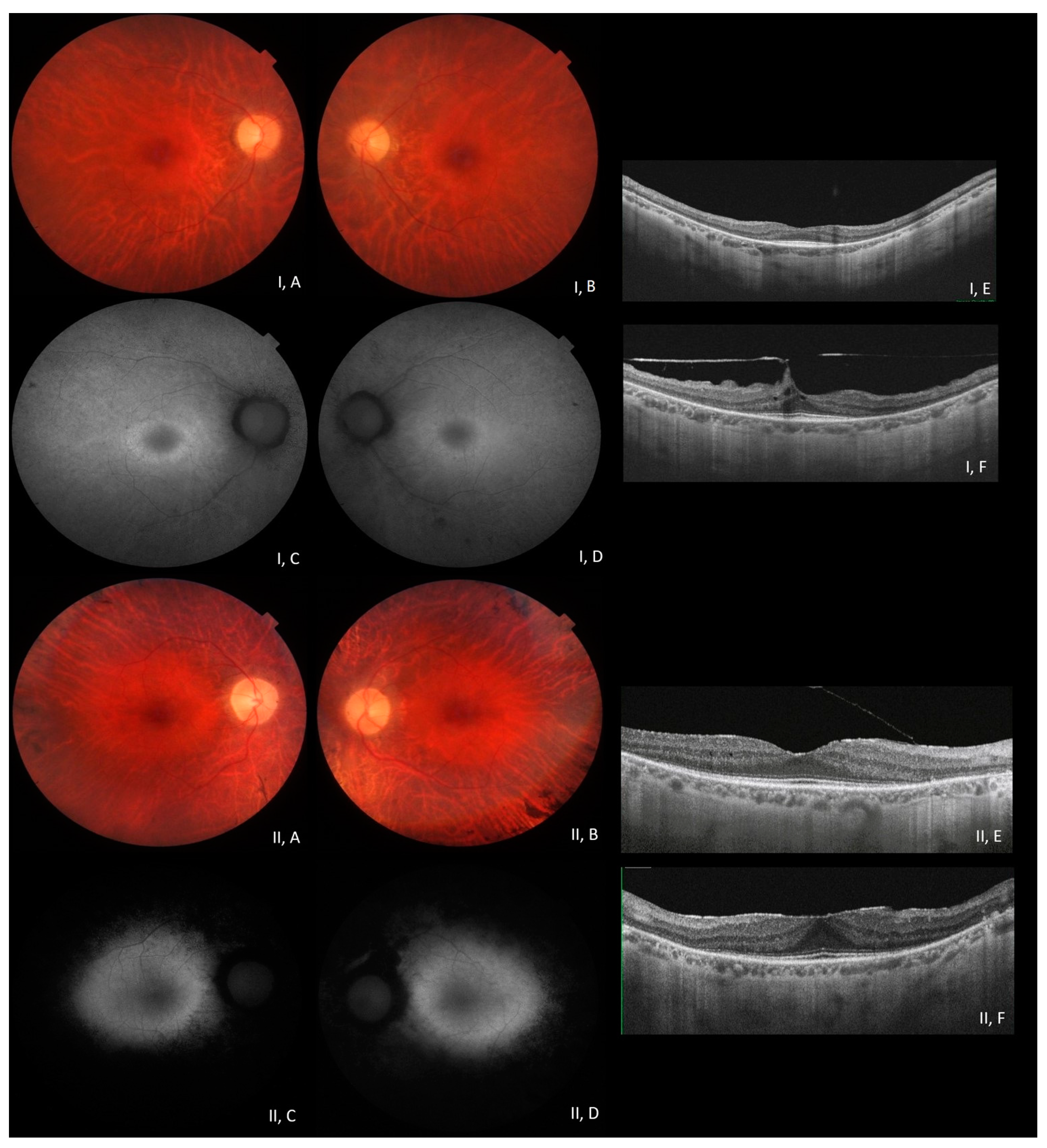

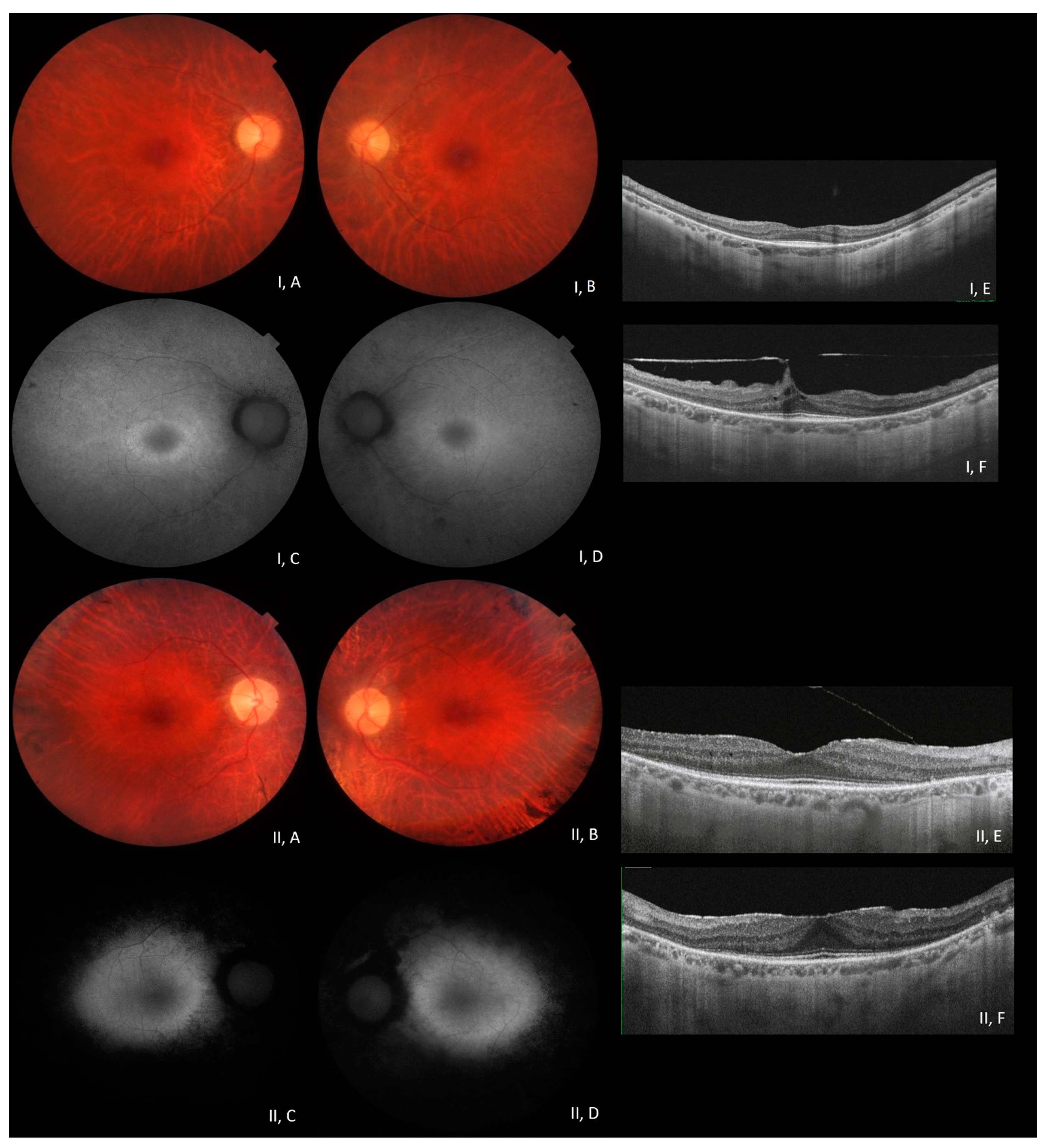{kind=link}
| Transmission Mode | RP General Population Transmission Mode (%) # | RP General Population Transmission Mode (Average %) | Described RP Cohort Transmission Mode (%) | p-Value |
|---|---|---|---|---|
| ADRP | 15–25% | 20.0 | 41.0 | 0.0181 * |
| ARRP | 5–20% | 12.5 | 24.0 | 0.1284 |
| XLRP | 5–15% | 10.0 | 5.0 | 0.1465 |
| SPORADIC RP | 40–50% | 45.0 | 30.0 | 0.0257 * |
| Pt ID | Sex | Age of Onset (Years) | Duration of Symptoms at Observation (Years) | BCVA; Associated Signs | Family History | Traceable Consanguinity | Other Variants (ACMG) |
|---|---|---|---|---|---|---|---|
| 1 | F | 40 | 6 | OD 20/20–OS 20/20; CME | + | ND | CA4: c.700G>A (B) |
| 2 | M | 30 | 35 | OD HM–OS HM | + | ND | USH2A: c.12112A>G (VUS) |
| 3 | F | 26 | 39 | OD 20/40–OS 20/40; PE OU | + | ND | ND |
| 4 | F | 21 | 18 | OD 20/25–OS 20/25; PE OU | + | Family CLB | CNGB1: c.3122G>A (VUS) |
| 5 | F | 45 | 20 | OD 20/25–OS 20/30; PE OU | + | Family CR | ND |
| 6 | M | 27 | <1 | OD 20/20–OS 20/20; R-CME OU | + | Family CLB | ND—RP1 sequencing |
| 7 | M | 50 | 25 | OD 20/100–OS 20/100 | + | Family CLB | ND |
| 8 | F | 40 | 10 | OD 20/25–OS 20/30; PE OU | + | Family CPL | ND |
| 9 | M | 40 | 23 | OD 20/25–OS 20/30; CAT/PRF OU | + | Family CLB | ND |
| 10 | F | 35 | 35 | OD 20/40–OS 20/100; PE/CD/CME OU | + | ND | ND—RP1 sequencing |
| 11 | F | 55 | 1 | OD 20/30–OS 20/20 | + | ND | ND |
| 12 | M | 26 | 7 | OD 20/20–OS 20/20 | + | ND | OFD1: c.815A>G (LB/VUS) |
| 13 | F | 45 | 1 | OD 20/25–OS 20/25; CAT OU; PRF OS | + | ND | ND |
| 14 | F | 31 | 8 | OD 20/25–OS 20/30 | + | ND | ND |
| 15 | M | 26 | 11 | OD 20/100–OS 20/50 | + | ND | ABCA4: c.6089G>A (P) |
| 16 | M | 20 | 24 | OD 20/20–OS 20/30; S-IOLO | + | ND | PROM1: c.652C>T (P) |
| 17 | M | 35 | 5 | OD20/25–OS 20/30; CME OU | + | Family CPL | ND |
| 18 | M | 50 | 23 | OD 20/70–OS 20/70; PE OU | + | ND | IMPDH1: c.189A>G (LB/VUS); CA4:c.700G>A (B) |
| 19 | F | 53 | 8 | OD 20/70–OS 20/200; PE OU | + | Family SMM | ND |
| 20 | F | 31 | 1 | OD 20/20–OS 20/20 | + | Family SMM | ND—RP1 sequencing |
| 21 | M | 40 | 7 | OD 20/25–OS 20/40; CAT OU | − | ND | RP1: c.6166G>A (LB/VUS); SNRNP200: c.1203+15G>A (LB); USH2A: c.12112A>G (VUS) |
| 22 | F | 37 | 15 | OD 20/25–OS 20/25; PE OU | + | Family SMM | ND |
| 23 | M | 48 | 4 | OD 20/20–OS 20/25; CAT OU | + | ND | PROM1:c.1345G>A (LB) |
| 24 | M | 10 | 15 | OD 20/100–OS 20/100 | − | ND | RP1: c.788-12A>G (VUS)PRPF6: c.2638_2639delinsGC (VUS)FSCN2: c.619C>T (LB) |
| 25 | M | 30 | 6 | OD 20/25–OS 20/25; R-CME OU | + | Family CPL | ND |
| 26 | M | 25 | 46 | OD 20/200–OS HM | + | ND | ND—RP1 sequencing |
| 27 | F | 30 | 19 | OD 20/20–OS 20/20 | + | Family CR | ND |
| 28 | F | 40 | 16 | OD 20/20–OS 20/20; PRF | + | ND | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Esposito, F.; Randazzo, V.; Vega, M.I.; Esposito, G.; Maltese, P.E.; Torregrossa, S.; Scibetta, P.; Listì, F.; Gagliano, C.; Scalia, L.; et al. RP1 Dominant p.Ser740* Pathogenic Variant in 20 Knowingly Unrelated Families Affected by Rod–Cone Dystrophy: Potential Founder Effect in Western Sicily. Medicina 2024, 60, 254. https://doi.org/10.3390/medicina60020254
D’Esposito F, Randazzo V, Vega MI, Esposito G, Maltese PE, Torregrossa S, Scibetta P, Listì F, Gagliano C, Scalia L, et al. RP1 Dominant p.Ser740* Pathogenic Variant in 20 Knowingly Unrelated Families Affected by Rod–Cone Dystrophy: Potential Founder Effect in Western Sicily. Medicina. 2024; 60(2):254. https://doi.org/10.3390/medicina60020254
Chicago/Turabian StyleD’Esposito, Fabiana, Viviana Randazzo, Maria Igea Vega, Gabriella Esposito, Paolo Enrico Maltese, Salvatore Torregrossa, Paola Scibetta, Florinda Listì, Caterina Gagliano, Lucia Scalia, and et al. 2024. "RP1 Dominant p.Ser740* Pathogenic Variant in 20 Knowingly Unrelated Families Affected by Rod–Cone Dystrophy: Potential Founder Effect in Western Sicily" Medicina 60, no. 2: 254. https://doi.org/10.3390/medicina60020254
APA StyleD’Esposito, F., Randazzo, V., Vega, M. I., Esposito, G., Maltese, P. E., Torregrossa, S., Scibetta, P., Listì, F., Gagliano, C., Scalia, L., Pioppo, A., Marino, A., Piergentili, M., Malvone, E., Fioretti, T., Vitrano, A., Piccione, M., Avitabile, T., Salvatore, F., ... D’Alcamo, E. (2024). RP1 Dominant p.Ser740* Pathogenic Variant in 20 Knowingly Unrelated Families Affected by Rod–Cone Dystrophy: Potential Founder Effect in Western Sicily. Medicina, 60(2), 254. https://doi.org/10.3390/medicina60020254