
Ferroptosis Involvement in Glioblastoma Treatment

 , and
, and 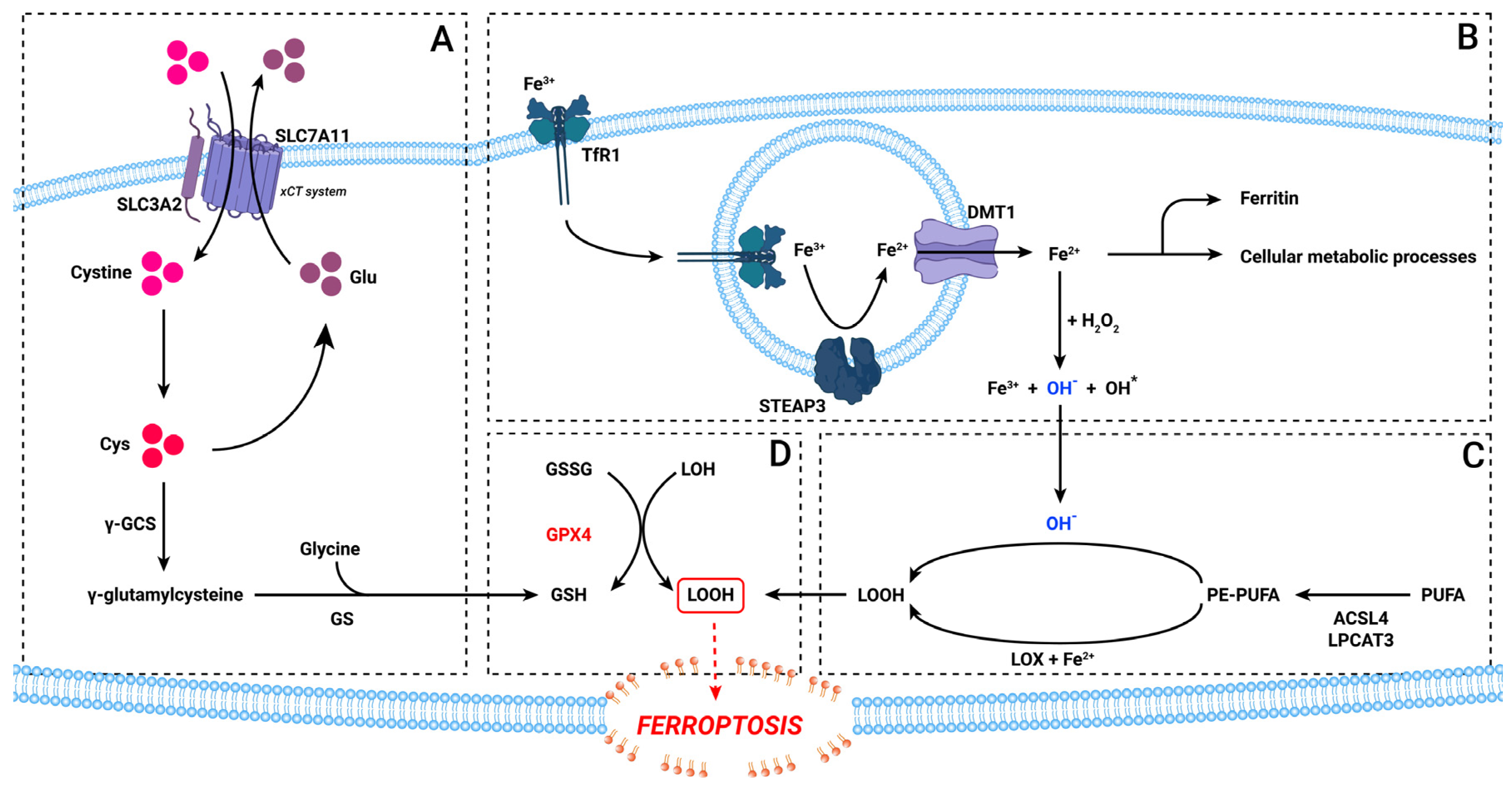{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. The Ferroptosis Pathway
4. Ferroptosis in Cancer Treatment
5. Ferroptosis in Glioblastoma
5.1. Iron Metabolism
5.2. The xCT System
5.3. Lipid Peroxidation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Batash, R.; Asna, N.; Schaffer, P.; Francis, N.; Schaffer, M. Glioblastoma Multiforme, Diagnosis and Treatment; Recent Literature Review. Curr. Med. Chem. 2017, 24, 3002–3009. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Alifieris, C.; Trafalis, D.T. Glioblastoma Multiforme: Pathogenesis and Treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; van den Bent, M.J.; Mason, W.P.; Weller, M.; Mirimanoff, R.O.; Cairncross, J.G.; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Changing Paradigms—An Update on the Multidisciplinary Management of Malignant Glioma. Oncologist 2006, 11, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Barani, I.J.; Larson, D.A. Radiation Therapy of Glioblastoma. Cancer Treat. Res. 2015, 163, 49–73. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2021, 13, 47. [Google Scholar] [CrossRef]
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Esposito, D.; Dorado, P.; Manuel Barbera, V.; Saceda, M. Radiotherapy Resistance Acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581. [Google Scholar] [CrossRef]
- Safari, M.; Khoshnevisan, A. Cancer Stem Cells and Chemoresistance in Glioblastoma Multiform: A Review Article. J. Stem Cells 2015, 10, 271–285. [Google Scholar]
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival Analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. 2018, 19, 2613–2617. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Labagnara, M.; Friedman, M.; Kwasnicki, A.; Murali, R. Glioblastoma: Molecular Pathways, Stem Cells and Therapeutic Targets. Cancers 2015, 7, 538. [Google Scholar] [CrossRef] [PubMed]
- Susman, S.; Pîrlog, R.; Leucuța, D.; Mitre, A.O.; Padurean, V.A.; Melincovici, C.; Moldovan, I.; Crișan, D.; Florian, S.I. The Role of P-Stat3 Y705 Immunohistochemistry in Glioblastoma Prognosis. Diagn. Pathol. 2019, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149, 1060. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 Exacerbates Ischemic Stroke by Promoting Ferroptosis-Induced Brain Injury and Neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Magtanong, L.; Dixon, S.J. Ferroptosis and Brain Injury. Dev. Neurosci. 2018, 40, 382–395. [Google Scholar] [CrossRef]
- Qiu, Y.; Cao, Y.; Cao, W.; Jia, Y.; Lu, N. The Application of Ferroptosis in Diseases. Pharmacol. Res. 2020, 159, 104919. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef]
- Gao, X.; Guo, N.; Xu, H.; Pan, T.; Lei, H.; Yan, A.; Mi, Y.; Xu, L. Ibuprofen Induces Ferroptosis of Glioblastoma Cells via Downregulation of Nuclear Factor Erythroid 2-Related Factor 2 Signaling Pathway. Anticancer Drugs 2020, 31, 27–34. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742. [Google Scholar] [CrossRef]
- De Brabander, M.; Van Belle, H.; Aerts, F.; Van De Veire, R.; Geuens, G. Protective Effect of Levamisole and Its Sulfhydryl Metabolite OMPI against Cell Death Induced by Glutathione Depletion. Int. J. Immunopharmacol. 1979, 1, 93–100. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, J.; Zhang, Y.; Chang, Y.-Z. Cellular Iron Metabolism and Regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each Member of the Poly-r(C)-Binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [PubMed]
- Geng, N.; Shi, B.-J.; Li, S.-L.; Zhong, Z.-Y.; Li, Y.-C.; Xua, W.-L.; Zhou, H.; Cai, J.-H. Knockdown of Ferroportin Accelerates Erastin-Induced Ferroptosis in Neuroblastoma Cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of Iron and Hydrogen Peroxide: The Fenton Reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhu, W.; Pei, D. System Xc−: A Key Regulatory Target of Ferroptosis in Cancer. Invest. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The Oxidative Stress-Inducible Cystine/Glutamate Antiporter, System x (c) (-): Cystine Supplier and Beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- Flohé, L. The Fairytale of the GSSG/GSH Redox Potential. Biochim. Biophys. Acta 2013, 1830, 3139–3142. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic/Adrenic Phosphatidylethanolamines Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. Acsl4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Dai, E.; Zhang, W.; Cong, D.; Kang, R.; Wang, J.; Tang, D. AIFM2 Blocks Ferroptosis Independent of Ubiquinol Metabolism. Biochem. Biophys. Res. Commun. 2020, 523, 966–971. [Google Scholar] [CrossRef]
- Shi, Z.-Z.; Tao, H.; Fan, Z.-W.; Song, S.-J.; Bai, J. Prognostic and Immunological Role of Key Genes of Ferroptosis in Pan-Cancer. Front. Cell Dev. Biol. 2021, 9, 748925. [Google Scholar] [CrossRef]
- Jin, Z.; El-Deiry, W.S. Overview of Cell Death Signaling Pathways. Cancer Biol. Ther. 2005, 4, 139–163. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495. [Google Scholar] [CrossRef]
- Herceg, Z.; Wang, Z.-Q. Functions of Poly(ADP-Ribose) Polymerase (PARP) in DNA Repair, Genomic Integrity and Cell Death. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2001, 477, 97–110. [Google Scholar] [CrossRef]
- Wu, X.Y.; Zhang, Y.L.; Xia, H.L.; Guan, Z.M.; Liu, Z.Y.; Wang, W.X.; Liu, Y. LIMK1 Attenuates Sevoflurane-Induced Neurodevelopmental Toxicity through Caspase-3/ Cofilin/PARP-1 Pathway. J. Biol. Regul. Homeost. Agents 2020, 34, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.-Y. Caspase-3 Regulates the Migration, Invasion and Metastasis of Colon Cancer Cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-J.; Lei, Y.-H.; Yao, N.; Wang, C.-R.; Hu, N.; Ye, W.-C.; Zhang, D.-M.; Chen, Z.-S. Autophagy and Multidrug Resistance in Cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Wang, X.; Simpson, E.R.; Brown, K.A. P53: Protection against Tumor Growth beyond Effects on Cell Cycle and Apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef]
- Bedi, A.; Barber, J.P.; Bedi, G.C.; el-Deiry, W.S.; Sidransky, D.; Vala, M.S.; Akhtar, A.J.; Hilton, J.; Jones, R.J. BCR-ABL-Mediated Inhibition of Apoptosis with Delay of G2/M Transition after DNA Damage: A Mechanism of Resistance to Multiple Anticancer Agents. Blood 1995, 86, 1148–1158. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad Targeting of Resistance to Apoptosis in Cancer. Semin. Cancer Biol. 2015, 35, S78. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis Is Induced Following Siramesine and Lapatinib Treatment of Breast Cancer Cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef]
- Woo, S.M.; Seo, S.U.; Min, K.; Im, S.-S.; Nam, J.-O.; Chang, J.-S.; Kim, S.; Park, J.-W.; Kwon, T.K. Corosolic Acid Induces Non-Apoptotic Cell Death through Generation of Lipid Reactive Oxygen Species Production in Human Renal Carcinoma Caki Cells. Int. J. Mol. Sci. 2018, 19, 1309. [Google Scholar] [CrossRef]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of Artesunate as a Specific Activator of Ferroptosis in Pancreatic Cancer Cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of Ferroptosis in Hepatocellular Carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Wang, H.; Deng, C.; Wang, X.; Yao, L.; Niu, W.; Fei, M.; Zhaba, W. Dihydroartemisinin Initiates Ferroptosis in Glioblastoma through GPX4 Inhibition. Biosci. Rep. 2020, 40, BSR20193314. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature 2007, 447, 864. [Google Scholar] [CrossRef]
- Zhang, Y.; Kong, Y.; Ma, Y.; Ni, S.; Wikerholmen, T.; Xi, K.; Zhao, F.; Zhao, Z.; Wang, J.; Huang, B.; et al. Loss of COPZ1 Induces NCOA4 Mediated Autophagy and Ferroptosis in Glioblastoma Cell Lines. Oncogene 2021, 40, 1425–1439. [Google Scholar] [CrossRef]
- Fan, Z.; Wirth, A.-K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 Pathway Promotes Cell Proliferation and Diminishes Ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef]
- Yuan, F.; Sun, Q.; Zhang, S.; Ye, L.; Xu, Y.; Xu, Z.; Liu, B.; Zhang, S.; Chen, Q. HSP27 Protects against Ferroptosis of Glioblastoma Cells. Hum. Cell 2021, 35, 238–249. [Google Scholar] [CrossRef]
- Qiu, C.; Zhang, X.; Huang, B.; Wang, S.; Zhou, W.; Li, C.; Li, X.; Wang, J.; Yang, N. Disulfiram, a Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization by Up-Regulating ROS in Glioblastoma. Onco. Targets Ther. 2020, 13, 10631–10640. [Google Scholar] [CrossRef]
- Wen, J.; Chen, H.; Ren, Z.; Zhang, P.; Chen, J.; Jiang, S. Ultrasmall Iron Oxide Nanoparticles Induced Ferroptosis via Beclin1/ATG5-Dependent Autophagy Pathway. Nano. Converg. 2021, 8, 10. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, W.; Wu, Z.; Chen, S.; Chen, X.; Zhuang, S.; Song, G.; Lv, Y.; Lin, Y. Over-Expression of LncRNA TMEM161B-AS1 Promotes the Malignant Biological Behavior of Glioma Cells and the Resistance to Temozolomide via up-Regulating the Expression of Multiple Ferroptosis-Related Genes by Sponging Hsa-MiR-27a-3p. Cell Death Discov. 2021, 7, 311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fu, X.; Jia, J.; Wikerholmen, T.; Xi, K.; Kong, Y.; Wang, J.; Chen, H.; Ma, Y.; Li, Z.; et al. Glioblastoma Therapy Using Codelivery of Cisplatin and Glutathione Peroxidase Targeting SiRNA from Iron Oxide Nanoparticles. ACS Appl. Mater. Interfaces 2020, 12, 43408–43421. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of Autophagy Increases Susceptibility of Glioblastoma Stem Cells to Temozolomide by Igniting Ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Guo, Y.; Li, H.; Han, L. FIN56, a Novel Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization in a TFEB-Dependent Manner in Glioblastoma. J. Cancer 2021, 12, 6610–6619. [Google Scholar] [CrossRef]
- Deng, S.; Zheng, Y.; Mo, Y.; Xu, X.; Li, Y.; Zhang, Y.; Liu, J.; Chen, J.; Tian, Y.; Ke, Y. Ferroptosis Suppressive Genes Correlate with Immunosuppression in Glioblastoma. World Neurosurg. 2021, 152, e436–e448. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The Emerging Role of Ferroptosis in Inflammation. Biomed. Pharmacother. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Jiang, R.; Xue, R.; Yin, X.; Wu, M.; Meng, Q. Ferroptosis in Liver Disease: New Insights into Disease Mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-Induced ACSL4 Activation Contributes to Ferroptosis-Mediated Tissue Injury in Intestinal Ischemia/Reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef]
- Proneth, B.; Conrad, M. Ferroptosis and Necroinflammation, a yet Poorly Explored Link. Cell Death Differ. 2019, 26, 14–24. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.d.L.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Park, K.-J.J.; Kim, J.; Testoff, T.; Adams, J.; Poklar, M.; Zborowski, M.; Venere, M.; Chalmers, J.J. Quantitative Characterization of the Regulation of Iron Metabolism in Glioblastoma Stem-like Cells Using Magnetophoresis. Biotechnol. Bioeng. 2019, 116, 1644–1655. [Google Scholar] [CrossRef]
- Legendre, C.; Garcion, E. Iron Metabolism: A Double-Edged Sword in the Resistance of Glioblastoma to Therapies. Trends Endocrinol. Metab. 2015, 26, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Hänninen, M.M.; Haapasalo, J.; Haapasalo, H.; Fleming, R.E.; Britton, R.S.; Bacon, B.R.; Parkkila, S. Expression of Iron-Related Genes in Human Brain and Brain Tumors. BMC Neurosci. 2009, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Voth, B.; Nagasawa, D.T.; Pelargos, P.E.; Chung, L.K.; Ung, N.; Gopen, Q.; Tenn, S.; Kamei, D.T.; Yang, I. Transferrin Receptors and Glioblastoma Multiforme: Current Findings and Potential for Treatment. J. Clin. Neurosci. 2015, 22, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Recht, L.; Torres, C.O.; Smith, T.W.; Raso, V.; Griffin, T.W. Transferrin Receptor in Normal and Neoplastic Brain Tissue: Implications for Brain-Tumor Immunotherapy. J. Neurosurg. 1990, 72, 941–945. [Google Scholar] [CrossRef]
- Calzolari, A.; Larocca, L.M.; Deaglio, S.; Finisguerra, V.; Boe, A.; Raggi, C.; Ricci-Vitani, L.; Pierconti, F.; Malavasi, F.; Maria, R.D.; et al. Transferrin Receptor 2 Is Frequently and Highly Expressed in Glioblastomas. Transl. Oncol. 2010, 3, 123. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap Proteins Are Metalloreductases. Blood 2006, 108, 1388. [Google Scholar] [CrossRef]
- Zhang, F.; Tao, Y.; Zhang, Z.; Guo, X.; An, P.; Shen, Y.; Wu, Q.; Yu, Y.; Wang, F. Metalloreductase Steap3 Coordinates the Regulation of Iron Homeostasis and Inflammatory Responses. Haematologica 2012, 97, 1826. [Google Scholar] [CrossRef]
- Li, P.-L.; Liu, H.; Chen, G.-P.; Li, L.; Shi, H.-J.; Nie, H.-Y.; Liu, Z.; Hu, Y.-F.; Yang, J.; Zhang, P.; et al. STEAP3 (Six-Transmembrane Epithelial Antigen of Prostate 3) Inhibits Pathological Cardiac Hypertrophy. Hypertension 2020, 76, 1219–1230. [Google Scholar] [CrossRef]
- Chen, H.; Xu, C.; Yu, Q.; Zhong, C.; Peng, Y.; Chen, J.; Chen, G. Comprehensive Landscape of STEAP Family Functions and Prognostic Prediction Value in Glioblastoma. J. Cell Physiol. 2021, 236, 2988–3000. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhou, Y.; Wang, X.; Zhao, H.; Nie, C.; Jiang, X. A Ferroptosis-Related Prognostic Risk Score Model to Predict Clinical Significance and Immunogenic Characteristics in Glioblastoma Multiforme. Oxid. Med. Cell Longev. 2021, 2021, 9107857. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Xu, R.; Wang, S.; Yang, N.; Ni, S.; Zhang, Q.; Xu, Y.; Zhang, X.; Zhang, C.; Wei, Y.; et al. Six-Transmembrane Epithelial Antigen of Prostate 3 Predicts Poor Prognosis and Promotes Glioblastoma Growth and Invasion. Neoplasia 2018, 20, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, R.; Garavaglia, B.; Memo, M. DMT1 Expression and Iron Levels at the Crossroads Between Aging and Neurodegeneration. Front. Neurosci. 2019, 13, 575. [Google Scholar] [CrossRef]
- Yang, C.; Xia, Z.; Li, T.; Chen, Y.; Zhao, M.; Sun, Y.; Ma, J.; Wu, Y.; Wang, X.; Wang, P.; et al. Antioxidant Effect of Propofol in Gliomas and Its Association With Divalent Metal Transporter 1. Front. Oncol. 2020, 10, 590931. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Peng, S.; Sun, Z.; Heng, X.; Zhu, X. Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells. Yonsei Med. J. 2021, 62, 843–849. [Google Scholar] [CrossRef]
- Han, W.; Xin, Z.; Zhao, Z.; Bao, W.; Lin, X.; Yin, B.; Zhao, J.; Yuan, J.; Qiang, B.; Peng, X. RNA-Binding Protein PCBP2 Modulates Glioma Growth by Regulating FHL3. J. Clin. Invest. 2013, 123, 2103–2118. [Google Scholar] [CrossRef]
- Tang, S.-L.; Gao, Y.-L.; Chen, X.-B. MicroRNA-214 Targets PCBP2 to Suppress the Proliferation and Growth of Glioma Cells. Int. J. Clin. Exp. Pathol. 2015, 8, 12571. [Google Scholar]
- Alkhateeb, A.A.; Connor, J.R. The Significance of Ferritin in Cancer: Anti-Oxidation, Inflammation and Tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2013, 1836, 245–254. [Google Scholar] [CrossRef]
- Sato, Y.; Sato, Y.; Hayashi, T.; Shojima, K.; Kaji, M. Cerebrospinal Fluid Ferritin in Patients with Central Nervous System Tumors. Kurume Med. J. 1985, 32, 229–235. [Google Scholar] [CrossRef][Green Version]
- Hayashima, K.; Kimura, I.; Katoh, H. Role of Ferritinophagy in Cystine Deprivation-Induced Cell Death in Glioblastoma Cells. Biochem. Biophys. Res. Commun. 2021, 539, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine Transporter SLC7A11/XCT in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Hu, W.-H.; Zhou, S.-L.; Yang, Z.; Liang, W.-L.; Yang, R.-Y.; Li, M.-H.; Jing, Z.; Li, Z.-A.; Fu, X.-D.; et al. SLC7A11 Negatively Associates with Mismatch Repair Gene Expression and Endows Glioblastoma Cells Sensitive to Radiation under Low Glucose Conditions. Neoplasma 2021, 68, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Wada, K.; Toyooka, T.; Shinomiya, N.; Shimazaki, H.; Nakanishi, K.; Nagatani, K.; Otani, N.; Osada, H.; Uozumi, Y.; et al. Increased XCT Expression Correlates with Tumor Invasion and Outcome in Patients with Glioblastomas. Neurosurgery 2013, 72, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The P53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Umans, R.A.; Martin, J.; Harrigan, M.E.; Patel, D.C.; Chaunsali, L.; Roshandel, A.; Iyer, K.; Powell, M.D.; Oestreich, K.; Sontheimer, H. Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme. Cancers 2021, 13, 6169. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, I.; Yoshimura, S.H.; Katoh, H. High Cell Density Increases Glioblastoma Cell Viability under Glucose Deprivation via Degradation of the Cystine/Glutamate Transporter XCT (SLC7A11). J. Biol. Chem. 2020, 295, 6936–6945. [Google Scholar] [CrossRef]
- Yamamoto, M.; Teramoto, K.; Katoh, H. Epidermal Growth Factor Promotes Glioblastoma Cell Death under Glucose Deprivation via Upregulation of XCT (SLC7A11). Cell. Signal. 2021, 78, 109874. [Google Scholar] [CrossRef]
- Goji, T.; Takahara, K.; Negishi, M.; Katoh, H. Cystine Uptake through the Cystine/Glutamate Antiporter XCT Triggers Glioblastoma Cell Death under Glucose Deprivation. J. Biol. Chem. 2017, 292, 19721–19732. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, K.; Katoh, H. The Cystine/Glutamate Antiporter XCT Is a Key Regulator of EphA2 S897 Phosphorylation under Glucose-Limited Conditions. Cell Signal 2019, 62, 109329. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino Acid Transporter SLC7A11/XCT at the Crossroads of Regulating Redox Homeostasis and Nutrient Dependency of Cancer. Cancer Commun. 2018, 38, 12–13. [Google Scholar] [CrossRef]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased Expression of System Xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. Mol. Cancer Res. 2016, 14, 1229–1242. [Google Scholar] [CrossRef]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. Cell Res. 2020, 30, 146. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T Cells Regulate Tumor Ferroptosis during Cancer Immunotherapy. Nature 2019, 569, 270. [Google Scholar] [CrossRef] [PubMed]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Førde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.-H.; Wang, J.; et al. Drug Repurposing: Sulfasalazine Sensitizes Gliomas to Gamma Knife Radiosurgery by Blocking Cystine Uptake through System Xc-, Leading to Glutathione Depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef]
- Garcia, C.G.; Kahn, S.A.; Geraldo, L.H.M.; Romano, I.; Domith, I.; Silva, D.C.L.E.; Dos Santos Assunção, F.; Ferreira, M.J.; Portugal, C.C.; de Souza, J.M.; et al. Combination Therapy with Sulfasalazine and Valproic Acid Promotes Human Glioblastoma Cell Death Through Imbalance of the Intracellular Oxidative Response. Mol. Neurobiol. 2018, 55, 6816–6833. [Google Scholar] [CrossRef]
- Ignarro, R.S.; Facchini, G.; Vieira, A.S.; De Melo, D.R.; Lopes-Cendes, I.; Castilho, R.F.; Rogerio, F. Sulfasalazine Intensifies Temozolomide Cytotoxicity in Human Glioblastoma Cells. Mol. Cell Biochem. 2016, 418, 167–178. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin Sensitizes Glioblastoma Cells to Temozolomide by Restraining XCT and Cystathionine-γ-Lyase Function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef]
- Takeuchi, S.; Wada, K.; Nagatani, K.; Otani, N.; Osada, H.; Nawashiro, H. Sulfasalazine and Temozolomide with Radiation Therapy for Newly Diagnosed Glioblastoma. Neurol. India 2014, 62, 42–47. [Google Scholar] [CrossRef]
- Nehser, M.; Dark, J.; Schweitzer, D.; Campbell, M.; Zwicker, J.; Hitt, D.M.; Little, H.; Diaz-Correa, A.; Holley, D.C.; Patel, S.A.; et al. System Xc- Antiporter Inhibitors: Azo-Linked Amino-Naphthyl-Sulfonate Analogues of Sulfasalazine. Neurochem. Res. 2020, 45, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Aboody, K.S. SLC7A11 Overexpression in Glioblastoma Is Associated with Increased Cancer Stem Cell-Like Properties. Stem Cells Dev. 2017, 26, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.; Hartmann, R.; Suwala, A.K.; Rios, D.H.; Kamp, M.A.; Sabel, M.; Steiger, H.-J.; Willbold, D.; Sharma, A.; Kahlert, U.D.; et al. Overexpression of Cystine/Glutamate Antiporter XCT Correlates with Nutrient Flexibility and ZEB1 Expression in Highly Clonogenic Glioblastoma Stem-like Cells (GSCs). Cancers 2021, 13, 6001. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive Oxygen Species-Mediated Therapeutic Response and Resistance in Glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef] [PubMed]
- Küch, E.-M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brügger, B.; Stremmel, W.; Ehehalt, R.; Poppelreuther, M.; Füllekrug, J. Differentially Localized Acyl-CoA Synthetase 4 Isoenzymes Mediate the Metabolic Channeling of Fatty Acids towards Phosphatidylinositol. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, Molecular Mechanisms and Markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.-Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-Induced Ferroptosis Promotes Tumor Necrosis in Glioblastoma Progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Bao, C.; Zhang, J.; Xian, S.-Y.; Chen, F. MicroRNA-670-3p Suppresses Ferroptosis of Human Glioblastoma Cells through Targeting ACSL4. Free Radic. Res. 2021, 55, 853–864. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The Role of Lipoxygenases in Pathophysiology; New Insights and Future Perspectives. Redox Biol. 2015, 6, 297. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966. [Google Scholar] [CrossRef]
- Zuo, X.; Morris, J.S.; Broaddus, R.; Shureiqi, I. 15-LOX-1 Transcription Suppression via the NuRD Complex in Colon Cancer Cells. Oncogene 2009, 28, 1496. [Google Scholar] [CrossRef] [PubMed]
- Wolff, C.; Zoschke, C.; Kalangi, S.K.; Reddanna, P.; Schäfer-Korting, M. Tumor Microenvironment Determines Drug Efficacy in Vitro—Apoptotic and Anti-Inflammatory Effects of 15-Lipoxygenase Metabolite, 13-HpOTrE. Eur. J. Pharm. Biopharm. 2019, 142, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Clemente, S.M.; Martínez-Costa, O.H.; Monsalve, M.; Samhan-Arias, A.K. Targeting Lipid Peroxidation for Cancer Treatment. Molecules 2020, 25, 5144. [Google Scholar] [CrossRef] [PubMed]
- Orafaie, A.; Matin, M.M.; Sadeghian, H. The Importance of 15-Lipoxygenase Inhibitors in Cancer Treatment. Cancer Metastasis Rev. 2018, 37, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hsi, L.C.; Kundu, S.; Palomo, J.; Xu, B.; Ficco, R.; Vogelbaum, M.A.; Cathcart, M.K. Silencing IL-13Rα2 Promotes Glioblastoma Cell Death via Endogenous Signaling. Mol. Cancer Ther. 2011, 10, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, O.; Honarmand, K.; Nateghinia, S.; Taheri, M.; Ghafouri-Fard, S. MiRNA Signature in Glioblastoma: Potential Biomarkers and Therapeutic Targets. Exp. Mol. Pathol. 2020, 117, 104550. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, P.; Zhao, W.; Yao, Y.; Liu, X.; Ma, J.; Xue, Y.; Liu, Y. MiR-18a Regulates the Proliferation, Migration and Invasion of Human Glioblastoma Cell by Targeting Neogenin. Exp. Cell Res. 2014, 324, 54–64. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.; Wang, C.; Cheng, K.K.-Y.; Xu, H.; Li, Q.; Hua, T.; Jiang, X.; Sheng, L.; Mao, J.; et al. MiR-18a Promotes Glioblastoma Development by down-Regulating ALOXE3-Mediated Ferroptotic and Anti-Migration Activities. Oncogenesis 2021, 10, 15. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-ΚB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxid Med. Cell Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Wortmann, M.; Mandal, P.K.; Arpornchayanon, W.; Jannasch, K.; Alves, F.; Strieth, S.; Conrad, M.; Beck, H. Absence of Glutathione Peroxidase 4 Affects Tumor Angiogenesis through Increased 12/15-Lipoxygenase Activity. Neoplasia 2010, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, X.; Wang, F.; Gao, H.; Hu, W. Dihydroartemisinin Suppresses Glioma Proliferation and Invasion via Inhibition of the ADAM17 Pathway. Neurol. Sci. 2015, 36, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-S.; Wang, J.; Shen, Y.-B.; Guo, C.-C.; Sai, K.; Chen, F.-R.; Mei, X.; Han, F.; Chen, Z.-P. Dihydroartemisinin Increases Temozolomide Efficacy in Glioma Cells by Inducing Autophagy. Oncol. Lett. 2015, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-C.; Chuang, J.-Y.; Ko, C.-Y.; Kao, T.-J.; Yang, P.-Y.; Yu, C.-H.; Liu, M.-S.; Hu, S.-L.; Tsai, Y.-T.; Chan, H.; et al. AR Ubiquitination Induced by the Curcumin Analog Suppresses Growth of Temozolomide-Resistant Glioblastoma through Disrupting GPX4-Mediated Redox Homeostasis. Redox Biol. 2020, 30, 101413. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitre, A.-O.; Florian, A.I.; Buruiana, A.; Boer, A.; Moldovan, I.; Soritau, O.; Florian, S.I.; Susman, S. Ferroptosis Involvement in Glioblastoma Treatment. Medicina 2022, 58, 319. https://doi.org/10.3390/medicina58020319
Mitre A-O, Florian AI, Buruiana A, Boer A, Moldovan I, Soritau O, Florian SI, Susman S. Ferroptosis Involvement in Glioblastoma Treatment. Medicina. 2022; 58(2):319. https://doi.org/10.3390/medicina58020319
Chicago/Turabian StyleMitre, Andrei-Otto, Alexandru Ioan Florian, Andrei Buruiana, Armand Boer, Ioana Moldovan, Olga Soritau, Stefan Ioan Florian, and Sergiu Susman. 2022. "Ferroptosis Involvement in Glioblastoma Treatment" Medicina 58, no. 2: 319. https://doi.org/10.3390/medicina58020319
APA StyleMitre, A.-O., Florian, A. I., Buruiana, A., Boer, A., Moldovan, I., Soritau, O., Florian, S. I., & Susman, S. (2022). Ferroptosis Involvement in Glioblastoma Treatment. Medicina, 58(2), 319. https://doi.org/10.3390/medicina58020319