
Alpha-1 Antitrypsin Augmentation Inhibits Proteolysis of Neutrophil Membrane Voltage-Gated Proton Channel-1 in Alpha-1 Deficient Individuals


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Study Design
2.3. Neutrophil Isolation and Assays
2.4. SDS-Polyacrylamide Gel Electrophoresis and Western Blotting
2.5. Flow Cytometry Experiments
2.6. Statistical Analysis
3. Results
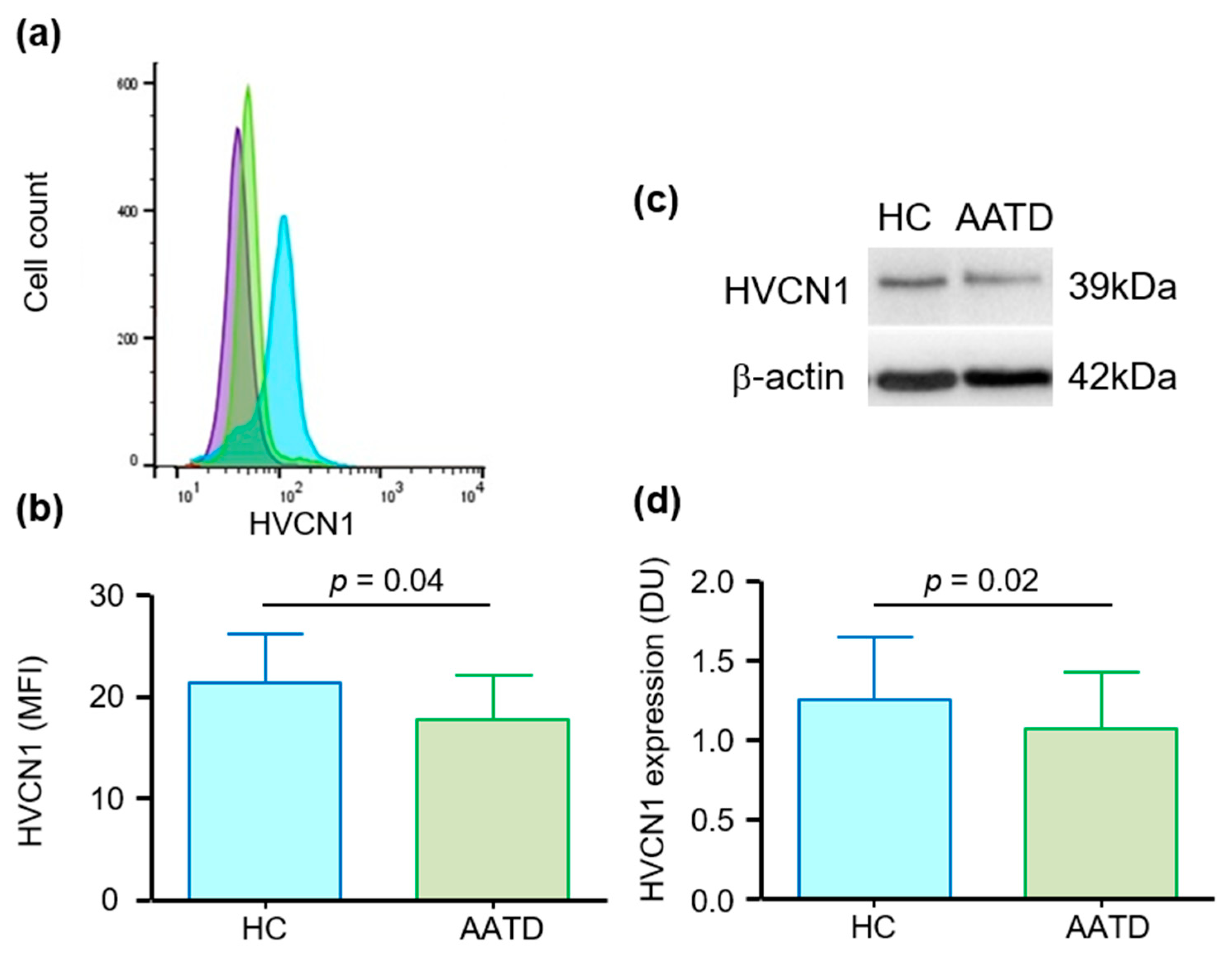
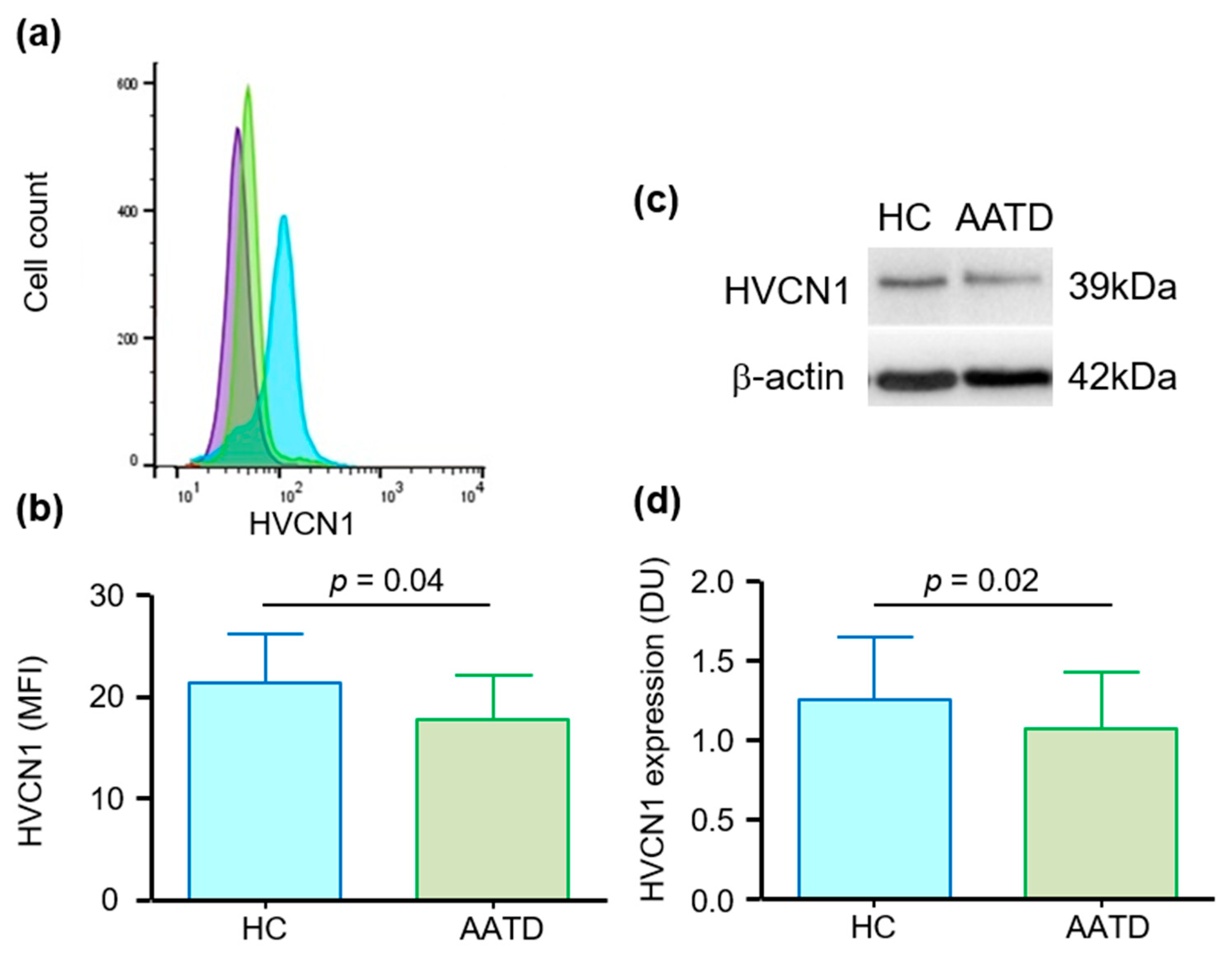
3.1. Cell Membrane Expression of HVCN1 in Neutrophils of AATD Is Decreased
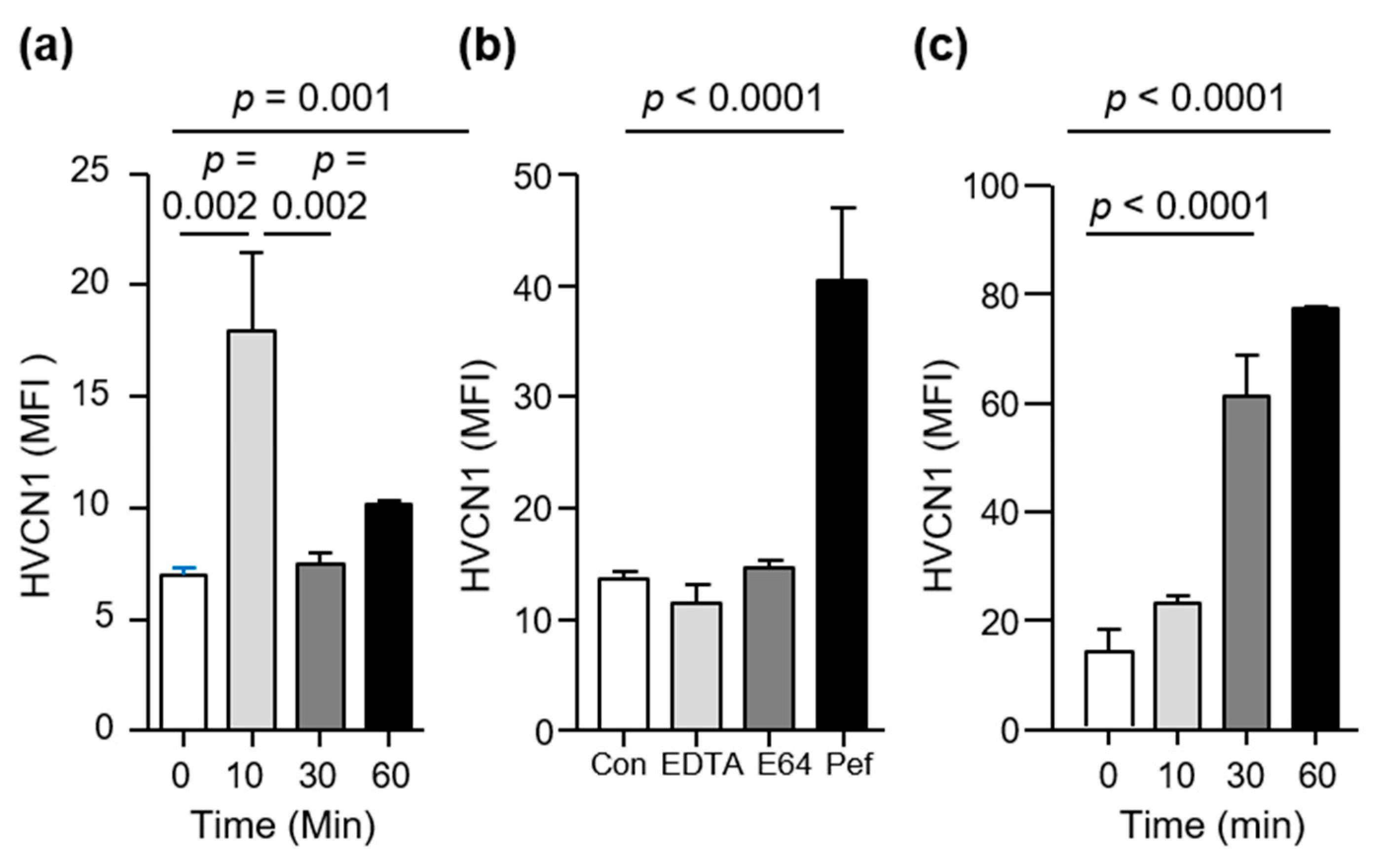
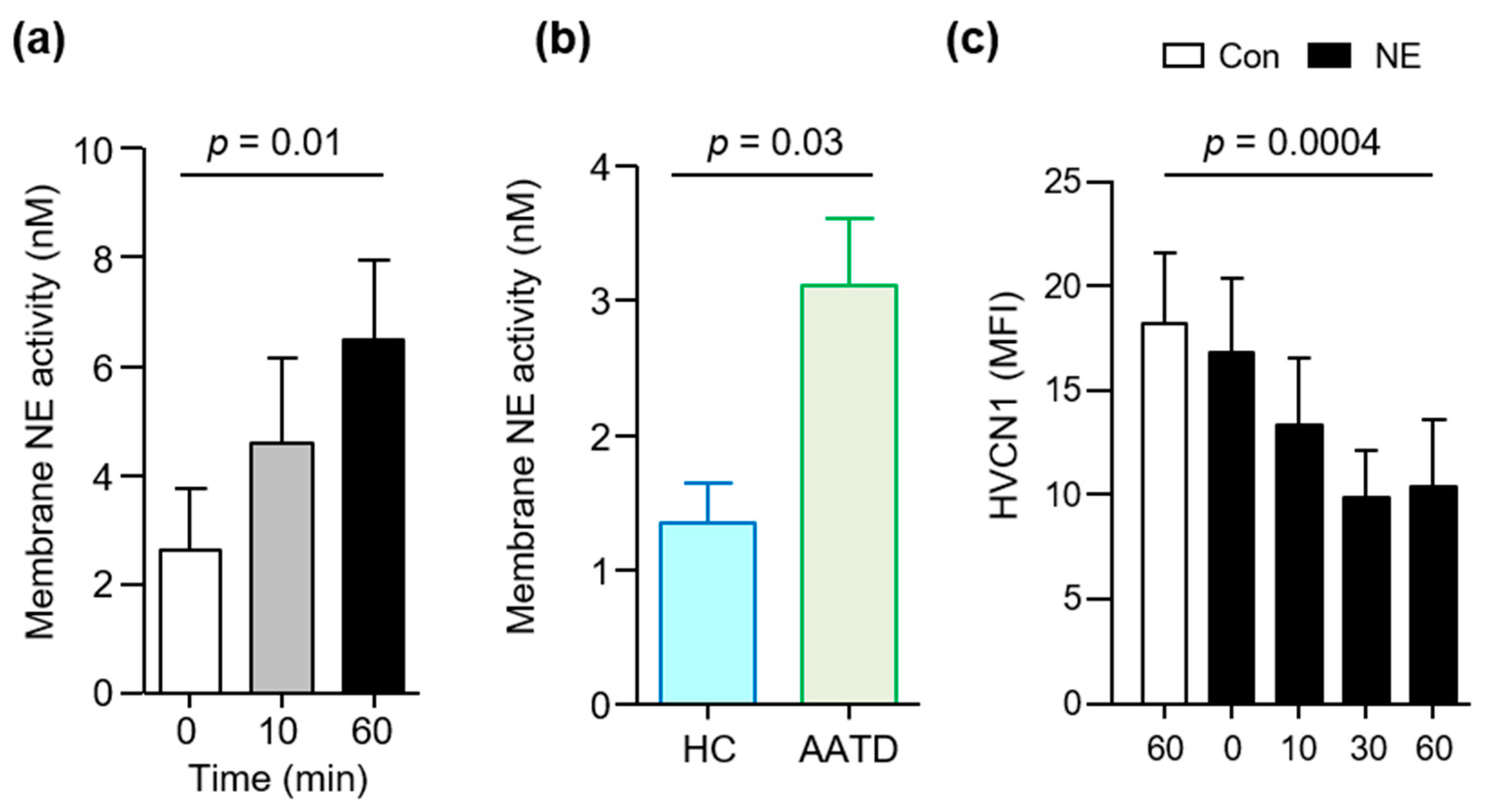
3.2. HVCN1 Is Proteolytically Cleaved from Neutrophil Plasma Membranes in AATD
3.3. HVCN1 Is Proteolytically Cleaved by Neutrophil Elastase
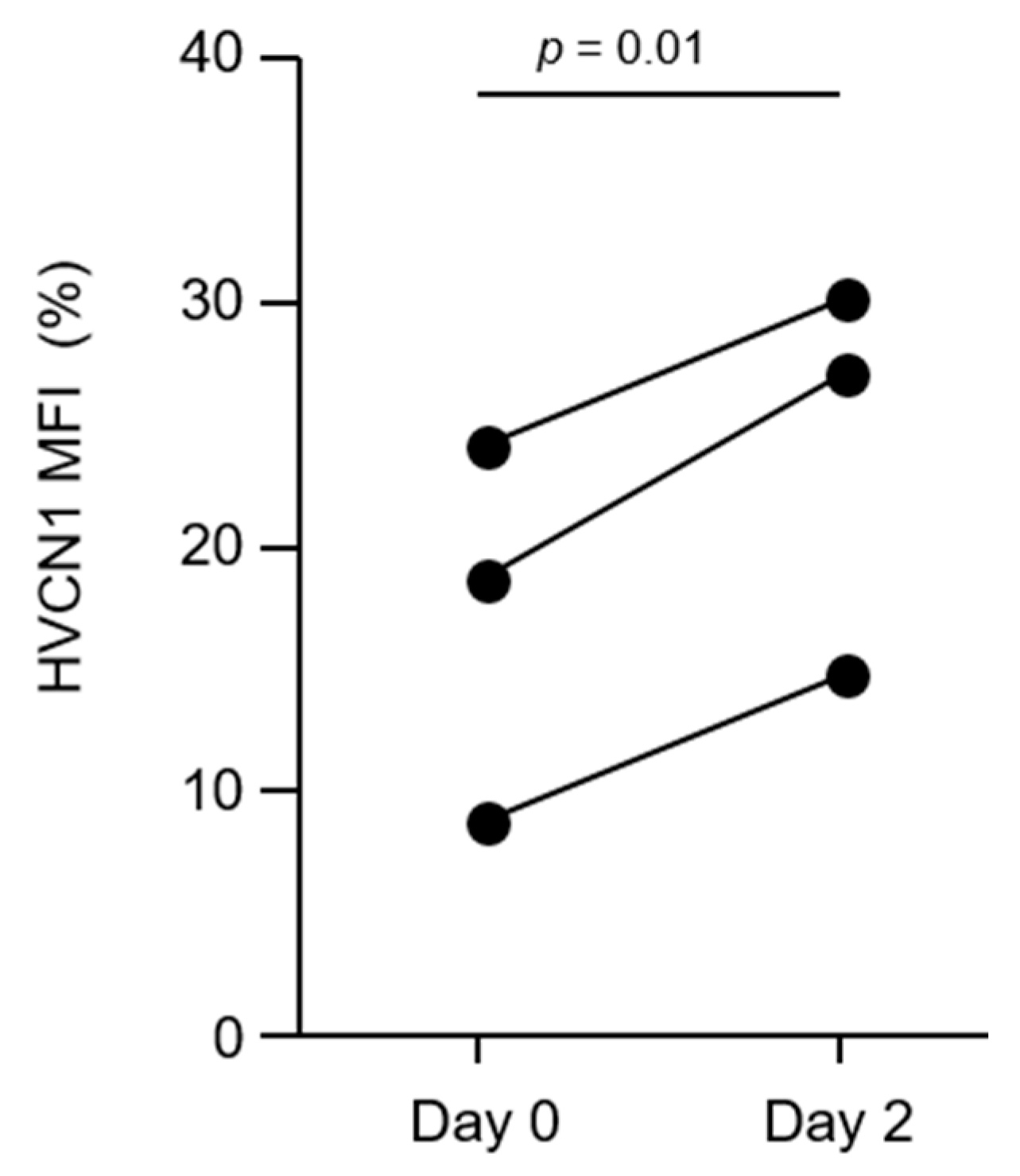
3.4. AAT Augmentation Therapy Increases HVCN1 Expression on AATD Neutrophils
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Stoller, J.K.; Aboussouan, L.S. A review of α1-antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Brantly, M.; Nukiwa, T.; Crystal, R.G. Molecular basis of alpha-1-antitrypsin deficiency. Am. J. Med. 1988, 84, 13–31. [Google Scholar] [CrossRef]
- Perlino, E.; Cortese, R.; Ciliberto, G. The human alpha 1-antitrypsin gene is transcribed from two different promoters in macrophages and hepatocytes. EMBO J. 1987, 6, 2767–2771. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, J.C.; Oton-Gonzalez, L.; Selvatici, R.; Rizzo, P.; Pavasini, R.; Campo, G.C.; Lanzillotti, C.; Mazziotta, C.; De Mattei, M.; Tognon, M.; et al. SERPINA1 Gene Promoter Is Differentially Methylated in Peripheral Blood Mononuclear Cells of Pregnant Women. Front. Cell Dev. Biol. 2020, 8, 550543. [Google Scholar] [CrossRef]
- Nyasae, L.K.; Hubbard, A.L.; Tuma, P.L. Transcytotic efflux from early endosomes is dependent on cholesterol and glycosphingolipids in polarized hepatic cells. Mol. Biol. Cell 2003, 14, 2689–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janciauskiene, S. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim. Biophys. Acta 2001, 1535, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Bergin, D.A.; Hurley, K.; McElvaney, N.G.; Reeves, E.P. Alpha-1 antitrypsin: A potent anti-inflammatory and potential novel therapeutic agent. Arch. Immunol. Ther. Exp. 2012, 60, 81–97. [Google Scholar] [CrossRef]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Stolk, J.; Aggarwal, N.; Hochnadel, I.; Wrenger, S.; Martinez-Delgado, B.; Welte, T.; Yevsa, T.; Janciauskiene, S. Blood monocyte profiles in COPD patients with PiMM and PiZZ α1-antitrypsin. Respir. Med. 2019, 148, 60–62. [Google Scholar] [CrossRef]
- Sandstrom, C.S.; Novoradovskaya, N.; Cilio, C.M.; Piitulainen, E.; Sveger, T.; Janciauskiene, S. Endotoxin receptor CD14 in PiZ alpha-1-antitrypsin deficiency individuals. Respir. Res. 2008, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Nita, I.M.; Serapinas, D.; Janciauskiene, S.M. α1-Antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int. J. Biochem. Cell Biol. 2007, 39, 1165–1176. [Google Scholar] [CrossRef]
- Lohrisch, I.; Scherbaum, I.; Gruhn, R.; Ambrosius, H.; Haustein, U.F.; Herrmann, K. Alpha 1-antitrypsin: Production and binding of in vitro stimulated peripheral blood lymphocytes. Acta Biol. Med. Ger. 1981, 40, 1767–1773. [Google Scholar]
- Janciauskiene, S.; Tumpara, S.; Wiese, M.; Wrenger, S.; Vijayan, V.; Gueler, F.; Chen, R.; Welte, T.; Mahadeva, R.; Immenschuh, S.; et al. Alpha1-antitrypsin binds hemin and prevents oxidative activation of human neutrophils: Putative pathophysiological significance. J. Leukoc. Biol. 2017, 102, 1127–1141. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Chattopadhaya, D.; Bhadoria, D.P.; Qadar Pasha, M.A.; Gupta, V.K.; Kumar, M.; Dabur, R.; Yadav, V.; Sharma, G.L. T lymphocyte subset profile and serum alpha-1-antitrypsin in pathogenesis of chronic obstructive pulmonary disease. Clin. Exp. Immunol. 2007, 149, 463–469. [Google Scholar] [CrossRef]
- Geraghty, P.; Eden, E.; Pillai, M.; Campos, M.; McElvaney, N.G.; Foronjy, R.F. α1-Antitrypsin activates protein phosphatase 2A to counter lung inflammatory responses. Am. J. Respir. Crit. Care Med. 2014, 190, 1229–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bata, J.; Deviller, P.; Vallier, P.; Revillard, J.P. Modification of lymphocyte DNA synthesis by alpha 1-antitrypsin. Ann. Immunol. 1981, 132C, 275–286. [Google Scholar]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. α1-Antitrypsin deficiency. Nat. Rev. Dis. Primers 2016, 2, 16051. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Aboussouan, L.S. Alpha1-antitrypsin deficiency. Lancet 2005, 365, 2225–2236. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society Statement. Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2003, 168, 818–900. [Google Scholar] [CrossRef] [PubMed]
- Abboud, R.T.; Nelson, T.N.; Jung, B.; Mattman, A. Alpha1-antitrypsin deficiency: A clinical-genetic overview. Appl. Clin. Genet. 2011, 4, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashiro, T.; Matsuoka, S.; Estépar, R.S.; Diaz, A.; Newell, J.D.; Sandhaus, R.A.; Mergo, P.J.; Brantly, M.L.; Murayama, S.; Reilly, J.J.; et al. Quantitative airway assessment on computed tomography in patients with alpha1-antitrypsin deficiency. COPD 2009, 6, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Molloy, K.; Hersh, C.P.; Morris, V.B.; Carroll, T.P.; O’Connor, C.A.; Lasky-Su, J.A.; Greene, C.M.; O’Neill, S.J.; Silverman, E.K.; McElvaney, N.G. Clarification of the Risk of COPD in α-1 Antitrypsin Deficiency PiMZ Heterozygotes. Am. J. Respir. Crit. Care Med. 2014, 189, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Franciosi, A.N.; Alkhunaizi, M.A.; Woodsmith, A.; Aldaihani, L.; Alkandari, H.; Lee, S.E.; Fee, L.T.; McElvaney, N.G.; Carroll, T.P. Alpha-1 Antitrypsin Deficiency and Tobacco Smoking: Exploring Risk Factors and Smoking Cessation in a Registry Population. COPD J. Chron. Obstr. Pulm. Dis. 2021, 18, 76–82. [Google Scholar] [CrossRef]
- Hubbard, R.C.; Fells, G.; Gadek, J.; Pacholok, S.; Humes, J.; Crystal, R.G. Neutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophages. J. Clin. Investig. 1991, 88, 891–897. [Google Scholar] [CrossRef]
- O’Dwyer, C.A.; O’Brien, M.E.; Wormald, M.R.; White, M.M.; Banville, N.; Hurley, K.; McCarthy, C.; McElvaney, N.G.; Reeves, E.P. The BLT1 Inhibitory Function of alpha-1 Antitrypsin Augmentation Therapy Disrupts Leukotriene B4 Neutrophil Signaling. J. Immunol. 2015, 195, 3628–3641. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.M.; Carroll, T.P.; Smith, S.G.; Taggart, C.C.; Devaney, J.; Griffin, S.; O’Neill, S.J.; McElvaney, N.G. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 2005, 174, 1638–1646. [Google Scholar] [CrossRef]
- Tosi, M.F.; Zakem, H.; Berger, M. Neutrophil elastase cleaves C3bi on opsonized pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J. Clin. Investig. 1990, 86, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Hartl, D.; Latzin, P.; Hordijk, P.; Marcos, V.; Rudolph, C.; Woischnik, M.; Koller, B.; Reinhardt, D.; Roscher, A.A.; Roos, D.; et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat. Med. 2007, 13, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; McEnery, T.; McQuillan, K.; McElvaney, O.F.; McElvaney, O.J.; Landers, S.; Coleman, O.; Hawkins, P.; Henry, M.; Meleady, P.; et al. α1 Antitrypsin therapy modulates the neutrophil membrane proteome and secretome. Eur. Respir. J. 2020, 55, 1901678. [Google Scholar] [CrossRef]
- McElvaney, N.G.; Burdon, J.; Holmes, M.; Glanville, A.; Wark, P.A.; Thompson, P.J.; Hernandez, P.; Chlumsky, J.; Teschler, T.; Ficker, J.H.; et al. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: An open-label extension trial (RAPID-OLE). Lancet Respir. Med. 2017, 5, 51–60. [Google Scholar] [CrossRef]
- Chapman, K.R.; Burdon, J.G.; Piitulainen, E.; Sandhaus, R.A.; Seersholm, N.; Stocks, J.M.; Stoel, B.C.; Huang, L.; Yao, Z.; Edelman, J.M.; et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 360–368. [Google Scholar] [CrossRef]
- Bergin, D.A.; Reeves, E.P.; Meleady, P.; Henry, M.; McElvaney, O.J.; Carroll, T.P.; Condron, C.; Chotirmall, S.H.; Clynes, M.; O’Neill, S.J.; et al. Alpha-1 Antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J. Clin. Investig. 2010, 120, 4236–4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, K.; Lacey, N.; O’Dwyer, C.A.; Bergin, D.A.; McElvaney, O.J.; O’Brien, M.E.; McElvaney, O.F.; Reeves, E.P.; McElvaney, N.G. Alpha-1 antitrypsin augmentation therapy corrects accelerated neutrophil apoptosis in deficient individuals. J. Immunol. 2014, 193, 3978–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.P.; Duchen, M.R.; de Villiers, S.; Rich, P.R.; Segal, A.W. Alkalinity of neutrophil phagocytic vacuoles is modulated by HVCN1 and has consequences for myeloperoxidase activity. PLoS ONE 2015, 10, e0125906. [Google Scholar] [CrossRef] [Green Version]
- Okochi, Y.; Aratani, Y.; Adissu, H.A.; Miyawaki, N.; Sasaki, M.; Suzuki, K.; Okamura, Y. The voltage-gated proton channel Hv1/VSOP inhibits neutrophil granule release. J. Leukoc. Biol. 2016, 99, 7–19. [Google Scholar] [CrossRef]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002, 416, 291–297. [Google Scholar] [CrossRef]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; White, M.M.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Janusonis, S. Comparing two small samples with an unstable, treatment-independent baseline. J. Neurosci. Methods 2009, 179, 173–178. [Google Scholar] [CrossRef]
- Petheo, G.L.; Orient, A.; Barath, M.; Kovacs, I.; Rethi, B.; Lanyi, A.; Rajki, A.; Rajnavölgyi, É.; Geiszt, M. Molecular and functional characterization of Hv1 proton channel in human granulocytes. PLoS ONE 2010, 5, e14081. [Google Scholar] [CrossRef] [Green Version]
- Hayes, E.; Murphy, M.P.; Pohl, K.; Browne, N.; McQuillan, K.; Saw, L.E.; Foley, C.; Gargoum, F.; Hawkins, P.; McElvaney, O.J.; et al. Altered Degranulation and pH of Neutrophil Phagosomes Impacts Antimicrobial Efficiency in Cystic Fibrosis. Front. Immunol. 2020, 11, 600033. [Google Scholar] [CrossRef]
- Bergin, D.A.; Reeves, E.P.; Hurley, K.; Wolfe, R.; Jameel, R.; Fitzgerald, S.; McElvaney, N.G. The Circulating Proteinase Inhibitor alpha-1 Antitrypsin Regulates Neutrophil Degranulation and Autoimmunity. Sci. Transl. Med. 2014, 6, 217ra1. [Google Scholar] [CrossRef]
- Vega-Carrascal, I.; Reeves, E.P.; Niki, T.; Arikawa, T.; McNally, P.; O’Neill, S.J.; Hirashima, M.; McElvaney, N.G. Dysregulation of TIM-3-galectin-9 pathway in the cystic fibrosis airways. J. Immunol. 2011, 186, 2897–2909. [Google Scholar] [CrossRef] [Green Version]
- Celli, B.R.; Wedzicha, J.A. Update on Clinical Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Dowson, L.J.; Guest, P.J.; Stockley, R.A. Longitudinal changes in physiological, radiological, and health status measurements in alpha(1)-antitrypsin deficiency and factors associated with decline. Am. J. Respir. Crit. Care Med. 2001, 164, 1805–1809. [Google Scholar] [CrossRef]
- Needham, M.; Stockley, R.A. Exacerbations in α1-antitrypsin deficiency. Eur. Respir. J. 2005, 25, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Hiller, A.M.; Piitulainen, E.; Jehpsson, L.; Tanash, H. Decline in FEV1 and hospitalized exacerbations in individuals with severe alpha-1 antitrypsin deficiency. Int. J. Chron. Obstr. Pulm. Dis. 2019, 14, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Rouhani, F.; Paone, G.; Smith, N.K.; Krein, P.; Barnes, P.; Brantly, M.L. Lung neutrophil burden correlates with increased pro-inflammatory cytokines and decreased lung function in individuals with alpha(1)-antitrypsin deficiency. Chest 2000, 117, 250S. [Google Scholar] [CrossRef] [PubMed]
- Malerba, M.; Ricciardolo, F.; Radaeli, A.; Torregiani, C.; Ceriani, L.; Mori, E.; Bontempelli, M.; Tantucci, C.; Grassi, V. Neutrophilic inflammation and IL-8 levels in induced sputum of alpha-1-antitrypsin PiMZ subjects. Thorax 2006, 61, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: A new hypothesis with supporting data. Chest 2000, 118, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, I.S.; Ruchti, E.; Kaczmarek, J.S.; Clapham, D.E. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc. Natl. Acad. Sci. USA 2009, 106, 7642–7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okochi, Y.; Umemoto, E.; Okamura, Y. Hv1/VSOP regulates neutrophil directional migration and ERK activity by tuning ROS production. J. Leukoc. Biol. 2020, 107, 819–831. [Google Scholar] [CrossRef]
- Okochi, Y.; Okamura, Y. Regulation of Neutrophil Functions by Hv1/VSOP Voltage-Gated Proton Channels. Int. J. Mol. Sci. 2021, 22, 2620. [Google Scholar] [CrossRef] [PubMed]
- El Chemaly, A.; Okochi, Y.; Sasaki, M.; Arnaudeau, S.; Okamura, Y.; Demaurex, N. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J. Exp. Med. 2010, 207, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Attucci, S.; Juliano, M.A.; Kalupov, T.; Jourdan, M.L.; Juliano, L.; Gauthier, F. Measuring elastase, proteinase 3 and cathepsin G activities at the surface of human neutrophils with fluorescence resonance energy transfer substrates. Nat. Protoc. 2008, 3, 991–1000. [Google Scholar] [CrossRef]
- Sawyer, J.G.; Martin, N.L.; Hancock, R.E. Interaction of macrophage cationic proteins with the outer membrane of Pseudomonas aeruginosa. Infect. Immun. 1988, 56, 693–698. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, R.I.; Lichtenstein, A.K.; Ganz, T. Defensins: Antimicrobial and cytotoxic peptides of mammalian cells. Annu. Rev. Immunol. 1993, 11, 105–128. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawkins, P.; Sya, J.; Hup, N.K.; Murphy, M.P.; McElvaney, N.G.; Reeves, E.P. Alpha-1 Antitrypsin Augmentation Inhibits Proteolysis of Neutrophil Membrane Voltage-Gated Proton Channel-1 in Alpha-1 Deficient Individuals. Medicina 2021, 57, 814. https://doi.org/10.3390/medicina57080814
Hawkins P, Sya J, Hup NK, Murphy MP, McElvaney NG, Reeves EP. Alpha-1 Antitrypsin Augmentation Inhibits Proteolysis of Neutrophil Membrane Voltage-Gated Proton Channel-1 in Alpha-1 Deficient Individuals. Medicina. 2021; 57(8):814. https://doi.org/10.3390/medicina57080814
Chicago/Turabian StyleHawkins, Padraig, Julian Sya, Nee Kee Hup, Mark P. Murphy, Noel G. McElvaney, and Emer P. Reeves. 2021. "Alpha-1 Antitrypsin Augmentation Inhibits Proteolysis of Neutrophil Membrane Voltage-Gated Proton Channel-1 in Alpha-1 Deficient Individuals" Medicina 57, no. 8: 814. https://doi.org/10.3390/medicina57080814
APA StyleHawkins, P., Sya, J., Hup, N. K., Murphy, M. P., McElvaney, N. G., & Reeves, E. P. (2021). Alpha-1 Antitrypsin Augmentation Inhibits Proteolysis of Neutrophil Membrane Voltage-Gated Proton Channel-1 in Alpha-1 Deficient Individuals. Medicina, 57(8), 814. https://doi.org/10.3390/medicina57080814