
Clinical Characterization and Predictive Factors for Progression in a Cohort of Patients with Interstitial Lung Disease and Features of Autoimmunity: The Need for a Revision of IPAF Classification Criteria

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
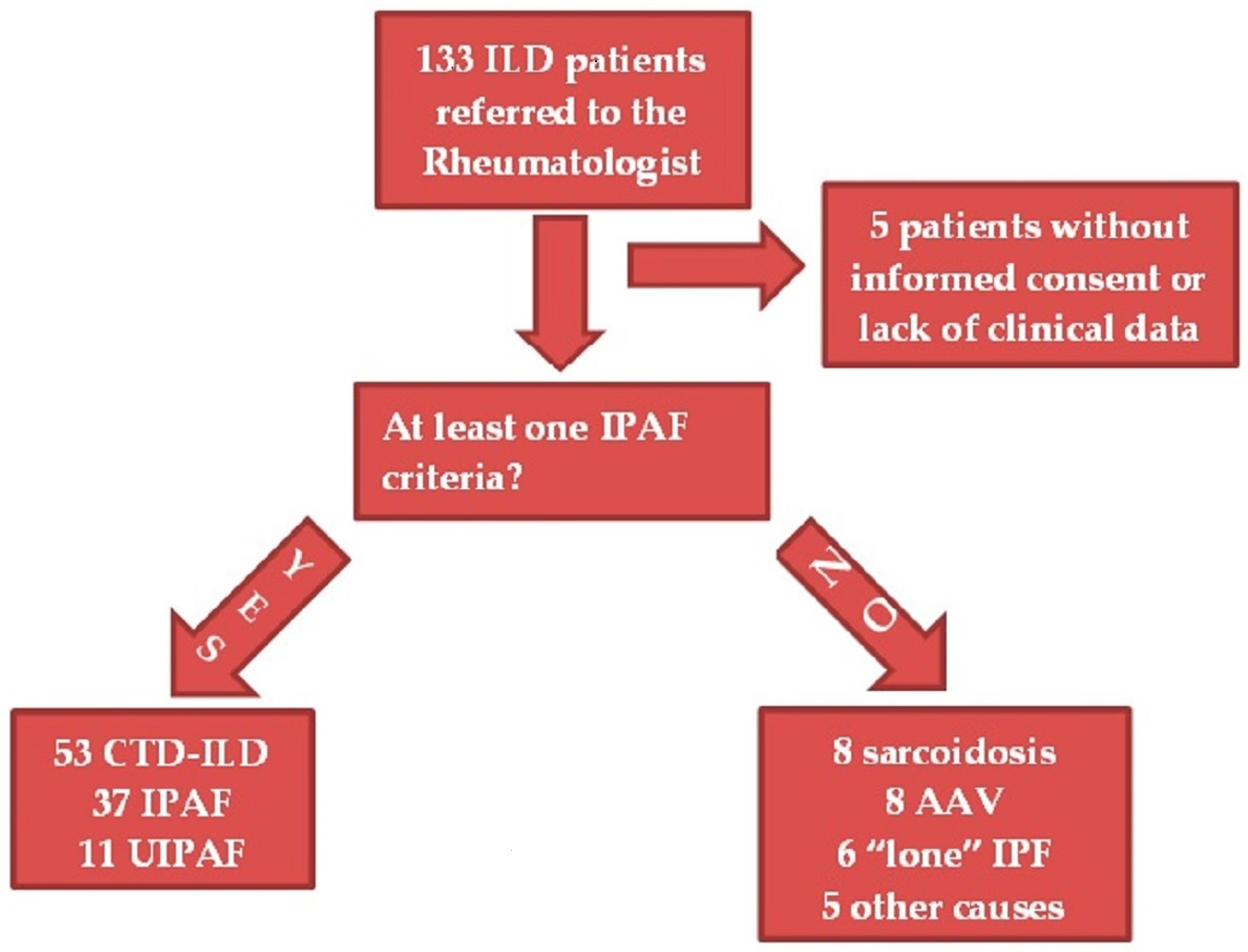
2.1. Patients
2.2. Diagnostic Process
2.2.1. Clinical Evaluation
2.2.2. Laboratory
2.2.3. Chest High-Resolution Computed Tomography (HRCT)
2.2.4. Pulmonary Function Tests (PFTs)
2.2.5. Transthoracic Echocardiography
2.3. Patient Classification
2.4. Follow-Up
- Deterioration of lung function on PFTs, defined by an absolute decline in forced vital capacity (FVC) of ≥5% predicted or in DLCO of 10% predicted [31].
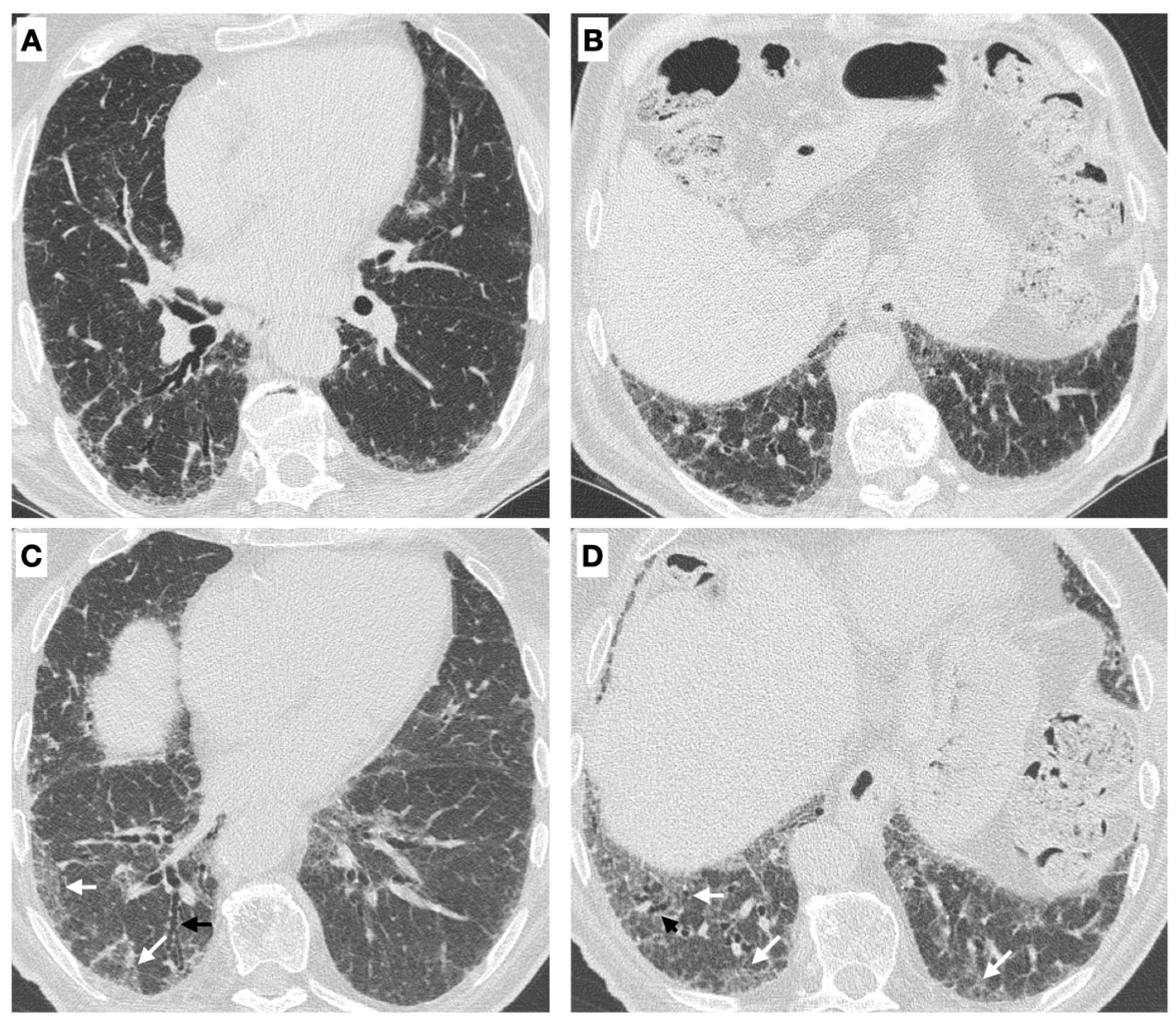
- Radiological progression on HRCT: the trend of ILD (stable, deteriorated, or ameliorated) was established by the most expert thoracic radiologist, blinded to the clinical and functional evaluation, through a semi-quantitative analysis. Particularly evaluated were the presence or increase of the extent of traction bronchiectasis/bronchiolectasis, ground glass opacities, reticulation, and honeycombing as well as the increase of the lobar volume loss [31]. Figure 2 reports an example of ILD progression on HRCT.
- New chronic need for supplemental oxygen.
- Death due to ILD.
- Acute exacerbations, determining an acute respiratory insufficiency and/or a hospital admission and/or a broad-spectrum antibiotic therapy.
2.5. Statistical Analysis
3. Results
3.1. Baseline Characteristics
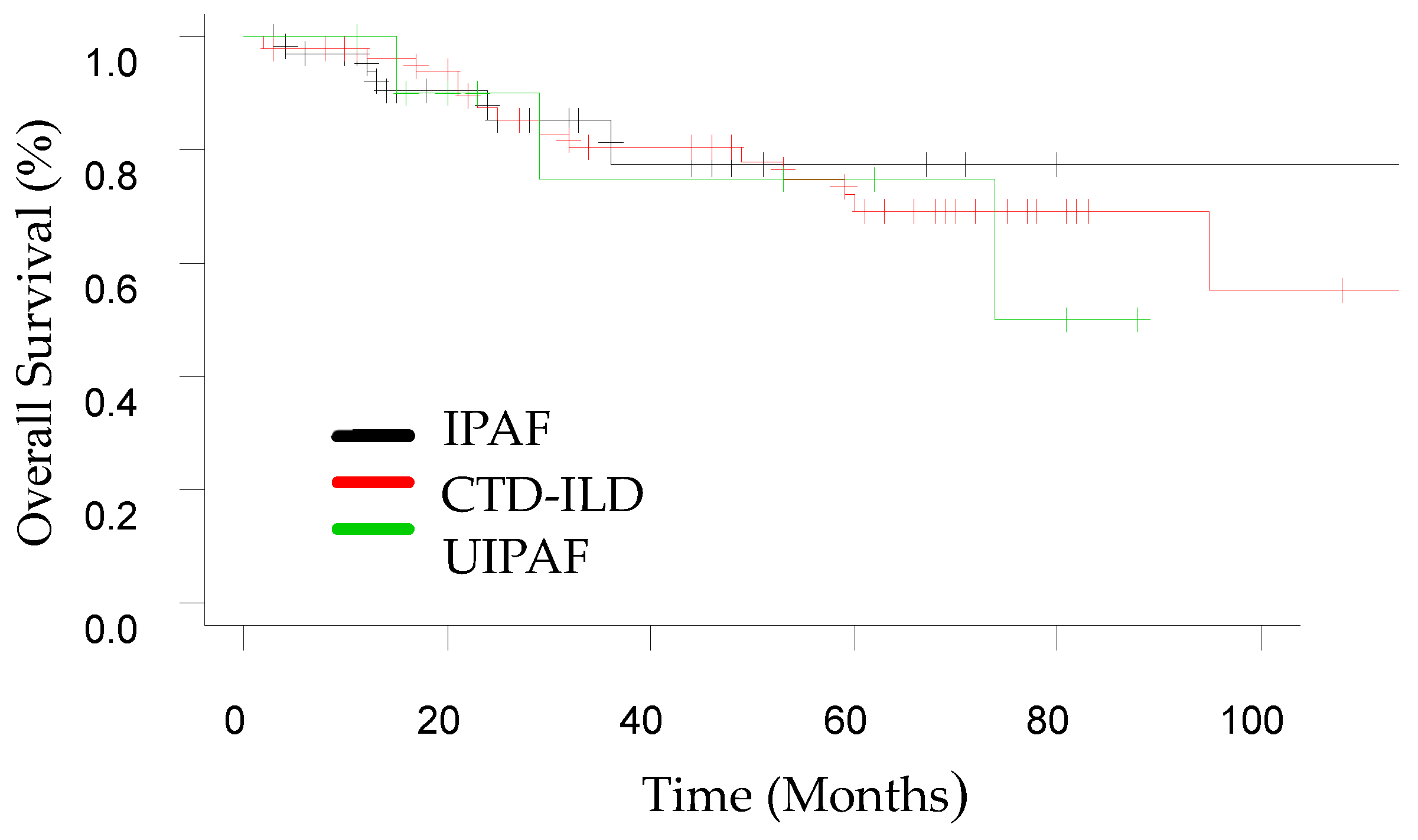
3.2. Follow-Up
3.3. Predictive Factors for ILD Progression at T1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olson, A.L.; Brown, K.K.; Fischer, A. Connective Tissue Disease–Associated Lung Disease. Immunol. Allergy Clin. N. Am. 2012, 32, 513–536. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Fischer, A.; Distler, J.; Distler, J. Progressive fibrosing interstitial lung disease associated with systemic autoimmune diseases. Clin. Rheumatol. 2019, 38, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Shao, T.; Shi, X.; Yang, S.; Zhang, W.; Li, X.; Shu, J.; Alqalyoobi, S.; Zeki, A.A.; Leung, P.S.; Shuai, Z. Interstitial Lung Disease in Connective Tissue Disease: A Common Lesion With Heterogeneous Mechanisms and Treatment Considerations. Front. Immunol. 2021, 12, 684699. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, L.-S.; Wei, Y.-R.; Du, S.-S.; Du, Y.-K.; He, X.; Li, N.; Zhou, Y.; Li, Q.-H.; Su, Y.-L.; et al. Clinical Characteristics of Connective Tissue Disease-Associated Interstitial Lung Disease in 1044 Chinese Patients. Chest 2016, 149, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Hino, T.; Han, J.; Franks, T.J.; Im, Y.; Hatabu, H.; Chung, M.P.; Lee, K.S. RETRACTED: Connective tissue disease-related interstitial lung disease (CTD-ILD) and interstitial lung abnormality (ILA): Evolving concept of CT findings, pathology and management. Eur. J. Radiol. Open 2020, 8, 100311. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Antoniou, K.M.; Brown, K.K.; Cadranel, J.; Corte, T.J.; du Bois, R.M.; Lee, J.S.; Leslie, K.O.; Lynch, D.A.; Matteson, E.L.; et al. An official European Respiratory Society/American Thoracic Society research statement: Interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015, 46, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Graney, B.A.; Fischer, A. Interstitial Pneumonia with Autoimmune Features. Ann. Am. Thorac. Soc. 2019, 16, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, G.; Vancheri, C.; Sambataro, D. Interstitial Pneumonia with Autoimmune Features (IPAF): Time to redefine the classification criteria. Expert Rev. Clin. Immunol. 2022, 19, 131–133. [Google Scholar] [CrossRef]
- Cavagna, L.; Trallero-Araguás, E.; Meloni, F.; Cavazzana, I.; Rojas-Serrano, J.; Feist, E.; Zanframundo, G.; Morandi, V.; Meyer, A.; da Silva, J.A.P.; et al. Influence of Antisynthetase Antibodies Specificities on Antisynthetase Syndrome Clinical Spectrum Time Course. J. Clin. Med. 2019, 8, 2013. [Google Scholar] [CrossRef]
- Sambataro, G.; Ferro, F.; Orlandi, M.; Sambataro, D.; Torrisi, S.E.; Quartuccio, L.; Vancheri, C.; Baldini, C.; Cerinic, M.M. Clinical, morphological features and prognostic factors associated with interstitial lung disease in primary Sjӧgren’s syndrome: A systematic review from the Italian Society of Rheumatology. Autoimmun. Rev. 2020, 19, 102447. [Google Scholar] [CrossRef]
- Sambataro, G.; Ferrara, C.A.; Spadaro, C.; Torrisi, S.E.; Vignigni, G.; Vancheri, A.; Muscato, G.; Del Papa, N.; Colaci, M.; Malatino, L.; et al. A New Method for the Assessment of Myalgia in Interstitial Lung Disease: Association with Positivity for Myositis-Specific and Myositis-Associated Antibodies. Diagnostics 2022, 12, 1139. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.F.; Spiekerman, C.F.; Shaw, M.A.; Ho, L.A.; Hayes, J.; Spada, C.A.; Stamato, C.M.; Raghu, G. Idiopathic Interstitial Pneumonia Associated With Autoantibodies. Chest 2017, 152, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, G.; Ferrara, C.A.; Torrisi, S.E.; Spadaro, C.; Vignigni, G.; Vancheri, A.; Del Papa, N.; Orlandi, M.; Colaci, M.; Malatino, L.; et al. “Usual” interstitial pneumonia with autoimmune features: A prospective study on a cohort of idiopathic pulmonary fibrosis patients. Clin. Exp. Rheumatol. 2022, 40, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Cutolo, M.; Sulli, A.; Smith, V. How to perform and interpret capillaroscopy. Best Pract. Res. Clin. Rheumatol. 2013, 27, 237–248. [Google Scholar] [CrossRef]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome. Ann. Rheum. Dis. 2017, 76, 9–16. [Google Scholar] [CrossRef]
- Daniels, T.E.; Cox, D.; Shiboski, C.H.; Schiødt, M.; Wu, A.; Lanfranchi, H.; Umehara, H.; Zhao, Y.; Challacombe, S.; Lam, M.Y.; et al. Associations between salivary gland histopathologic diagnoses and phenotypic features of Sjögren’s syndrome among 1726 registry participants. Arthritis Rheum. 2011, 63, 2021–2030. [Google Scholar] [CrossRef]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Zavorsky, G.S.; Hsia, C.C.; Hughes, J.M.B.; Borland, C.D.; Guénard, H.; van der Lee, I.; Steenbruggen, I.; Naeije, R.; Cao, J.; Dinh-Xuan, A.T. Standardisation and application of the single-breath determination of nitric oxide uptake in the lung. Eur. Respir. J. 2017, 49, 1600962. [Google Scholar] [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; Van Der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef]
- ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. American Thoracic Society. ATS statement: Guidelines for the six-minute walk test. Am. J. Respir. Crit. Care Med. 2002, 166, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Rudski, L.G.; Lai, W.W.; Afilalo, J.; Hua, L.; Handschumacher, M.D.; Chandrasekaran, K.; Solomon, S.D.; Louie, E.K.; Schiller, N.B. Guidelines for the Echocardiographic Assessment of the Right Heart in Adults: A Report from the American Society of Echocardiography: Endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J. Am. Soc. Echocardiogr. 2010, 23, 685–713. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Orbai, A.-M.; Alarcón, G.S.; Gordon, C.; Merrill, J.T.; Fortin, P.R.; Bruce, I.N.; Isenberg, D.; Wallace, D.J.; Nived, O.; et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2677–2686. [Google Scholar] [CrossRef]
- Van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis. 2013, 72, 1747–1755. [Google Scholar] [CrossRef]
- Bohan, A.; Peter, J.B. Polymyositis and Dermatomyositis (First of Two Parts). N. Engl. J. Med. 1975, 292, 344–347. [Google Scholar] [CrossRef]
- Bohan, A.; Peter, J.B. Polymyositis and Dermatomyositis (Second of Two Parts). N. Engl. J. Med. 1975, 292, 403–407. [Google Scholar] [CrossRef]
- Sambataro, G.; Sambataro, D.; Torrisi, S.E.; Vancheri, A.; Colaci, M.; Pavone, M.; Pignataro, F.; Del Papa, N.; Palmucci, S.; Vancheri, C. Clinical, serological and radiological features of a prospective cohort of Interstitial Pneumonia with Autoimmune Features (IPAF) patients. Respir. Med. 2019, 150, 154–160. [Google Scholar] [CrossRef]
- Vij, R.; Noth, I.; Strek, M.E. Autoimmune-Featured Interstitial Lung Disease: A Distinct Entity. Chest 2011, 140, 1292–1299. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Hagiwara, E.; Kitamura, H.; Iwasawa, T.; Otoshi, R.; Aiko, N.; Katano, T.; Shintani, R.; Ikeda, S.; Okuda, R.; et al. Predictive Factors for the Long-Term Deterioration of Pulmonary Function in Interstitial Lung Disease Associated with Anti-Aminoacyl-tRNA Synthetase Antibodies. Respiration 2018, 96, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.W.; Ryerson, C.J.; Guler, S.A. Progression of fibrosing interstitial lung disease. Respir. Res. 2020, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef]
- Furini, F.; Carnevale, A.; Casoni, G.L.; Guerrini, G.; Cavagna, L.; Govoni, M.; Sciré, C.A. The Role of the Multidisciplinary Evaluation of Interstitial Lung Diseases: Systematic Literature Review of the Current Evidence and Future Perspectives. Front. Med. 2019, 6, 246. [Google Scholar] [CrossRef]
- Sebastiani, M.; Faverio, P.; Manfredi, A.; Cassone, G.; Vacchi, C.; Stainer, A.; Pozzi, M.R.; Salvarani, C.; Pesci, A.; Luppi, F. Interstitial Pneumonia with Autoimmune Features: Why Rheumatologist–Pulmonologist Collaboration Is Essential. Biomedicines 2020, 9, 17. [Google Scholar] [CrossRef]
- Levi, Y.; Israeli-Shani, L.; Kuchuk, M.; Shochet, G.E.; Koslow, M.; Shitrit, D. Rheumatological Assessment Is Important for Interstitial Lung Disease Diagnosis. J. Rheumatol. 2018, 45, 1509–1514. [Google Scholar] [CrossRef]
- Chernau, A.K.; Leone, P.M.; Swigris, J.J. Interstitial Pneumonia with Autoimmune Features and Undifferentiated Connective Tissue Disease. Semin. Respir. Crit. Care Med. 2019, 40, 271–277. [Google Scholar] [CrossRef]
- Sebastiani, M.; Cassone, G.; De Pasquale, L.; Cerri, S.; Della Casa, G.; Vacchi, C.; Luppi, F.; Salvarani, C.; Manfredi, A. Interstitial pneumonia with autoimmune features: A single center prospective follow-up study. Autoimmun. Rev. 2020, 19, 102451. [Google Scholar] [CrossRef]
- Oldham, J.M.; Adegunsoye, A.; Valenzi, E.; Lee, C.; Witt, L.; Chen, L.; Husain, A.N.; Montner, S.; Chung, J.H.; Cottin, V.; et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur. Respir. J. 2016, 47, 1767–1775. [Google Scholar] [CrossRef]
- Kelly, B.T.; Moua, T. Overlap of interstitial pneumonia with autoimmune features with undifferentiated connective tissue disease and contribution of UIP to mortality. Respirology 2018, 23, 600–605. [Google Scholar] [CrossRef]
- Ferri, C.; Manfredi, A.; Sebastiani, M.; Colaci, M.; Giuggioli, D.; Vacchi, C.; Della Casa, G.; Cerri, S.; Torricelli, P.; Luppi, F. Interstitial Pneumonia with Autoimmune Features and Undifferentiated Connective Tissue Disease: Our Interdisciplinary Rheumatology-Pneumology Experience, and Review of the Literature. Autoimmun. Rev. 2016, 15, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, G.; Sambataro, D.; Spicuzza, L.; Meloni, F.; Lorini, G.; Malatino, L.; Colaci, M.; Sebastiani, G.; Iuliano, A.; Canofari, C.; et al. Progression and prognosis of interstitial pneumonia with autoimmune features: A longitudinal, prospective, multi-centre study [published online ahead of print, 2022 Oct 3]. Clin. Exp. Rheumatol. 2022. [Google Scholar] [CrossRef]
- Jiwrajka, N.; Loizidis, G.; Patterson, K.C.; Kreider, M.E.M.; Johnson, C.R.M.; Miller, W.T.J.; Barbosa, E.J.M.J.; Patel, N.; Beers, M.F.; Litzky, L.A.; et al. Identification and Prognosis of Patients with Interstitial Pneumonia with Autoimmune Features. Am. J. Clin. Oncol. 2022, 28, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Scirè, C.A.; Gonzalez-Gay, M.A.; Selva-O’callaghan, A.; Cavagna, L. Clinical spectrum time course of interstitial pneumonia with autoimmune features in patients positive for antisynthetase antibodies. Respir. Med. 2017, 132, 265–266. [Google Scholar] [CrossRef]
- Zanframundo, G.; Faghihi-Kashani, S.; Scirè, C.A.; Bonella, F.; Corte, T.J.; Doyle, T.J.; Fiorentino, D.; Gonzalez-Gay, M.A.; Hudson, M.; Kuwana, M.; et al. Defining anti-synthetase syndrome: A systematic literature review. Ann. Rheum. Dis. 2022, 40, 309–319. [Google Scholar] [CrossRef]
- Chung, J.H.; Cox, C.W.; Montner, S.M.; Adegunsoye, A.; Oldham, J.M.; Husain, A.N.; Vij, R.; Noth, I.; Lynch, D.A.; Strek, M.E. CT Features of the Usual Interstitial Pneumonia Pattern: Differentiating Connective Tissue Disease–Associated Interstitial Lung Disease from Idiopathic Pulmonary Fibrosis. Am. J. Roentgenol. 2018, 210, 307–313. [Google Scholar] [CrossRef]
- Ghang, B.; Lee, J.; Kwon, O.C.; Ahn, S.M.; Oh, J.S.; Hong, S.; Kim, Y.-G.; Yoo, B.; Jeong, W.S.; Kim, J.; et al. Clinical significance of autoantibody positivity in idiopathic pulmonary fibrosis. Respir. Med. 2019, 155, 43–48. [Google Scholar] [CrossRef]
- Ghang, B.; Nam, S.H.; Lee, J.; Lim, D.-H.; Ahn, S.M.; Oh, J.S.; Hong, S.; Kim, Y.-G.; Yoo, B.; Kim, J.; et al. Risk of progression of idiopathic pulmonary fibrosis to connective tissue disease: A long-term observational study in 527 patients. Clin. Rheumatol. 2021, 40, 2447–2456. [Google Scholar] [CrossRef]
- Lim, J.U.; Gil, B.M.; Kang, H.S.; Oh, J.; Kim, Y.H.; Kwon, S.S. Interstitial pneumonia with autoimmune features show better survival and less exacerbations compared to idiopathic pulmonary fibrosis. BMC Pulm. Med. 2019, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Lee, J.H.; Chae, E.J.; Song, J.S.; Song, J.W. Long-term clinical course and outcome of interstitial pneumonia with autoimmune features. Respirology 2020, 25, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Panlaqui, O.M. Systematic review and meta-analysis of the prognosis and prognostic factors of interstitial pneumonia with autoimmune features. BMJ Open 2019, 9, e031444. [Google Scholar] [CrossRef] [PubMed]
- Adegunsoye, A.; Oldham, J.M.; Valenzi, E.; Lee, C.; Witt, L.J.; Chen, L.; Montner, S.; Chung, J.H.; Noth, I.; Vij, R.; et al. Interstitial Pneumonia With Autoimmune Features: Value of Histopathology. Arch. Pathol. Lab. Med. 2017, 141, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Park, J.K.; Roh, J.H.; Song, J.W.; Lee, C.K.; Kim, M.; Jang, S.J.; Colby, T.V.; Kim, D.S. Clinical Significance of Serum Autoantibodies in Idiopathic Interstitial Pneumonia. J. Korean Med. Sci. 2013, 28, 731–737. [Google Scholar] [CrossRef]
- Ajeganova, S.; Humphreys, J.; Verheul, M.K.; Van Steenbergen, H.W.; Nies, J.A.B.V.; Hafström, I.; Svensson, B.; Huizinga, T.W.J.; Trouw, L.; Verstappen, S.; et al. Anticitrullinated protein antibodies and rheumatoid factor are associated with increased mortality but with different causes of death in patients with rheumatoid arthritis: A longitudinal study in three European cohorts. Ann. Rheum. Dis. 2016, 75, 1924–1932. [Google Scholar] [CrossRef]
- Hyldgaard, C.; Ellingsen, T.; Hilberg, O.; Bendstrup, E. Rheumatoid Arthritis-Associated Interstitial Lung Disease: Clinical Characteristics and Predictors of Mortality. Respiration 2019, 98, 455–460. [Google Scholar] [CrossRef]
- Xie, S.; Li, S.; Chen, B.; Zhu, Q.; Xu, L.; Li, F. Serum anti-citrullinated protein antibodies and rheumatoid factor increase the risk of rheumatoid arthritis–related interstitial lung disease: A meta-analysis. Clin. Rheumatol. 2021, 40, 4533–4543. [Google Scholar] [CrossRef]
- Johnson, S.R.; Fransen, J.; Khanna, D.; Baron, M.; Hoogen, F.V.D.; Medsger, T.A., Jr.; Peschken, C.A.; Carreira, P.E.; Riemekasten, G.; Tyndall, A.; et al. Validation of potential classification criteria for systemic sclerosis. Arthritis Care Res. 2011, 64, 358–367. [Google Scholar] [CrossRef]
- Kovacs, S.O. Dermatomyositis. J. Am. Acad. Dermatol. 1998, 39, 899–922. [Google Scholar] [CrossRef]
- Jee, A.S.; Parker, M.J.; Bleasel, J.F.; Troy, L.K.; Lau, E.M.; Jo, H.E.; Teoh, A.K.; Adelstein, S.; Webster, S.; Corte, T.J. Baseline Characteristics and Survival of an Australian Interstitial Pneumonia with Autoimmune Features Cohort. Respiration 2021, 100, 853–864. [Google Scholar] [CrossRef] [PubMed]
- La Corte, R.; Monaco, A.L.; Locaputo, A.; Dolzani, F.; Trotta, F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006, 39, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Hudson, M.; Pope, J.; Mahler, M.; Tatibouet, S.; Steele, R.; Baron, M.; Fritzler, M.J.; Canadian Scleroderma Research Group (CSRG). Clinical significance of antibodies to Ro52/TRIM21 in systemic sclerosis. Thromb. Haemost. 2012, 14, R50. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Li, S.; Yang, H.; Zhang, Y.; Peng, Q.; Lu, X.; Wang, G. Clinical Profiles and Prognosis of Patients with Distinct Antisynthetase Autoantibodies. J. Rheumatol. 2017, 44, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Alba, M.A.; Flores-Suárez, L.F.; Henderson, A.G.; Xiao, H.; Hu, P.; Nachman, P.H.; Falk, R.J.; Charles Jennette, J. Interstital lung disease in ANCA vasculitis. Autoimmun. Rev. 2017, 16, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Gannon, W.D.; Anderson, M.R.; Podolanczuk, A.J.; Kawut, S.M.; Michos, E.D.; Cottin, V.; Kreuter, M.; Raghu, G.; Barr, R.G.; Lederer, D.J. Angiotensin Receptor Blockers and Subclinical Interstitial Lung Disease: The MESA Study. Ann. Am. Thorac. Soc. 2019, 16, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Noth, I.; Valenzuela, C.; Frankenstein, L.; Weycker, D.; Atwood, M.; Kirchgaessler, K.-U.; Cottin, V. Association of Angiotensin Modulators with the Course of Idiopathic Pulmonary Fibrosis. Chest 2019, 156, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.D.; Panza, G.; Zaleski, A.; Taylor, B. Statin-Associated Side Effects. J. Am. Coll. Cardiol. 2016, 67, 2395–2410. [Google Scholar] [CrossRef]
- Kim, J.S.; Podolanczuk, A.J.; Borker, P.; Kawut, S.M.; Raghu, G.; Kaufman, J.D.; Stukovsky, K.D.H.; Hoffman, E.A.; Barr, R.G.; Gottlieb, D.J.; et al. Obstructive Sleep Apnea and Subclinical Interstitial Lung Disease in the Multi-Ethnic Study of Atherosclerosis (MESA). Ann. Am. Thorac. Soc. 2017, 14, 1786–1795. [Google Scholar] [CrossRef]
- Koteci, A.; Morgan, A.D.; Portas, L.; Whittaker, H.R.; Kallis, C.; George, P.M.; Quint, J.K. Left-sided heart failure burden and mortality in idiopathic pulmonary fibrosis: A population-based study. BMC Pulm. Med. 2022, 22, 190. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Post-Hoc Analysis | |||||||
|---|---|---|---|---|---|---|---|
| IPAF | CTD-ILD | UIPAF | p-Value | IPAF vs. CTD-ILD | IPAF vs. UIPAF | CTD-ILD vs. UIPAF | |
| No. of subjects | 37 (36.6) | 53 (52.5) | 11 (10.9) | ||||
| Age (years) | 71 (62.5–75.5) | 69 (60.5–74) | 68 (66–79) | 0.64 | |||
| Female | 20 (54.1) | 37 (69.8) | 8 (72.7) | 0.27 | |||
| Never-smokers | 13 (35.1) | 23 (43.4) | 4 (36.4) | 0.77 | |||
| Months from symptoms’ onset | 15 (7–39.5) | 11 (6–34.5) | 12 (6–17) | 0.64 | |||
| Systemic hypertension | 31 (83.8) | 30 (56.6) | 5 (45.5) | <0.01 | 0.011 | 0.017 | 0.526 |
| Left-sided heart failure | 9 (24.3) | 3 (5.7) | 2 (18.2) | 0.03 | 0.024 | 1.00 | 0.201 |
| Type 2 diabetes | 7 (18.9) | 10 (18.9) | 2 (18.2) | 1 | |||
| Hypercholesterolemia | 17 (45.9) | 17 (32.1) | 4 (36.4) | 0.41 | |||
| BMI (kg/m2) | 27.5 (24.8–31) | 25.6 (22.9–29) | 25.5 (24–28.9) | 0.09 | |||
| OSAS | 6 (16.2) | 3 (5.7) | 1 (9.1) | 0.25 | |||
| GERD | 12 (32.4) | 17 (32.1) | 5 (45.5) | 0.68 | |||
| Concurrent autoimmune disease | 12 (32.4) | 19 (35.8) | 0 (0) | 0.04 | 0.824 | 0.041 | <0.01 |
| Autoimmune thyroiditis | 7 (18.9) | 11 (20.8) | 0 (0) | ||||
| Pulmonary hypertension (PH) | 12 (32.4) | 13 (24.5) | 2 (18.2) | 0.61 | |||
| Pre-capillary PH | 3 (8.1) | 6 (11.3) | 0 (0) | ||||
| ARDS at onset | 2 (5.4) | 3 (5.6) | 0 (0) | 1 | |||
| Corticosteroid therapy | 31 (83.8) | 49 (92.5) | 8 (72.7) | 0.12 | |||
| With an immunosuppressant | 18 (48.6) | 26 (49.1) | 1 (9.1) | 0.04 | 0.970 | 0.032 | 0.018 |
| With an anti-fibrotic agent | 4 (10.8) | 6 (11.3) | 8 (72.7) | <0.01 | 0.940 | <0.01 | <0.01 |
| Post-Hoc Analysis | |||||||
|---|---|---|---|---|---|---|---|
| IPAF | CTD-ILD | UIPAF | p-Value | IPAF vs. CTD-ILD | IPAF vs. UIPAF | CTD-ILD vs. UIPAF | |
| Pattern | |||||||
| UIP | 4 (10.8) | 17 (32.1) | 11 (100) | <0.01 | 0.023 | <0.01 | <0.01 |
| NSIP | 21 (56.8) | 23 (43.4) | 0 (0) | <0.01 | 0.212 | <0.01 | <0.01 |
| OP | 4 (10.8) | 2 (3.8) | 0 (0) | 0.3 | |||
| ILD + Multi-compartment involvement ꝉ | 7 (18.9) | 10 (18.9) | 0 (0) | 0.35 | |||
| Other patterns | 1 (2.7) | 1 (1.9) | 0 (0) | 1 | |||
| PFTs | |||||||
| FEV1 (% predicted) | 83 (76–102.5) | 84 (75–101) | 92 (76–115) | 0.55 | |||
| FVC (% predicted) | 86 (73–97) | 91 (80–110) | 94 (69–106) | 0.3 | |||
| DLCO (% predicted) | 61 (49–70) | 49.5 (34.8–75) | 46 (36.5–60.5) | 0.36 | |||
| DLCO <35% and/or FVC <50% | 5 (13.5) | 14 (26.4) | 3 (27.7) | 0.28 | |||
| 6MWT | |||||||
| Distance (m) | 495 (378–540) | 459 (394–539) | 461 (351–500) | 0.53 | |||
| Lowest SpO2 (%) | 92 (89–96) | 91 (83–96) | 86 (76–93) | 0.12 | |||
| Post-Hoc Analysis | |||||||
|---|---|---|---|---|---|---|---|
| IPAF | CTD-ILD | UIPAF | p-Value | IPAF vs. CTD-ILD | IPAF vs. UIPAF | CTD-ILD vs. UIPAF | |
| No. of subjects | 35 (36.1) | 51 (52.6) | 11 (11.3) | ||||
| PFTs | |||||||
| progression | 9 (26.5) | 22 (43.1) | 7 (63.6) | 0.07 | |||
| improvement | 9 (25.7) | 9 (17.6) | 0 (0) | 0.14 | |||
| stable | 16 (47.1) | 20 (39.2) | 4 (36.4) | 0.72 | |||
| DLCO < 35% and/or FVC < 50% | 4 (11.4) | 20 (39.2) | 3 (27.3) | 0.01 | <0.01 | 0.337 | 0.516 |
| HRCT | |||||||
| progression | 6 (17.1) | 18 (35.3) | 5 (45.5) | 0.09 | |||
| improvement | 9 (24.3) | 4 (7.8) | 0 (0) | 0.02 | 0.029 | 0.087 | 1.00 |
| stable | 19 (54.3) | 29 (56.9) | 6 (54.5) | 0.95 | |||
| Supplementary oxygen | 3 (8.6) | 16 (31.4) | 4 (36.4) | 0.01 | 0.017 | 0.050 | 0.735 |
| At least one exacerbation | 7 (20.0) | 17 (33.3) | 2 (18.2) | 0.35 | |||
| Composite endpoint of ILD progression | 11 (32.3) | 30 (58.8) | 8 (72.7) | 0.02 | 0.017 | 0.033 | 0.461 |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| Characteristics | OR (95% CI) | p-Value | OR (95% CI) | p-Value |
| Age (years) | 1.00 (0.97–1.04) | 0.91 | 0.98 (0.94–1.03) | 0.53 |
| Female sex | 0.81 (0.34–1.88) | 0.62 | 0.96 (0.34–2.74) | 0.94 |
| Never-smoker | 0.85 (0.37–1.95) | 0.71 | ||
| Pulmonary hypertension | 2.36 (0.92–6.48) | 0.12 | ||
| ARDS at onset | 1.14 (0.13–9.83) | 0.91 | ||
| IPAF | 0.30 (0.12–0.72) | <0.01 | 0.28 (0.09–0.79) | 0.02 |
| UIP pattern | 4.31 (1.73–11.7) | <0.01 | 3.80 (1.33–11.9) | 0.01 |
| ANA | 0.76 (0.33–1.22) | 0.51 | ||
| RF ≥ 2 times above the upper limit | 5.77 (1.41–39.0) | 0.03 | 9.13 (1.8–53.3) | 0.02 |
| Anti-CCP | 0.96 (0.17–5.41) | 0.96 | ||
| Anti-Ro (SSA) | 0.48 (0.14–1.51) | 0.22 | ||
| Anti-Scl-70 | 0.96 (0.04–24.7) | 0.98 | ||
| Anti-tRNA synthetase | 0.68 (0.21–2.13) | 0.51 | ||
| Anti-PM/Scl | 0.70 (0.13–3.36) | 0.65 | ||
| Mechanic’s hand | 0.47 (0.21–5.06) | 0.54 | ||
| Arthritis or joint stiffness ≥ 60 min | 1.02 (0.45–2.30) | 0.97 | ||
| Palmar teleangiectasia | 2.06 (0.76–6.03) | 0.17 | ||
| Raynaud phenomenon | 1.74 (0.72–4.35) | 0.23 | ||
| Sicca syndrome | 0.49 (0.19–1.23) | 0.14 | ||
| Corticosteroid therapy | 1.81 (0.56–6.41) | 0.33 | ||
| With immunosuppressant | 1.00 (0.45–2.26) | 0.98 | ||
| With an anti-fibrotic agent ꝉ | 9.00 (2.32–59.8) | <0.01 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozzao, F.; Tomietto, P.; Baratella, E.; Kodric, M.; Cifaldi, R.; Della Porta, R.; Prearo, I.; Pirronello, S.M.G.; Confalonieri, P.; Ruaro, B.; et al. Clinical Characterization and Predictive Factors for Progression in a Cohort of Patients with Interstitial Lung Disease and Features of Autoimmunity: The Need for a Revision of IPAF Classification Criteria. Medicina 2023, 59, 794. https://doi.org/10.3390/medicina59040794
Bozzao F, Tomietto P, Baratella E, Kodric M, Cifaldi R, Della Porta R, Prearo I, Pirronello SMG, Confalonieri P, Ruaro B, et al. Clinical Characterization and Predictive Factors for Progression in a Cohort of Patients with Interstitial Lung Disease and Features of Autoimmunity: The Need for a Revision of IPAF Classification Criteria. Medicina. 2023; 59(4):794. https://doi.org/10.3390/medicina59040794
Chicago/Turabian StyleBozzao, Francesco, Paola Tomietto, Elisa Baratella, Metka Kodric, Rossella Cifaldi, Rossana Della Porta, Ilaria Prearo, Silvia Maria Grazia Pirronello, Paola Confalonieri, Barbara Ruaro, and et al. 2023. "Clinical Characterization and Predictive Factors for Progression in a Cohort of Patients with Interstitial Lung Disease and Features of Autoimmunity: The Need for a Revision of IPAF Classification Criteria" Medicina 59, no. 4: 794. https://doi.org/10.3390/medicina59040794
APA StyleBozzao, F., Tomietto, P., Baratella, E., Kodric, M., Cifaldi, R., Della Porta, R., Prearo, I., Pirronello, S. M. G., Confalonieri, P., Ruaro, B., Fischetti, F., & Fabris, B. (2023). Clinical Characterization and Predictive Factors for Progression in a Cohort of Patients with Interstitial Lung Disease and Features of Autoimmunity: The Need for a Revision of IPAF Classification Criteria. Medicina, 59(4), 794. https://doi.org/10.3390/medicina59040794