Abstract
Psychomotor developmental delay is a disorder with a prevalence of 12–18% in the pediatric population, characterized by the non-acquisition of motor, cognitive and communication skills during the child’s development, in relation to chronological age. An appropriate neuropsychomotor evaluation and the use of new technologies, such as Array Comparative Genomic Hybridization (a-CGH) and Next-generation sequencing (NGS), can contribute to early diagnosis and improving the quality of life. In this case, we have analyzed a boy aged 2 years and 8 months, with a diagnosis of psychomotor developmental delay, mainly in the area of communication and language. The a-CGH analysis identified three de novo deletions of uncertain clinical significance, involving PLXNA2 (1q32.2), PRELID2, GRXCR2 and SH3RF2 (5q32), RIMS1 (6q13), and a heterozygous duplication of maternal origin involved three genes: HELZ, PSMD12 and PITPNC1 (17q24.2). Among all these alterations, our attention focused on the PLXNA2 gene because of the central function that plexin 2 carries out in the development of the central nervous system. However, all genes detected in the analysis could contribute to the phenotypic characteristics of the patient.
1. Introduction
Child development is a gradual and continuous process, with different levels of complexity, with a similar sequence in all children but with a variable rhythm. The different stages of growth include the development of body systems that are responsible for cognitive, physical, and social-emotional skills. Psychomotor developmental delay has a negative impact on social interactions and the performance of daily activities [1]. In fact, psychomotor developmental delay is characterized by the lack of delayed acquisition of motor, cognitive and communication skills during the child’s development, in relation to chronological age. Compared to simple motor retardation, which refers only to global motor skills, psychomotor developmental delay compromises all adaptive functions. The prevalence is 12–18% in developed countries [2,3,4,5,6], where 75% of total cases are genetic and the rest depend on family and social factors [7]. In fact, during their development, children are not passive subjects but actively participate in this process by exploring the environment that surrounds them. In particular, social relationships are fundamental for healthy development, so that the absence of adequate stimulation from the family and social environment could be one of the causes of the delay. Moreover, a delay may be due to the effect of an isolated sensory deficit, such as congenital sensorineural hearing loss or mental deficiency, usually not noticeable until the end of preschool age. Neuropsychomotor evaluation is of fundamental importance to define the level of child development, in order to identify areas of strength and weakness within his development profile and to undertake early rehabilitation treatment [8]. In fact, a study showed that more than 60% of the timely interventions in the psychomotor developmental delay result in a statistically significant improvement [9]. New technologies, such as Array Comparative Genomic Hybridization (a-CGH) and Next-generation sequencing (NGS), can contribute to the identification of variants associated with this delay, thus, representing a great contribution to early diagnosis [10,11,12,13,14,15,16].
2. Case Report
We report the case of a child aged 2 years and 8 months, born from healthy unrelated parents, with a diagnosis of psychomotor developmental delay, mainly in the area of communication and language. The proband was born in the thirty-seventh week of gestation by caesarean section, and the mother reports a threat of abortion in the first trimester and a miscarriage that occurred in a previous pregnancy. The mother was affected by pituitary microadenoma and the grandmother by ovarian cancer at 29 years. The parents report the presence in the family of a maternal uncle with psychiatric problems, who died of acute myocardial infarction (Figure 1). The patient began to take their first steps at the age of 11 months and currently has not yet acquired control of the sphincters and does not say a word. From the age of 18 months, the child has been followed by pediatric neuropsychiatry due to the absence of language and hyperactivity. The electroencephalogram (EEG) reveals brain electrical activity in sleep within normal limits, as well as the ABR (Auditory Brainstem Response) and echo-abdomen test. Physical examination revealed low hairline, wide auricles and feet: clinodactyly of the 4th and 5th toes bilaterally, syndactyly of the 2nd and 3rd toes, large hallux, bilateral flat foot and valgus knees. Furthermore, tone and trophism are normal, and the cranial nerves are unaffected. Currently, the child practices psychomotor and speech therapy twice a week and attends kindergarten without school support and a good interaction with his peers is reported.
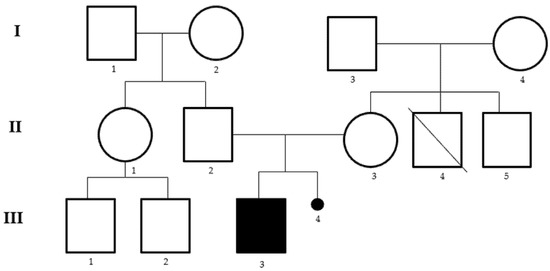
Figure 1.
Pedigree of the family. I.2 grandmother ovarian cancer at 29 years, II.3 mother with pituitary microadenoma, II.4 uncle died of acute myocardial infarction at 31 years of age with psychiatric problems, III.3 proband, III.4 miscarriage in a previous pregnancy.
Molecular analysis was conducted on DNA of the proband, and his parents extracted this from peripheral blood, with the extraction kit, Maxwell RSC Blood DNA Kit (Promega, Madison, Wisconsin, USA), using the a-CGH platform 4 × 180 K SurePrint G3 Human CGH Microarray (Agilent Technologies, Santa Clara, CA, USA), with an average spacing of 13 kb, allowing an average resolution of 25 kb. The microarray was scanned on an Agilent G2600D scanner. Image files were quantified, and data were visualized by using Agilent’s Cytogenomics software version 4.0.3.12 (Agilent Technologies, Santa Clara, CA, USA). The human assembly utilized was GRCh37, hg19 (http://www.ensembl.org/, last accession 1 April 2022). The a-CGH analysis showed the presence of a heterozygous de novo deletion on chromosome 1, in the q32.2 region, ranging from position 208236806 to 208521372, with an extension of 284.57 kb, partially involving the Plexin A2 (PLXNA2) (RefSeq # NC_000001.11) gene (Figure 2A), the presence of a heterozygous de novo deletion on chromosome 5, in the q32 region, ranging from position 145191813 to 145342040, with an extension of 150.23 kb, partially involving PRELI Domain Containing 2 (PRELID2), Glutaredoxin and Cysteine Rich Domain Containing 2 (GRXCR2) and partially SH3 Domain Containing Ring Finger 2 (SH3RF2) (RefSeq # NC_000005.10) genes (Figure 2B). Furthermore, we also observed the presence of a heterozygous de novo deletion on chromosome 6, in the q13 region, ranging from position 72605565 to 72651412, with an extension of 45.85 kb, involving the intron 1 of Regulating Synaptic Membrane Exocytosis 1 (RIMS1) (RefSeq # NC_000006.12) gene (Figure 2C) and the presence of a heterozygous duplication of maternal origin on chromosome 17, in the q24.2 region, ranging from position 65214693 to 65596540, with an extension of 381.85 kb, partially involving Helicase With Zinc Finger (HELZ), Proteasome 26S Subunit, Non-ATPase 12 (PSMD12), partially Phosphatidylinositol Transfer Protein Cytoplasmic 1 (PITPNC1) (RefSeq # NC_000017.11) genes (Figure 2D).
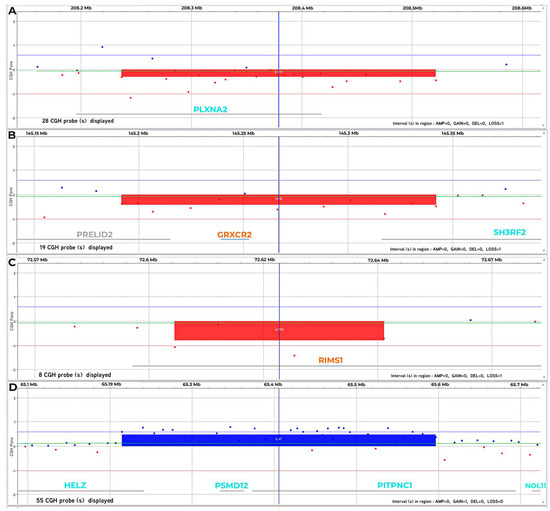
Figure 2.
(A) a-CGH profile of chromosome 1. This analysis shows a heterozygous de novo deletion in 1q32.2 region of 284.57 kb involving PLXNA2. (B) a-CGH profile of chromosome 5. The analysis shows a heterozygous de novo deletion in 5q32 region of 150.23 kb, partially involving PRELID2, GRXCR2 and partially SH3RF2. (C) a-CGH profile of chromosome 6. This analysis shows a heterozygous de novo deletion in 6q13 region of 45.85 kb, involving RIMS1. (D) a-CGH profile of chromosome 17. The analysis shows a heterozygous duplication of maternal origin in the 17q24.2 region of 381.85 kb partially involving HELZ, PSMD12, partially PITPNC1. Results are interpreted as log2 ratio of test vs. control. The deletion and duplication, when present, is indicated by a rectangle.
3. Discussion
The PLXNA2 gene, located on chromosome 1 cytoband q32.2 (208195588–208417665 (GRCh37/hg19), is characterized by 32 exons, for a total length of about 222,078 bp and mainly expressed in neural tissue. PLXNA2 belongs to the family of plexin genes, which encode several type 1 transmembrane proteins that function as semaphorin receptors. Semaphorins are plexin ligands and the plexin-semaphorin signaling system is extensively involved in many neuronal events, including driving axons, cell migration, axon pruning and synaptic plasticity [17]. In particular, plexin A2 is thought to bind the secreted proteins semaphorin-3A, -3C, or -5A, thereby precipitating plexin A2 dimerization and the activation of its intrinsic GTPase activating protein domain to act as a negative regulator to Rap1B GTPase. This initiates a signal transduction cascade that mediates axonal repulsion and guidance, dendritic guidance, and neuronal migration during nervous system development [18,19]. Defects in dendritic spine density during development and in adult-born hippocampus [17] and perturbation in migration were shown in a knock-out mouse model for PlxnA2 (PlxnA2−/−) of hippocampal granules [20]. Behavioral studies in PlxnA2−/− mice revealed deficits in associative learning, sociability and sensorimotor gating that are common features of neuropsychiatric disorders [18]. All these data support that PlxnA2 expression is relevant to the structure and function of the nervous system. For this reason, PlxnA2 dysfunction appears to contribute to the development of neurological disorders, such as intellectual disability (ID), retardation and schizophrenia [19]. Differences in phenotypes may be explained by the incomplete penetrance of the gene. Only in very rare cases, mutations in the PLXNA2 gene can be associated with cardiac abnormalities [21]. The gene is not present in the Simons Foundation Autism Research Initiative (SFARI) database (https://gene.sfari.org/, last accession 1 April 2022), unlike the other genes in the same family, PLXNA3 and PLXNA4, which appear to be closely related to ASD. Indeed, a de novo variant of the junction site in the PLXNA3 gene was identified in a proband with ASD [22] and targeted sequencing of 3195 Chinese probands [23] identified three rare deleterious variants in PLXNA3 in Chinese ASD probands. To date, most of the studies on the PLXNA2 gene identify single nucleotide polymorphisms (SNPs) and missense point mutations that contribute to altering the function of the protein and, thus, contribute to candidate PLXNA2 for the development of intellectual disability.
With the implementation of techniques, such as a-CGH and NGS, it has been observed that chromosomal aberrations are found in the genome of 15% of patients with ID, while in 36% of cases, the disorder is related to single-gene variations. Finally, single-gene variations are de novo, while in only 4% of cases, they are inherited in an autosomic recessive manner [19]. In this case, a de novo deletion was identified in the heterozygosity of the PLXNA2 gene, extending from exon 1 to exon 12, partially including intron 12. This deletion is reported of uncertain clinical significance on the consulted databases Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 1 April 2022), Decipher (https://decipher.sanger.ac.uk/, accessed on 1 April 2022), Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home, accessed on 1 April 2022), GeneCards (http://www.genecards.org/, accessed on 1 April 2022), and OMIM (https://www.omim.org/, accessed on 1 April 2022). The alteration involves the binding site of plexin with semaphorin and the absence of this site alters the entire cellular pathway, in which the PLXNA2 gene is involved. In light of the observations in the literature, we can hypothesize a possible role of the altered PLXNA2 gene in the development of phenotypic characteristics detected in the proband and in other patients with dysmorphism and cardiac anomalies. Further studies are needed to elucidate the role of the gene in the pathogenesis of psychomotor developmental delay.
In addition, we found a de novo heterozygous deletion that includes the GRXCR2 and SH3RF2 genes located on chromosome 5, cytoband q32. The GRXCR2 gene (145239296–145252531 (GRCh37/hg19) consists of three exons, for a total length of about 13,236 bp and encodes a protein, which causes the loss of hearing. GRXCR2 and its paralogue GRXCR1 have a size of ~30 kDa and are highly conserved cytosolic proteins. The mutation of GRXCR2 has been associated with autosomal recessive non-syndromic deafness [24], characterized by moderate to severe bilateral hearing loss. In particular, GRXCR2 is concentrated in the basal region of the stereocilia and is essential for the localization of taperin. In Grxcr2-deficient hair cells, taperin is reduced at the base and is found along the length of the stereocilia, which are elongated and disorganized; therefore, GRXCR2 would seem to limit the taperin at the base of the stereocilia, causing deafness [25,26].
The SH3 protein, encoded by the SH3RF2 gene, is an E3 ubiquitin-protein ligase [27]. It acts as an anti-apoptotic regulator of the JNK pathway by ubiquitinating and promoting the degradation of SH3RF1, a scaffold protein required for the pro-apoptotic activation of JNK [28]. Studies using SH3RF2 knockout mice show that haploinsufficiency of the gene is related to unilateral disorders of hippocampal function and autistic behaviors, social and communication interactions, repetitive behaviors and seizures [29].
Finally, a maternal duplication involves the PSMD12 gene. The PSMD12 gene is located on chromosome 17 of the cytoband q24.2 (65336619–65362721 (GRCh37/hg19), for a length of about 26.103 bp and 11 component exons of the proteasome regulatory subunit, a large multi-subunit enzyme complex responsible for ATP-dependent and ubiquitin-mediated degradation of proteins and is highly conserved. In particular, PSMD12 is required for proper assembly and localization of the proteasome [30] and is highly expressed in the brain. Recently, de novo mutations in PSMD12 have been identified in individuals with ID and ASD [31].
4. Conclusions
The a-CGH technique represents an important advance in the screening of unbalanced rearrangements, caused by the loss and/or duplication of small genomic material, not detectable by other cytogenetic methods, acting as a fundamental diagnostic tool in the identification of the molecular component underlying multiple genotype–phenotype relationships [32,33,34,35,36]. Due the significant increase in resolution, it is possible to identify the presence of causative or potentially causative chromosomal abnormalities on the whole genome, in a great variety of cases, from the most common neurological and congenital disorders to multifactorial syndromes, with advantages in terms of time, precision and standardization [37,38,39].
In this study, the a-CGH detected copy number variations (CNVs) containing genes potentially related to the clinical characteristics of the patient. In particular, the involvement of the PLXNA2 gene in the structure and function of the central nervous system and its implication in neurological disorders make it an interesting candidate gene for neurodevelopmental disorders. Furthermore, the other alterations identified in the patient could contribute to the severity of the phenotype, with roles not yet defined.
For this reason, the progressive evolution of the a-CGH, through the use of higher resolution platforms, such as the SNP-array technology, may lead, in the near future, to its increasingly massive use for the benefit of understanding the disease’s origin of still unknown etiology. Finally, a combination between the a-CGH and NGS could represent an added value to the diagnosis, providing a complete diagnosis of the patients and helping to provide further information on the causal mechanisms involved in neurodevelopmental disorders and congenital anomalies.
Author Contributions
Conceptualization L.P. and B.L.; methodology N.F., A.R. and A.V.; data analysis N.F., A.R. and A.V.; validation L.P. and B.L.; writing—original draft preparation N.F., A.R. and A.V.; writing—review and editing L.P. and B.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data will be available by contacting the corresponding authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Piccolo, L.D.R.; Segabinazi, J.D.; Falceto, O.G.; Fernandes, C.L.C.; Bandeira, D.R.; Trentini, C.M.; Hutz, C.S.; Salles, J.F. Developmental Delay in Early Childhood Is Associated with Visual-Constructive Skills at School Age in a Brazilian Cohort. Psicol. Reflex. Crit. 2016, 29, 41. [Google Scholar] [CrossRef]
- Vericat, A.; Orden, A.B. Psychomotor Development and Its Disorders: Between Normal and Pathological Development. Cien Saude Colet 2013, 18, 2977–2984. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Avaria, M.d.l.A. Pediatria Del Desarrollo y Comportamiento. Rev. Pediatría Electrón. 2005, 2, 88–91. [Google Scholar]
- Glascoe, F.P. Early Detection of Developmental and Behavioral Problems Epidemiology and Issues for Clinicians. Pediatr. Rev. 2000, 21, 272–280. [Google Scholar] [CrossRef]
- Bornstein, M.; Hendricks, C. Screening for Developmental Disabilities in Developing Countries. Soc. Sci. Med. 2013, 97, 307–315. [Google Scholar] [CrossRef]
- Lecannelier, F.; Ewert, J.C.P.; Groissman, S.; Gallardo, D.; Bardet, A.M.; Bascuñan, A.; Rodríguez, J. Validación Del Inventario de Conductas Infantiles Para Niños de Entre 11/2-5 Años (CBCL 11/2-5) En La Ciudad de Santiago de Chile. Univ. Psychol. 2014, 13, 491–500. [Google Scholar] [CrossRef]
- Delgado, L.; Montes, R.; Saborit, J.A.P. Prevalence of Psychomotor Retardation and Its Relation to the Sensory Profile in Preschool Children. J. Hum. Growth Dev. 2016, 26, 323–330. [Google Scholar] [CrossRef]
- Zhurba, L.T.; Mastiukova, E.M.; Aĭngorn, E.D. Retardation of Psychomotor Development in Children during the First Year of Life. Comp. Study 1980, 80, 1475–1479. [Google Scholar]
- Riethmuller, A.M.; Jones, R.A.; Okely, A.D. Efficacy of Interventions to Improve Motor Development in Young Children: A Systematic Review. Pediatrics 2009, 124, e782–e792. [Google Scholar] [CrossRef]
- Brancaccio, M.; Mennitti, C.; Cesaro, A.; Monda, E.; D’argenio, V.; Casaburi, G.; Mazzaccara, C.; Ranieri, A.; Fimiani, F.; Barretta, F.; et al. Multidisciplinary In-Depth Investigation in a Young Athlete Suffering from Syncope Caused by Myocardial Bridge. Diagnostics 2021, 11, 2144. [Google Scholar] [CrossRef]
- De Angelis, C.; Nardelli, C.; Concolino, P.; Pagliuca, M.; Setaro, M.; De Paolis, E.; De Placido, P.; Forestieri, V.; Scaglione, G.L.; Ranieri, A.; et al. Case Report: Detection of a Novel Germline PALB2 Deletion in a Young Woman With Hereditary Breast Cancer: When the Patient’s Phenotype History Doesn’t Lie. Front. Oncol. 2021, 11, 602523. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic Analysis Resolves Differential Diagnosis of a Familial Syndromic Dilated Cardiomyopathy: A New Case of Alström Syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar] [CrossRef] [PubMed]
- Nunziato, M.; Starnone, F.; Lombardo, B.; Pensabene, M.; Condello, C.; Verdesca, F.; Carlomagno, C.; De Placido, S.; Pastore, L.; Salvatore, F.; et al. Fast Detection of a BRCA2 Large Genomic Duplication by next Generation Sequencing as a Single Procedure: A Case Report. Int. J. Mol. Sci. 2017, 18, 2487. [Google Scholar] [CrossRef] [PubMed]
- Mozzillo, E.; Cozzolino, C.; Genesio, R.; Melis, D.; Frisso, G.; Orrico, A.; Lombardo, B.; Fattorusso, V.; Discepolo, V.; Della Casa, R.; et al. Mulibrey Nanism: Two Novel Mutations in a Child Identified by Array CGH and DNA Sequencing. Am. J. Med. Genet. Part A 2016, 170, 2196–2199. [Google Scholar] [CrossRef] [PubMed]
- Zebisch, A.; Schulz, E.; Grosso, M.; Lombardo, B.; Acierno, G.; Sill, H.; Iolascon, A. Identification of a Novel Variant of Epsilon-Gamma-Delta-Beta Thalassemia Highlights Limitations of next Generation Sequencing. Am. J. Hematol. 2015, 90, E52–E54. [Google Scholar] [CrossRef]
- Sanna, V.; Ceglia, C.; Tarsitano, M.; Lombardo, B.; Coppola, A.; Zarrilli, F.; Castaldo, G.; Di Minno, G. Aberrant F8 Gene Intron 1 Inversion with Concomitant Duplication and Deletion in a Severe Hemophilia A Patient from Southern Italy. J. Thromb. Haemost. 2013, 11, 195–197. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, S.H.; Song, J.; Mironova, Y.; Ming, G.-L.; Kolodkin, A.L.; Giger, R.J. Semaphorin 5A Inhibits Synaptogenesis in Early Postnatal- and Adult-Born Hippocampal Dentate Granule Cells. eLife 2014, 3, e04390. [Google Scholar] [CrossRef]
- Zhao, X.F.; Kohen, R.; Parent, R.; Duan, Y.; Fisher, G.L.; Korn, M.J.; Ji, L.; Wan, G.; Jin, J.; Püschel, A.W.; et al. PlexinA2 Forward Signaling through Rap1 GTPases Regulates Dentate Gyrus Development and Schizophrenia-like Behaviors. Cell Rep. 2018, 22, 456–470. [Google Scholar] [CrossRef]
- Altuame, F.D.; Shamseldin, H.E.; Albatti, T.H.; Hashem, M.; Ewida, N.; Abdulwahab, F.; Alkuraya, F.S. PLXNA2 as a Candidate Gene in Patients with Intellectual Disability. Am. J. Med. Genet. Part A 2021, 185, 3859–3865. [Google Scholar] [CrossRef]
- Hatanaka, Y.; Kawasaki, T.; Abe, T.; Shioi, G.; Kohno, T.; Hattori, M.; Sakakibara, A.; Kawaguchi, Y.; Hirata, T. Semaphorin 6A–Plexin A2/A4 Interactions with Radial Glia Regulate Migration Termination of Superficial Layer Cortical Neurons. iScience 2019, 21, 359–374. [Google Scholar] [CrossRef]
- Epstein, J.A.; Aghajanian, H.; Singh, M.K. Semaphorin Signaling in Cardiovascular Development. Cell Metab. 2015, 21, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Krumm, N.; Turner, T.N.; Baker, C.; Vives, L.; Mohajeri, K.; Witherspoon, K.; Raja, A.; Coe, B.P.; Stessman, H.A.; He, Z.X.; et al. Excess of Rare, Inherited Truncating Mutations in Autism. Nat. Genet. 2015, 47, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, Y.; Liu, L.; Wang, Y.; Chen, H.; Fan, T.; Li, J.; Xia, K.; Sun, Z. Targeted Sequencing and Integrative Analysis of 3,195 Chinese Patients with Neurodevelopmental Disorders Prioritized 26 Novel Candidate Genes. J. Genet. Genom. 2021, 48, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Luo, N.; Tung, C.Y.; Perrin, B.J.; Zhao, B. GRXCR2 Regulates Taperin Localization Critical for Stereocilia Morphology and Hearing. Cell Rep. 2018, 25, 1268–1280.e4. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, C.; Zhao, B. N-Terminus of GRXCR2 Interacts With CLIC5 and Is Essential for Auditory Perception. Front. Cell Dev. Biol. 2021, 9, 671364. [Google Scholar] [CrossRef]
- Iossa, S.; Costa, V.; Corvino, V.; Auletta, G.; Barruffo, L.; Cappellani, S.; Ceglia, C.; Cennamo, G.; D’Adamo, A.P.; D’Amico, A.; et al. Phenotypic and Genetic Characterization of a Family Carrying Two Xq21.1-21.3 Interstitial Deletions Associated with Syndromic Hearing Loss. Mol. Cytogenet. 2015, 8, 18. [Google Scholar] [CrossRef]
- Kim, T.W.; Kang, Y.K.; Park, Z.Y.; Kim, Y.H.; Hong, S.W.; Oh, S.J.; Sohn, H.A.; Yang, S.J.; Jang, Y.J.; Lee, D.C.; et al. SH3RF2 Functions as an Oncogene by Mediating PAK4 Protein Stability. Carcinogenesis 2014, 35, 624–634. [Google Scholar] [CrossRef]
- Wilhelm, M.; Kukekov, N.V.; Schmit, T.L.; Biagas, K.V.; Sproul, A.A.; Gire, S.; Maes, M.E.; Xu, Z.; Greene, L.A. Sh3rf2/POSHER Protein Promotes Cell Survival by Ring-Mediated Proteasomal Degradation of the c-Jun N-Terminal Kinase Scaffold POSH (Plenty of SH3s) Protein. J. Biol. Chem. 2012, 287, 2247–2256. [Google Scholar] [CrossRef]
- Wang, S.; Tan, N.; Zhu, X.; Yao, M.; Wang, Y.; Zhang, X.; Xu, Z. Sh3rf2 Haploinsufficiency Leads to Unilateral Neuronal Development Deficits and Autistic-Like Behaviors in Mice. Cell Rep. 2018, 25, 2963–2971.e6. [Google Scholar] [CrossRef]
- Küry, S.; Besnard, T.; Ebstein, F.; Khan, T.N.; Gambin, T.; Douglas, J.; Bacino, C.A.; Sanders, S.J.; Lehmann, A.; Latypova, X.; et al. De Novo Disruption of the Proteasome Regulatory Subunit PSMD12 Causes a Syndromic Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017, 100, 352–363. [Google Scholar] [CrossRef]
- Khalil, R.; Kenny, C.; Hill, R.S.; Mochida, G.H.; Nasir, R.; Partlow, J.N.; Barry, B.J.; Al-Saffar, M.; Egan, C.; Stevens, C.R.; et al. PSMD12 Haploinsufficiency in a Neurodevelopmental Disorder with Autistic Features. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2018, 177, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; Ceglia, C.; Verdesca, F.; Vitale, A.; Perrotta, C.; Leggiero, E.; Pastore, L. CGH Array for the Identification of a Compound Heterozygous Mutation in the CYP1B1 Gene in a Patient with Bilateral Anterior Segment Dysgenesis. Clin. Chem. Lab. Med. 2019, 57, E63–E66. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; Zarrilli, F.; Ceglia, C.; Vitale, A.; Keller, S.; Sarchiapone, M.; Carli, V.; Stuppia, L.; Chiariotti, L.; Castaldo, G.; et al. Two Novel Genomic Rearrangements Identified in Suicide Subjects Using A-CGH Array. Clin. Chem. Lab. Med. 2015, 53, e245–e248. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, C.; Lombardo, B.; Fabbricatore, C.; Munno, C.; Caliendo, I.; Gallo, F.; Pastore, L. Oculo-Facio-Cardio-Dental (OFCD) Syndrome: The First Italian Case of BCOR and Co-Occurring OTC Gene Deletion. Gene 2015, 559, 203–206. [Google Scholar] [CrossRef]
- Tarsitano, M.; Ceglia, C.; Novelli, A.; Capalbo, A.; Lombardo, B.; Pastore, L.; Fioretti, G.; Vicari, L.; Pisanti, M.A.; Friso, P.; et al. Microduplications in 22q11.2 and 8q22.1 Associated with Mild Mental Retardation and Generalized Overgrowth. Gene 2014, 536, 213–216. [Google Scholar] [CrossRef]
- Lombardo, B.; Ceglia, C.; Tarsitano, M.; Pierucci, I.; Salvatore, F.; Pastore, L. Identification of a Deletion in the NDUFS4 Gene Using Array-Comparative Genomic Hybridization in a Patient with Suspected Mitochondrial Respiratory Disease. Gene 2014, 535, 376–379. [Google Scholar] [CrossRef]
- Iafusco, F.; De Sanctis, P.; Pirozzi, D.; Capone, S.; Lombardo, B.; Gambale, A.; Confetto, S.; Zanfardino, A.; Iolascon, A.; Pastore, L.; et al. Molecular Diagnosis of MODY3 Permitted to Reveal a de Novo 12q24.31 Deletion and to Explain a Complex Phenotype in a Young Diabetic Patient. Clin. Chem. Lab. Med. 2019, 57, e306–e331. [Google Scholar] [CrossRef]
- Lombardo, B.; Esposito, D.; Iossa, S.; Vitale, A.; Verdesca, F.; Perrotta, C.; Di Leo, L.; Costa, V.; Pastore, L.; Franzé, A. Intragenic Deletion in Macrod2: A Family with Complex Phenotypes Including Microcephaly, Intellectual Disability, Polydactyly, Renal and Pancreatic Malformations. Cytogenet. Genome Res. 2019, 158, 25–31. [Google Scholar] [CrossRef]
- Vitale, A.; Labruna, G.; Mancini, A.; Alfieri, A.; Iaffaldano, L.; Nardelli, C.; Pasanisi, F.; Pastore, L.; Buono, P.; Lombardo, B. 3q29 Microduplication in a Small Family with Complex Metabolic Phenotype from Southern Italy. Clin. Chem. Lab. Med. 2018, 56, e167–e170. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).