
Epigenetic Regulation of Obesity-Associated Type 2 Diabetes

Abstract
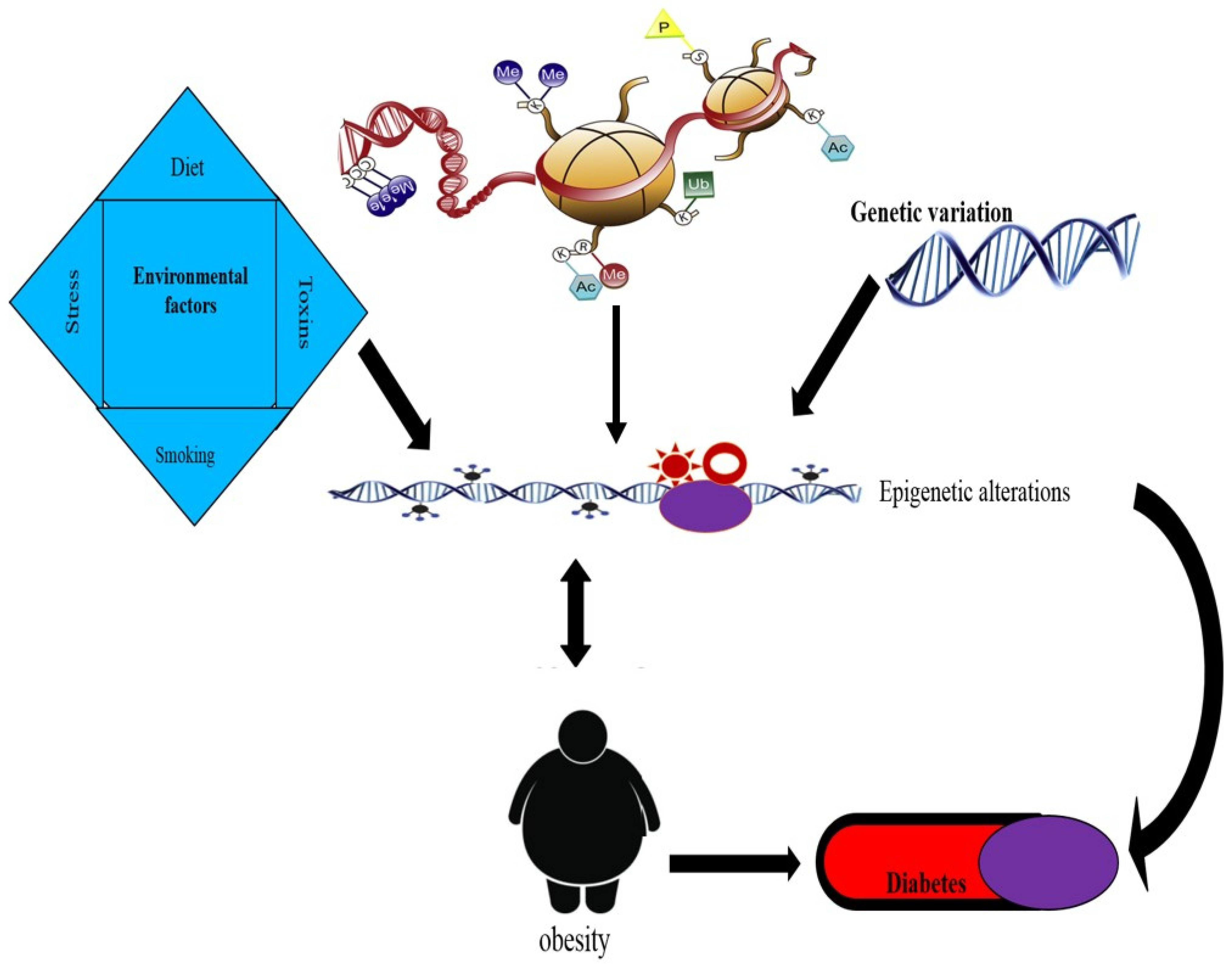
1. Introduction
2. Epigenetic Process: Epigenators, Initiators, and Maintainers
2.1. Epigenator
2.2. Epigenetic Initiator
2.3. Epigenetic Maintainer
3. Epigenetics
4. Epigenetic Regulation of Insulin Resistance
5. Epigenetics in Gestational Diabetes Mellitus (GDM)
6. Role of Diet in Epigenetics of Obesity and T2DM
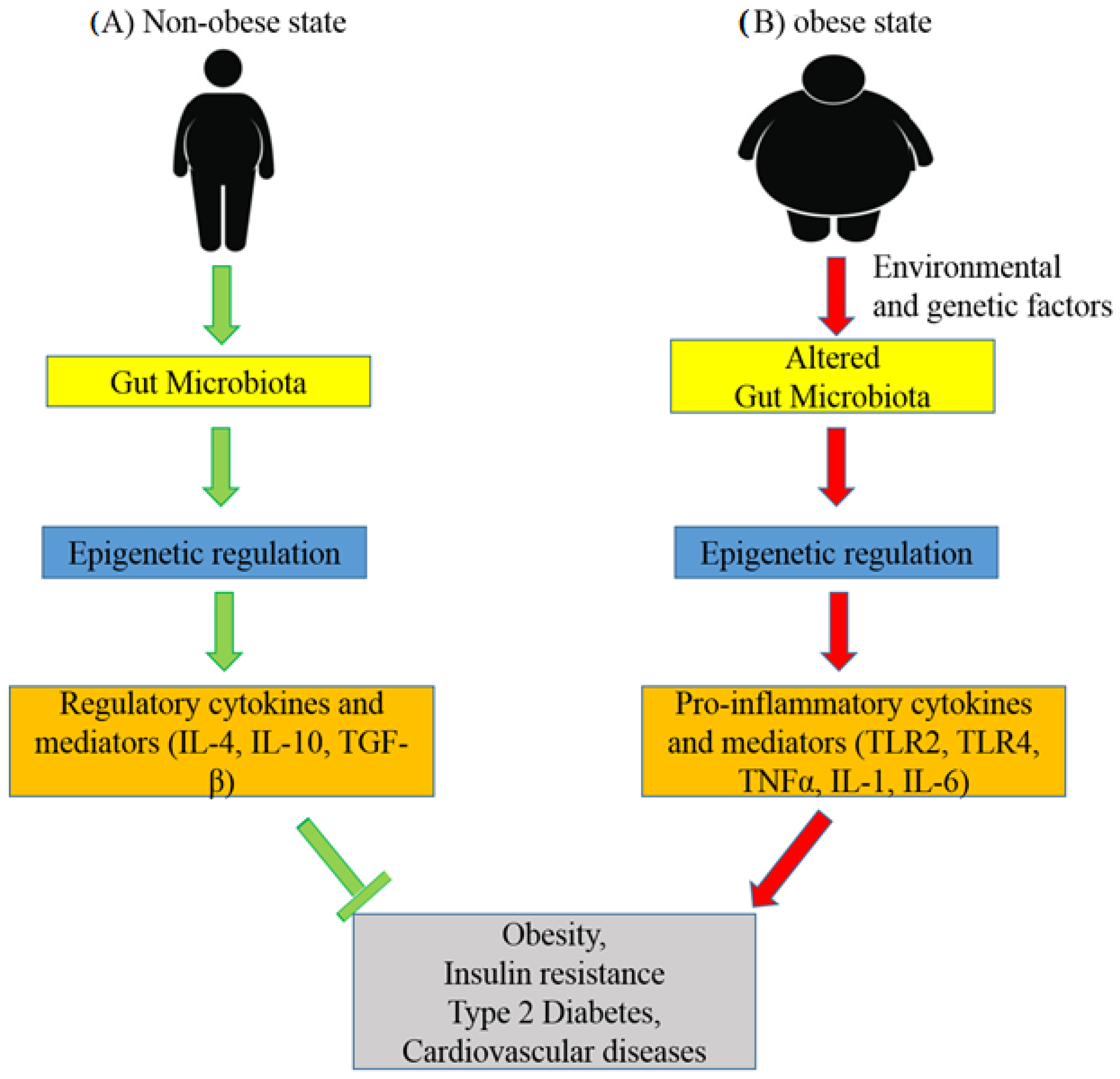
7. Role of the Gut Microbiota in the Epigenetics of Obesity and Type 2 Diabetes
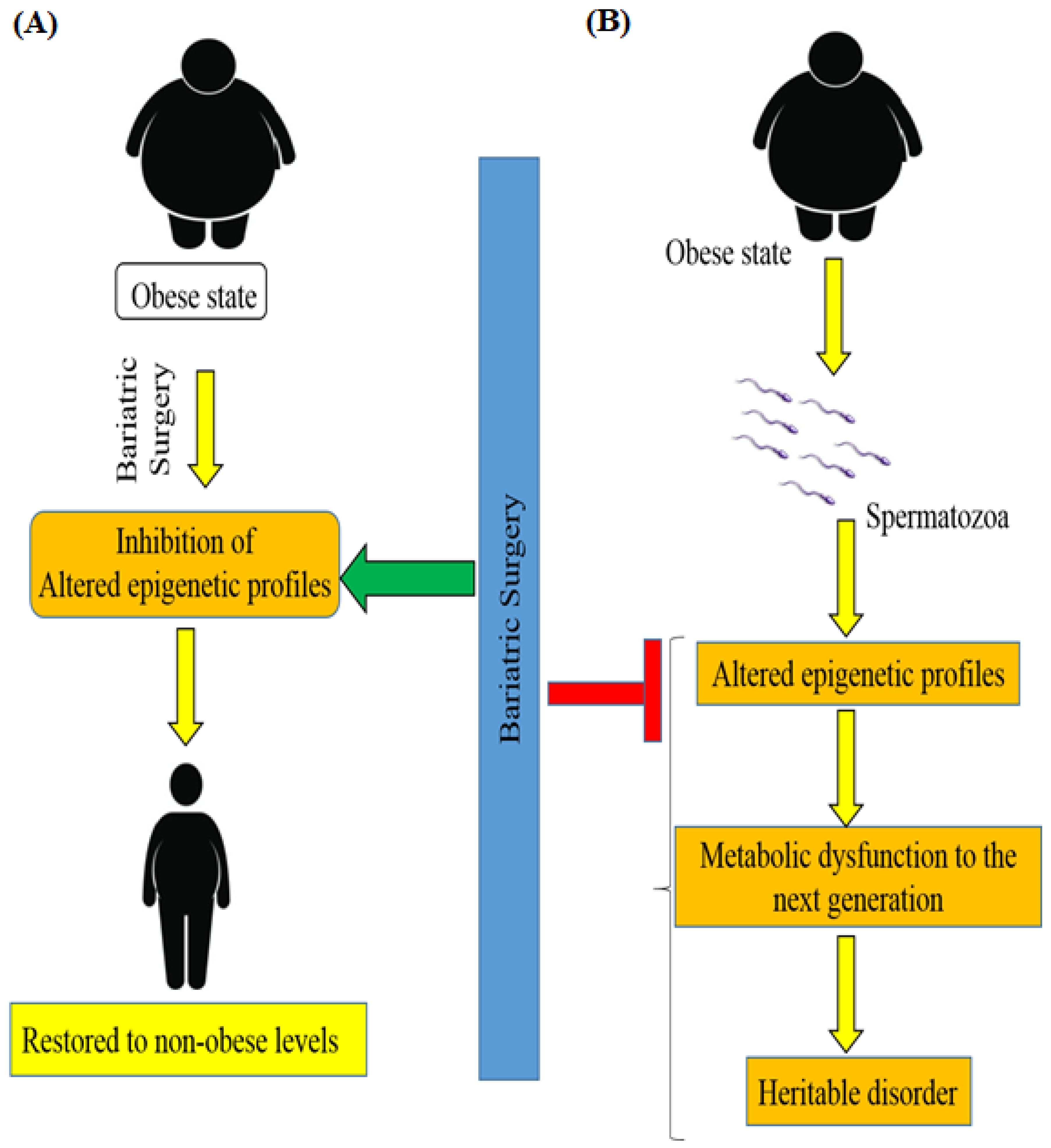
8. Bariatric Surgery and Epigenetics of Patients with Obesity-Associated T2DM
9. Links to Other Diseases
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF diabetes atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.A. Obesity and the built environment: Changes in environmental cues cause energy imbalances. Int. J. Obes. 2008, 32 (Suppl. S7), S137–S142. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Pallati, P.K.; Sharma, P.; Agrawal, D.K.; Nandipati, K.C. TREM-1 associated macrophage polarization plays a significant role in inducing insulin resistance in obese population. J. Transl. Med. 2017, 15, 85. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Pallati, P.K.; Rai, V.; Sharma, P.; Agrawal, D.K.; Nandipati, K.C. Increased expression of triggering receptor expressed on myeloid cells-1 in the population with obesity and insulin resistance. Obesity 2017, 25, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases: Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell. Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Morange, P.E.; Alessi, M.-C. The insulin resistance syndrome: Implications for thrombosis and cardiovascular disease. Pathophysiol. Haemost. Thromb. 2002, 32, 269–273. [Google Scholar] [CrossRef]
- Krause, C.; Geißler, C.; Tackenberg, H.; El Gammal, A.T.; Wolter, S.; Spranger, J.; Mann, O.; Lehnert, H.; Kirchner, H. Multi-layered epigenetic regulation of IRS2 expression in the liver of obese individuals with type 2 diabetes. Diabetologia 2020, 63, 2182–2193. [Google Scholar] [CrossRef]
- Herrera, B.M.; Keildson, S.; Lindgren, C.M. Genetics and epigenetics of obesity. Maturitas 2011, 69, 41–49. [Google Scholar] [CrossRef]
- McCarthy, M.I. Genomics, Type 2 Diabetes, and Obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef]
- Almgren, P.; Lehtovirta, M.; Isomaa, B.; Sarelin, L.; Taskinen, M.R.; Lyssenko, V.; Tuomi, T.; Groop, L.; Botnia Study Group. Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia Study. Diabetologia 2011, 54, 2811–2819. [Google Scholar] [CrossRef]
- Lai, C.Q.; Parnell, L.D.; Smith, C.E.; Guo, T.; Sayols-Baixeras, S.; Aslibekyan, S.; Tiwari, H.K.; Irvin, M.R.; Bender, C.; Fei, D.; et al. Carbohydrate and fat intake associated with risk of metabolic diseases through epigenetics of CPT1A. Am. J. Clin. Nutr. 2020, 112, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Retinopathy in a Diet-Induced Type 2 Diabetic Rat Model and Role of Epigenetic Modifications. Diabetes 2020, 69, 689–698. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, S.J.; Tellam, R.L.; Morrison, J.L.; Muhlhausler, B.S.; Molloy, P.L. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin. Epigenet. 2015, 7, 66. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.A.H.; Ansari, S.A.; Mensah-Brown, E.P.K.; Emerald, B.S. The role of DNA methylation in the pathogenesis of type 2 diabetes mellitus. Clin. Epigenet. 2020, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Pinney, S.E. DNA methylation and its role in the pathogenesis of diabetes. Pediatr. Diabetes 2017, 18, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, H.T.; Fallin, M.D.; Feinberg, A.P. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004, 20, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar]
- Turner, B.M. Epigenetic responses to environmental change and their evolutionary implications. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 3403–3418. [Google Scholar] [CrossRef] [PubMed]
- Multhaup, M.L.; Seldin, M.M.; Jaffe, A.E.; Lei, X.; Kirchner, H.; Mondal, P.; Feinberg, A.P. Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab. 2015, 21, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Karachanak-Yankova, S.; Dimova, R.; Nikolova, D.; Nesheva, D.; Koprinarova, M.; Maslyankov, S.; Tafradjiska, R.; Gateva, P.; Velizarova, M.; Hammoudeh, Z.; et al. Epigenetic alterations in patients with type 2 diabetes mellitus. Balk. J. Med. Genet. 2015, 18, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-C.; Wang, J.-S.; Lee, I.-T.; Sheu, W.H.-H.; Tan, T.-H. Epigenetic regulation of HGK/MAP4K4 in T cells of type 2 diabetes patients. Oncotarget 2016, 7, 10976–10989. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, T.; Volkov, P.; Salö, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.; Wollheim, C.B.; Eliasson, L.; Rönn, T.; et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef]
- Zhang, Y.; Kent, J.W.; Lee, A.; Cerjak, D.; Ali, O.; Diasio, R.; Olivier, M.; Blangero, J.; A Carless, M.; Kissebah, A.H. Fatty acid binding protein 3 (fabp3) is associated with insulin, lipids and cardiovascular phenotypes of the metabolic syndrome through epigenetic modifications in a Northern European family population. BMC Med. Genom. 2013, 6, 9. [Google Scholar] [CrossRef]
- Barrès, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Zierath, J.R. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Jonas, W.; Ernst, S.B.; Kammel, A.; Jähnert, M.; Schürmann, A. Diet-dependent alterations of hepatic Scd1 expression are accompanied by differences in promoter methylation. Horm. Metab. Res. 2013, 45, 786–794. [Google Scholar] [CrossRef]
- Kirchner, H.; Nylen, C.; Laber, S.; Barrès, R.; Yan, J.; Krook, A.; Näslund, E. Altered promoter methylation of PDK4, IL1 B, IL6, and TNF after Roux-En Y gastric bypass. Surg. Obes. Relat. Dis. 2014, 10, 671–678. [Google Scholar] [CrossRef]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Natarajan, R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc. Res. 2011, 90, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Kim Chung le, T.; Hosaka, T.; Yoshida, M.; Harada, N.; Sakaue, H.; Sakai, T.; Nakaya, Y. Exendin-4, a GLP-1 receptor agonist, directly induces adiponectin expression through protein kinase A pathway and prevents inflammatory adipokine expression. Biochem. Biophys. Res. Commun. 2009, 390, 613–618. [Google Scholar] [CrossRef]
- Ginés, I.; Gil-Cardoso, K.; D’Addario, C.; Falconi, A.; Bellia, F.; Blay, M.T.; Terra, X.; Ardévol, A.; Pinent, M.; Beltrán-Debón, R. Long-Lasting Effects of GSPE on Ileal GLP-1R Gene Expression Are Associated with a Hypomethylation of the GLP-1R Promoter in Female Wistar Rats. Biomolecules 2019, 9, 865. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, S.; Hu, Y.; Cao, G.; Wang, S.; Rai, P.; Wang, X.; Sun, K. The Expression Level of mRNA, Protein, and DNA Methylation Status of FOSL2 of Uyghur in XinJiang in Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 5957404. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, S.; Sun, J.; Huang, Z.; Zhu, T.; Zhang, H.; Gu, J.; He, Y.; Wang, W.; Ma, K.; et al. Methylcap-seq reveals novel DNA methylation markers for the diagnosis and recurrence prediction of bladder cancer in a Chinese population. PLoS ONE 2012, 7, e35175. [Google Scholar] [CrossRef]
- Huang, Q.; Han, L.; Liu, Y.; Wang, C.; Duan, D.; Lu, N.; Wang, K.; Zhang, L.; Gu, K.; Duan, S.; et al. Elevation of PTPN1 promoter methylation is a significant risk factor of type 2 diabetes in the Chinese population. Exp. Ther. Med. 2017, 14, 2976–2982. [Google Scholar] [CrossRef]
- Picardi, P.K.; Caricilli, A.M.; Abreu, L.L.F.D.; Carvalheira, J.B.C.; Velloso, L.A.; Saad, M.J.A. Modulation of hypothalamic PTP1B in the TNF-α-induced insulin and leptin resistance. FEBS Lett. 2010, 584, 3179–3184. [Google Scholar] [CrossRef]
- Simar, D.; Versteyhe, S.; Donkin, I.; Liu, J.; Hesson, L.; Nylander, V.; Fossum, A.; Barrès, R. DNA methylation is altered in B and NK lymphocytes in obese and type 2 diabetic human. Metabolism 2014, 63, 1188–1197. [Google Scholar] [CrossRef]
- Panee, J. Monocyte Chemoattractant Protein 1 (MCP-1) in obesity and diabetes. Cytokine 2012, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rönn, T.; Ling, C. DNA methylation as a diagnostic and therapeutic target in the battle against Type 2 diabetes. Epigenomics 2015, 7, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Morera, J.L.; Rodríguez-Rodero, S.; Menéndez-Torre, E.; Fraga, M.F. The possible role of epigenetics in gestational diabetes: Cause, consequence, or both. Obstet. Gynecol. Int. 2010, 2010, 605163. [Google Scholar] [CrossRef] [PubMed]
- Haertle, L.; El Hajj, N.; Dittrich, M.; Müller, T.; Nanda, I.; Lehnen, H.; Haaf, T. Epigenetic signatures of gestational diabetes mellitus on cord blood methylation. Clin. Epigenet. 2017, 9, 28. [Google Scholar] [CrossRef]
- Bouchard, L.; Thibault, S.; Guay, S.-P.; Santure, M.; Monpetit, A.; St-Pierre, J.; Perron, P.; Brisson, D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010, 33, 2436–2441. [Google Scholar] [CrossRef]
- Bouchard, L.; Hivert, M.-F.; Guay, S.-P.; St-Pierre, J.; Perron, P.; Brisson, D. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes 2012, 61, 1272–1280. [Google Scholar] [CrossRef]
- Novakovic, B.; Gordon, L.; Robinson, W.P.; Desoye, G.; Saffery, R. Glucose as a fetal nutrient: Dynamic regulation of several glucose transporter genes by DNA methylation in the human placenta across gestation. J. Nutr. Biochem. 2013, 24, 282–288. [Google Scholar] [CrossRef]
- Rong, C.; Cui, X.; Chen, J.; Qian, Y.; Jia, R.; Hu, Y. DNA methylation profiles in placenta and its association with gestational diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2015, 123, 282–288. [Google Scholar] [CrossRef]
- McGee, S.L.; Hargreaves, M. Exercise and skeletal muscle glucose transporter 4 expression: Molecular mechanisms. Clin. Exp. Pharmacol. Physiol. 2006, 33, 395–399. [Google Scholar] [CrossRef]
- Houde, A.A.; Guay, S.P.; Desgagné, V.; Hivert, M.F.; Baillargeon, J.P.; St-Pierre, J.; Bouchard, L. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics 2013, 8, 1289–1302. [Google Scholar] [CrossRef]
- del Rosario, M.C.; Ossowski, V.; Knowler, W.C.; Bogardus, C.; Baier, L.J.; Hanson, R.L. Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: An analysis of genome-wide DNA methylation. Metabolism 2014, 63, 654–660. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, N.; Pliushch, G.; Schneider, E.; Dittrich, M.; Müller, T.; Korenkov, M.; Haaf, T. Metabolic programming of MEST DNA methylation by intrauterine exposure to gestational diabetes mellitus. Diabetes 2013, 62, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, A.A.; Dunbar, J.A.; Janus, E.D.; Best, J.D.; Ebeling, P.R.; Ackland, M.J.; Asproloupos, D.; Ackland, M.L. Epigenetic Markers to Predict Conversion From Gestational Diabetes to Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.A.; Milagro, F.I.; Claycombe, K.J.; Schalinske, K.L. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv. Nutr. 2014, 5, 71–81. [Google Scholar] [CrossRef]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Rando, O.J. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef]
- Begum, G.; Stevens, A.; Smith, E.B.; Connor, K.; Challis, J.R.G.; Bloomfield, F.; White, A. Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. 2012, 26, 1694–1703. [Google Scholar] [CrossRef]
- Milagro, F.I.; Campión, J.; García-Díaz, D.F.; Goyenechea, E.; Paternain, L.; Martínez, J.A. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J. Physiol. Biochem. 2009, 65, 1–9. [Google Scholar] [CrossRef]
- Lomba, A.; Martínez, J.A.; García-Díaz, D.F.; Paternain, L.; Marti, A.; Campión, J.; Milagro, F.I. Weight gain induced by an isocaloric pair-fed high fat diet: A nutriepigenetic study on FASN and NDUFB6 gene promoters. Mol. Genet. Metab. 2010, 101, 273–278. [Google Scholar] [CrossRef]
- Cauchi, S.; Choquet, H.; Gutiérrez-Aguilar, R.; Capel, F.; Grau, K.; Proença, C.; Dina, C.; Duval, A.; Balkau, B.; Marre, M.; et al. Effects of TCF7L2 polymorphisms on obesity in European populations. Obesity 2008, 16, 476–482. [Google Scholar] [CrossRef]
- Jacobsen, S.C.; Brøns, C.; Bork-Jensen, J.; Ribel-Madsen, R.; Yang, B.; Lara, E.; Vaag, A. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia 2012, 55, 3341–3349. [Google Scholar] [CrossRef]
- Toperoff, G.; Aran, D.; Kark, J.D.; Rosenberg, M.; Dubnikov, T.; Nissan, B.; Wainstein, J.; Friedlander, Y.; Levy-Lahad, E.; Glaser, B.; et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum. Mol. Genet. 2012, 21, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Uriarte, G.; Paternain, L.; Milagro, F.I.; Martínez, J.A.; Campion, J. Shifting to a control diet after a high-fat, high-sucrose diet intake induces epigenetic changes in retroperitoneal adipocytes of Wistar rats. J. Physiol. Biochem. 2013, 69, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Hernández, D.L. Letter to the Editor: Use of Antibiotics, Gut Microbiota, and Risk of Type 2 Diabetes: Epigenetics Regulation. J. Clin. Endocrinol. Metab. 2016, 101, L62–L63. [Google Scholar] [CrossRef] [PubMed]
- Baothman, O.A.; Zamzami, M.A.; Taher, I.; Abubaker, J.; Abu-Farha, M. The role of Gut Microbiota in the development of obesity and Diabetes. Lipids Health Dis. 2016, 15, 108. [Google Scholar] [PubMed]
- Remely, M.; Aumueller, E.; Jahn, D.; Hippe, B.; Brath, H.; Haslberger, A.G. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef. Microbes 2014, 5, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Sook Lee, E.; Ji Song, E.; Do Nam, Y. Dysbiosis of Gut Microbiome and Its Impact on Epigenetic Regulation. J. Clin. Epigenet. 2017, 3, 14. [Google Scholar] [CrossRef]
- Remely, M.; Aumueller, E.; Merold, C.; Dworzak, S.; Hippe, B.; Zanner, J.; Pointner, A.; Brath, H.; Haslberger, A.G. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene 2014, 537, 85–92. [Google Scholar] [CrossRef]
- Kumar, H.; Lund, R.; Laiho, A.; Lundelin, K.; Ley, R.; Isolauri, E.; Salminen, S. Gut microbiota as an epigenetic regulator: Pilot study based on whole-genome methylation analysis. mBio 2014, 5, e02113-14. [Google Scholar] [CrossRef]
- Tachibana, K.; Sakurai, K.; Watanabe, M.; Miyaso, H.; Mori, C. Associations between changes in the maternal gut microbiome and differentially methylated regions of diabetes-associated genes in fetuses: A pilot study from a birth cohort study. J. Diabetes Investig. 2017, 8, 550–553. [Google Scholar] [CrossRef]
- National Institue of Health. Gastrointestinal surgery for severe obesity: National Institutes of Health Consensus Development Conference Statement. Am. J. Clin. Nutr. 1992, 55 (Suppl. S2), 615S–619S. [Google Scholar] [CrossRef]
- Zerrweck, C.; Sánchez, E.; Martinez, G.; Baca, P.; Maydón, H.; Sepúlveda, E.; Martinez, A.; Barajas-Olmos, F.; Centeno, F.; Orozco, L. The Impact of Bariatric Surgery in the Epigenetics of Patients with Obesity and Type 2 Diabetes Mellitus: A Prospective Study of the DNA Methylation Remodeling in Adipose Tissue. Surg. Obes. Relat. Dis. 2016, 12, S24–S25. [Google Scholar] [CrossRef]
- Vohl, M.C.; Guénard, F.; Tchernof, A.; Deshaies, Y.; Cianflone, K.M.; Kral, J.G.; Marceau, P. Differential Methylation of Inflammatory and Insulinotropic Genes after Metabolic Surgery in Women. J. Clin. Epigenet. 2015, 1, 1–9. [Google Scholar]
- de Toro-Martín, J.; Frédéric, G.; André, T. Yves Deshaies PM and M-CV. Bariatric Surgery Induces Hypomethylation of Genes Related to Type 2 Diabetes and Insulin Resistance. Int. J. Mol. Biol. 2017, 2, 1–4. [Google Scholar]
- Kleinberger, J.W.; Copeland, K.C.; Gandica, R.G.; Haymond, M.W.; Levitsky, L.L.; Linder, B.; Shuldiner, A.R.; Tollefsen, S.; White, N.H.; Pollin, T.I. Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes: The TODAY clinical trial. Genet. Med. 2018, 20, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Kirchner, H.; Rasmussen, M.; Yan, J.; Kantor, F.R.; Krook, A.; Zierath, J.R. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef]
- Nilsson, E.K.; Ernst, B.; Voisin, S.; Almén, M.S.; Benedict, C.; Mwinyi, J.; Fredriksson, R.; Schultes, B.; Schiöth, H.B. Roux-En Y gastric bypass surgery induces genome-wide promoter-specific changes in DNA methylation in whole blood of obese patients. PLoS ONE 2015, 10, e0115186. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C. Epigenetics: Weight change alters the spermatozoal epigenome. Nat. Rev. Endocrinol. 2016, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.R.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef]
- Eckel, R.H. Obesity and heart disease: A statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation 1997, 96, 3248–3250. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; Dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5·24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008, GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, B.; Fabius, A.W.; Wu, W.H.; Pedraza, A.; Brennan, C.W.; Schultz, N.; Pitter, K.L.; Bromberg, J.F.; Huse, J.T.; Holland, E.C.; et al. Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 8149–8154. [Google Scholar] [CrossRef] [PubMed]
- Sudarsan, S.S.; Paramasivan, V.K.; Arumugam, S.V.; Murali, S.; Kameswaran, M. Comparison of treatment modalities in syndromic children with obstructive sleep apnea--a randomized cohort study. Int J. Pediatr. Otorhinolaryngol. 2014, 78, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Manco, M.; Alterio, A.; Bugianesi, E.; Ciampalini, P.; Mariani, P.; Finocchi, M.; Agostoni, C.; Nobili, V. Insulin dynamics of breast- or formula-fed overweight and obese children. J. Am. Coll. Nutr. 2011, 30, 29–38. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No. | Gene | Epigenetic Change | Significance | Reference |
|---|---|---|---|---|
| 1 | Peroxiredoxin-2 (Prdx2) and Scavenger Receptor Class A Member 3 (SCARA3) | DNA methylation | Elevated methylation of Prdx2 and SCARA3 in T2DM | [23] |
| 2 | hepatocyte progenitor kinase-like/germinal center kinase-like kinase (HGK)/Mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) | DNA methylation | Upregulated expression of HGK promoter methylation frequencies in T2D patients | [24] |
| 3 | Insulin (INS) and pancreatic and duodenal homeobox 1 (PDX-1) | DNA methylation | DNA methylation was increased in the islets of T2DM patients as compared with normal control patients | [25] |
| 4 | fatty acid binding protein (FABP3) | DNA methylation | FABP3 (fatty acid binding protein 3) methylation process in peripheral white blood cells associated with plasma total cholesterol, insulin sensitivity, and blood pressure | [26] |
| 5 | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) –(PPARGC1A) | non-CpG methylation | Rapid epigenetic modulation of PGC-1a, which is involved in the development of T2DM and related metabolic disorders. | [27] |
| 6 | Stearoyl-CoA desaturase-1 (Scd1) | DNA methylation | Scd1 factors regulate expression in hepatocytes via altered promoter methylation through a habitual diet. | [28] |
| 7 | Pyruvate dehydrogenase lipoamide kinase isozyme 4 (PDK4) | DNA methylation | Roux-en Y gastric bypass increases methylation | [29] |
| 8 | PDX1 | Histone modification | Chromatin remodeling in the fetus is responsible for the development of T2DM-mediated Intrauterine growth deformities (IUGD) | [30] |
| 9 | histone acetylation (HAT) and deacetylation (HDAC) HATs/HDACs or Histone methyltransferases (HMTs) | Histone acetylation | Regulate the gene promoter of islet-specific insulin gene expression in response to changing glucose levels | [31] |
| 10 | Jumonji C domain-containing protein (JHDM2A) and Histone H3 lysine 9 dimethylation (H3k9me2) | Histone acetylation | The histone demethylase has been reported to lead to obesity and hyperlipidemia, implying an important role of histone modifications in diabetes | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, H.I.M. Epigenetic Regulation of Obesity-Associated Type 2 Diabetes. Medicina 2022, 58, 1366. https://doi.org/10.3390/medicina58101366
Ibrahim HIM. Epigenetic Regulation of Obesity-Associated Type 2 Diabetes. Medicina. 2022; 58(10):1366. https://doi.org/10.3390/medicina58101366
Chicago/Turabian StyleIbrahim, Hairul Islam Mohamed. 2022. "Epigenetic Regulation of Obesity-Associated Type 2 Diabetes" Medicina 58, no. 10: 1366. https://doi.org/10.3390/medicina58101366
APA StyleIbrahim, H. I. M. (2022). Epigenetic Regulation of Obesity-Associated Type 2 Diabetes. Medicina, 58(10), 1366. https://doi.org/10.3390/medicina58101366