
The Application of Control Materials for Ongoing Quality Management of Next-Generation Sequencing in a Clinical Genetic Laboratory

Abstract
1. Introduction
2. Considerations of Control Material Selection
2.1. Sample Characteristics
2.2. Variant Types
2.3. Variant Allele Frequency Range
3. Materials and Applications for Quality Control
3.1. Controls for Germline Variants
3.2. Controls for Somatic Mutations
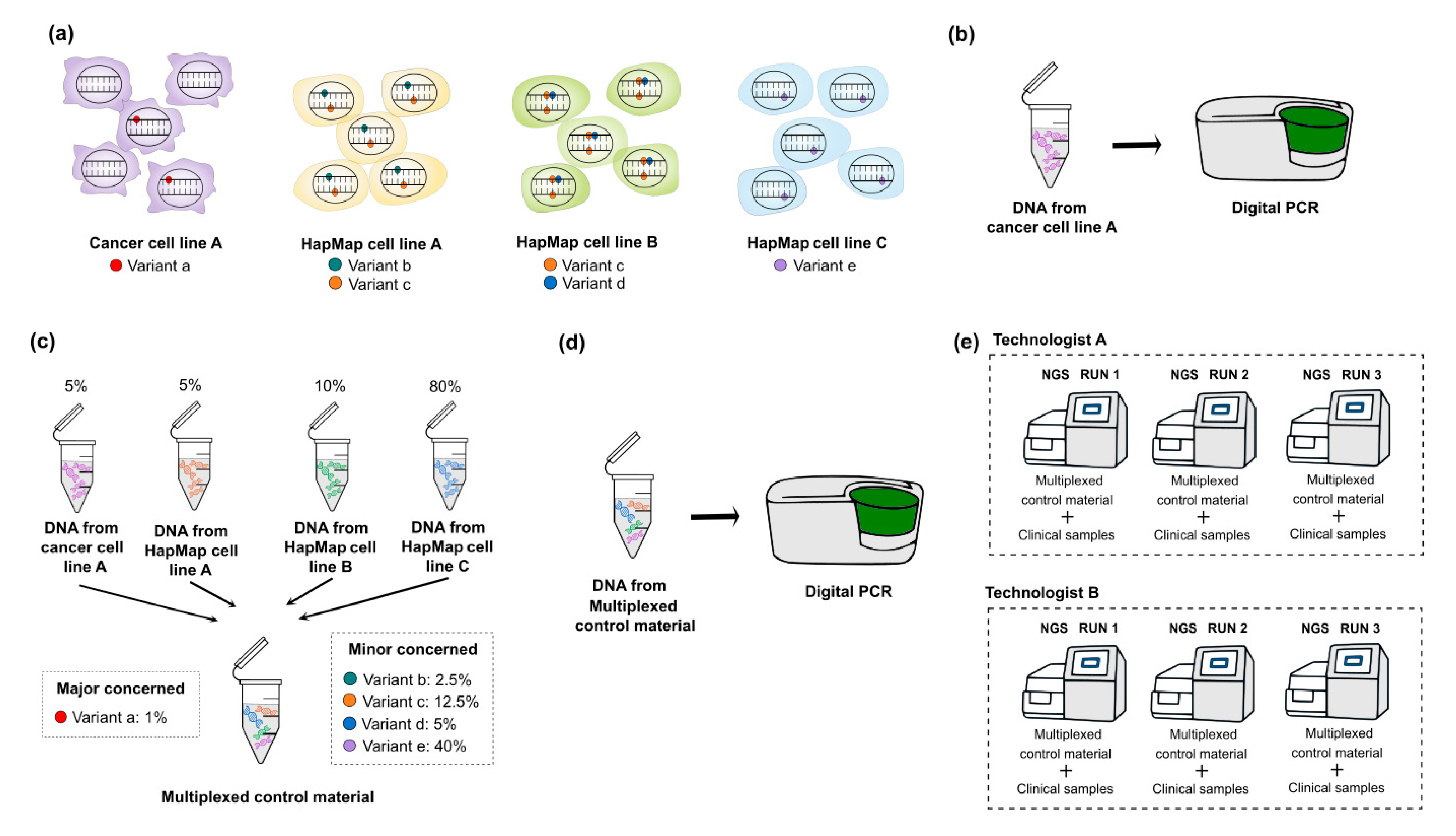
3.3. Multiplexing HapMap and Well-Characterized Cancer Cell Lines for Somatic Mutations
3.4. Mimicking Process
3.5. Synthetic DNA or Engineered Cell Line by Gene-Editing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.R. Next-Generation Sequencing in High-Sensitive Detection of Mutations in Tumors: Challenges, Advances, and Applications. J. Mol. Diagn. 2020, 22, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, I.; Aziz, N.; Farkas, D.H.; Furtado, M.; Gonzalez, A.F.; Greiner, T.C.; Grody, W.W.; Hambuch, T.; Kalman, L.; Kant, J.A.; et al. Opportunities and challenges associated with clinical diagnostic genome sequencing: A report of the Association for Molecular Pathology. J. Mol. Diagn. 2012, 14, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Yu, Y.; Qing, T.; Guo, L.; Shi, L. Next-generation sequencing in the clinic: Promises and challenges. Cancer Lett. 2013, 340, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, S.A.; Deveson, I.W.; Mercer, T.R. Reference standards for next-generation sequencing. Nat. Rev. Genet. 2017, 18, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Aziz, N.; Zhao, Q.; Bry, L.; Driscoll, D.K.; Funke, B.; Gibson, J.S.; Grody, W.W.; Hegde, M.R.; Hoeltge, G.A.; Leonard, D.G.; et al. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch. Pathol. Lab. Med. 2015, 139, 481–493. [Google Scholar] [CrossRef]
- Rehm, H.L.; Bale, S.J.; Bayrak-Toydemir, P.; Berg, J.S.; Brown, K.K.; Deignan, J.L.; Friez, M.J.; Funke, B.H.; Hegde, M.R.; Lyon, E. ACMG clinical laboratory standards for next-generation sequencing. Genet. Med. 2013, 15, 733–747. [Google Scholar] [CrossRef]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef]
- Pisapia, P.; Pepe, F.; Sgariglia, R.; Nacchio, M.; Russo, G.; Conticelli, F.; Girolami, I.; Eccher, A.; Bellevicine, C.; Vigliar, E.; et al. Next generation sequencing in cytology. Cytopathology 2021. [Google Scholar] [CrossRef]
- Arreaza, G.; Qiu, P.; Pang, L.; Albright, A.; Hong, L.Z.; Marton, M.J.; Levitan, D. Pre-Analytical Considerations for Successful Next-Generation Sequencing (NGS): Challenges and Opportunities for Formalin-Fixed and Paraffin-Embedded Tumor Tissue (FFPE) Samples. Int. J. Mol. Sci. 2016, 17, 1579. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, D.W.; Chiu, R.W.K. Sequencing of Circulating Cell-free DNA during Pregnancy. N. Engl. J. Med. 2018, 379, 464–473. [Google Scholar] [CrossRef]
- Norton, M.E.; Jacobsson, B.; Swamy, G.K.; Laurent, L.C.; Ranzini, A.C.; Brar, H.; Tomlinson, M.W.; Pereira, L.; Spitz, J.L.; Hollemon, D.; et al. Cell-free DNA analysis for noninvasive examination of trisomy. N. Engl. J. Med. 2015, 372, 1589–1597. [Google Scholar] [CrossRef]
- Goldfeder, R.L.; Priest, J.R.; Zook, J.M.; Grove, M.E.; Waggott, D.; Wheeler, M.T.; Salit, M.; Ashley, E.A. Medical implications of technical accuracy in genome sequencing. Genome Med. 2016, 8, 24. [Google Scholar] [CrossRef]
- Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome Med. 2020, 12, 91. [Google Scholar] [CrossRef]
- Nagy, R.; Sweet, K.; Eng, C. Highly penetrant hereditary cancer syndromes. Oncogene 2004, 23, 6445–6470. [Google Scholar] [CrossRef]
- Velázquez, C.; Lastra, E.; Avila Cobos, F.; Abella, L.; de la Cruz, V.; Hernando, B.A.; Hernández, L.; Martínez, N.; Infante, M.; Durán, M. A comprehensive custom panel evaluation for routine hereditary cancer testing: Improving the yield of germline mutation detection. J. Transl. Med. 2020, 18, 232. [Google Scholar] [CrossRef]
- Strom, S.P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef]
- Abecasis, G.; Altshuler, D.; Auton, A.; Brooks, L.; Durbin, R. 1000 Genomes Project Consortium. Nature 2010, 467, 1061–1073. [Google Scholar] [PubMed]
- Kumaran, M.; Subramanian, U.; Devarajan, B. Performance assessment of variant calling pipelines using human whole exome sequencing and simulated data. BMC Bioinform. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, E.; Lee, I.; Marcotte, E.M. Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 2015, 5, 17875. [Google Scholar] [CrossRef] [PubMed]
- Zook, J.M.; Hansen, N.F.; Olson, N.D.; Chapman, L.; Mullikin, J.C.; Xiao, C.; Sherry, S.; Koren, S.; Phillippy, A.M.; Boutros, P.C.; et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol. 2020, 38, 1347–1355. [Google Scholar] [CrossRef]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef]
- Lee, A.Y.; Ewing, A.D.; Ellrott, K.; Hu, Y.; Houlahan, K.E.; Bare, J.C.; Espiritu, S.M.G.; Huang, V.; Dang, K.; Chong, Z.; et al. Combining accurate tumor genome simulation with crowdsourcing to benchmark somatic structural variant detection. Genome Biol. 2018, 19, 188. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.W.; Nasser, S.; Corbett, R.; Chan, S.K.; Murray, L.; Legendre, C.; Tembe, W.; Adkins, J.; Kim, N.; Wong, S.; et al. A somatic reference standard for cancer genome sequencing. Sci. Rep. 2016, 6, 24607. [Google Scholar] [CrossRef]
- Zook, J.M.; Catoe, D.; McDaniel, J.; Vang, L.; Spies, N.; Sidow, A.; Weng, Z.; Liu, Y.; Mason, C.E.; Alexander, N.; et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 2016, 3, 160025. [Google Scholar] [CrossRef] [PubMed]
- Zook, J.M.; McDaniel, J.; Olson, N.D.; Wagner, J.; Parikh, H.; Heaton, H.; Irvine, S.A.; Trigg, L.; Truty, R.; McLean, C.Y.; et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 2019, 37, 561–566. [Google Scholar] [CrossRef]
- Wrzeszczynski, K.O.; Felice, V.; Abhyankar, A.; Kozon, L.; Geiger, H.; Manaa, D.; London, F.; Robinson, D.; Fang, X.; Lin, D.; et al. Analytical Validation of Clinical Whole-Genome and Transcriptome Sequencing of Patient-Derived Tumors for Reporting Targetable Variants in Cancer. J. Mol. Diagn. 2018, 20, 822–835. [Google Scholar] [CrossRef]
- Hardwick, S.A.; Chen, W.Y.; Wong, T.; Deveson, I.W.; Blackburn, J.; Andersen, S.B.; Nielsen, L.K.; Mattick, J.S.; Mercer, T.R. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods 2016, 13, 792–798. [Google Scholar] [CrossRef]
- Chen, K.; Meric-Bernstam, F.; Zhao, H.; Zhang, Q.; Ezzeddine, N.; Tang, L.Y.; Qi, Y.; Mao, Y.; Chen, T.; Chong, Z.; et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin. Chem. 2015, 61, 544–553. [Google Scholar] [CrossRef]
- Huggett, J.F.; Cowen, S.; Foy, C.A. Considerations for digital PCR as an accurate molecular diagnostic tool. Clin. Chem. 2015, 61, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Sreejith, K.R.; Ooi, C.H.; Jin, J.; Dao, D.V.; Nguyen, N.T. Digital polymerase chain reaction technology-recent advances and future perspectives. Lab. Chip. 2018, 18, 3717–3732. [Google Scholar] [CrossRef]
- Cao, L.; Cui, X.; Hu, J.; Li, Z.; Choi, J.R.; Yang, Q.; Lin, M.; Ying Hui, L.; Xu, F. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosens. Bioelectron. 2017, 90, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.K.; Lee, Y.K.; Nam, S.H.; Kim, J.K.; Park, K.S.; Kim, J.W. Quantitative and Qualitative QC of Next-Generation Sequencing for Detecting Somatic Variants: An Example of Detecting Clonal Hematopoiesis of Indeterminate Potential. Clin. Chem. 2020, 66, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, J.R.; Opdam, F.J.; Boonyaratanakornkit, J.; Schönbrunner, E.R.; Shahbazian, M.; Edsjö, A.; Hoefler, G.; Jung, A.; Kotsinas, A.; Gorgoulis, V.G.; et al. Implementation of formalin-fixed, paraffin-embedded cell line pellets as high-quality process controls in quality assessment programs for KRAS mutation analysis. J. Mol. Diagn. 2012, 14, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhou, H.; Shi, D.; Shen, S.; Tian, Y.; Wang, L.; Lou, J.; Cong, R.; Lu, J.; Zhang, H.; et al. Quality Control of Next-generation Sequencing-based In vitro Diagnostic Test for Onco-relevant Mutations Using Multiplex Reference Materials in Plasma. J. Cancer 2018, 9, 1680–1688. [Google Scholar] [CrossRef]
- Lih, C.J.; Sims, D.J.; Harrington, R.D.; Polley, E.C.; Zhao, Y.; Mehaffey, M.G.; Forbes, T.D.; Das, B.; Walsh, W.D.; Datta, V.; et al. Analytical Validation and Application of a Targeted Next-Generation Sequencing Mutation-Detection Assay for Use in Treatment Assignment in the NCI-MPACT Trial. J. Mol. Diagn. 2016, 18, 51–67. [Google Scholar] [CrossRef]
- Kudalkar, E.M.; Almontashiri, N.A.; Huang, C.; Anekella, B.; Bowser, M.; Hynes, E.; Garlick, R.; Funke, B.H. Multiplexed Reference Materials as Controls for Diagnostic Next-Generation Sequencing: A Pilot Investigating Applications for Hypertrophic Cardiomyopathy. J. Mol. Diagn. 2016, 18, 882–889. [Google Scholar] [CrossRef]
- Sims, D.J.; Harrington, R.D.; Polley, E.C.; Forbes, T.D.; Mehaffey, M.G.; McGregor, P.M., 3rd; Camalier, C.E.; Harper, K.N.; Bouk, C.H.; Das, B.; et al. Plasmid-Based Materials as Multiplex Quality Controls and Calibrators for Clinical Next-Generation Sequencing Assays. J. Mol. Diagn. 2016, 18, 336–349. [Google Scholar] [CrossRef]
- Suzuki, T.; Tsukumo, Y.; Furihata, C.; Naito, M.; Kohara, A. Preparation of the standard cell lines for reference mutations in cancer gene-panels by genome editing in HEK 293 T/17 cells. Genes Environ. 2020, 42, 8. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Purpose | Control Materials | Characteristics | Sources |
|---|---|---|---|
| Detecting germline variants | Standard reference materials (e.g., HapMap reference materials) |
|
|
| Reference materials for hereditary genetic disorders or conditions |
|
| |
| Detecting somatic mutations | Cancer cell lines |
|
|
| Multiplexing reference materials (multiplexing HapMap and well-characterized cancer cell lines) |
| ||
| Detecting germline variants or Somatic mutations | Synthetic DNA or engineered cell line by gene-editing |
| |
| Commercial reference materials |
|
| |
| Patient specimens |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, Y.-K.; Park, K.-S. The Application of Control Materials for Ongoing Quality Management of Next-Generation Sequencing in a Clinical Genetic Laboratory. Medicina 2021, 57, 543. https://doi.org/10.3390/medicina57060543
Min Y-K, Park K-S. The Application of Control Materials for Ongoing Quality Management of Next-Generation Sequencing in a Clinical Genetic Laboratory. Medicina. 2021; 57(6):543. https://doi.org/10.3390/medicina57060543
Chicago/Turabian StyleMin, Young-Kyu, and Kyung-Sun Park. 2021. "The Application of Control Materials for Ongoing Quality Management of Next-Generation Sequencing in a Clinical Genetic Laboratory" Medicina 57, no. 6: 543. https://doi.org/10.3390/medicina57060543
APA StyleMin, Y.-K., & Park, K.-S. (2021). The Application of Control Materials for Ongoing Quality Management of Next-Generation Sequencing in a Clinical Genetic Laboratory. Medicina, 57(6), 543. https://doi.org/10.3390/medicina57060543