Preliminary Work Towards Finding Proteins as Potential Vaccine Candidates for Vibrio cholerae Pakistani Isolates through Reverse Vaccinology



Abstract
1. Introduction
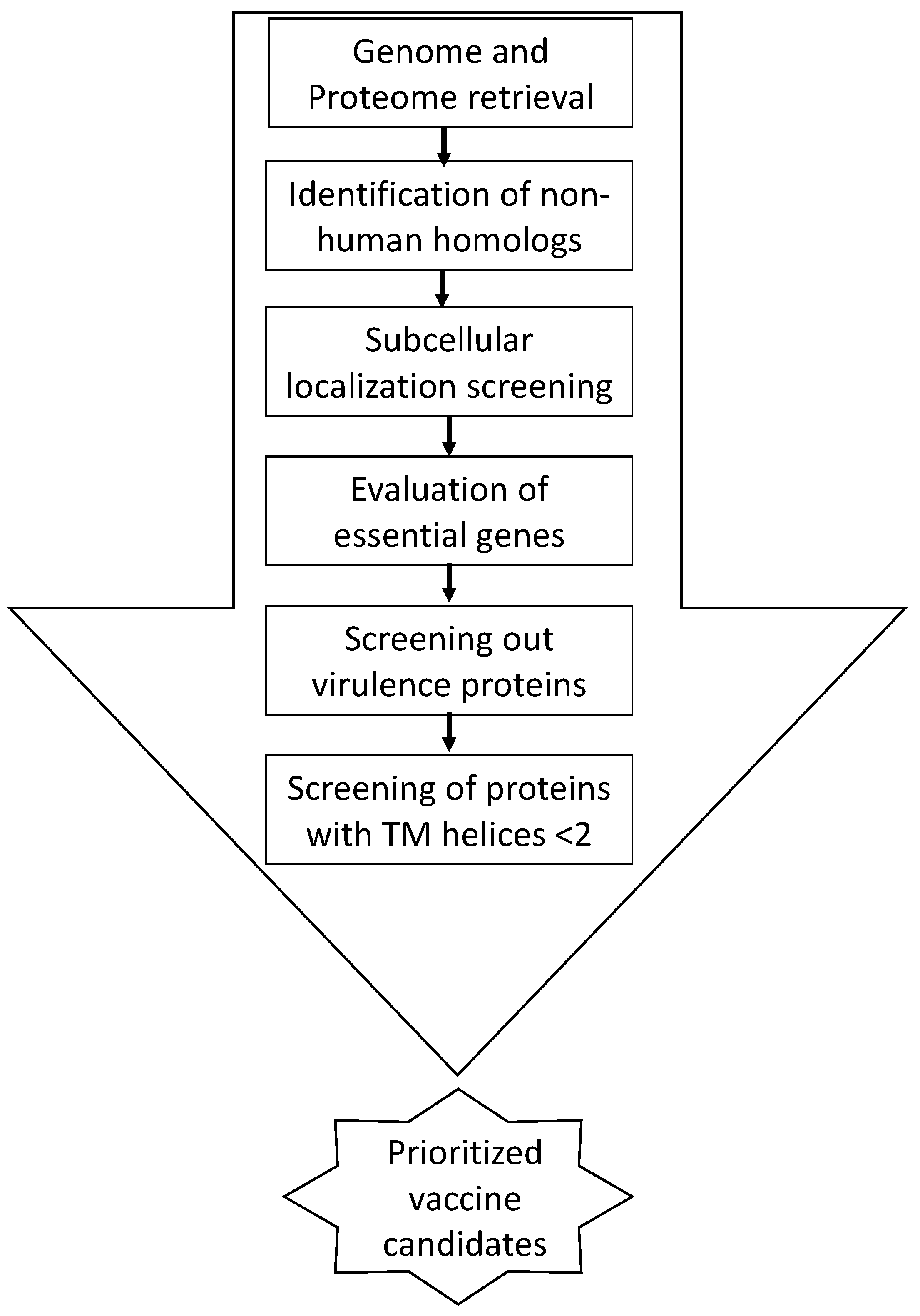
2. Material and Methods
3. Results
4. Discussion on the Results
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Almagro-Moreno, S.; Taylor, R.K. Cholera: Environmental Reservoirs and Impact on Disease Transmission. Microbiol. Spectr. 2012, 1. [Google Scholar] [CrossRef]
- Faruque, S.M.; Albert, M.J.; Mekalanos, J.J. Epidemiology, Genetics, and Ecology of Toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 1998, 62, 1301–1314. [Google Scholar] [PubMed]
- Reidl, J.; Klose, K.E. Vibrio cholerae and cholera: Out of the water and into the host. FEMS Microbiol. Rev. 2002, 26, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Noureen, N.; Tariq, M.; Farooq, A.; Arif, A.; Bokhari, H. In silico identification of receptor specific epitopes as potential vaccine candidates from Vibrio cholerae strains. Gene Rep. 2016, 4, 222–232. [Google Scholar] [CrossRef]
- Baselski, V.S.; Medina, R.; Parker, C. In vivo and in vitro characterization of virulence-deficient mutants of Vibrio cholerae. Infect. Immun. 1979, 24, 111–116. [Google Scholar] [PubMed]
- Faruque, S.M.; Kamruzzaman, M.; Meraj, I.M.; Chowdhury, N.; Nair, G.B.; Sack, R.B.; Colwell, R.R.; Sack, D.A. Pathogenic potential of environmental Vibrio cholerae strains carrying genetic variants of the toxin-coregulated pilus pathogenicity island. Infect. Immun. 2003, 71, 1020–1025. [Google Scholar] [CrossRef]
- Waldor, M.K.; Tschape, H.; Mekalanos, J.J. A new type of conjugative transposon encodes resistance to sulfamethoxazole, trimethoprim, and streptomycin in Vibrio cholerae O139. J. Bacteriol. 1996, 178, 4157–4165. [Google Scholar] [CrossRef]
- Karaolis, D.K.; Johnson, J.A.; Bailey, C.C.; Boedeker, E.C.; Kaper, J.B.; Reeves, P.R. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc. Natl. Acad. Sci. USA 1998, 95, 3134–3139. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Katzianer, D.S.; Zhong, Z.; Zhu, J. LysR family activator-regulated major facilitator superfamily transporters are involved in Vibrio cholerae antimicrobial compound resistance and intestinal colonisation. Int. J. Antimicrob. Agents 2013, 41, 188–192. [Google Scholar] [CrossRef]
- Faruque, S.M.; Islam, M.J.; Ahmad, Q.S.; Biswas, K.; Faruque, A.; Nair, G.B.; Sack, R.B.; Sack, D.A.; Mekalanos, J.J. An improved technique for isolation of environmental Vibrio cholerae with epidemic potential: Monitoring the emergence of a multiple-antibiotic–resistant epidemic strain in Bangladesh. J. Infect. Dis. 2006, 193, 1029–1036. [Google Scholar] [CrossRef]
- Kim, H.B.; Wang, M.; Ahmed, S.; Park, C.H.; LaRocque, R.C.; Faruque, A.S.; Salam, M.A.; Khan, W.A.; Qadri, F.; Calderwood, S.B.; et al. Transferable quinolone resistance in Vibrio cholerae. Antimicrob. Agents Chemother. 2010, 54, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Tarca, A.L.; Carey, V.J.; Chen, X.W.; Romero, R.; Drăghici, S. Machine learning and its applications to biology. PLoS Comput. Biol. 2007, 3, e116. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, B.; Serruto, D.; Adu-Bobie, J.; Rappuoli, R.; Pizza, M. The genome revolution in vaccine research. Curr. Issues Mol. Biol. 2004, 6, 17–28. [Google Scholar] [PubMed]
- Kimman, T. Risks connected with the use of conventional and genetically engineerd vaccines. Vet. Q. 1992, 14, 110–118. [Google Scholar] [CrossRef]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Sette, A.; Rappuoli, R. Reverse vaccinology: Developing vaccines in the era of genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B.; et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef]
- Barh, D.; Tiwari, S.; Jain, N.; Ali, A.; Santos, A.R.; Misra, A.N.; Azevedo, V.; Kumar, A. In silico subtractive genomics for target identification in human bacterial pathogens. Drug Dev. Res. 2011, 72, 162–177. [Google Scholar] [CrossRef]
- Maione, D.; Margarit, I.; Rinaudo, C.D.; Masignani, V.; Mora, M.; Scarselli, M.; Tettelin, H.; Brettoni, C.; Iacobini, E.T.; Rosini, R.; et al. Identification of a universal Group B streptococcus vaccine by multiple genome screen. Science 2005, 309, 148–150. [Google Scholar] [CrossRef]
- Giuliani, M.M.; Adu-Bobie, J.; Comanducci, M.; Aricò, B.; Savino, S.; Santini, L.; Brunelli, B.; Bambini, S.; Biolchi, A.; Capecchi, B.; et al. A universal vaccine for serogroup B meningococcus. Proc. Natl. Acad. Sci. USA 2006, 103, 10834–10839. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.; Edwards, L.; Snelgrove, R.; Finco, O.; Rae, A.; Grandi, G.; Guilio, R.; Hussell, T. Discovery of a vaccine antigen that protects mice from Chlamydia pneumoniae infection. Vaccine 2007, 25, 2252–2260. [Google Scholar] [CrossRef]
- Schild, S.; Nelson, E.J.; Camilli, A. Immunization with Vibrio cholerae outer membrane vesicles induces protective immunity in mice. Infect. Immun. 2008, 76, 4554–4563. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Mutreja, A.; Thomson, N.; Baker, S.; Parkhill, J.; Dougan, G.; Bokhari, H.; Wren, B.W. Genomic epidemiology of Vibrio cholerae O1 associated with floods, Pakistan, 2010. Emerg. Infect. Dis. 2014, 20, 13. [Google Scholar] [CrossRef]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef] [PubMed]
- Meiler, J.; Baker, D. Coupled prediction of protein secondary and tertiary structure. Proc. Natl. Acad. Sci. USA 2003, 100, 12105–12110. [Google Scholar] [CrossRef]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein–ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef]
- Ryan, E.T. The cholera pandemic, still with us after half a century: Time to rethink. PLoS Negl. Trop. Dis. 2011, 5, e1003. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y.H.; Fan, M.W.; Jia, R.; Xu, Q.A.; Guo, J.H.; Yu, F.; Tian, Q.W. Enhanced efficacy of CTLA-4 fusion anti-caries DNA vaccines in gnotobiotic hamsters. Acta Pharmacol. Sin. 2007, 28, 1236–1242. [Google Scholar] [CrossRef][Green Version]
- Karalus, R.J.; Murphy, T.F. Purification and characterization of outer membrane protein P6, a vaccine antigen of non-typeable Haemophilus influenzae. FEMS Immunol. Med. Microbiol. 1999, 26, 159–166. [Google Scholar] [CrossRef]
- Liang, H.; Xia, L.; Wu, Z.; Jian, J.; Lu, Y. Expression, purification and antibody preparation of flagellin FlaA from Vibrio alginolyticus strain HY9901. Lett. Appl. Microbiol. 2010, 50, 181–186. [Google Scholar] [CrossRef]
- Bren, A.; Eisenbach, M. Changing the direction of flagellar rotation in bacteria by modulating the ratio between the rotational states of the switch protein FliM1. J. Mol. Biol. 2001, 312, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, M.M.; Van de Verg, L.L. Combination vaccines against diarrheal diseases. Hum. Vaccines Immunother. 2015, 11, 1434–1448. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
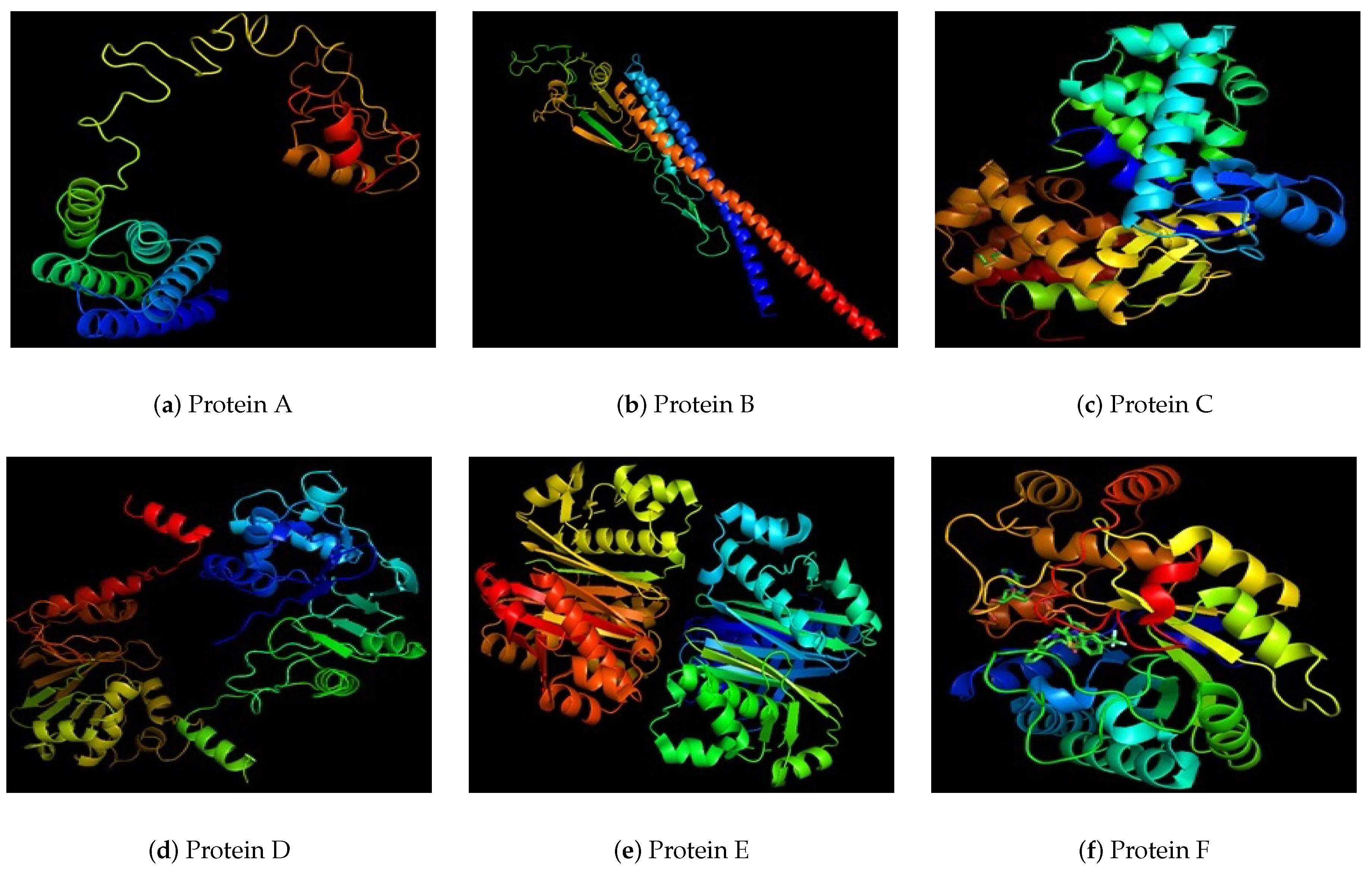
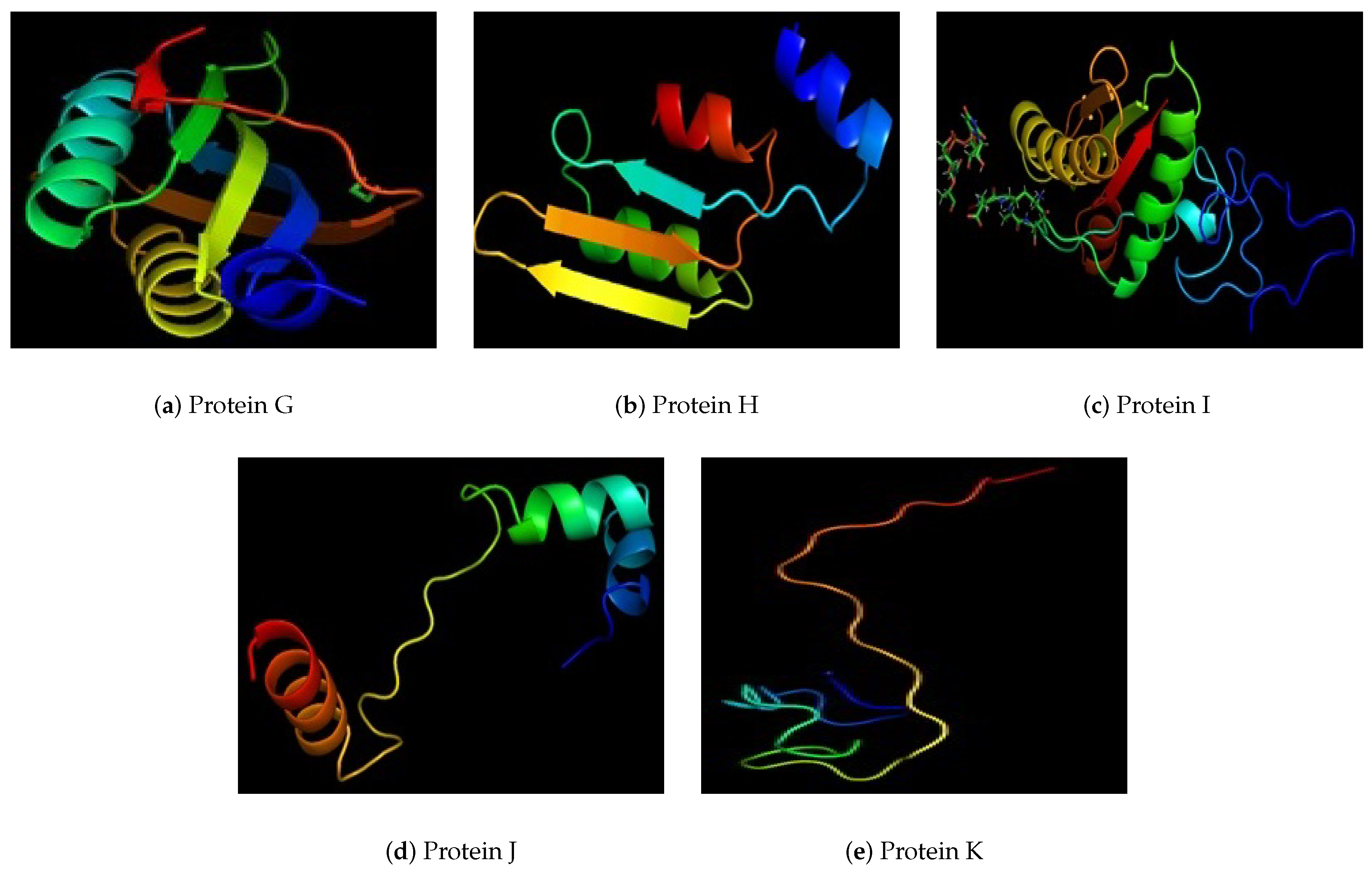
| Protein Id | Uniport Blast Similarity % | Number of Amino Acids | Helices | Strands | Localization |
|---|---|---|---|---|---|
| A | 97.9% to flagellin A (N16961) | 760 | 30 | 19 | Outer Membrane Protein |
| B | 100% Zn transport system binding protein | 380 | 12 | 12 | Extracellular |
| C | 100% to beta lactamase (Vibrio cholerae) | 290 | 10 | 7 | Periplasmic |
| D | 100% beta lactamase (Vibrio cholerae) | 280 | 7 | 12 | Unknown |
| E | 99.6% to tRNA Pseuriudine synthase A | 280 | 8 | 12 | Unknown |
| F | 100% outer membrane lipocarrier protein | 270 | 5 | 10 | Extracellular |
| G | 100% uncharacterized protein (V. albensis) | 198 | 3 | 15 | Periplasmic |
| H | 100% peptidoglycan-associated lipo protein | 180 | 6 | 5 | Extracellular |
| I | 100% DNA binding protein | 170 | 6 | 4 | Outer Membrance Protein |
| J | 100% uncharacterized protein (Enterobacter sp. R4-368) | 70 | 3 | 2 | Unknown |
| K | 100% chemotaxis protein che A (Vibrio cholerae) | 40 | 2 | 5 | Unknown |
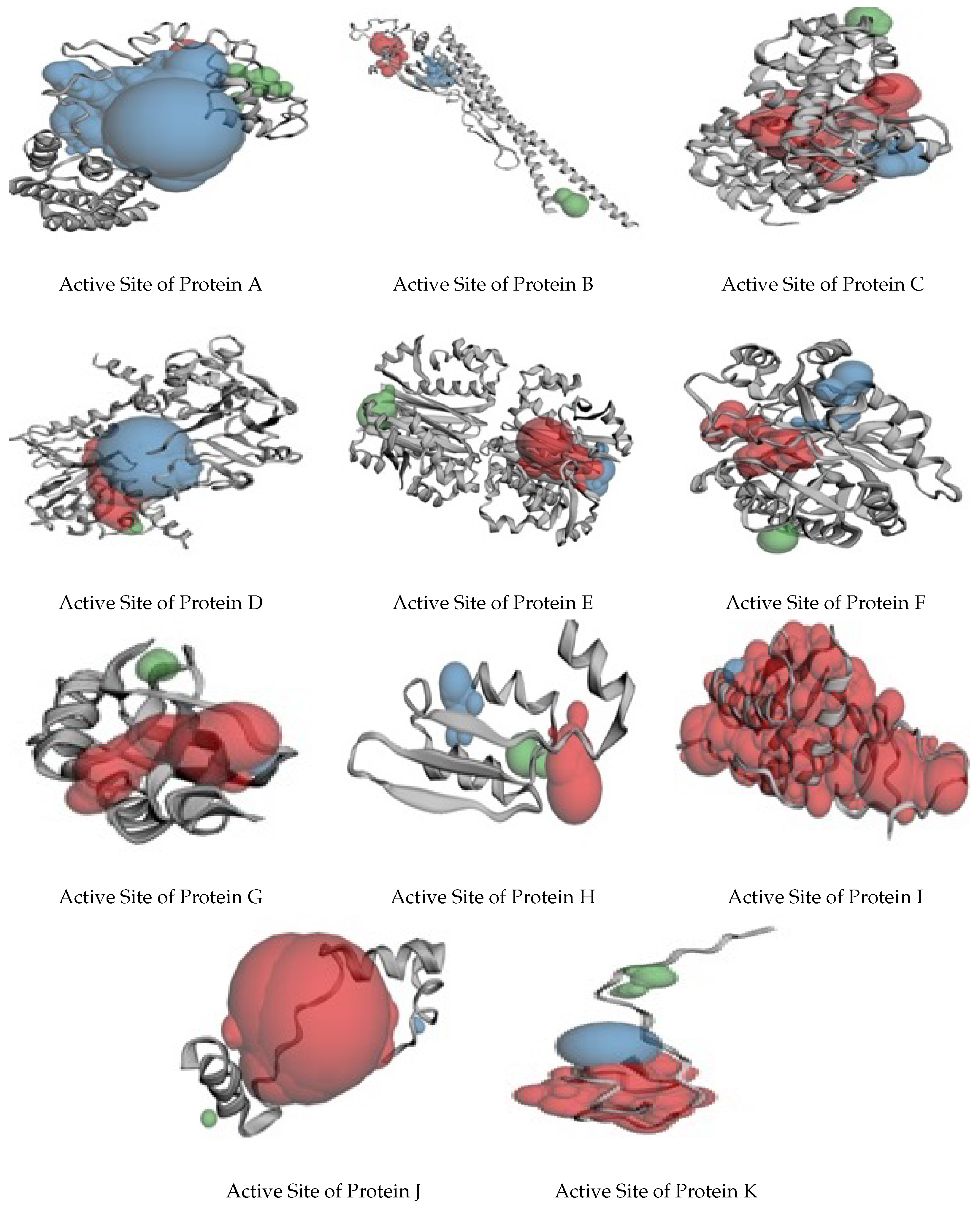
| Proteins | Number of Pockets | Number of Amino Acids Rich in Active Sites |
|---|---|---|
| A | 38 | LEU, ARG, PHE |
| B | 55 | LYS, SER, GLN, PRO |
| C | 32 | GLU, LEU, THR, ARG |
| D | 110 | LYS, ALA, SER, PHE, LEU |
| E | 66 | PRO, LYS, GLU |
| F | 42 | PHE, HIS, LYS, SER |
| G | 3 | PHE, HIS, LYS, SER |
| H | 17 | ILE, LEU, SER |
| I | 12 | THR, PHE, LEU |
| J | 3 | ASP, ALA, LEU |
| K | 10 | ARG, ASP, LYS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeb, S.; Ali, A.; Gulfam, S.M.; Bokhari, H. Preliminary Work Towards Finding Proteins as Potential Vaccine Candidates for Vibrio cholerae Pakistani Isolates through Reverse Vaccinology. Medicina 2019, 55, 195. https://doi.org/10.3390/medicina55050195
Zeb S, Ali A, Gulfam SM, Bokhari H. Preliminary Work Towards Finding Proteins as Potential Vaccine Candidates for Vibrio cholerae Pakistani Isolates through Reverse Vaccinology. Medicina. 2019; 55(5):195. https://doi.org/10.3390/medicina55050195
Chicago/Turabian StyleZeb, Samia, Amjad Ali, Sardar Muhammad Gulfam, and Habib Bokhari. 2019. "Preliminary Work Towards Finding Proteins as Potential Vaccine Candidates for Vibrio cholerae Pakistani Isolates through Reverse Vaccinology" Medicina 55, no. 5: 195. https://doi.org/10.3390/medicina55050195
APA StyleZeb, S., Ali, A., Gulfam, S. M., & Bokhari, H. (2019). Preliminary Work Towards Finding Proteins as Potential Vaccine Candidates for Vibrio cholerae Pakistani Isolates through Reverse Vaccinology. Medicina, 55(5), 195. https://doi.org/10.3390/medicina55050195