
Philadelphia-Negative MPN: A Molecular Journey, from Hematopoietic Stem Cell to Clinical Features

Abstract
:1. Introduction
2. MPN Hematopoietic Stem Cells
3. Molecular Pathogenesis
3.1. Germline Predisposition
3.2. Additional Somatic Mutations
4. Clinical Implications
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Moulard, O.; Mehta, J.; Fryzek, J.; Olivares, R.; Iqbal, U.; Mesa, R.A. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur. J. Haematol. 2014, 92, 289–297. [Google Scholar] [CrossRef]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef]
- Jain, T.; Mesa, R.A.; Palmer, J.M. Allogeneic stem cell transplantation in myelofibrosis. Biol. Blood Marrow Transplant. 2017, 23, 1429–1436. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef]
- Spivak, J.L. Myeloproliferative neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.-S.; Lai, D.-M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 1–17. [Google Scholar] [CrossRef]
- Spivak, J.L.; Considine, M.; Williams, D.M.; Talbot, C.C.; Rogers, O.; Moliterno, A.R.; Jie, C.; Ochs, M.F.N. Two clinical phenotypes in polycythemia vera. N. Engl. J. Med. 2014, 371, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Jasper, H.; Rudolph, K.L. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Stem Cell 2015, 16, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Moliterno, A.R.; Hankins, W.D.; Spivak, J.L. Impaired expression of the thrombopoietin receptor by platelets from patients with polycythemia vera. N. Engl. J. Med. 1998, 338, 572–580. [Google Scholar] [CrossRef]
- Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Bogani, C.; Verrucci, M.; Ponziani, V.; Longo, G.; Bosi, A.; Vannucchi, A.M. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005, 19, 1847–1849. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Gerber, J.M.; Ngwang, B.; Zhang, H.; Williams, D.M. The Leukemic Stem Cell in Polycythemia Vera and Primary Myelofibrosis Is Distinct From the Initiating JAK2 V617F-Positive Hematopoietic Stem Cell. Blood 2011, 118, 613. [Google Scholar] [CrossRef]
- Wang, X.; Prakash, S.; Lu, M.; Tripodi, J.; Ye, F.; Najfeld, V.; Li, Y.; Schwartz, M.; Weinberg, R.; Roda, P.; et al. Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. J. Clin. Investig. 2012, 122, 3888–3899. [Google Scholar] [CrossRef]
- Anand, S.; Stedham, F.; Beer, P.; Gudgin, E.; Ortmann, C.A.; Bench, A.; Erber, W.; Green, A.R.; Huntly, B.J.P. Effects of the JAK2 mutation on the hematopoietic stem and progenitor compartment in human myeloproliferative neoplasms. Blood 2011, 118, 177–181. [Google Scholar] [CrossRef]
- Masarova, L.; Patel, K.P.; Newberry, K.J.; Cortes, J.; Borthakur, G.; Konopleva, M.; Estrov, Z.; Kantarjian, H.; Verstovsek, S. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: A post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017, 4, e165–e175. [Google Scholar] [CrossRef] [Green Version]
- Manz, M.G.; Miyamoto, T.; Akashi, K.; Weissman, I.L. Prospective isolation of human clonogenic common myeloid progenitors. Proc. Natl. Acad. Sci. USA 2002, 99, 11872–11877. [Google Scholar] [CrossRef] [Green Version]
- Pang, W.W.; Pluvinage, J.V.; Price, E.A.; Sridhar, K.; Arber, D.A.; Greenberg, P.L.; Schrier, S.L.; Park, C.Y.; Weissman, I.L. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc. Natl. Acad. Sci. USA 2013, 110, 3011–3016. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Hutchison, R.E.; Mohi, G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood 2012, 119, 3539–3549. [Google Scholar] [CrossRef] [Green Version]
- Duek, A.; Lundberg, P.; Shimizu, T.; Grisouard, J.; Karow, A.; Kubovcakova, L.; Hao-Shen, H.; Dirnhofer, S.; Skoda, R.C. Loss of Stat1 decreases megakaryopoiesis and favors erythropoiesis in a JAK2-V617F–driven mouse model of MPNs. Blood 2014, 123, 3943–3950. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C.N. A gain-of-function mutation of JAK2 in myeloproliferative disorders. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Pieri, L.; Guglielmelli, P.; Park, J.; Jurcic, J.G.; Rosenblat, T.; Tallman, M.S. JAK2 allele burden in the myeloproliferative neoplasms: Effects on phenotype, prognosis and change with treatment. Ther. Adv. Hematol. 2011, 2, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Varghese, L.N.; Defour, J.-P.; Pecquet, C.; Constantinescu, S.N. The thrombopoietin receptor: Structural basis of traffic and activation by ligand, mutations, agonists, and mutated calreticulin. Front. Endocrinol. 2017, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Vannucchi, A.M.; Tefferi, A. Rationale for revision and proposed changes of the WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Blood Cancer J. 2015, 5, e337. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, K.; Warsch, W.; Gonzalez-Arias, C.; Nice, F.L.; Avezov, E.; Milburn, J.; Li, J.; Dimitropoulou, D.; Biddie, S.; Wang, M.; et al. A novel signalling screen demonstrates that CALR mutations activate essential MAPK signalling and facilitate megakaryocyte differentiation. Leukemia 2017, 31, 934–944. [Google Scholar] [CrossRef]
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J. Clin. Investig. 2019, 129, 1596–1611. [Google Scholar] [CrossRef]
- Qi, M.; Elion, E.A. MAP kinase pathways. J. Cell Sci. 2005, 118, 3569–3572. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Cantley, L.C. Ras, PI (3) K and MTOR Signalling Controls Tumour Cell Growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.-P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef]
- Slezak, S.; Jin, P.; Caruccio, L.; Ren, J.; Bennett, M.; Zia, N.; Adams, S.; Wang, E.; Ascensao, J.; Schechter, G.; et al. Gene and microRNA analysis of neutrophils from patients with polycythemia vera and essential thrombocytosis: Down-regulation of micro RNA-1 and -133a. J. Transl. Med. 2009, 7, 17–39. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Zini, R.; Bogani, C.; Salati, S.; Pancrazzi, A.; Bianchi, E.; Mannelli, F.; Ferrari, S.; Le Bousse-Kerdilès, M.-C.; Bosi, A.; et al. Molecular profiling of CD34+ cells in idiopathic myelofibrosis identifies a set of disease-associated genes and reveals the clinical significance of Wilms’ tumor gene 1 (WT1). Stem Cells 2007, 25, 165–173. [Google Scholar] [CrossRef]
- Berkofsky-Fessler, W.; Buzzai, M.; Kim, M.K.-H.; Fruchtman, S.; Najfeld, V.; Min, D.-J.; Costa, F.F.; Bischof, J.M.; Soares, M.B.; McConnell, M.J.; et al. Transcriptional profiling of polycythemia vera identifies gene expression patterns both dependent and independent from the action of JAK2V617F. Clin. Cancer Res. 2010, 16, 4339–4352. [Google Scholar] [CrossRef] [Green Version]
- Catani, L.; Zini, R.; Sollazzo, D.; Ottaviani, E.; Vannucchi, A.M.; Ferrari, S.; Baccarani, M.; Vianelli, N.; Lemoli, R.M.; Manfredini, R. Molecular profile of CD34+ stem/progenitor cells according to JAK2V617F mutation status in essential thrombocythemia. Leukemia 2009, 23, 997–1000. [Google Scholar] [CrossRef]
- Rumi, E.; Cazzola, M. Advances in understanding the pathogenesis of familial myeloproliferative neoplasms. Br. J. Haematol. 2017, 178, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasho, T.L.; Jimma, T.; Finke, C.M.; Patnaik, M.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012, 120, 4168–4171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010, 24, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Triviai, I.; Zeschke, S.; Rentel, J.; Spanakis, M.; Scherer, T.; Gabdoulline, R.; Panagiota, V.; Thol, F.; Heuser, M.; Stocking, C.; et al. ASXL1/EZH2 mutations promote clonal expansion of neoplastic HSC and impair erythropoiesis in PMF. Leukemia 2019, 33, 99–109. [Google Scholar] [CrossRef]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.-H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, L.; Datta, D.; Serrat, J.; Morey, L.; Solanas, G.; Avgustinova, A.; Blanco, E.; Pons, J.I.; Matallanas, D.; von Kriegsheim, A.; et al. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell 2016, 19, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.-C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Sashida, G.; Wang, C.; Tomioka, T.; Oshima, M.; Aoyama, K.; Kanai, A.; Mochizuki-Kashio, M.; Harada, H.; Shimoda, K.; Iwama, A. The loss of Ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J. Exp. Med. 2016, 213, 1459–1477. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; LaFave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Dey, A. The ASXL–BAP1 axis: New factors in myelopoiesis, cancer and epigenetics. Leukemia 2013, 27, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef]
- Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Szuber, N.; Begna, K.H.; Patnaik, M.M.; Gangat, N.; Pardanani, A.; et al. Targeted next-generation sequencing in blast phase myeloproliferative neoplasms. Blood Adv. 2018, 2, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef]
- Chowdhury, O.; O’Sullivan, J.; Barkas, N.; Wang, G.; Buck, G.; Hamblin, A.; Tefferi, A.; Al-Ali, H.K.; Barosi, G.; Devos, T.; et al. Spliceosome mutations are common in persons with myeloproliferative neoplasm-associated myelofibrosis with RBC-transfusion-dependence and correlate with response to pomalidomide. Leukemia 2021, 35, 1197–1202. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Sanada, M.; Suzuki, T.; Shih, L.-Y.; Otsu, M.; Kato, M.; Yamazaki, S.; Tamura, A.; Honda, H.; Sakata-Yanagimoto, M.; Kumano, K.; et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 2009, 460, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef] [PubMed]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Gagelmann, N.; Ditschkowski, M.; Bogdanov, R.; Thol, F.; et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood 2019, 133, 2233–2242. [Google Scholar]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Biamonte, F.; Rotunno, G.; Artusi, V.; Artuso, L.; Bernardis, I.; Tenedini, E.; Pieri, L.; Paoli, C.; Mannarelli, C.; et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood 2014, 123, 2157–2160. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Verstovsek, S.; Guglielmelli, P.; Griesshammer, M.; Burn, T.C.; Naim, A.; Paranagama, D.; Marker, M.; Gadbaw, B.; Kiladjian, J.J. Ruxolitinib reduces JAK2 p. V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann. Hematol. 2017, 96, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Guglielmelli, P. Traffic lights for ruxolitinib. Blood 2017, 130, 1075–1077. [Google Scholar] [CrossRef] [PubMed]
- Pacilli, A.; Rotunno, G.; Mannarelli, C.; Fanelli, T.; Pancrazzi, A.; Contini, E.; Mannelli, F.; Gesullo, F.; Bartalucci, N.; Fattori, G.C.; et al. Mutation landscape in patients with myelofibrosis receiving ruxolitinib or hydroxyurea. Blood Cancer J. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Talpaz, M.; Kiladjian, J.J. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia 2020, 35, 1–17. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs. best available therapy, including ruxolitinib, in patients with myelofibrosis: A randomized clinical trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2018, 5, e73–e81. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Komrokji, R.S.; Palandri, F.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Hoffman, R.; Odenike, O.; et al. Randomized, Single-Blind, Multicenter Phase II Study of Two Doses of Imetelstat in Relapsed or Refractory Myelofibrosis. J. Clin. Oncol. 2021, 39, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Masarova, L.; Verstovsek, S. Novel Concepts of Treatment for Patients with Myelofibrosis and Related Neoplasms. Cancers 2020, 12, 2891. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Quintás-Cardama, A.; Kantarjian, H.; Manshouri, T.; Luthra, R.; Estrov, Z.; Pierce, S.; Richie, M.A.; Borthakur, G.; Konopleva, M.; Cortes, J.; et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. Clin. Oncol. 2009, 27, 5418–5424. [Google Scholar] [CrossRef] [Green Version]
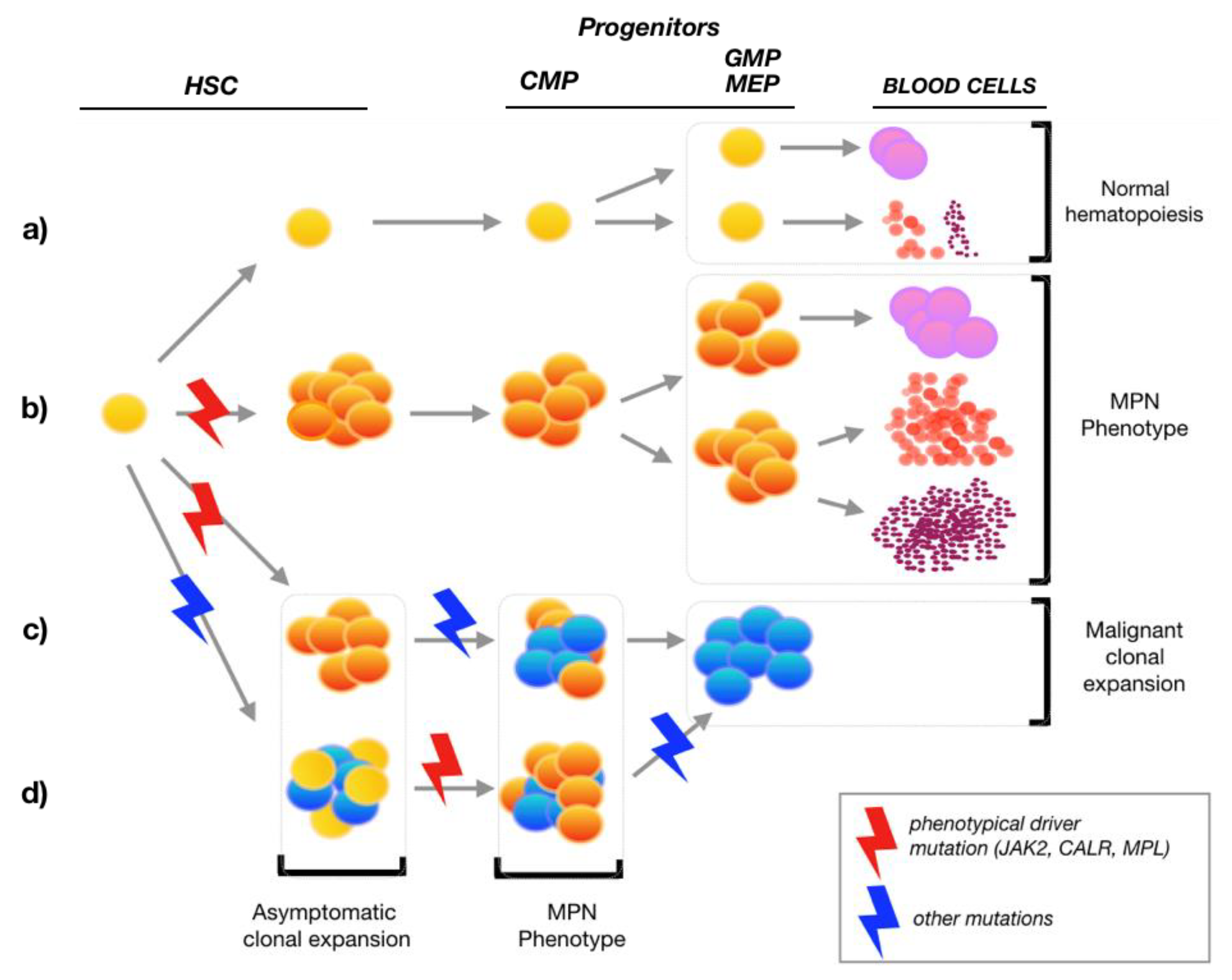

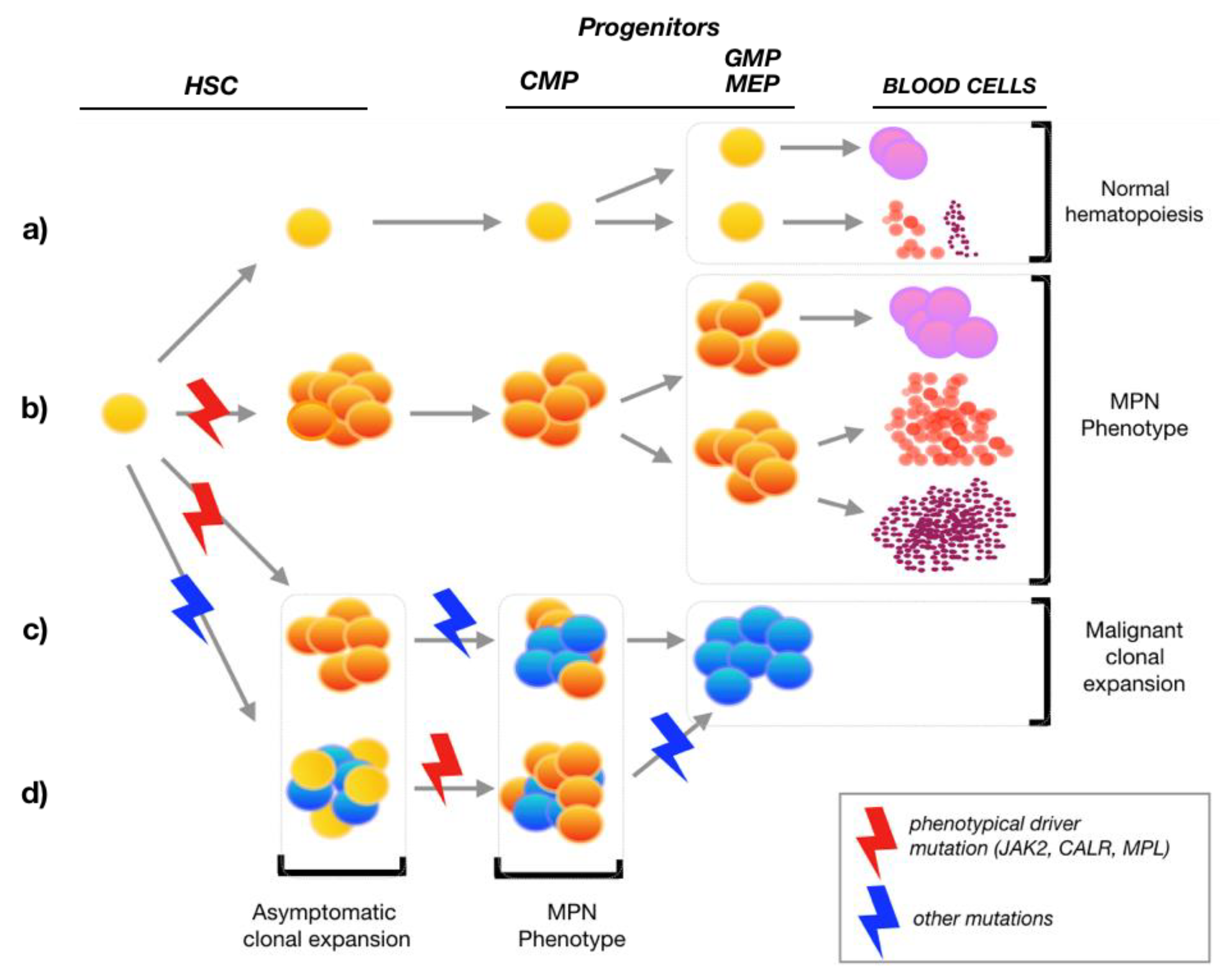{kind=link}
| Category | Gene | Function/Mutation Effect | Effects on Prognosis | REF | |
|---|---|---|---|---|---|
| Histone modification | ASXL1 | Demethylation and transcription repression by heterozygous mutations. | Increased AML evolution and fibrosis development. | [49,51,55] | |
| Histone modification | EZH2 | Histone methyltransferase and transcription repression by heterozygous and homozygous mutation. | Increased AML evolution and fibrosis development. | [46,56] | |
| DNA Methylation regulation | DNMT3A | Reduced methyltransferase activity in DNA and histone methylation. | Reduced OS in MF. | [55,57] | |
| DNA Methylation regulation | IDH1/2 | Epigenetic dysregulation influencing leukemogenesis. Heterozygous mutation. | Reduced OS in MF. | [14,58] | |
| Splicing machinery | SRSF2 | Needed for splicing of pre-mRNA. Heterozygous mutations. | Reduced OS in MPN and increased risk of AML evolution. | [44,51] | |
| Splicing machinery | U2AF1 | Needed for splicing of pre-mRNA. Heterozygous mutations. | Disease progression and reduced OS in MF. | [59,60] | |
| Signalling | CBL | Increased STAT5 signalling. Homozygous mutations. | Reduced OS in MF. Resistance to JAK inhibitors. | [61,62] | |
| Signalling | NRAS/KRAS | Increased proliferation. Heterozygous mutations. | Reduced OS in MF. Resistance to JAK inhibitors. | [57,61] | |
| Signalling | PTPNI1 | Activation of signalling. | Reduced OS in blastic phase. | [57] | |
| Transcription | RUNX1 | Role in regulation of normal hematopoiesis. | Reduced OS in blastic phase. | [36] | |
| Transcription | TP53 | Regulation of apoptosis and cell cycle arrest. | Reduced OS in MPN and increase of disease progression. | [57,61] | |
| Splicing machinery | SF3B1 | Member of the splicing machinery. | Increased risk of fibrotic evolution. | [55] |
| Prognostic Score | Variables (Points) | Points | Risk Categories (Points) | Median Survival (Years) |
|---|---|---|---|---|
| MIPSS-PV [63] | Leukocyte count ≥ 15 × 109/L Thrombosis history Age > 67 years SRSF2 mutation | 1 1 2 3 | Low (0–1) Intermediate (2–3) High (4–7) | 24 13.1 3.2 |
| MIPSS-ET [63] | Leukocyte count ≥ 11 × 109/L Age > 60 years Male sex SRSF2, SF3B1, U2AF1 and TP53 mutation | 1 4 1 2 | Low (0–1) Intermediate (2–5) High (6–8) | 34.3 14.1 7.9 |
| Prognostic Score | Variables (Points) | Points | Risk Categories (Points) | Median Survival (Years) |
|---|---|---|---|---|
| MIPSS70 [64] | Hemoglobin < 10 g/dL Blasts > 2% Constitutional symptoms Leukocytes > 25 × 109/L Platelet count < 100 × 109/L BM fibrosis ≥ 2 Non CALR type-1 HMR = 1 HMR ≥ 2 | 1 1 1 2 2 1 1 1 2 | Low (0–1) Intermediate (2–4) High (5–12) | 27.7 7.1 2.3 |
| MIPSS70 plus [64] | Hemoglobin < 10 g/dL Blasts > 2% Constitutional symptoms Non CALR type-1 HMR = 1 HMR ≥ 2 Unfavourable karyotype | 1 1 1 2 1 2 3 | Low (0–2) Intermediate (3) High (4–6) Very high (7–11) | 20.0 6.3 3.9 1.7 |
| MIPSS70 plus v2.0 [65] | Hemoglobin 8–10 g/dL Hemoglobin < 8 g/dL Blasts > 2% Constitutional symptoms Non CALR type-1 HMR+U2AF1 Q157 = 1 HMR+U2AF1 Q157 ≥ 2 HR Karyotype VHR Karyotype | 1 2 1 2 2 2 3 3 4 | Very low (0) Low (1–2) Intermediate (3–4) High (5–8) Very high (9–14) | Not reached 10.3 7 3.5 1.8 |
| GIPSS [66] | Non CALR type-1 ASXL1 mutation SRSF2 mutation U2AF1 Q157 HR karyotype VHR karyotype | 1 1 1 1 1 2 | Low (0) Intermediate-1 (1) Intermediate-2 (2) High (3–6) | 26.4 8.0 4.2 2.0 |
| MYSEC-PM [67] | Hemoglobin < 11 g/dL Blasts ≥ 3% Platelets < 150 × 109/L Constitutional symptoms Age at secondary MF (0.15 point/year) CALR not mutated genotype | 1 1 1 2 2 | Low (<11) Intermediate-1 (11-14) Intermediate-2 (14-16) High (≥16) | Not reached 9.3 4.4 2.0 |
| MTSS [68] | Platelets < 150 × 109/L Leukocytes > 25 × 109/L Karnofsky PS < 90% Age ≥ 57 years HLA-mismatched unrelated donor Non CALR/MPL mutation ASXL1 mutation | 1 1 1 1 2 2 1 | Low (0–2) Intermediate (3–4) High (5) Very high (6–9) | 5-years OS 83% 5-years OS 64% 5-years OS 37% 5-years OS 22% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giai, V.; Secreto, C.; Freilone, R.; Pregno, P. Philadelphia-Negative MPN: A Molecular Journey, from Hematopoietic Stem Cell to Clinical Features. Medicina 2021, 57, 1043. https://doi.org/10.3390/medicina57101043
Giai V, Secreto C, Freilone R, Pregno P. Philadelphia-Negative MPN: A Molecular Journey, from Hematopoietic Stem Cell to Clinical Features. Medicina. 2021; 57(10):1043. https://doi.org/10.3390/medicina57101043
Chicago/Turabian StyleGiai, Valentina, Carolina Secreto, Roberto Freilone, and Patrizia Pregno. 2021. "Philadelphia-Negative MPN: A Molecular Journey, from Hematopoietic Stem Cell to Clinical Features" Medicina 57, no. 10: 1043. https://doi.org/10.3390/medicina57101043
APA StyleGiai, V., Secreto, C., Freilone, R., & Pregno, P. (2021). Philadelphia-Negative MPN: A Molecular Journey, from Hematopoietic Stem Cell to Clinical Features. Medicina, 57(10), 1043. https://doi.org/10.3390/medicina57101043