
Deciphering Respiratory-Virus-Associated Interferon Signaling in COPD Airway Epithelium

 ,
,  ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virus Sensing Pathways in Airway Epithelium
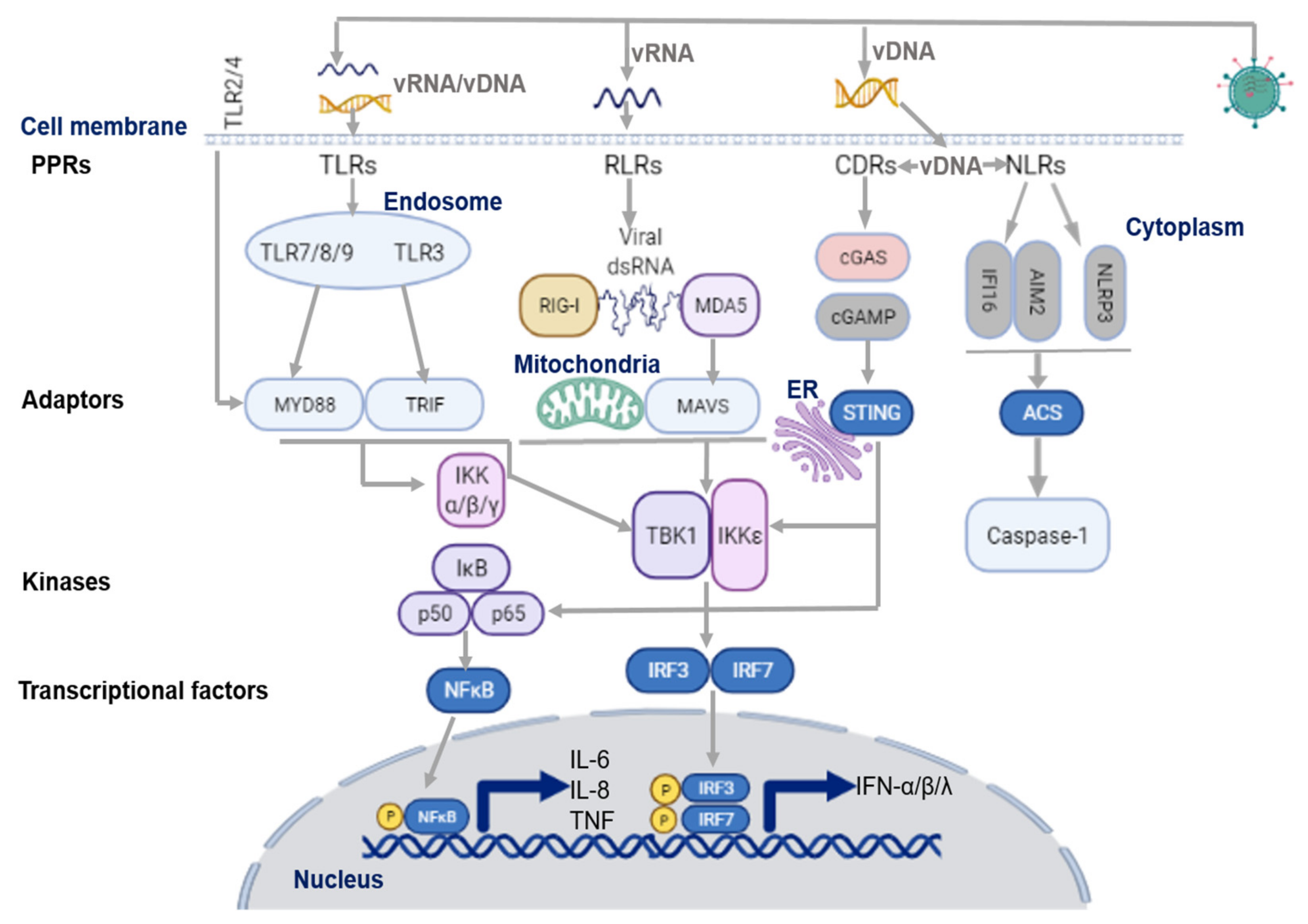
2.1. Virus Recognition by TLRs
2.2. Virus Recognition by RLRs
2.3. Virus Recognition by NLRs
2.4. Viral Evasion during Virus Sensing
3. Type I and Type III Interferon Signaling Pathways in Airway Epithelium
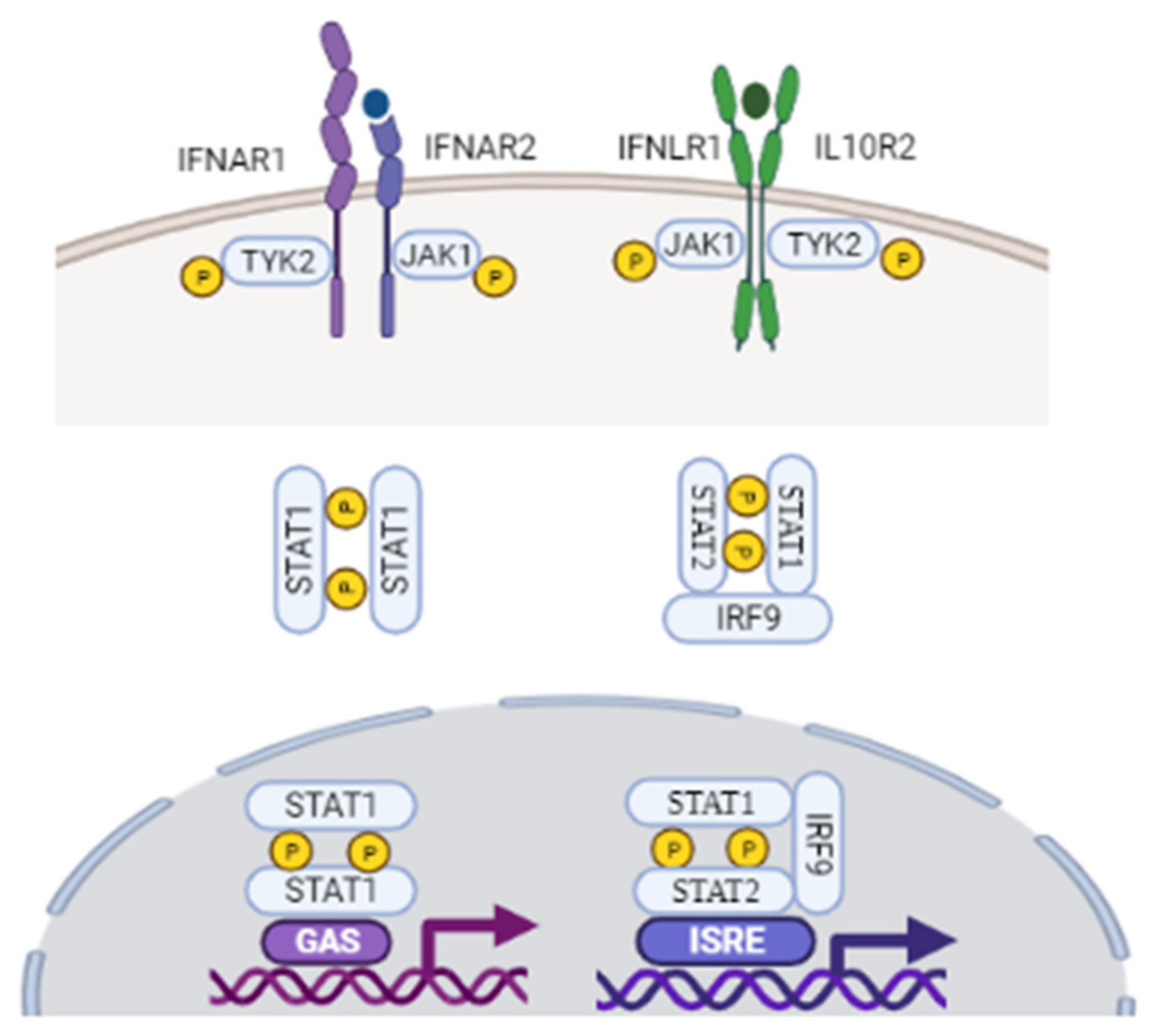
3.1. Type I Interferon Signaling
3.2. Type III Interferon Signaling
3.3. ISGs Induce an Antiviral State in Airway Epithelium
4. SARS-CoV-2 Associated IFN Signaling and Implications for COPD
5. Therapeutic Implications of IFN Signalling Cascades in COPD Exacerbations: Current Development and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.; Guo-Parke, H.; Coyle, P.V.; Fairley, D.; McAuley, D.F.; Taggart, C.C.; Kidney, J. Respiratory viral infection: A potential “missing link” in the pathogenesis of COPD. Eur. Respir. Rev. 2019, 28, 180063. [Google Scholar] [CrossRef] [Green Version]
- Guo-Parke, H.; Linden, D.; Weldon, S.; Kidney, J.C.; Taggart, C.C. Mechanisms of Virus-Induced Airway Immunity Dysfunction in the Pathogenesis of COPD Disease, Progression, and Exacerbation. Front. Immunol. 2020, 11, 1205. [Google Scholar] [CrossRef] [PubMed]
- Vareille, M.; Kieninger, E.; Edwards, M.R.; Regamey, N. The airway epithelium: Soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 2011, 24, 210–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, R.N.; Brighton, L.E.; Mueller, L.; Xiang, Z.; Rager, J.E.; Fry, R.C.; Peden, D.B.; Jaspers, I. Influenza enhances caspase-1 in bronchial epithelial cells from asthmatic volunteers and is associated with pathogenesis. J. Allergy Clin. Immunol. 2012, 130, 958–967.e14. [Google Scholar] [CrossRef] [Green Version]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and immune response in COPD: Where do we stand? Mediators Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef] [Green Version]
- Alexander-Brett, J.; Holtzman, M.J. Chapter 53—Virus Infection of Airway Epithelial Cells, in Mucosal Immunology, 4th ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 1013–1021. [Google Scholar]
- The Global Initiative for Chronic Obstructive Lung Disease GOLD. 2021. Available online: https://goldcopd.org/2021-gold-reports (accessed on 10 October 2021).
- Singanayagam, A.; Loo, S.L.; Calderazzo, M.; Finney, L.J.; Torralbo, M.B.T.; Bakhsoliani, E.; Girkin, J.; Veerati, P.; Pathinayake, P.S.; Nichol, K.S.; et al. Antiviral immunity is impaired in COPD patients with frequent exacerbations. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L893–L903. [Google Scholar] [CrossRef]
- Veerati, P.C.; Troy, N.M.; Reid, A.T.; Li, N.F.; Nichol, K.S.; Kaur, P.; Maltby, S.; Wark, P.A.B.; Knight, D.A.; Bosco, A.; et al. Airway Epithelial Cell Immunity Is Delayed During Rhinovirus Infection in Asthma and COPD. Front. Immunol. 2020, 11, 974. [Google Scholar] [CrossRef]
- Hsu, A.C.; Starkey, M.R.; Hanish, I.; Parsons, K.; Haw, T.J.; Howland, L.J.; Barr, I.; Mahony, J.B.; Foster, P.S.; Knight, D.A.; et al. Targeting PI3K-p110α Suppresses Influenza Virus Infection in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1012–1023. [Google Scholar] [CrossRef]
- Hsu, A.C.; Dua, K.; Starkey, M.R.; Haw, T.J.; Nair, P.M.; Nichol, K.; Zammit, N.; Grey, S.T.; Baines, K.J.; Foster, P.S.; et al. MicroRNA-125a and -b inhibit A20 and MAVS to promote inflammation and impair antiviral response in COPD. JCI Insight. 2017, 2, e90443. [Google Scholar] [CrossRef] [Green Version]
- Finney, L.J.; Glanville, N.; Farne, H.; Aniscenko, J.; Fenwick, P.; Kemp, S.V.; Trujillo-Torralbo, M.B.; Loo, S.L.; Calderazzo, M.A.; Wedzicha, J.A.; et al. Inhaled corticosteroids downregulate the SARS-CoV-2 receptor ACE2 in COPD through suppression of type I interferon. J. Allergy Clin. Immunol. 2021, 147, 510–519.e5. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNAsensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Sousa, C.R.E. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signaling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Sartorius, R.; Trovato, M.; Manco, R.; D’Apice, L.; De Berardinis, P. Exploiting viral sensing mediated by Toll-like receptors to design innovative vaccines. NPJ Vaccines 2021, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, I.; Ye, F.; McNally, B.; Willette, M.; Flano, E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J. Virol. 2013, 87, 3261–3270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovach, M.A.; Standiford, T.J. Toll like receptors in diseases of the lung. Int. Immunopharmacol. 2011, 11, 1399–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sioud, M. Innate sensing of self and non-self RNAs by Toll-like receptors. Trends Mol. Med. 2006, 12, 167–176. [Google Scholar] [CrossRef]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.T.; Flavell, R.A.; Liljeström, P.; Sousa, C.R.E. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef]
- Volkova, M.; Zhang, Y.; Shaw, A.C.; Lee, P.J. The role of Toll-like receptors in age-associated lung diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 247–253. [Google Scholar] [CrossRef]
- Pace, E.; Giarratano, A.; Ferraro, M.; Bruno, A.; Siena, L.; Mangione, S.; Johnson, M.; Gjomarkaj, M. TLR4 upregulation underpins airway neutrophilia in smokers with chronic obstructive pulmonary disease and acute respiratory failure. Hum. Immunol. 2011, 72, 54–62. [Google Scholar] [CrossRef]
- Wang, R.; Ahmed, J.; Wang, G.; Hassan, I.; Strulovici-Barel, Y.; Salit, J.; Mezey, J.G.; Crysta, R.G. Airway epithelial expression of TLR5 is downregulated in healthy smokers and smokers with chronic obstructive pulmonary disease. J. Immunol. 2012, 189, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Haw, T.J.; Starkey, M.R.; Pavlidis, S.; Fricker, M.; Arthurs, A.L.; Nair, P.M.; Liu, G.; Hanish, I.; Kim, R.Y.; Foster, P.S.; et al. Toll-like receptor 2 and 4 have opposing roles in the pathogenesis of cigarette smoke-induced chronic obstructive pulmonary disease. Am. J. Physiol. Lung. Cell Mol. Physiol. 2018, 314, L298–L317. [Google Scholar] [CrossRef]
- Onofrio, L.; Caraglia, M.; Facchini, G.; Margherita, V.; Placido, S.; Buonerba, C. Toll-like receptors and COVID-19: A two-faced story with an exciting ending. Future Sci. OA 2020, 6, FSO605. [Google Scholar] [CrossRef]
- Koff, J.L.; Shao, M.X.; Ueki, I.F.; Nadel, J.A. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1068–L1075. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Karki, R.; William, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, H.K. Innate immune recognition of respiratory syncytial virus infection. BMB Rep. 2014, 47, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murawski, M.R.; Bowen, G.N.; Cerny, A.M.; Anderson, L.J.; Haynes, L.M.; Tripp, R.A.; Kurt-Jones, E.A.; Finberg, R.W. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 2009, 83, 1492–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshaghdali, K.; Saeed, M.; Kamal, M.A.; Saeed, A. Interaction of ectodomain of Respiratory Syncytial Virus G protein with TLR2/TLR6 heterodimer: An in vitro and in silico approach to decipher the role of RSV G protein in pro-inflammatory response against the virus. Curr. Pharm. Des. 2021, 27, 4464–4476. [Google Scholar] [CrossRef]
- Pace, E.; Ferraro, M.; Siena, L.; Melis, M.; Montalbano, A.M.; Johnson, M.; Bonsignore, M.R.; Bonsignore, G.; Gjomarkaj, M. Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology 2008, 124, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Maes, T.; Bracke, K.R.; Vermaelen, K.Y.; Demedts, I.K.; Joos, G.F.; Pauwels, R.A.; Brusselle, G.G. Murine TLR4 is implicated in cigarette smoke-induced pulmonary inflammation. Int. Arch. Allergy Immunol. 2006, 141, 354–368. [Google Scholar] [CrossRef]
- Simpson, J.L.; McDonald, V.M.; Baines, K.J.; Oreo, K.M.; Wang, F.; Hansbro, P.M.; Gibson, P.G. Influence of age, past smoking, and disease severity on TLR2, neutrophilic inflammation, and MMP-9 levels in COPD. Mediat. Inflamm. 2013, 2013, 462934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidletskaya, K.; Vitkina, T.; Denisenko, Y. The Role of Toll-Like Receptors 2 and 4 in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Int. J. Chron. Obstr. Pulmon. Dis. 2020, 15, 1481–1493. [Google Scholar] [CrossRef]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Valero, J.; Olloquequi, J.; Montes, J.F.; Rodríguez, E.; Martín-Satué, M.; Texidó, L.; Ferrer Sancho, J. Deficient pulmonary IFN-β expression in COPD patients. PLoS ONE 2019, 14, e0217803. [Google Scholar]
- Nascimento, M.; Gombault, A.; Lacerda-Queiroz, N.; Panek, C.; Savigny, F.; Sbeity, M.; Bourinet, M.; Le Bert, M.; Riteau, N.; Ryffel, B.; et al. Self-DNA release and STING-dependent sensing drives inflammation to cigarette smoke in mice. Sci. Rep. 2019, 9, 14848. [Google Scholar] [CrossRef]
- Avriel, A.; Rozenberg, D.; Raviv, Y.; Heimer, D.; Bar-Shai, A.; Gavish, R.; Sheynin, J.; Douvdevani, A. Prognostic utility of admission cell-free DNA levels in patients with chronic obstructive pulmonary disease exacerbations. Int. J. Chron. Obstr. Pulmon. Dis. 2016, 11, 3153–3161. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Ortiz Serrano, T.P.; Davis, J.; Prigge, A.D.; Ridge, K.M. The cGAS-STING pathway: The role of self-DNA sensing in inflammatory lung disease. FASEB J. 2020, 34, 13156–13170. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Damania, B. NLRs, inflammasomes, and viral infection. J. Leukoc. Biol. 2012, 92, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, G.; León-Juárez, M.; García-Cordero, J.; Meza-Sánchez, D.E.; Cedillo-Barrón, L. Inflammasomes and its importance in viral infections. Immunol. Res. 2016, 64, 1101–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.; Bose, S. TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komune, N.; Ichinohe, T.; Ito, M.; Yanagi, Y. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1β secretion. J Virol. 2011, 85, 13019–13026. [Google Scholar] [CrossRef] [Green Version]
- Rajan, J.V.; Rodriguez, D.; Miao, E.A.; Aderem, A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol. 2011, 85, 4167–4172. [Google Scholar] [CrossRef] [Green Version]
- Burdette, D.; Haskett, A.; Presser, L.; McRae, S.; Iqbal, J.; Waris, G. Hepatitis C virus activates interleukin-1β via caspase-1-inflammasome complex. J. Gen. Virol. 2012, 93 Pt 2, 235–246. [Google Scholar] [CrossRef]
- Holley, C.L.; Schroder, K. The rOX-stars of inflammation: Links between the inflammasome and mitochondrial meltdown. Clin. Transl. Immunol. 2020, 9, e01109. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.P.; Amar, S.; Gehlot, P.; Patra, S.K.; Kanwar, N.; Kanwal, A. Mitochondrial Modulations, Autophagy Pathways Shifts in Viral Infections: Consequences of COVID-19. Int. J. Mol. Sci. 2021, 22, 8180. [Google Scholar] [CrossRef]
- Nachmias, N.; Langier, S.; Brzezinski, R.Y.; Siterman, M.; Stark, M.; Etkin, S.; Avriel, A.; Schwarz, Y.; Shenhar-Tsarfaty, S.; Bar-Shai, A. NLRP3 inflammasome activity is upregulated in an in-vitro model of COPD exacerbation. PLoS ONE 2019, 14, e0214622. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lv, C.; Wang, S.; Ying, H.; Weng, Y.; Yu, W. NLRP3 Inflammasome Involves in the Acute Exacerbation of Patients with Chronic Obstructive Pulmonary Disease. Inflammation 2018, 41, 1321–1333. [Google Scholar] [CrossRef]
- Yoon, C.M.; Nam, M.; Oh, Y.M.; Dela Cruz, C.S.; Kang, M.J. Mitochondrial Regulation of Inflammasome Activation in Chronic Obstructive Pulmonary Disease. J. Innate Immun. 2016, 8, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Zhang, M.Y.; Qu, Y.Q. The Underlying Role of Mitophagy in Different Regulatory Mechanisms of Chronic Obstructive Pulmonary Disease. Int. J. Chron. Obstr. Pulmon. Dis. 2020, 15, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome—A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michi, A.N.; Yipp, B.G.; Dufour, A.; Lopes, F.; Proud, D. PGC-1α mediates a metabolic host defense response in human airway epithelium during rhinovirus infections. Nat. Commun. 2021, 12, 3669. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Shcherbik, N.; Sawaya, B.E. Why do SARS-CoV-2 NSPs rush to the ER? J. Neurol. 2021, 268, 2013–2022. [Google Scholar] [CrossRef]
- Roulin, P.S.; Lötzerich, M.; Torta, F.; Tanner, L.B.; van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E., Jr.; Santangelo, P.J. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef] [Green Version]
- Oudshoorn, D.; Rijs, K.; Limpens, R.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Expression and Cleavage of Middle East Respiratory Syndrome Coronavirus nsp3-4 Polyprotein Induce the Formation of Double-Membrane Vesicles That Mimic Those Associated with Coronaviral RNA Replication. mBio 2017, 8, e01658-17. [Google Scholar] [CrossRef] [Green Version]
- Kikkert, M. Innate Immune Evasion by Human Respiratory RNA Viruses. J. Innate Immun. 2020, 12, 4–20. [Google Scholar] [CrossRef]
- Scutigliani, E.M.; Kikkert, M. Interaction of the innate immune system with positive-strand RNA virus replication organelles. Cytokine Growth Factor Rev. 2017, 37, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Praefcke, G.J.K. Regulation of innate immune functions by guanylate-binding proteins. Int. J. Med. Microbiol. 2018, 308, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Graf, L.; Chen, T.; Liao, Q.; Bai, T.; Petric, P.P.; Zhu, W.; Yang, L.; Dong, J.; Lu, J.; et al. Rare variant MX1 alleles increase human susceptibility to zoonotic H7N9 influenza virus. Science 2021, 373, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, J.; Hulpiau, P.; Saelens, X. Mx proteins: Antiviral gatekeepers that restrain the uninvited. Microbiol. Mol. Biol. Rev. 2013, 77, 551–566. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, P.B. Metastable biomolecular condensates of interferon-inducible antiviral Mx-family GTPases: A paradigm shift in the last three years. J. Biosci. 2021, 46, 72. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lu, Y.; Thulasi Raman, S.N.; Xu, F.; Wu, Q.; Li, Z.; Brownlie, R.; Liu, Q.; Zhou, Y. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 2018, 9, 3199. [Google Scholar] [CrossRef]
- Nordmann, A.; Wixler, L.; Boergeling, Y.; Wixler, V.; Ludwig, S. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. FASEB J. 2012, 26, 1290–1300. [Google Scholar] [CrossRef]
- Flanegan, J.B.; Petterson, R.F.; Ambros, V.; Hewlett, N.J.; Baltimore, D. Covalent linkage of a protein to a defined nucleotide sequence at the 5′-terminus of virion and replicative intermediate RNAs of poliovirus. Proc. Natl. Acad. Sci. USA 1977, 74, 961–965. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.F.; Nomoto, A.; Detjen, B.M.; Wimmer, E. A protein covalently linked to poliovirus genome RNA. Proc. Natl. Acad. Sci. USA 1977, 74, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barral, P.M.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. RIG-I is cleaved during picornavirus infection. Virology 2009, 391, 171–176. [Google Scholar] [CrossRef] [Green Version]
- De Vlugt, C.; Sikora, D.; Pelchat, M. Insight into Influenza: A Virus Cap-Snatching. Viruses 2018, 10, 641. [Google Scholar] [CrossRef] [Green Version]
- Barik, S. The structure of the 5′ terminal cap of the respiratory syncytial virus mRNA. J. Gen. Virol. 1993, 74, 485–490. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindler, E.; Gil-Cruz, C.; Spanier, J.; Li, Y.; Wilhelm, J.; Rabouw, H.H.; Züst, R.; Hwang, M.; V’kovski, P.; Stalder, H.; et al. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog. 2017, 13, e1006195. [Google Scholar] [CrossRef]
- Yuan, P.; Bartlam, M.; Lou, Z.; Chen, S.; Zhou, J.; He, X.; Lv, Z.; Ge, R.; Li, X.; Deng, T.; et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 2009, 458, 909–913. [Google Scholar] [CrossRef]
- Nogales, A.; Villamayor, L.; Utrilla-Trigo, S.; Ortego, J.; Martinez-Sobrido, L.; De Diego, M.L. Natural Selection of H5N1 Avian Influenza A Viruses with Increased PA-X and NS1 Shutoff Activity. Viruses 2021, 13, 1760. [Google Scholar] [CrossRef]
- Kong, J.Q.; Shen, J.H.; Huang, Y.; Ruan, R.Y.; Xiang, B.; Zheng, X.D.; Cheng, K.D.; Wang, W. Development of a yeast two-hybrid screen for selection of A/H1N1 influenza NS1 non-structural protein and human CPSF30 protein interaction inhibitors. Yao Xue Xue Bao 2010, 45, 388–394. [Google Scholar] [PubMed]
- Terada, Y.; Kawachi, K.; Matsuura, Y.; Kamitani, W. MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology 2017, 511, 95–105. [Google Scholar] [CrossRef]
- Petersen, J.F.; Cherney, M.M.; Liebig, H.D.; Skern, T.; Kuechler, E.; James, M.N. The structure of the 2A proteinase from a common cold virus: A proteinase responsible for the shut-off of host-cell protein synthesis. EMBO J. 1999, 18, 5463–5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zheng, Y.; Huang, J.; Li, J. Mitophagy in Antiviral Immunity. Front. Cell Dev. Biol. 2021, 9, 723108. [Google Scholar] [CrossRef] [PubMed]
- Eiermann, N.; Haneke, K.; Sun, Z.; Stoecklin, G.; Ruggieri, A. Dance with the Devil: Stress Granules and Signaling in Antiviral Responses. Viruses 2020, 12, 984. [Google Scholar] [CrossRef] [PubMed]
- Montero, H.; Trujillo-Alonso, V. Stress granules in the viral replication cycle. Viruses 2011, 3, 2328–2338. [Google Scholar] [CrossRef] [Green Version]
- Lindquist, M.E.; Lifland, A.W.; Utley, T.J.; Santangelo, P.; Crowe, J.E., Jr. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J. Virol. 2010, 84, 12274–12284. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Langereis, M.A.; van Kuppeveld, F.J. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev. 2014, 25, 577–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, N.; Prasad, K.; AlAsmari, A.F.; Alharbi, M.; Rashid, S.; Kumar, V. Genomics-guided targeting of stress granule proteins G3BP1/2 to inhibit SARS-CoV-2 propagation. Int. J. Biol. Macromol. 2021, 10, 636–648. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Heaton, S.M.; Borg, N.A.; Dixit, V.M. Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 2016, 213, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Müller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Ban, J.; Lee, N.R.; Lee, N.J.; Lee, J.K.; Quan, F.S.; Inn, K.S. Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling. Viruses 2018, 10, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzorno, A.; Dubois, J.; Machado, D.; Cartet, G.; Traversier, A.; Julien, T.; Lina, B.; Bourdon, J.C.; Rosa-Calatrava, M.; Terrier, O. Influenza A viruses alter the stability and antiviral contribution of host E3-ubiquitin ligase Mdm2 during the time-course of infection. Sci. Rep. 2018, 8, 3746. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.C.; Sridhar, P.R.; Baldridge, M.T. Differential roles of interferons in innate responses to mucosal viral infections. Trends Immunol. 2021, 42, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Mazewski, C.; Perez, R.E.; Fish, E.N.; Platanias, L.C. Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 2020, 11, 606456. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 21, 1445. [Google Scholar] [CrossRef] [Green Version]
- Khaitov, M.R.; Laza-Stanca, V.; Edwards, M.R.; Walton, R.P.; Rohde, G.; Contoli, M.; Papi, A.; Stanciu, L.A.; Kotenko, S.V.; Johnston, S.L. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy 2009, 64, 375–386. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, W.; Verhaegh, W.; Holtzer, L.; van de Stolpe, A. Measurement of Cellular Immune Response to Viral Infection and Vaccination. Front. Immunol. 2020, 11, 575074. [Google Scholar] [CrossRef]
- Moore, E.C.; Barber, J.; Trip, R.A. Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol. J. 2008, 5, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, D.K.; Forero, A. Mechanisms of Antiviral Immune Evasion of SARS-CoV-2. J. Mol. Biol. 2021, 22, 167265. [Google Scholar] [CrossRef] [PubMed]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokugamage, K.G.; Hage, A.; de Vries, M.; Valero-Jimenez, A.M.; Schindewolf, C.; Dittmann, M.; Rajsbaum, R.; Menachery, V.D. Type I Interferon Susceptibility Distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020, 94, e01410-20. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Gielen, V.; Sykes, A.; Zhu, J.; Chan, B.; Macintyre, J.; Regamey, N.; Kieninger, E.; Gupta, A.; Shoemark, A.; Bossley, C.; et al. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J. Allergy Clin. Immunol. 2015, 136, 177–188.e11. [Google Scholar] [CrossRef] [Green Version]
- Pauli, E.K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008, 4, e1000196. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Ueki, I.F.; Min-Oo, G.; Kalinowski, A.; Ballon-Landa, E.; Lanier, L.L.; Nadel, J.A.; Koff, J.L. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J. Exp. Med. 2013, 210, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- McGillivary, G.; Jordan, Z.B.; Peeples, M.E.; Bakaletz, L.O. Replication of respiratory syncytial virus is inhibited by the host defense molecule viperin. J. Innate Immun. 2013, 5, 60–71. [Google Scholar] [CrossRef]
- Proud, D.; Turner, R.B.; Winther, B.; Wiehler, S.; Tiesman, J.P.; Reichling, T.D.; Juhlin, K.D.; Fulmer, A.W.; Ho, B.Y.; Walanski, A.A.; et al. Gene expression profiles during in vivo human rhinovirus infection: Insights into the host response. Am. J. Respir. Crit. Care Med. 2008, 178, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.S.; Olfat, F.; Phoon, M.C.; Hsu, J.P.; Howe, J.L.C.; Seet, J.E.; Chin, K.C.; Chow, V.T.K. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J. Gen. Virol. 2012, 93, 1269–1277. [Google Scholar] [CrossRef]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhang, W.; Booth, J.L.; Hutchings, D.C.; Wang, X.; White, V.L.; Youness, H.; Cross, C.D.; Zou, M.H.; Burian, D.; et al. Human primary airway epithelial cells isolated from active smokers have epigenetically impaired antiviral responses. Respir. Res. 2016, 17, 111. [Google Scholar] [CrossRef] [Green Version]
- Niwa, M.; Fujisawa, T.; Mori, K.; Yamanaka, K.; Yasui, H.; Suzuki, Y.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; et al. IL-17A Attenuates IFN-λ Expression by Inducing Suppressor of Cytokine Signaling Expression in Airway Epithelium. J. Immunol. 2018, 201, 2392–2402. [Google Scholar] [CrossRef] [Green Version]
- Hilzendeger, C.; da Silva, J.; Henket, M.; Schleich, F.; Corhay, J.L.; Kebadze, T.; Edwards, M.R.; Mallia, P.; Johnston, S.L.; Louis, R. Reduced sputum expression of interferon-stimulated genes in severe COPD. Int. J. Chron. Obstr. Pulmon. Dis. 2016, 11, 1485–1494. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course 881 and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, 882 retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.M.; Niikura, M.; Yang, C.W.T.; Sin, D.D. COVID-19 and COPD. Eur. Respir. J. 2020, 56, 2002108. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D. COVID-19 in COPD: A growing concern. E Clin. Med. 2020, 26, 100546. [Google Scholar] [CrossRef]
- Leung, J.M.; Yang, C.X.; Tam, A.; Shaipanich, T.; Hackett, T.L.; Singhera, G.K.; Dorscheid, D.R.; Sin, D.D. ACE-2 expression in the small airway epithelia of smokers and COPD patients: Implications for COVID-19. Eur. Respir. J. 2020, 55, 2000688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.; Öberg, L.; Angermann, B.; Spalluto, C.M.; Hühn, M.; Burke, H.; Cellura, D.; Freeman, A.; Muthas, D.; Etal, D.; et al. Dysregulation of COVID-19 related gene expression in the COPD lung. Respir. Res. 2021, 22, 164. [Google Scholar] [CrossRef] [PubMed]
- Osan, J.K.; Talukdar, S.N.; Feldmann, F.; DeMontigny, B.A.; Jerome, K.; Bailey, K.L.; Feldmann, H.; Mehedi, M. Goblet Cell Hyperplasia Increases SARS-CoV-2 Infection in COPD. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ziegler, C.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef] [PubMed]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021. [Google Scholar] [CrossRef] [PubMed]
- Taefehshokr, N.; Taefehshokr, S.; Hemmat, N.; Heit, B. COVID-19: Perspectives on Innate Immune Evasion. Front. Immunol. 2020, 11, 580641. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.K.; Lam, J.Y.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Cai, J.P.; Jin, D.Y.; To, K.K.; Chan, J.F.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Liu, G.; Lee, J.H.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; van Gent, M.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-dependent activation of the sensor MDA5 is antagonized by the SARS-CoV-2 papain-like protease to evade host innate immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infec. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomerenke, A.; Lea, S.R.; Herrick, S.; Lindsay, M.A.; Singh, D. Characterization of TLR-induced inflammatory responses in COPD and control lung tissue explants. Int. J. Chron. Obstr. Pulmon. Dis. 2016, 11, 2409–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djukanović, R.; Harrison, T.; Johnston, S.L.; Gabbay, F.; Wark, P.; Thomson, N.C.; Niven, R.; Singh, D.; Reddel, H.K.; Davies, D.E.; et al. The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am. J. Respir. Crit. Care Med. 2014, 190, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb-Mäurer, A.; Goebeler, M.; Mäurer, M. Cutaneous Adverse Events Associated with Interferon-β Treatment of Multiple Sclerosis. Int. J. Mol. Sci. 2015, 16, 14951–14960. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.; Spalluto, C.M.; McCrae, C.; Cellura, D.; Burke, H.; Cunoosamy, D.; Freeman, A.; Hicks, A.; Hühn, M.; Ostridge, K.; et al. Dynamics of IFN-β Responses during Respiratory Viral Infection. Insights for Therapeutic Strategies. Am. J. Respir. Crit. Care Med. 2020, 201, 83–94. [Google Scholar] [CrossRef]
- McCrae, C.; Olsson, M.; Gustafson, P.; Malmgren, A.; Aurell, M.; Fagerås, M.; Da Silva, C.A.; Cavallin, A.; Paraskos, J.; Karlsson, K.; et al. INEXAS: A Phase 2 Randomized Trial of On-demand Inhaled Interferon Beta-1a in Severe Asthmatics. Clin. Exp. Allergy 2021, 51, 273–283. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo-Parke, H.; Linden, D.; Weldon, S.; Kidney, J.C.; Taggart, C.C. Deciphering Respiratory-Virus-Associated Interferon Signaling in COPD Airway Epithelium. Medicina 2022, 58, 121. https://doi.org/10.3390/medicina58010121
Guo-Parke H, Linden D, Weldon S, Kidney JC, Taggart CC. Deciphering Respiratory-Virus-Associated Interferon Signaling in COPD Airway Epithelium. Medicina. 2022; 58(1):121. https://doi.org/10.3390/medicina58010121
Chicago/Turabian StyleGuo-Parke, Hong, Dermot Linden, Sinéad Weldon, Joseph C. Kidney, and Clifford C. Taggart. 2022. "Deciphering Respiratory-Virus-Associated Interferon Signaling in COPD Airway Epithelium" Medicina 58, no. 1: 121. https://doi.org/10.3390/medicina58010121
APA StyleGuo-Parke, H., Linden, D., Weldon, S., Kidney, J. C., & Taggart, C. C. (2022). Deciphering Respiratory-Virus-Associated Interferon Signaling in COPD Airway Epithelium. Medicina, 58(1), 121. https://doi.org/10.3390/medicina58010121