
Clinical and Radiological Features of Interstitial Lung Diseases Associated with Polymyositis and Dermatomyositis

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Methods
3. Clinical Features
3.1. Laboratory Findings
3.2. Classification Criteria
3.3. Pulmonary Involvement in IIMs
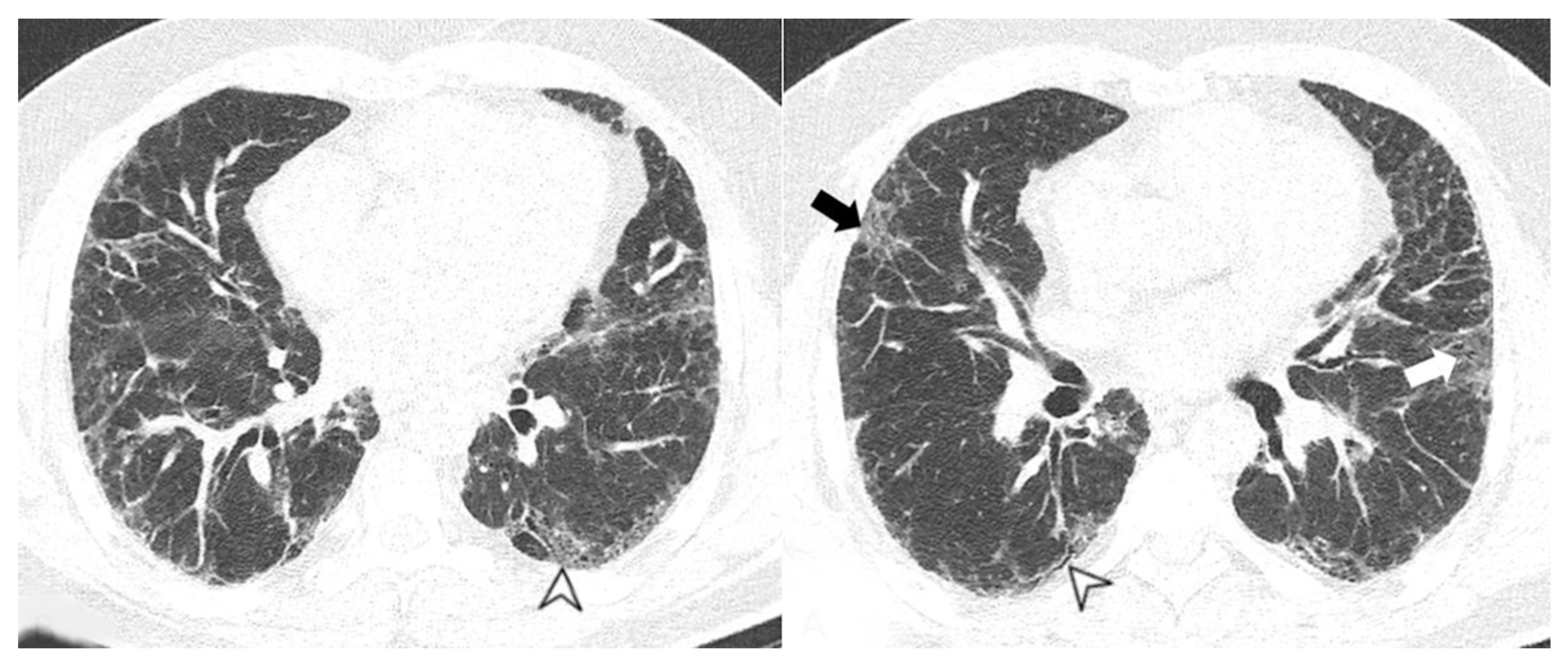
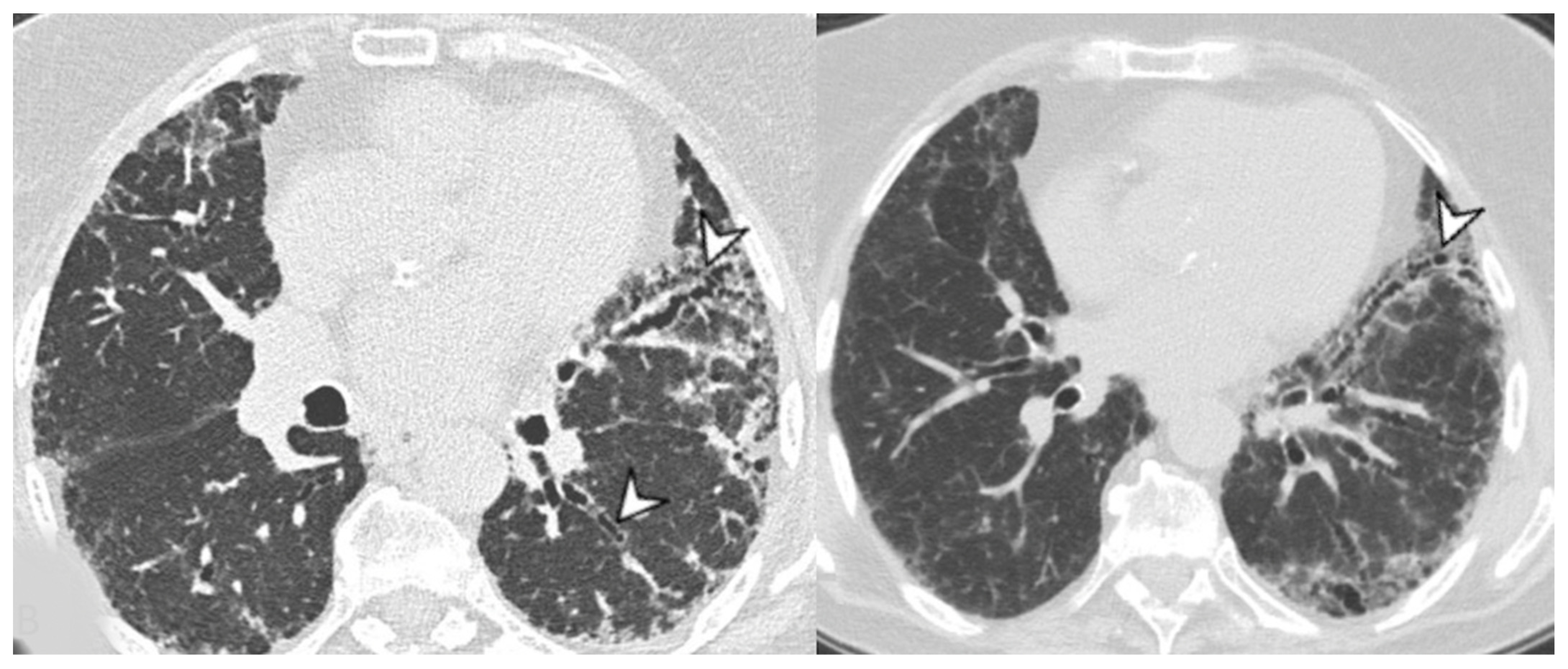
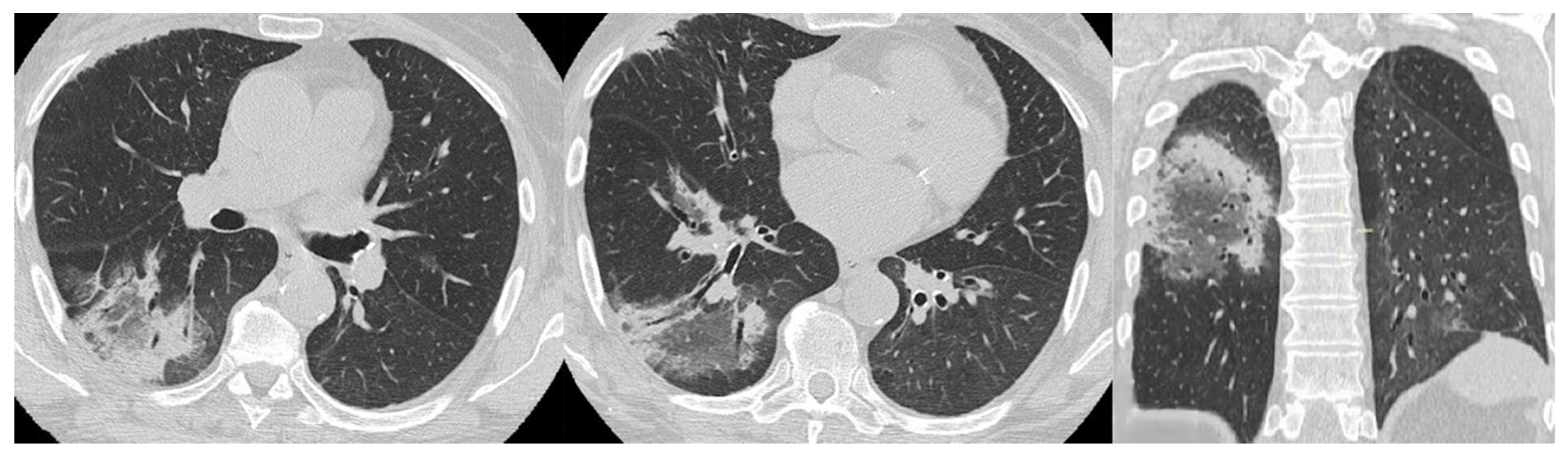
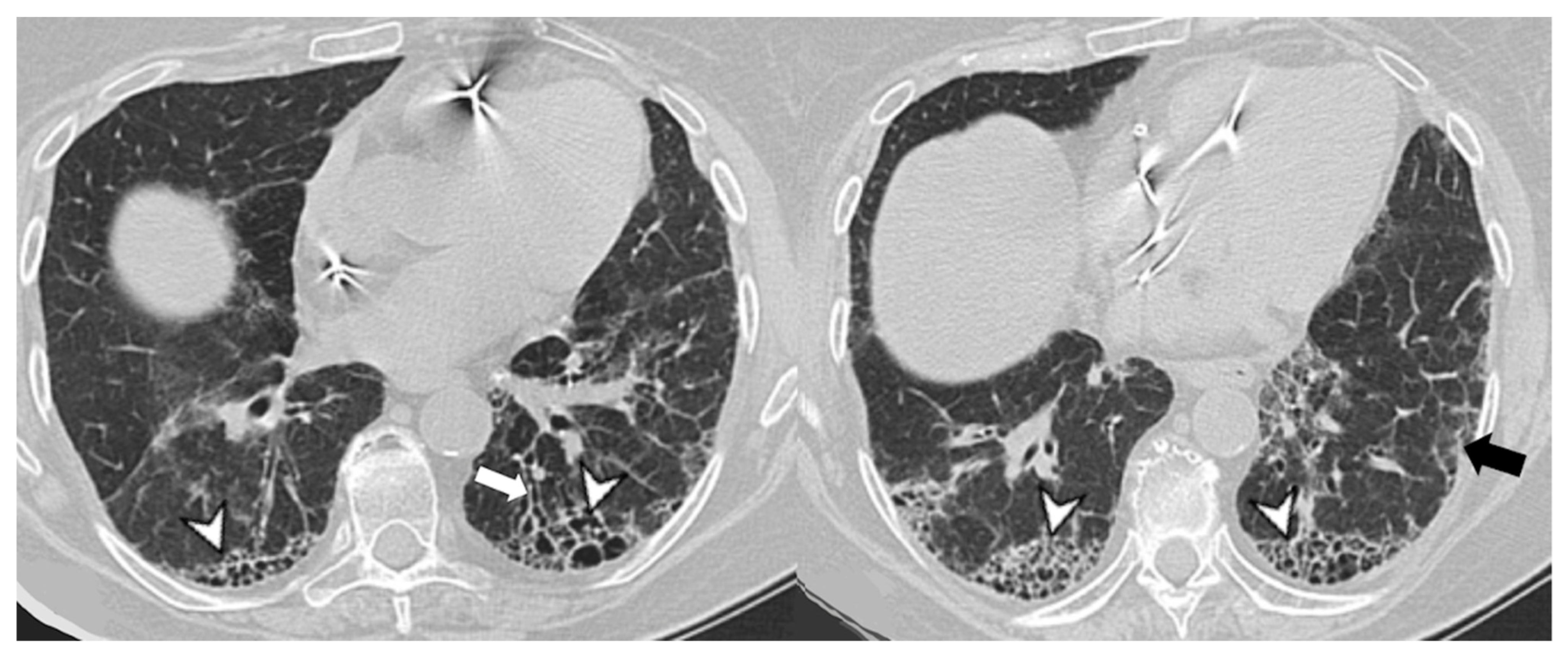
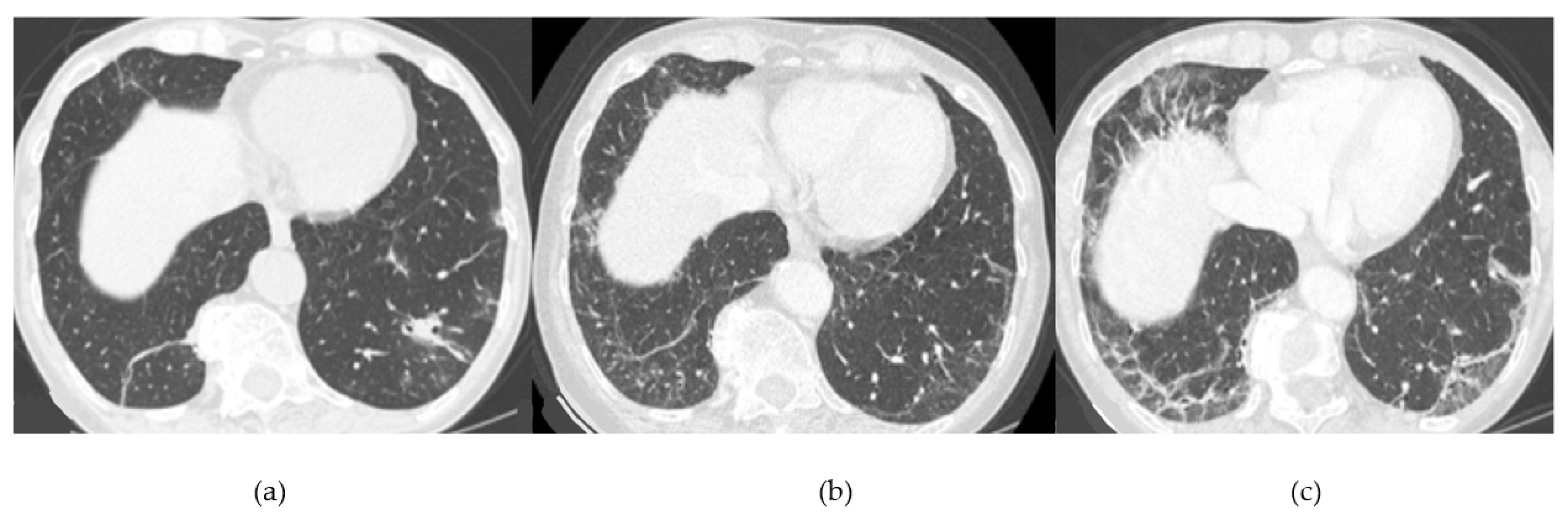
4. Radiological Features
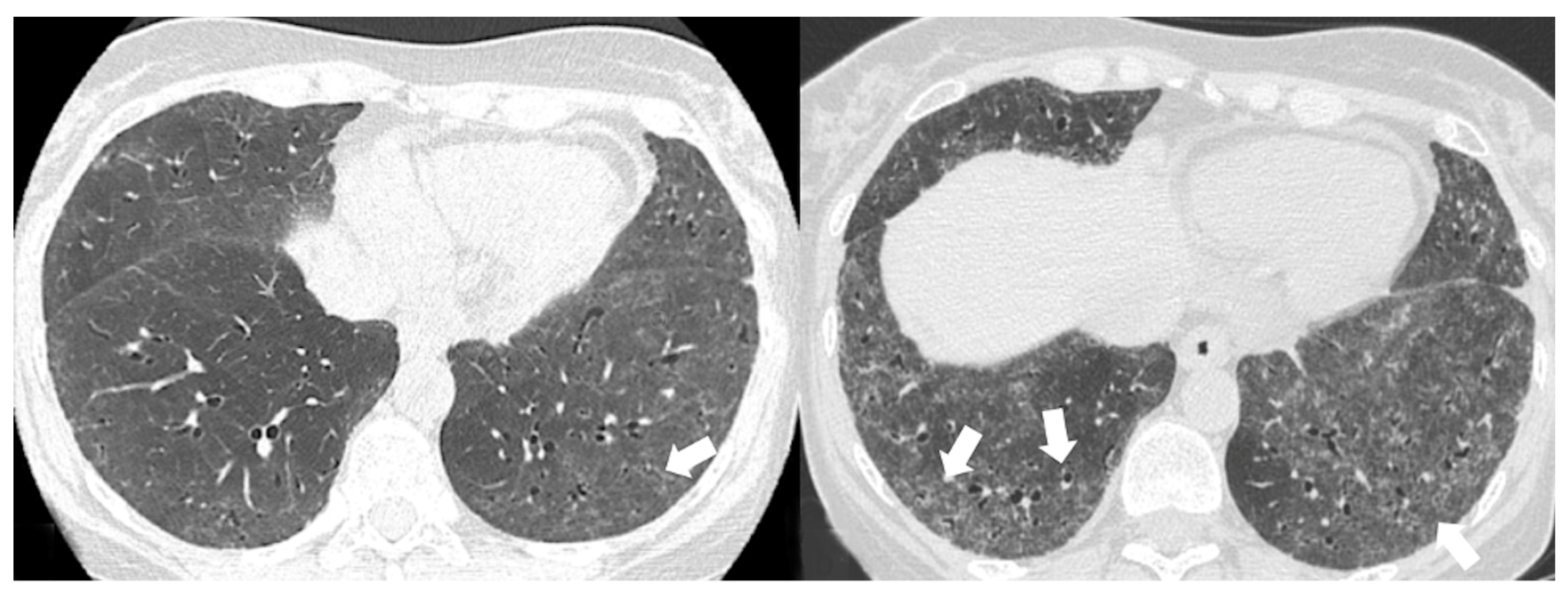
4.1. Radiological Features of ILD Presentation/Progression in DM/PM
- ▪
- Asymptomatic/occult ILD (minimal or absent changes seen on HRCT) [43];
- ▪
- Acute or rapidly progressive ILD (acute interstitial pneumonia and radiological evidence of acute respiratory distress syndrome (ARDS) due to diffuse alveolar damage) [44];
- ▪
- ▪
- Chronic progressive fibrosing ILD (reticulation and honeycombing with minimal GGOs on HRCT scans, related to fibrotic NSIP or UIP) [43];
Acute Type
4.2. Relationship between Myositis-Specific Antibodies and ILD
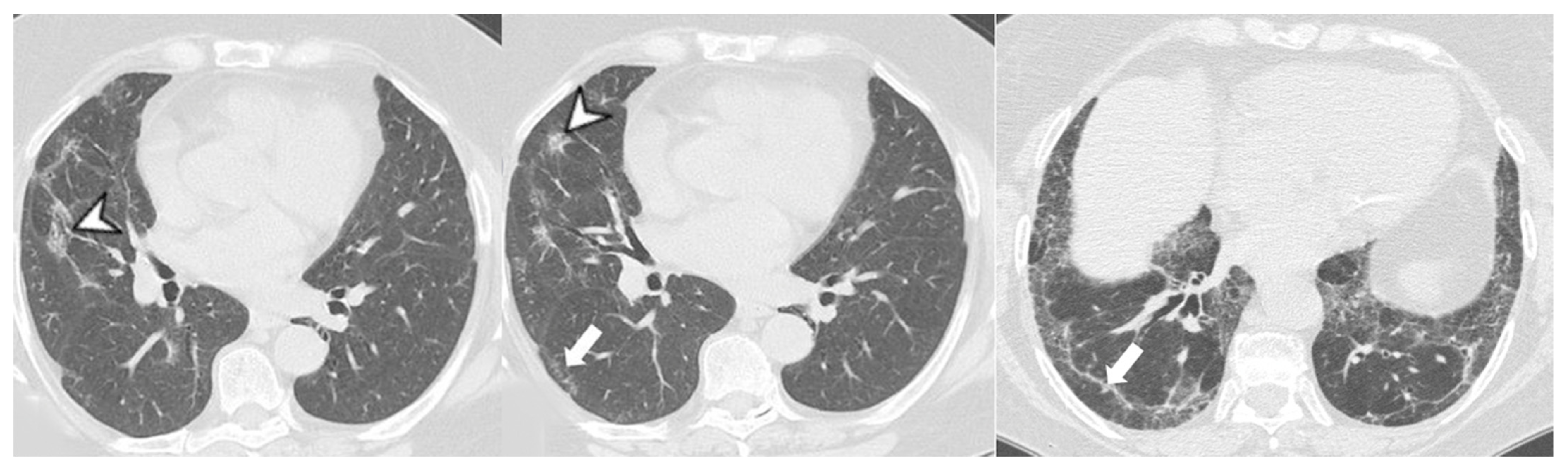
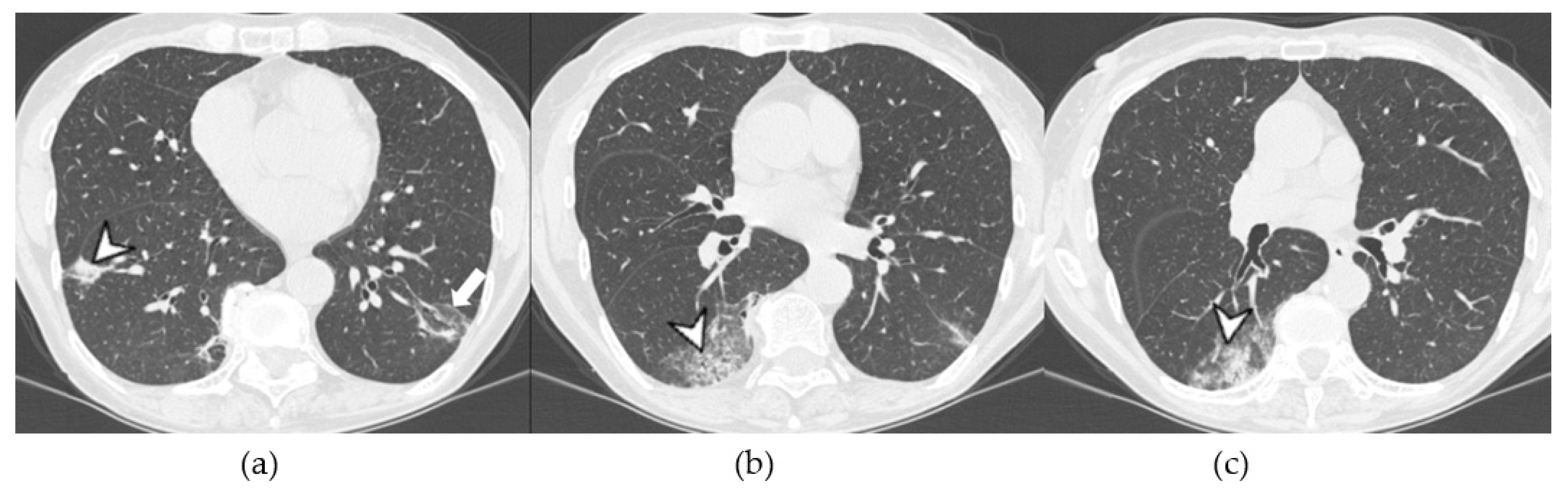
4.2.1. HRCT Findings of Patients with Anti-ARS Abs
4.2.2. HRCT Findings of Patients with Anti-MDA-5 Abs
5. Pulmonary Comorbidities of ILD in Patients with PM/DM
5.1. Pulmonary Ipertension
5.2. Pneumonia Secondary to Pulmonary Muscular Weakness
5.3. Drug-Induced Pneumonitis
6. Cancer-Associated Myositis
7. Treatment
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayashi, S.; Tanaka, M.; Kobayashi, H.; Nakazono, T.; Satoh, T.; Fukuno, Y.; Aragane, N.; Tada, Y.; Koarada, S.; Ohta, A.; et al. High-resolution computed tomography characterization of interstitial lung diseases in polymyositis/dermatomyositis. J. Rheumatol. 2008, 35, 260–269. [Google Scholar] [PubMed]
- Furst, D.E.; Amato, A.A.; Iorga, Ş.R.; Gajria, K.; Fernandes, A.W. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle Nerve 2012, 45, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Lega, J.C.; Reynaud, Q.; Belot, A.; Fabien, N.; Durieu, I.; Cottin, V. Idiopathic inflammatory myopathies and the lung. Eur. Respir. Rev. 2015, 24, 545. [Google Scholar] [CrossRef]
- Mills, E.S.; Mathews, W.H. Interstitial pneumonitis in dermatomyositis. J. Am. Med. Assoc. 1956, 160, 1467–1470. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Yoshimi, R.; Tamura, M.; Takeno, M.; Kunishita, Y.; Kishimoto, D.; Yoshioka, Y.; Kobayashi, K.; Takase-Minegishi, K.; Watanabe, T.; et al. The predictive prognostic factors for polymyositis/dermatomyositis-associated interstitial lung disease. Arthritis Res. Ther. 2018, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Ciancio, N.; Pavone, M.; Torrisi, S.E.; Vancheri, A.; Sambataro, D.; Palmucci, S.; Vancheri, C.; Di Marco, F.; Sambataro, G. Contribution of pulmonary function tests (PFTs) to the diagnosis and follow up of connective tissue diseases. Multidiscip. Respir. Med. 2019, 14, 17. [Google Scholar] [CrossRef]
- Zuo, Y.; Ye, L.; Liu, M.; Li, S.; Liu, W.; Chen, F.; Lu, X.; Gordon, P.; Wang, G.; Shu, X. Clinical significance of radiological patterns of HRCT and their association with macrophage activation in dermatomyositis. Rheumatology 2020, 59, 2829–2837. [Google Scholar] [CrossRef]
- Jeganathan, N.; Sathananthan, M. Connective Tissue Disease-Related Interstitial Lung Disease: Prevalence, Patterns, Predictors, Prognosis, and Treatment. Lung 2020, 198, 735–759. [Google Scholar] [CrossRef]
- Yang, S.H.; Chang, C.; Lian, Z.X. Polymyositis and dermatomyositis—Challenges in diagnosis and management. J. Transl. Autoimmun. 2019, 2, 100018. [Google Scholar] [CrossRef]
- Long, K.; Danoff, S.K. Interstitial Lung Disease in Polymyositis and Dermatomyositis. Clin. Chest Med. 2019, 40, 561–572. [Google Scholar] [CrossRef]
- Qudsiya, Z.; Waseem, M. Dermatomyositis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Vojinovic, T.; Cavazzana, I.; Ceruti, P.; Fredi, M.; Modina, D.; Berlendis, M.; Franceschini, F. Predictive Features and Clinical Presentation of Interstitial Lung Disease in Inflammatory Myositis. Clin. Rev. Allergy Immunol. 2021, 60, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, D.; Sambataro, G.; Pignataro, F.; Zanframundo, G.; Codullo, V.; Fagone, E.; Martorana, E.; Ferro, F.; Orlandi, M.; Del Papa, N.; et al. Patients with Interstitial Lung Disease Secondary to Autoimmune Diseases: How to Recognize Them? Diagnostics 2020, 10, 208. [Google Scholar] [CrossRef] [PubMed]
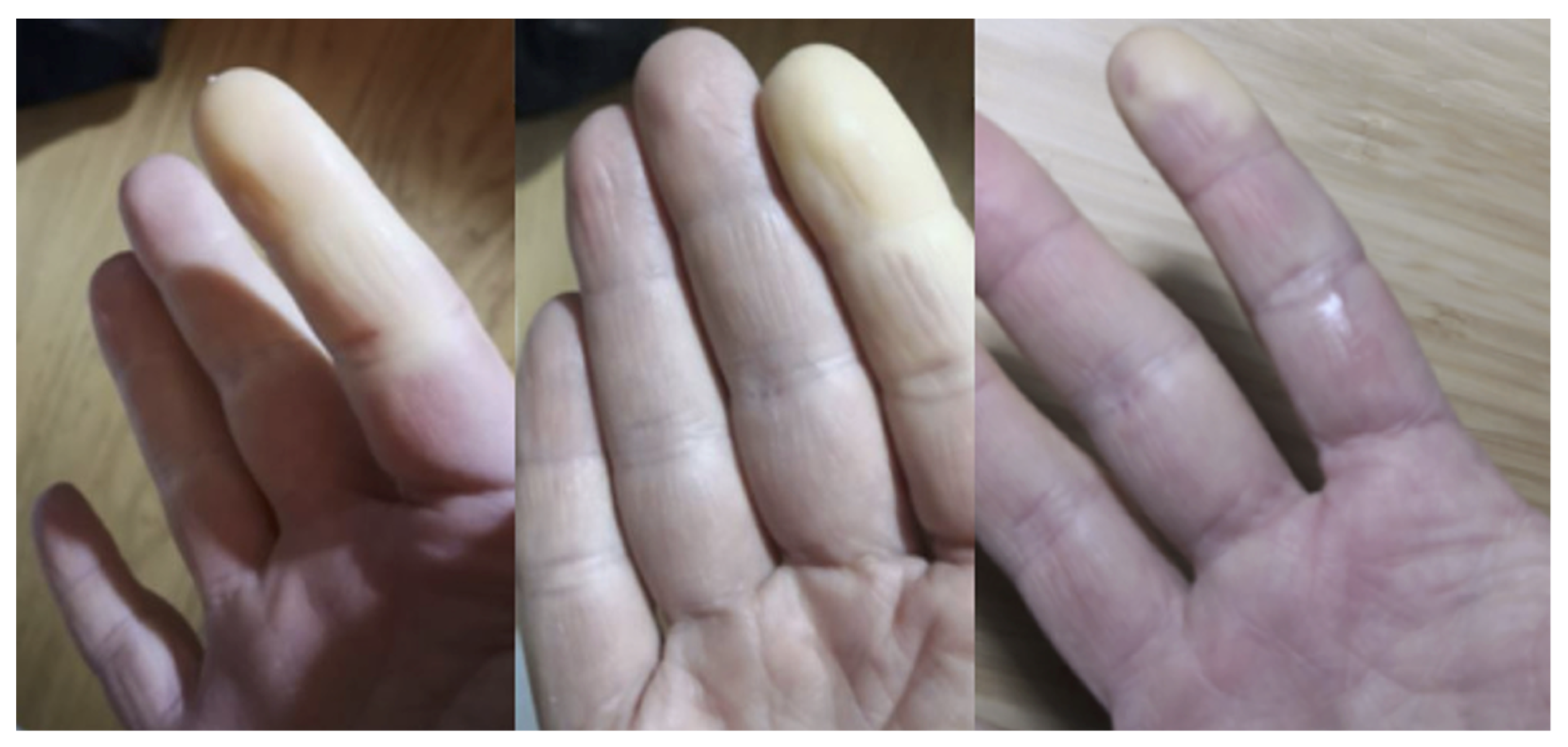
- Sambataro, D.; Sambataro, G.; Libra, A.; Vignigni, G.; Pino, F.; Fagone, E.; Fruciano, M.; Gili, E.; Pignataro, F.; Del Papa, N.; et al. Nailfold Videocapillaroscopy is a Useful Tool to Recognize Definite Forms of Systemic Sclerosis and Idiopathic Inflammatory Myositis in Interstitial Lung Disease Patients. Diagnostics 2020, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- González-Gay, M.A.; Montecucco, C.; Selva-O’Callaghan, A.; Trallero-Araguas, E.; Molberg, O.; Andersson, H.; Rojas-Serrano, J.; Perez-Roman, D.I.; Bauhammer, J.; Fiehn, C.; et al. Timing of onset affects arthritis presentation pattern in antisyntethase syndrome. Clin. Exp. Rheumatol. 2018, 36, 44–49. [Google Scholar] [PubMed]
- Mielnik, P.; Wiesik-Szewczyk, E.; Olesinska, M.; Chwalinska-Sadowska, H.; Zabek, J. Clinical features and prognosis of patients with idiopathic inflammatory myopathies and anti-Jo-1 antibodies. Autoimmunity 2006, 39, 243–247. [Google Scholar] [CrossRef]
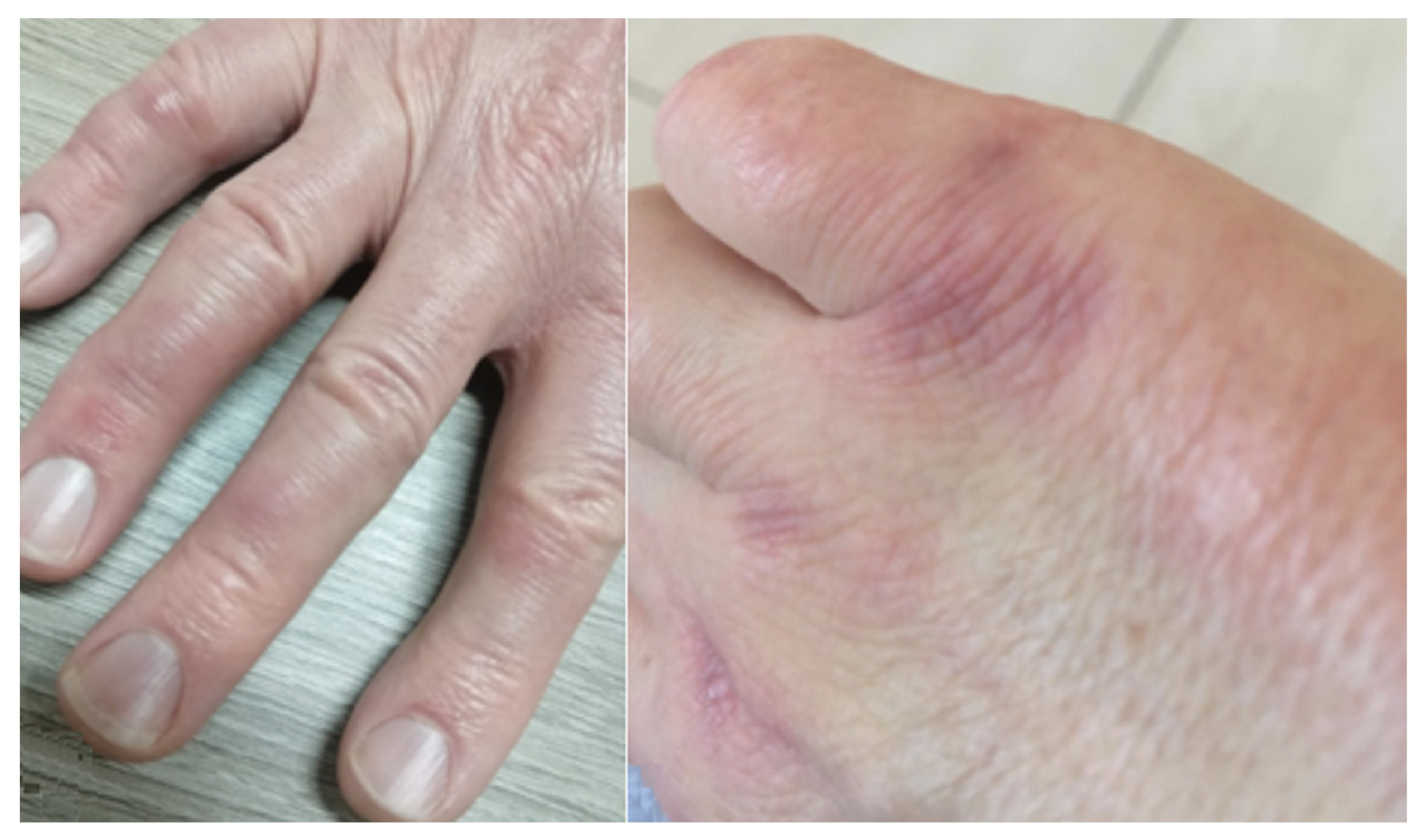
- Manfredi, A.; Sebastiani, M.; Cassone, G.; Pipitone, N.; Giuggioli, D.; Colaci, M.; Salvarani, C.; Ferri, C. Nailfold capillaroscopic changes in dermatomyositis and polymyositis. Clin. Rheumatol. 2015, 34, 279–284. [Google Scholar] [CrossRef]
- Sebastiani, M.; Triantafyllias, K.; Manfredi, A.; González-Gay, M.A.; Palmou-Fontana, N.; Cassone, G.; Drott, U.; Delbrück, C.; Rojas-Serrano, J.; Bertolazzi, C.; et al. Nailfold Capillaroscopy Characteristics of Antisynthetase Syndrome and Possible Clinical Associations: Results of a Multicenter International Study. J. Rheumatol. 2019, 46, 279–284. [Google Scholar] [CrossRef]
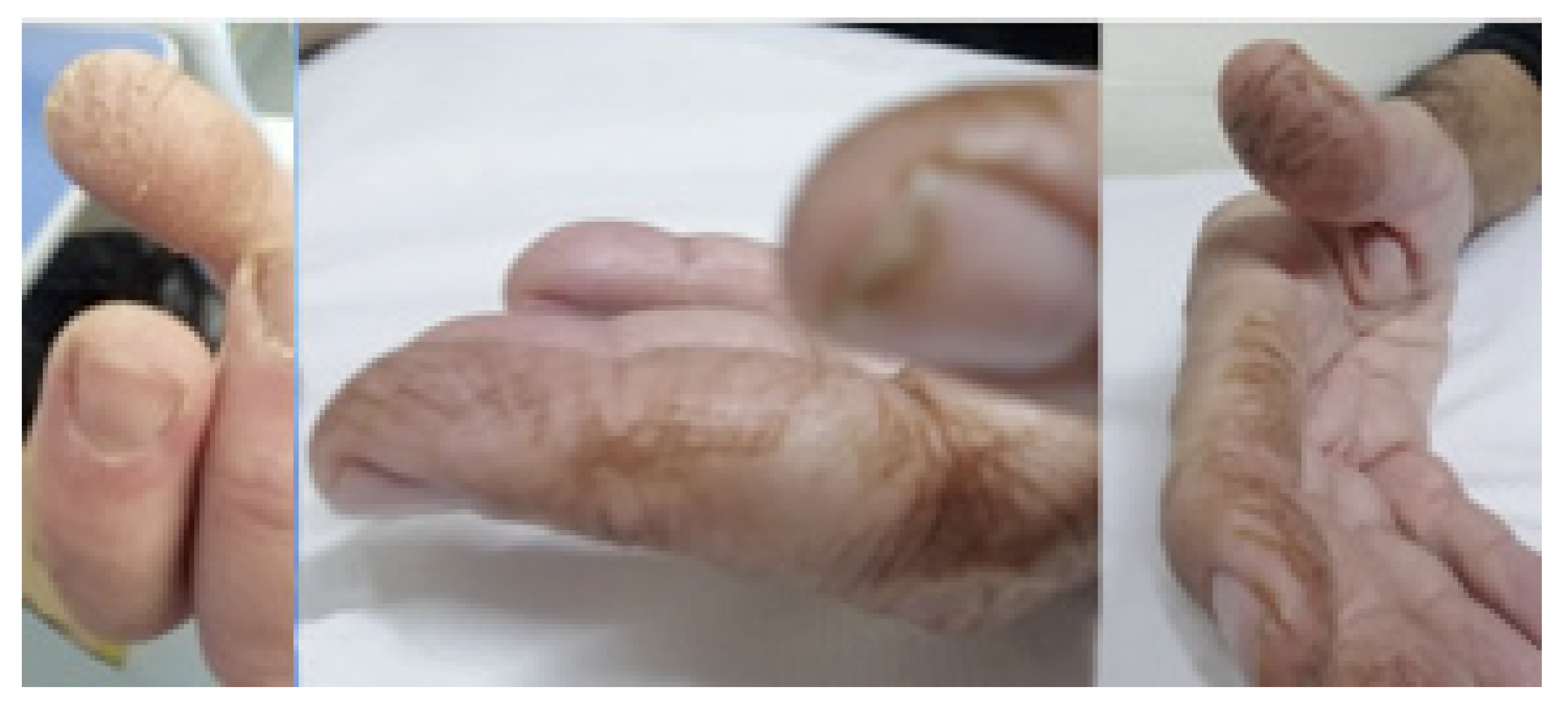
- Shiraishi, K.; Jinta, T.; Nishimura, N.; Nakaoka, H.; Tsugitomi, R.; Okafuji, K.; Kitamura, A.; Tomishima, Y.; Deshpande, G.A.; Tamura, T. Digital Clubbing Is Associated with Higher Serum KL-6 Levels and Lower Pulmonary Function in Patients with Interstitial Lung Disease. Can. Respir. J. 2018, 2018, 3640967. [Google Scholar] [CrossRef]
- Grathwohl, K.W.; Thompson, J.W.; Riordan, K.K.; Roth, B.J.; Dillard, T.A. Digital clubbing associated with polymyositis and interstitial lung disease. Chest 1995, 108, 1751–1752. [Google Scholar] [CrossRef]
- Van Manen, M.J.G.; Vermeer, L.C.; Moor, C.C.; Vrijenhoeff, R.; Grutters, J.C.; Veltkamp, M.; Wijsenbeek, M.S. Clubbing in patients with fibrotic interstitial lung diseases. Respir. Med. 2017, 132, 226–231. [Google Scholar] [CrossRef]
- Essouma, M.; Noubiap, J.J.; Singwe-Ngandeu, M.; Hachulla, E. Epidemiology of Idiopathic Inflammatory Myopathies in Africa: A Contemporary Systematic Review. J. Clin. Rheumatol. 2022, 28, e552–e562. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Tanaka, S.; Ceribelli, A.; Calise, S.J.; Chan, E.K. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin. Rev. Allergy Immunol. 2017, 52, 1–19. [Google Scholar] [CrossRef] [PubMed]
- In Seol, Y.; Jinhyun, K. The Role of Autoantibodies in Idiopathic Inflammatory Myopathies. J. Rheum. Dis. 2019, 26, 165–178. [Google Scholar] [CrossRef]
- Bohan, A.; Peter, J.B. Polymyositis and dermatomyositis (first of two parts). N. Engl. J. Med. 1975, 292, 344–347. [Google Scholar] [CrossRef]
- Bohan, A.; Peter, J.B. Polymyositis and dermatomyositis (second of two parts). N. Engl. J. Med. 1975, 292, 403–407. [Google Scholar] [CrossRef]
- Love, L.A.; Leff, R.L.; Fraser, D.D.; Targoff, I.N.; Dalakas, M.; Plotz, P.H.; Miller, F.W. A new approach to the classification of idiopathic inflammatory myopathy: Myositis-specific autoantibodies define useful homogeneous patient groups. Medicine 1991, 70, 360–374. [Google Scholar] [CrossRef]
- Lundberg, I.E.; Tjärnlund, A.; Bottai, M.; Werth, V.P.; Pilkington, C.; Visser, M.; Alfredsson, L.; Amato, A.A.; Barohn, R.J.; Liang, M.H.; et al. European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann. Rheum. Dis. 2018, 77, e64. [Google Scholar] [CrossRef]
- Shappley, C.; Paik, J.J.; Saketkoo, L.A. Myositis-Related Interstitial Lung Diseases: Diagnostic Features, Treatment, and Complications. Curr. Treat. Opt. Rheumatol. 2019, 5, 56–83. [Google Scholar] [CrossRef]
- Kang, E.H.; Lee, E.B.; Shin, K.C.; Im, C.H.; Chung, D.H.; Han, S.K.; Song, Y.W. Interstitial lung disease in patients with polymyositis, dermatomyositis and amyopathic dermatomyositis. Rheumatology 2005, 44, 1282–1286. [Google Scholar] [CrossRef]
- Marie, I.; Ménard, J.F.; Hachulla, E.; Chérin, P.; Benveniste, O.; Tiev, K.; Hatron, P.Y. Infectious complications in polymyositis and dermatomyositis: A series of 279 patients. Semin. Arthritis Rheum. 2011, 41, 48–60. [Google Scholar] [CrossRef]
- Sun, K.Y.; Fan, Y.; Wang, Y.X.; Zhong, Y.J.; Wang, G.F. Prevalence of interstitial lung disease in polymyositis and dermatomyositis: A meta-analysis from 2000 to 2020. Semin. Arthritis Rheum. 2021, 51, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Hallowell, R.W.; Ascherman, D.P.; Danoff, S.K. Pulmonary manifestations of polymyositis/dermatomyositis. Semin. Respir. Crit. Care Med. 2014, 35, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, G.; Gao, D.; Liu, G.; Pan, L.; Ni, L.; Li, Z.; Wang, Q. Factors Associated with Interstitial Lung Disease in Patients with Polymyositis and Dermatomyositis: A Systematic Review and Meta-Analysis. PLoS One 2016, 11, e0155381. [Google Scholar] [CrossRef] [PubMed]
- Tillie-Leblond, I.; Wislez, M.; Valeyre, D.; Crestani, B.; Rabbat, A.; Israel-Biet, D.; Humbert, M.; Couderc, L.J.; Wallaert, B.; Cadranel, J. Interstitial lung disease and anti-Jo-1 antibodies: Difference between acute and gradual onset. Thorax 2008, 63, 53–59. [Google Scholar] [CrossRef]
- Woeltjen, M.M.; Niehoff, J.H.; Michael, A.E.; Horstmeier, S.; Moenninghoff, C.; Borggrefe, J.; Kroeger, J.R. Low-Dose High-Resolution Photon-Counting CT of the Lung: Radiation Dose and Image Quality in the Clinical Routine. Diagnostics 2022, 12, 1441. [Google Scholar] [CrossRef]
- Cottin, V.; Thivolet-Béjui, F.; Reynaud-Gaubert, M.; Cadranel, J.; Delaval, P.; Ternamian, P.J.; Cordier, J.F.; Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur. Respir. J. 2003, 22, 245–250. [Google Scholar] [CrossRef]
- Kim, T.S.; Lee, K.S.; Chung, M.P.; Han, J.; Park, J.S.; Hwang, J.H.; Kwon, O.J.; Rhee, C.H. Nonspecific interstitial pneumonia with fibrosis: High-resolution CT and pathologic findings. AJR Am. J. Roentgenol. 1998, 171, 1645–1650. [Google Scholar] [CrossRef]
- Solomon, J.; Swigris, J.J.; Brown, K.K. Myositis-related interstitial lung disease and antisynthetase syndrome. J. Bras. Pneumol. 2011, 37, 100–109. [Google Scholar] [CrossRef]
- Faria, I.M.; Zanetti, G.; Barreto, M.M.; Rodrigues, R.S.; Araujo-Neto, C.A.; Silva, J.L.; Escuissato, D.L.; Souza, A.S., Jr.; Irion, K.L.; Mançano, A.D.; et al. Organizing pneumonia: Chest HRCT findings. J. Bras. Pneumol. 2015, 41, 231–237. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Ye, S.; Chen, X.X.; Lu, X.Y.; Wu, M.F.; Deng, Y.; Huang, W.Q.; Guo, Q.; Yang, C.D.; Gu, Y.Y.; Bao, C.D.; et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: A retrospective cohort study. Clin. Rheumatol. 2007, 26, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Saketkoo, L.A.; Ascherman, D.P.; Cottin, V.; Christopher-Stine, L.; Danoff, S.K.; Oddis, C.V. Interstitial Lung Disease in Idiopathic Inflammatory Myopathy. Curr. Rheumatol. Rev. 2010, 6, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Polosa, R.; Di Mauro, C.; Spampinato, B.; Castelli, L.; D’Amico, G.; Edwards, C.J.; Russo, C. A patient with antihistidyl-tRNA synthetase positive polymyositis presenting as acute respiratory distress syndrome. J. Clin. Rheumatol. 2008, 14, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Hozumi, H.; Kono, M.; Enomoto, N.; Hashimoto, D.; Nakamura, Y.; Inui, N.; Yokomura, K.; Koshimizu, N.; Toyoshima, M.; et al. Prognostic factors for myositis-associated interstitial lung disease. PLoS ONE 2014, 9, e98824. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, H.; Fujisawa, T.; Nakashima, R.; Johkoh, T.; Sumikawa, H.; Murakami, A.; Enomoto, N.; Inui, N.; Nakamura, Y.; Hosono, Y.; et al. Comprehensive assessment of myositis-specific autoantibodies in polymyositis/dermatomyositis-associated interstitial lung disease. Respir. Med. 2016, 121, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Fujisawa, T.; Enomoto, N.; Nakamura, Y.; Inui, N.; Naito, T.; Hashimoto, D.; Sato, J.; Toyoshima, M.; Hashizume, H.; et al. Interstitial lung diseases associated with amyopathic dermatomyositis. Eur. Respir. J. 2006, 28, 1005–1012. [Google Scholar] [CrossRef]
- Sato, S.; Masui, K.; Nishina, N.; Kawaguchi, Y.; Kawakami, A.; Tamura, M.; Ikeda, K.; Nunokawa, T.; Tanino, Y.; Asakawa, K.; et al. Initial predictors of poor survival in myositis-associated interstitial lung disease: A multicentre cohort of 497 patients. Rheumatology 2018, 57, 1212–1221. [Google Scholar] [CrossRef]
- Bonnefoy, O.; Ferretti, G.; Calaque, O.; Coulomb, M.; Begueret, H.; Beylot-Barry, M.; Laurent, F. Serial chest CT findings in interstitial lung disease associated with polymyositis-dermatomyositis. Eur. J. Radiol. 2004, 49, 235–244. [Google Scholar] [CrossRef]
- Tanizawa, K.; Handa, T.; Nakashima, R.; Kubo, T.; Hosono, Y.; Aihara, K.; Ikezoe, K.; Watanabe, K.; Taguchi, Y.; Hatta, K.; et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir. Med. 2013, 107, 745–752. [Google Scholar] [CrossRef]
- Miller, S.A.; Glassberg, M.K.; Ascherman, D.P. Pulmonary complications of inflammatory myopathy. Rheum. Dis. Clin. 2015, 41, 249–262. [Google Scholar] [CrossRef]
- Pan, L.; Liu, Y.; Sun, R.; Fan, M.; Shi, G. Comparison of characteristics of connective tissue disease-associated interstitial lung diseases, undifferentiated connective tissue disease-associated interstitial lung diseases, and idiopathic pulmonary fibrosis in Chinese Han population: A retrospective study. Clin. Dev. Immunol. 2013, 2013, 121578. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Cox, C.W.; Montner, S.M.; Adegunsoye, A.; Oldham, J.M.; Husain, A.N.; Vij, R.; Noth, I.; Lynch, D.A.; Strek, M.E. CT Features of the Usual Interstitial Pneumonia Pattern: Differentiating Connective Tissue Disease-Associated Interstitial Lung Disease from Idiopathic Pulmonary Fibrosis. AJR Am. J. Roentgenol. 2018, 210, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Egashira, R. High-Resolution CT Findings of Myositis-Related Interstitial Lung Disease. Medicina 2021, 57, 692. [Google Scholar] [CrossRef] [PubMed]
- Lega, J.C.; Fabien, N.; Reynaud, Q.; Durieu, I.; Durupt, S.; Dutertre, M.; Cordier, J.F.; Cottin, V. The clinical phenotype associated with myositis-specific and associated autoantibodies: A meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun. Rev. 2014, 13, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Waseda, Y.; Johkoh, T.; Egashira, R.; Sumikawa, H.; Saeki, K.; Watanabe, S.; Matsunuma, R.; Takato, H.; Ichikawa, Y.; Hamaguchi, Y.; et al. Antisynthetase syndrome: Pulmonary computed tomography findings of adult patients with antibodies to aminoacyl-tRNA synthetases. Eur. J. Radiol. 2016, 85, 1421–1426. [Google Scholar] [CrossRef]
- Aggarwal, R.; Cassidy, E.; Fertig, N.; Koontz, D.C.; Lucas, M.; Ascherman, D.P.; Oddis, C.V. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann. Rheum. Dis. 2014, 73, 227–232. [Google Scholar] [CrossRef]
- Hallowell, R.W.; Paik, J.J. Myositis-associated interstitial lung disease: A comprehensive approach to diagnosis and management. Clin. Exp. Rheumatol. 2022, 40, 373–383. [Google Scholar] [CrossRef]
- Hamaguchi, Y.; Fujimoto, M.; Matsushita, T.; Kaji, K.; Komura, K.; Hasegawa, M.; Kodera, M.; Muroi, E.; Fujikawa, K.; Seishima, M.; et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: Heterogeneity within the syndrome. PLoS ONE 2013, 8, e60442. [Google Scholar] [CrossRef]
- Wu, W.; Guo, L.; Fu, Y.; Wang, K.; Zhang, D.; Xu, W.; Chen, Z.; Ye, S. Interstitial Lung Disease in Anti-MDA5 Positive Dermatomyositis. Clin. Rev. Allergy Immunol. 2021, 60, 293–304. [Google Scholar] [CrossRef]
- Hervier, B.; Uzunhan, Y. Inflammatory Myopathy-Related Interstitial Lung Disease: From Pathophysiology to Treatment. Front. Med. 2020, 6, 326. [Google Scholar] [CrossRef]
- Mehta, P.; Aggarwal, R.; Porter, J.C.; Gunawardena, H. Management of interstitial lung disease (ILD) in myositis syndromes: A practical guide for clinicians. Best Pract. Res. Clin. Rheumatol. 2022, 36, 101769. [Google Scholar] [CrossRef] [PubMed]
- Syue, S.H.; Chang, Y.H.; Shih, P.J.; Lin, C.L.; Yeh, J.J.; Kao, C.H. Polymyositis/dermatomyositis is a potential risk factor for acute respiratory failure: A pulmonary heart disease. Ann. Transl. Med. 2020, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, B. Rare complications of anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis: Time to nip them in the bud. Front. Immunol. 2022, 13, 1009546. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, T.; Cai, Y.Y.; Luo, L.; Wang, M.; Yang, M.; Cai, L. Pulmonary hypertension in polymyositis. Clin. Rheumatol. 2015, 34, 2105–2112. [Google Scholar] [CrossRef]
- Hervier, B.; Meyer, A.; Dieval, C.; Uzunhan, Y.; Devilliers, H.; Launay, D.; Canuet, M.; Têtu, L.; Agard, C.; Sibilia, J.; et al. Pulmonary hypertension in antisynthetase syndrome: Prevalence, aetiology and survival. Eur. Respir. J. 2013, 42, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Cavagna, L.; Nuño, L.; Scirè, C.A.; Govoni, M.; Longo, F.J.L.; Franceschini, F.; Neri, R.; Castañeda, S.; Giraldo, W.A.S.; Caporali, R.; et al. Clinical Spectrum Time Course in Anti Jo-1 Positive Antisynthetase Syndrome: Results from an International Retrospective Multicenter Study. Medicine 2015, 94, e1144. [Google Scholar] [CrossRef] [PubMed]
- Sanges, S.; Yelnik, C.M.; Sitbon, O.; Benveniste, O.; Mariampillai, K.; Phillips-Houlbracq, M.; Pison, C.; Deligny, C.; Inamo, J.; Cottin, V.; et al. Pulmonary arterial hypertension in idiopathic inflammatory myopathies: Data from the French pulmonary hypertension registry and review of the literature. Medicine 2016, 95, e4911. [Google Scholar] [CrossRef]
- Gasparotto, M.; Gatto, M.; Saccon, F.; Ghirardello, A.; Iaccarino, L.; Doria, A. Pulmonary involvement in antisynthetase syndrome. Curr. Opin. Rheumatol. 2019, 31, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, Y.; Makino, S.; Ogura, T.; Suda, T.; Tomioka, H.; Amano, H.; Anraku, M.; Enomoto, N.; Fujii, T.; Fujisawa, T.; et al. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir. Investig. 2021, 59, 709–740. [Google Scholar] [CrossRef]
- Callen, J.P.; Wortmann, R.L. Dermatomyositis. Clin. Dermatol. 2006, 24, 363–373. [Google Scholar] [CrossRef]
- Wu, C.; Wang, Q.; He, L.; Yang, E.; Zeng, X. Hospitalization mortality and associated risk factors in patients with polymyositis and dermatomyositis: A retrospective case-control study. PLoS ONE 2018, 13, e0192491. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Benito, A.; Curran, A.; Villar-Gomez, A.; Trallero-Araguas, E.; Fernández-Codina, A.; Pinal-Fernandez, I.; Rodrigo-Pendás, J.Á.; Selva-O’Callaghan, A. Opportunistic infections in patients with idiopathic inflammatory myopathies. Int. J. Rheum. Dis. 2018, 21, 487–496. [Google Scholar] [CrossRef]
- Tobin, R.W.; Pope, C.E., 2nd; Pellegrini, C.A.; Emond, M.J.; Sillery, J.; Raghu, G. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1998, 158, 1804–1808. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, S.; Colby, T.V.; Leslie, K.O.; Helmers, R.A. Methotrexate pneumonitis: Review of the literature and histopathological findings in nine patients. Eur. Respir. J. 2000, 15, 373–381. [Google Scholar] [CrossRef]
- Rossi, S.E.; Erasmus, J.J.; McAdams, H.P.; Sporn, T.A.; Goodman, P.C. Pulmonary drug toxicity: Radiologic and pathologic manifestations. Radiographics 2000, 20, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Yamasaki, M.; Kurihara, Y.; Yamada, H.; Nakajima, Y. Methotrexate-induced pulmonary injury: Serial CT findings. J. Thorac. Imaging 2003, 18, 231–236. [Google Scholar] [CrossRef]
- Malik, S.W.; Myers, J.L.; DeRemee, R.A.; Specks, U. Lung toxicity associated with cyclophosphamide use. Two distinct patterns. Am. J. Respir. Crit. Care Med. 1996, 154 Pt 1, 1851–1856. [Google Scholar] [CrossRef]
- Savage, R.L.; Highton, J.; Boyd, I.W.; Chapman, P. Pneumonitis associated with leflunomide: A profile of New Zealand and Australian reports. Intern. Med. J. 2006, 36, 162–169. [Google Scholar] [CrossRef]
- Chikura, B.; Lane, S.; Dawson, J.K. Clinical expression of leflunomide-induced pneumonitis. Rheumatology 2009, 48, 1065–1068. [Google Scholar] [CrossRef]
- Hadjinicolaou, A.V.; Nisar, M.K.; Bhagat, S.; Parfrey, H.; Chilvers, E.R.; Ostör, A.J. Non-infectious pulmonary complications of newer biological agents for rheumatic diseases—A systematic literature review. Rheumatology 2011, 50, 2297–2305. [Google Scholar] [CrossRef]
- Oldroyd, A.G.S.; Allard, A.B.; Callen, J.P.; Chinoy, H.; Chung, L.; Fiorentino, D.; George, M.D.; Gordon, P.; Kolstad, K.; Kurtzman, D.J.; et al. A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology 2021, 60, 5483. [Google Scholar] [CrossRef] [PubMed]
- Sterz, G. Polymyositis. In Berliner Klinische Wochenschrift; Verlag von August Hirschwald: Berlin, Germany, 1916; Volume 53, p. 489. Available online: https://scholar.google.com/scholar_lookup?journal=Berliner+Klinische+Wochenschrift&title=Polymyositis&author=G+Sterz&volume=53&publication_year=1916&pages=489& (accessed on 21 September 2022).
- Hill, C.L.; Zhang, Y.; Sigurgeirsson, B.; Pukkala, E.; Mellemkjaer, L.; Airio, A.; Evans, S.R.; Felson, D.T. Frequency of specific cancer types in dermatomyositis and polymyositis: A population-based study. Lancet 2001, 357, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Zahr, Z.A.; Baer, A.N. Malignancy in myositis. Curr. Rheumatol. Rep. 2011, 13, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, M.; Geng, J.; Ban, C.; Zhang, S.; Chen, W.; Ren, Y.; He, X.; Chen, W.; Dai, H. Incidence and radiologic-pathological features of lung cancer in idiopathic pulmonary fibrosis. Clin. Respir. J. 2018, 12, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Machado, P.M.; Gupta, L. Understanding and managing anti-MDA 5 dermatomyositis, including potential COVID-19 mimicry. Rheumatol. Int. 2021, 41, 1021–1036. [Google Scholar] [CrossRef]
- Somogyi, V.; Chaudhuri, N.; Torrisi, S.E.; Kahn, N.; Müller, V.; Kreuter, M. The therapy of idiopathic pulmonary fibrosis: What is next? Eur. Respir. Rev. 2019, 28, 190021. [Google Scholar] [CrossRef]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases—Subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PM-DM | |
|---|---|
| Laboratory findings | Muscular enzymes:
|
| Clinical features | Muscle weakness:
|
Cutaneous manifestation:
| |
Lung involvement:
| |
| Radiological features | ILD:
|
| Anti-ARS-Abs | Anti-MDA-5 Abs | ||
|---|---|---|---|
| Clinical course | subacute to chronic | acute rapidly progressive to subacute | |
| Lesions | GGO, reticulations, consolidations | consolidations, GGOs | |
| Distribution | Homogeneous; lower lung lobes, along bronchovascular bundles and lung periphery; loss of volume of lower lobes | Patchy; peripheral lower lobes or along the bronchovascular bundles | |
| CT pattern | NSIP OP NSIP-OP UIP DAD-unclassifiable | 50% 20% 25% 10% +/− | 20% 50% 25% <5% ++ |
| Prognosis | good response to treatment; possibility of relapses | Poorer prognosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmucci, S.; Di Mari, A.; Cancemi, G.; Pennisi, I.; Mauro, L.A.; Sambataro, G.; Sambataro, D.; Galioto, F.; Fazio, G.; Ferlito, A.; et al. Clinical and Radiological Features of Interstitial Lung Diseases Associated with Polymyositis and Dermatomyositis. Medicina 2022, 58, 1757. https://doi.org/10.3390/medicina58121757
Palmucci S, Di Mari A, Cancemi G, Pennisi I, Mauro LA, Sambataro G, Sambataro D, Galioto F, Fazio G, Ferlito A, et al. Clinical and Radiological Features of Interstitial Lung Diseases Associated with Polymyositis and Dermatomyositis. Medicina. 2022; 58(12):1757. https://doi.org/10.3390/medicina58121757
Chicago/Turabian StylePalmucci, Stefano, Alessia Di Mari, Giovanna Cancemi, Isabella Pennisi, Letizia Antonella Mauro, Gianluca Sambataro, Domenico Sambataro, Federica Galioto, Giulia Fazio, Agata Ferlito, and et al. 2022. "Clinical and Radiological Features of Interstitial Lung Diseases Associated with Polymyositis and Dermatomyositis" Medicina 58, no. 12: 1757. https://doi.org/10.3390/medicina58121757
APA StylePalmucci, S., Di Mari, A., Cancemi, G., Pennisi, I., Mauro, L. A., Sambataro, G., Sambataro, D., Galioto, F., Fazio, G., Ferlito, A., Pino, F., Basile, A., & Vancheri, C. (2022). Clinical and Radiological Features of Interstitial Lung Diseases Associated with Polymyositis and Dermatomyositis. Medicina, 58(12), 1757. https://doi.org/10.3390/medicina58121757