
The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies

 ,
,  ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Perfluorinated Compounds
3. Polybrominated Diphenyl Ethers (PBDE)
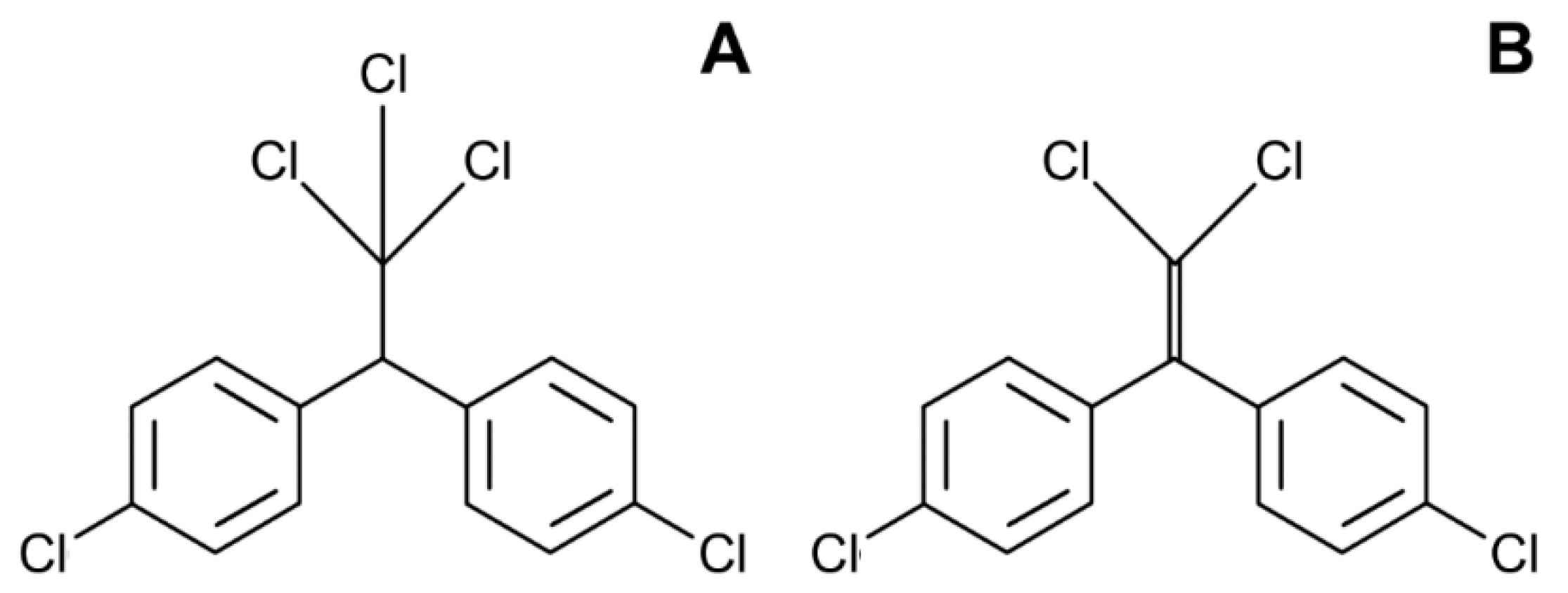
4. Dichlorodiphenyltrichloroethane (DDT)
5. Polychlorinated Biphenyls (PCBs)
6. Polycyclic Aromatic Hydrocarbons (PAHs)
7. Dioxin
8. Bisphenol A (BPA)
9. Phthalates
10. Diethylstilbestrol (DES)
11. POPs Mixtures and Obesity
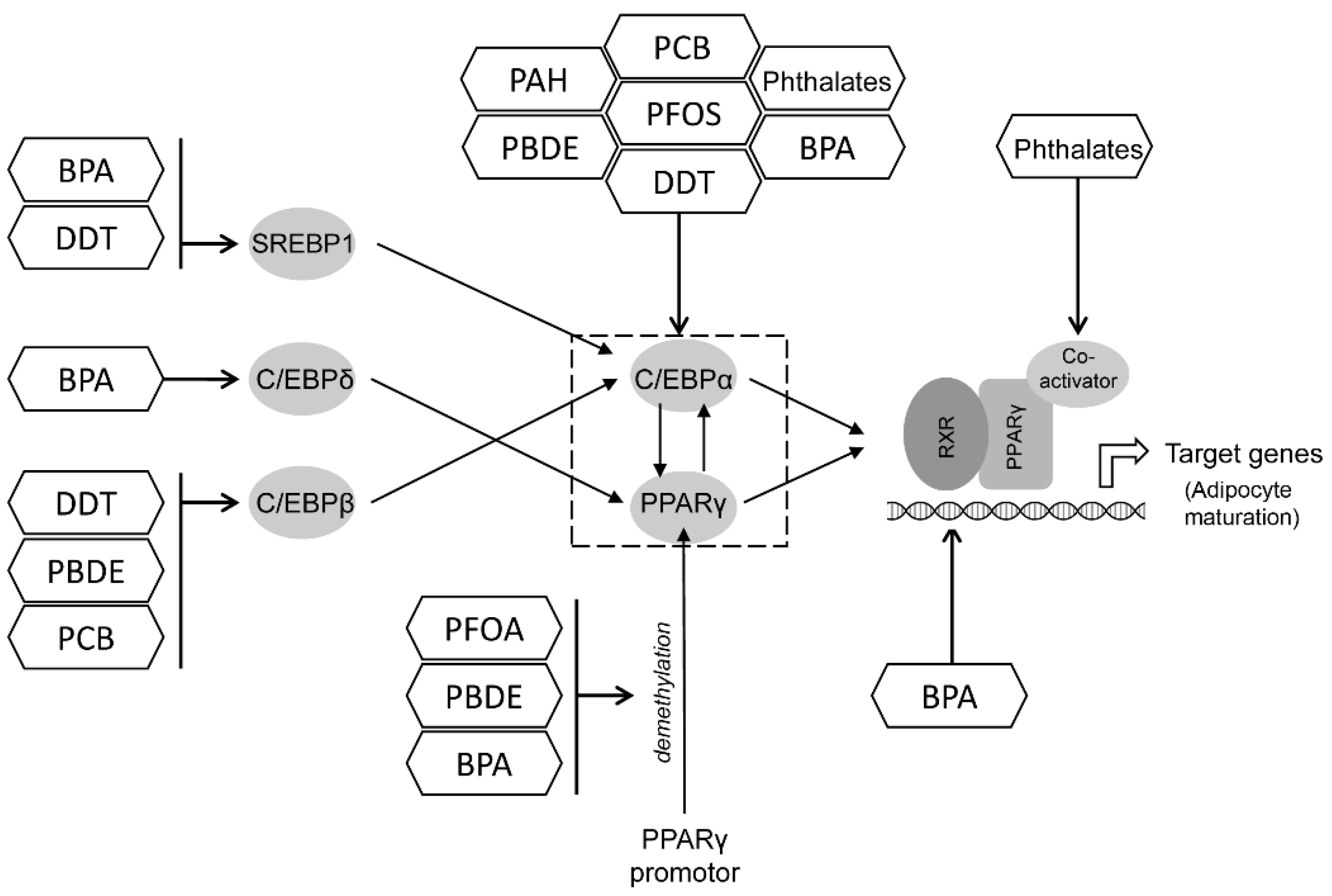
12. A Summary of Key Targets for POPs Obesogenic Effects
12.1. PPAR and C/EBP
12.2. Estrogen Receptor (ER)
12.3. Glucocorticoid Receptor
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Haththotuwa, R.N.; Wijeyaratne, C.N.; Senarath, U. Worldwide Epidemic of Obesity. In Obesity; Elsevier: Amsterdam, The Netherlands, 2013; pp. 3–11. ISBN 9780124160453. [Google Scholar]
- Blüher, M. Obesity: Global Epidemiology and Pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Martínez-Villanueva, J.; González-Leal, R.; Argente, J.; Martos-Moreno, G.Á. La Obesidad Parental Se Asocia Con La Gravedad de La Obesidad Infantil y de Sus Comorbilidades. An. Pediatr. 2019, 90, 224–231. [Google Scholar] [CrossRef]
- The GBD 2015 Obesity Collaborators. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Maury, E.; Brichard, S.M. Adipokine Dysregulation, Adipose Tissue Inflammation and Metabolic Syndrome. Mol. Cell. Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef]
- Mottillo, S.; Filion, K.B.; Genest, J.; Joseph, L.; Pilote, L.; Poirier, P.; Rinfret, S.; Schiffrin, E.L.; Eisenberg, M.J. The Metabolic Syndrome and Cardiovascular Risk. J. Am. Coll. Cardiol. 2010, 56, 1113–1132. [Google Scholar] [CrossRef]
- Zammit, C.; Liddicoat, H.; Moonsie, I.; Makker, H. Obesity and Respiratory Diseases. Am. J. Clin. Hypn. 2011, 53, 335–343. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Callaghan, B.C.; Feldman, E.L. Neurological Consequences of Obesity. Lancet Neurol. 2017, 16, 465–477. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and Cancer Risk: Emerging Biological Mechanisms and Perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef]
- Dietz, W.; Santos-Burgoa, C. Obesity and Its Implications for COVID-19 Mortality. Obesity 2020, 28, 1005. [Google Scholar] [CrossRef]
- Kadouh, H.C.; Acosta, A. Current Paradigms in the Etiology of Obesity. Tech. Gastrointest. Endosc. 2017, 19, 2–11. [Google Scholar] [CrossRef]
- Baillie-Hamilton, P.F. Chemical Toxins: A Hypothesis to Explain the Global Obesity Epidemic. J. Altern. Complement. Med. 2002, 8, 185–192. [Google Scholar] [CrossRef]
- Wang, Y.; Hollis-Hansen, K.; Ren, X.; Qiu, Y.; Qu, W. Do Environmental Pollutants Increase Obesity Risk in Humans? Obes. Rev. 2016, 17, 1179–1197. [Google Scholar] [CrossRef]
- Hatch, E.E.; Nelson, J.W.; Stahlhut, R.W.; Webster, T.F. Association of Endocrine Disruptors and Obesity: Perspectives from Epidemiological Studies. Int. J. Androl. 2010, 33, 324–332. [Google Scholar] [CrossRef]
- Ashraf, M.A. Persistent Organic Pollutants (POPs): A Global Issue, a Global Challenge. Environ. Sci. Pollut. Res. 2017, 24, 4223–4227. [Google Scholar] [CrossRef]
- Gaur, N.; Narasimhulu, K.; PydiSetty, Y. Recent Advances in the Bio-Remediation of Persistent Organic Pollutants and Its Effect on Environment. J. Clean. Prod. 2018, 198, 1602–1631. [Google Scholar] [CrossRef]
- World Health Organisation. Food Safety: Persistent Organic Pollutants (POPs); World Health Organisation: Geneva, Switzerland, 2020. [Google Scholar]
- González, N.; Domingo, J. Concentrations of Persistent Organic Pollutants in Blood of the Spanish Population: Temporal Trend. Arch. Farm. 2021, 71, 455–479. [Google Scholar] [CrossRef]
- Eskenazi, B.; Chevrier, J.; Rosas, L.G.; Anderson, H.A.; Bornman, M.S.; Bouwman, H.; Chen, A.; Cohn, B.A.; de Jager, C.; Henshel, D.S.; et al. The Pine River Statement: Human Health Consequences of DDT Use. Environ. Health Perspect 2009, 117, 1359–1367. [Google Scholar] [CrossRef]
- Panieri, E.; Buha-Đorđevic, A.; Saso, L. Endocrine Disruption by PFAS: A Major Concern Associated with Legacy and Replacement Substances. Arh. Farm. 2021, 71, 429–454. [Google Scholar] [CrossRef]
- Linares, V.; Perelló, G.; Nadal, M.; Gómez-Catalán, J.; Llobet, J.M.; Domingo, J.L. Environmental versus Dietary Exposure to POPs and Metals: A Probabilistic Assessment of Human Health Risks. J. Environ. Monit. 2010, 12, 681–688. [Google Scholar] [CrossRef]
- Jones, K.C.; de Voogt, P. Persistent Organic Pollutants (POPs): State of the Science. Environ. Pollut. 1999, 100, 209–221. [Google Scholar] [CrossRef]
- Kärrman, A.; van Bavel, B.; Järnberg, U.; Hardell, L.; Lindström, G. Perfluorinated Chemicals in Relation to Other Persistent Organic Pollutants in Human Blood. Chemosphere 2006, 64, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Davidsen, N.; Lauvås, A.J.; Myhre, O.; Ropstad, E.; Carpi, D.; Gyves, E.M.; Berntsen, H.F.; Dirven, H.; Paulsen, R.E.; Bal-Price, A.; et al. Exposure to Human Relevant Mixtures of Halogenated Persistent Organic Pollutants (POPs) Alters Neurodevelopmental Processes in Human Neural Stem Cells Undergoing Differentiation. Reprod. Toxicol. 2021, 100, 17–34. [Google Scholar] [CrossRef]
- La Merrill, M.; Emond, C.; Kim, M.J.; Antignac, J.-P.; Le Bizec, B.; Clément, K.; Birnbaum, L.S.; Barouki, R. Toxicological Function of Adipose Tissue: Focus on Persistent Organic Pollutants. Environ. Health Perspect. 2013, 121, 162–169. [Google Scholar] [CrossRef]
- Darbre, P.D. Endocrine Disruptors and Obesity. Curr. Obes. Rep. 2017, 6, 18–27. [Google Scholar] [CrossRef]
- Nappi, F.; Barrea, L.; Di Somma, C.; Savanelli, M.; Muscogiuri, G.; Orio, F.; Savastano, S. Endocrine Aspects of Environmental “Obesogen” Pollutants. Int. J. Environ. Res. Public Health 2016, 13, 765. [Google Scholar] [CrossRef]
- Heindel, J.J.; Blumberg, B. Environmental Obesogens: Mechanisms and Controversies. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 89–106. [Google Scholar] [CrossRef]
- Stel, J.; Legler, J. The Role of Epigenetics in the Latent Effects of Early Life Exposure to Obesogenic Endocrine Disrupting Chemicals. Endocrinology 2015, 156, 3466–3472. [Google Scholar] [CrossRef]
- Grün, F.; Blumberg, B. Endocrine Disrupters as Obesogens. Mol. Cell. Endocrinol. 2009, 304, 19–29. [Google Scholar] [CrossRef]
- Griffin, M.D.; Pereira, S.R.; DeBari, M.K.; Abbott, R.D. Mechanisms of Action, Chemical Characteristics, and Model Systems of Obesogens. BMC Biomed. Eng. 2020, 2, 6. [Google Scholar] [CrossRef]
- Inoue, K.; Goto, A.; Sugiyama, T.; Ramlau-Hansen, C.H.; Liew, Z. The Confounder-Mediator Dilemma: Should We Control for Obesity to Estimate the Effect of Perfluoroalkyl Substances on Health Outcomes? Toxics 2020, 8, 125. [Google Scholar] [CrossRef]
- Apelberg, B.J.; Goldman, L.R.; Calafat, A.M.; Herbstman, J.B.; Kuklenyik, Z.; Heidler, J.; Needham, L.L.; Halden, R.U.; Witter, F.R. Determinants of Fetal Exposure to Polyfluoroalkyl Compounds in Baltimore, Maryland. Environ. Sci. Technol. 2007, 41, 3891–3897. [Google Scholar] [CrossRef]
- Lauritzen, H.B.; Larose, T.L.; Øien, T.; Sandanger, T.M.; Odland, J.O.; Van De Bor, M.; Jacobsen, G.W. Prenatal Exposure to Persistent Organic Pollutants and Child Overweight/Obesity at 5-Year Follow-up: A Prospective Cohort Study. Environ. Health Glob. Access Sci. Source 2018, 17, 9. [Google Scholar] [CrossRef]
- Braun, J.M. Early-Life Exposure to EDCs: Role in Childhood Obesity and Neurodevelopment. Nat. Rev. Endocrinol. 2017, 13, 161–173. [Google Scholar] [CrossRef]
- Halldorsson, T.I.; Rytter, D.; Haug, L.S.; Bech, B.H.; Danielsen, I.; Becher, G.; Henriksen, T.B.; Olsen, S.F. Prenatal Exposure to Perfluorooctanoate and Risk of Overweight at 20 Years of Age: A Prospective Cohort Study. Environ. Health Perspect. 2012, 120, 668–673. [Google Scholar] [CrossRef]
- Domazet, S.L.; GrØntved, A.; Timmermann, A.G.; Nielsen, F.; Jensen, T.K. Longitudinal Associations of Exposure to Perfluoroalkylated Substances in Childhood and Adolescence and Indicators of Adiposity and Glucose Metabolism 6 and 12 Years Later: The European Youth Heart Study. Diabetes Care 2016, 39, 1745–1751. [Google Scholar] [CrossRef]
- Geiger, S.D.; Yao, P.; Vaughn, M.G.; Qian, Z. PFAS Exposure and Overweight/Obesity among Children in a Nationally Representative Sample. Chemosphere 2021, 268, 128852. [Google Scholar] [CrossRef]
- Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.; Leblanc, J.C.; Nebbia, C.S.; et al. Risk to Human Health Related to the Presence of Perfluoroalkyl Substances in Food. EFSA J. 2020, 18, e06223. [Google Scholar] [CrossRef]
- Watkins, A.M.; Wood, C.R.; Lin, M.T.; Abbott, B.D. The Effects of Perfluorinated Chemicals on Adipocyte Differentiation in Vitro. Mol. Cell. Endocrinol. 2015, 400, 90–101. [Google Scholar] [CrossRef]
- Liu, W.; Qin, H.; Pan, Y.; Luo, F.; Zhang, Z. Low Concentrations of Perfluorooctane Sulfonate Repress Osteogenic and Enhance Adipogenic Differentiation of Human Mesenchymal Stem Cells. Toxicol. Appl. Pharmacol. 2019, 367, 82–91. [Google Scholar] [CrossRef]
- Yamamoto, J.; Yamane, T.; Oishi, Y.; Kobayashi-Hattori, K. Perfluorooctanoic Acid Binds to Peroxisome Proliferator-Activated Receptor γ and Promotes Adipocyte Differentiation in 3T3-L1 Adipocytes. Biosci. Biotechnol. Biochem. 2015, 79, 636–639. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, J.; Wan, Y.; Peng, Y.; Ding, S.; Li, Y.; Xu, B.; Chen, X.; Xia, W.; Ke, Y.; et al. Low-Level Perfluorooctanoic Acid Enhances 3 T3-L1 Preadipocyte Differentiation via Altering Peroxisome Proliferator Activated Receptor Gamma Expression and Its Promoter DNA Methylation. J. Appl. Toxicol. 2018, 38, 398–407. [Google Scholar] [CrossRef]
- Li, Z.; Yu, Z.; Gao, P.; Yin, D. Multigenerational Effects of Perfluorooctanoic Acid on Lipid Metabolism of Caenorhabditis Elegans and Its Potential Mechanism. Sci. Total Environ. 2020, 703, 134762. [Google Scholar] [CrossRef]
- Van Esterik, J.C.J.; Sales, L.B.; Dollé, M.E.T.; Håkansson, H.; Herlin, M.; Legler, J.; van der Ven, L.T.M. Programming of Metabolic Effects in C57BL/6JxFVB Mice by in Utero and Lactational Exposure to Perfluorooctanoic Acid. Arch. Toxicol. 2016, 90, 701–715. [Google Scholar] [CrossRef]
- Xu, J.; Shimpi, P.; Armstrong, L.; Salter, D.; Slitt, A.L. PFOS Induces Adipogenesis and Glucose Uptake in Association with Activation of Nrf2 Signaling Pathway. Toxicol. Appl. Pharmacol. 2016, 290, 21–30. [Google Scholar] [CrossRef]
- Du, G.; Sun, J.; Zhang, Y. Perfluorooctanoic Acid Impaired Glucose Homeostasis through Affecting Adipose AKT Pathway. Cytotechnology 2018, 70, 479–487. [Google Scholar] [CrossRef]
- Gao, Y.; Guo, X.; Wang, S.; Chen, F.; Ren, X.; Xiao, H.; Wang, L. Perfluorooctane Sulfonate Enhances MRNA Expression of PPARγ and Ap2 in Human Mesenchymal Stem Cells Monitored by Long-Retained Intracellular Nanosensor. Environ. Pollut. 2020, 263, 114571. [Google Scholar] [CrossRef]
- Lu, Z.; Yifan, P.; Hui, Q.; Jiangyu, Z.; Wei, L. Interference of Perfluorooctane Sulfonate (PFOS) on PPARs Subtypes and Differentiation Potential in Human Bone Marrow Mesenchymal Stem Cells. Asian J. Ecotoxicol. 2021, 2, 151–157. [Google Scholar] [CrossRef]
- Van den Dungen, M.W.; Murk, A.J.; Kok, D.E.; Steegenga, W.T. Persistent Organic Pollutants Alter DNA Methylation during Human Adipocyte Differentiation. Toxicol. In Vitro 2017, 40, 79–87. [Google Scholar] [CrossRef]
- Qi, W.; Clark, J.M.; Timme-Laragy, A.R.; Park, Y. Perfluorobutanesulfonic Acid (PFBS) Potentiates Adipogenesis of 3T3-L1 Adipocytes. Food Chem. Toxicol. 2018, 120, 340–345. [Google Scholar] [CrossRef]
- Li, C.H.; Ren, X.M.; Ruan, T.; Cao, L.Y.; Xin, Y.; Guo, L.H.; Jiang, G. Chlorinated Polyfluorinated Ether Sulfonates Exhibit Higher Activity toward Peroxisome Proliferator-Activated Receptors Signaling Pathways than Perfluorooctanesulfonate. Environ. Sci. Technol. 2018, 52, 3232–3239. [Google Scholar] [CrossRef]
- Xie, Y.; Berntsen, H.F.; Zimmer, K.E.; Ropstad, E.; Verhaegen, S.; Connolly, L. Lipogenic Potency of Individual Perfluorinated Alkyl Acids (PFAAs) and Persistent Organic Pollutant (POP) Mixtures at Human Blood-Based Exposure Levels on Adipogenesis in 3T3-L1 Cells. Expo. Health 2021. [Google Scholar] [CrossRef]
- Ćurčić, M.; Janković, S.; Jaćević, V.; Stanković, S.; Vučinić, S.; Durgo, K.; Bulat, Z.; Antonijević, B. Combined Effects of Cadmium and Decabrominated Diphenyl Ether on Thyroid Hormones in Rats. Arhiv za Higijenu Rada i Toksikologiju 2012, 63, 255–262. [Google Scholar] [CrossRef]
- Yang, C.; Kong, A.P.S.; Cai, Z.; Chung, A.C.K. Persistent Organic Pollutants as Risk Factors for Obesity and Diabetes. Curr. Diabetes Rep. 2017, 17, 132. [Google Scholar] [CrossRef]
- Llm, J.S.; Lee, D.H.; Jacobs, D.R. Association of Brominated Flame Retardants with Diabetes and Metabolic Syndrome in the U.S. Population, 2003–2004. Diabetes Care 2008, 31, 1802–1807. [Google Scholar] [CrossRef]
- Helaleh, M.; Diboun, I.; Al-Tamimi, N.; Al-Sulaiti, H.; Al-Emadi, M.; Madani, A.; Mazloum, N.A.; Latiff, A.; Elrayess, M.A. Association of Polybrominated Diphenyl Ethers in Two Fat Compartments with Increased Risk of Insulin Resistance in Obese Individuals. Chemosphere 2018, 209, 268–276. [Google Scholar] [CrossRef]
- Erkin-Cakmak, A.; Harley, K.G.; Chevrier, J.; Bradman, A.; Kogut, K.; Huen, K.; Eskenazi, B. In Utero and Childhood Polybrominated Diphenyl Ether Exposures and Body Mass at Age 7 Years: The CHAMACOS Study. Environ. Health Perspect. 2015, 123, 636–642. [Google Scholar] [CrossRef]
- Pereira-Fernandes, A.; Dirinck, E.; Dirtu, A.C.; Malarvannan, G.; Covaci, A.; Van Gaal, L.; Vanparys, C.; Jorens, P.G.; Blust, R. Expression of Obesity Markers and Persistent Organic Pollutants Levels in Adipose Tissue of Obese Patients: Reinforcing the Obesogen Hypothesis? PLoS ONE 2014, 9, 84816. [Google Scholar] [CrossRef]
- Tung, E.W.Y.; Boudreau, A.; Wade, M.G.; Atlas, E. Induction of Adipocyte Differentiation by Polybrominated Diphenyl Ethers (PBDEs) in 3T3-L1 Cells. PLoS ONE 2014, 9, e94583. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Akinbo, S.; Slitt, A.L. 2,2′,4,4′,5-Pentabromodiphenyl Ether Induces Lipid Accumulation throughout Differentiation in 3T3-L1 and Human Preadipocytes in Vitro. J. Biochem. Mol. Toxicol. 2020, 34, e22485. [Google Scholar] [CrossRef]
- Wen, Q.; Xie, X.; Zhao, C.; Ren, Q.; Zhang, X.; Wei, D.; Emanuelli, B.; Du, Y. The Brominated Flame Retardant PBDE 99 Promotes Adipogenesis via Regulating Mitotic Clonal Expansion and PPARγ Expression. Sci. Total Environ. 2019, 670, 67–77. [Google Scholar] [CrossRef]
- Kamstra, J.H.; Hruba, E.; Blumberg, B.; Janesick, A.; Mandrup, S.; Hamers, T.; Legler, J. Transcriptional and Epigenetic Mechanisms Underlying Enhanced in Vitro Adipocyte Differentiation by the Brominated Flame Retardant BDE-47. Environ. Sci. Technol. 2014, 48, 4110–4119. [Google Scholar] [CrossRef]
- Yang, C.; Wong, C.M.; Wei, J.; Chung, A.C.K.; Cai, Z. The Brominated Flame Retardant BDE 47 Upregulates Purine Metabolism and Mitochondrial Respiration to Promote Adipocyte Differentiation. Sci. Total Environ. 2018, 644, 1312–1322. [Google Scholar] [CrossRef]
- Yang, C.; Zhu, L.; Kang, Q.; Lee, H.K.; Li, D.; Chung, A.C.K.; Cai, Z. Chronic Exposure to Tetrabromodiphenyl Ether (BDE-47) Aggravates Hepatic Steatosis and Liver Fibrosis in Diet-Induced Obese Mice. J. Hazard. Mater. 2019, 378, 120766. [Google Scholar] [CrossRef]
- Yang, C.; Wei, J.; Cao, G.; Cai, Z. Lipid Metabolism Dysfunction and Toxicity of BDE-47 Exposure in White Adipose Tissue Revealed by the Integration of Lipidomics and Metabolomics. Sci. Total Environ. 2022, 806, 150350. [Google Scholar] [CrossRef]
- Allgood, E.L.; Carey, G. The Effect of Diet and Polybrominated Diphenyl Ether Exposure on Adipocyte and Whole Body Metabolism in Male Wistar Rats. FASEB J. 2009, 23, 505.3. [Google Scholar] [CrossRef]
- Scoville, D.K.; Li, C.Y.; Wang, D.; Dempsey, J.L.; Raftery, D.; Mani, S.; Gu, H.; Cui, J.Y. Polybrominated Diphenyl Ethers and Gut Microbiome Modulate Metabolic Syndrome–Related Aqueous Metabolites in Mice. Drug Metab. Dispos. 2019, 47, 928–940. [Google Scholar] [CrossRef]
- Cano-Sancho, G.; Salmon, A.G.; La Merrill, M.A. Association between Exposure to p,p′-DDT and Its Metabolite p,p′-DDE with Obesity: Integrated Systematic Review and Meta-Analysis. Environ. Health Perspect. 2017, 125, 096002. [Google Scholar] [CrossRef]
- González-Casanova, J.E.; Pertuz-Cruz, S.L.; Caicedo-Ortega, N.H.; Rojas-Gomez, D.M. Adipogenesis Regulation and Endocrine Disruptors: Emerging Insights in Obesity. BioMed Res. Int. 2020, 2020, 7453786. [Google Scholar] [CrossRef]
- Arrebola, J.P.; Ocaña-Riola, R.; Arrebola-Moreno, A.L.; Fernández-Rodríguez, M.; Martin-Olmedo, P.; Fernández, M.F.; Olea, N. Associations of Accumulated Exposure to Persistent Organic Pollutants with Serum Lipids and Obesity in an Adult Cohort from Southern Spain. Environ. Pollut. 2014, 195, 9–15. [Google Scholar] [CrossRef]
- Dhooge, W.; Den Hond, E.; Koppen, G.; Bruckers, L.; Nelen, V.; Van De Mieroop, E.; Bilau, M.; Croes, K.; Baeyens, W.; Schoeters, G.; et al. Internal Exposure to Pollutants and Body Size in Flemish Adolescents and Adults: Associations and Dose–Response Relationships. Environ. Int. 2010, 36, 330–337. [Google Scholar] [CrossRef]
- Heggeseth, B.; Harley, K.; Warner, M.; Jewell, N.; Eskenazi, B. Detecting Associations between Early-Life DDT Exposures and Childhood Growth Patterns: A Novel Statistical Approach. PLoS ONE 2015, 10, e0131443. [Google Scholar] [CrossRef]
- Warner, M.; Schall, R.A.; Harley, K.G.; Bradman, A.; Barr, D.; Eskenazi, B. In Utero DDT and DDE Exposure and Obesity Status of 7-Year-Old Mexican-American Children in the CHAMACOS Cohort. Environ. Health Perspect. 2013, 121, 631–636. [Google Scholar] [CrossRef]
- Warner, M.; Wesselink, A.; Harley, K.G.; Bradman, A.; Kogut, K.; Eskenazi, B. Prenatal Exposure to Dichlorodiphenyltrichloroethane and Obesity at 9 Years of Age in the CHAMACOS Study Cohort. Am. J. Epidemiol. 2014, 179, 1312–1322. [Google Scholar] [CrossRef]
- Warner, M.; Ye, M.; Harley, K.; Kogut, K.; Bradman, A.; Eskenazi, B. Prenatal DDT Exposure and Child Adiposity at Age 12: The CHAMACOS Study. Environ. Res. 2017, 159, 606–612. [Google Scholar] [CrossRef]
- La Merrill, M.A.; Krigbaum, N.Y.; Cirillo, P.M.; Cohn, B.A. Association between Maternal Exposure to the Pesticide Dichlorodiphenyltrichloroethane (DDT) and Risk of Obesity in Middle Age. Int. J. Obes. 2020, 44, 1723–1732. [Google Scholar] [CrossRef]
- Verhulst, S.L.; Nelen, V.; Hond, E.D.; Koppen, G.; Beunckens, C.; Vael, C.; Schoeters, G.; Desager, K. Intrauterine Exposure to Environmental Pollutants and Body Mass Index during the First 3 Years of Life. Environ. Health Perspect. 2009, 117, 122–126. [Google Scholar] [CrossRef]
- Henríquez-Hernández, L.A.; Luzardo, O.P.; Valerón, P.F.; Zumbado, M.; Serra-Majem, L.; Camacho, M.; González-Antuña, A.; Boada, L.D. Persistent Organic Pollutants and Risk of Diabetes and Obesity on Healthy Adults: Results from a Cross-Sectional Study in Spain. Sci. Total Environ. 2017, 607–608, 1096–1102. [Google Scholar] [CrossRef]
- Tawar, N.; Banerjee, B.; Mishra, B.; Sharma, T.; Tyagi, S.; Madhu, S.; Agarwal, V.; Gupta, S. Adipose Tissue Levels of DDT as Risk Factor for Obesity and Type 2 Diabetes Mellitus. Indian J. Endocrinol. Metab. 2021, 25, 160–165. [Google Scholar] [CrossRef]
- Kim, J.; Sun, Q.; Yue, Y.; Yoon, K.S.; Whang, K.-Y.; Marshall Clark, J.; Park, Y. 4,4′-Dichlorodiphenyltrichloroethane (DDT) and 4,4′-Dichlorodiphenyldichloroethylene (DDE) Promote Adipogenesis in 3T3-L1 Adipocyte Cell Culture. Pestic. Biochem. Physiol. 2016, 131, 40–45. [Google Scholar] [CrossRef]
- Moreno-Aliaga, M.J.; Matsumura, F. Effects of 1,1,1-Trichloro-2,2-Bis(p-Chlorophenyl)-Ethane (p,P′-DDT) on 3T3-L1 and 3T3-F442A Adipocyte Differentiation. Biochem. Pharmacol. 2002, 63, 997–1007. [Google Scholar] [CrossRef]
- Pesta, M.; Cedikova, M.; Dvorak, P.; Dvorakova, J.; Kulda, V.; Srbecka, K.; Muller, L.; Bouchalova, V.; Kralickova, M.; Babuska, V.; et al. Trends in Gene Expression Changes during Adipogenesis in Human Adipose Derived Mesenchymal Stem Cells under Dichlorodiphenyldichloroethylene Exposure. Mol. Cell. Toxicol. 2018, 14, 369–379. [Google Scholar] [CrossRef]
- Strong, A.L.; Shi, Z.; Strong, M.J.; Miller, D.F.B.; Rusch, D.B.; Buechlein, A.M.; Flemington, E.K.; McLachlan, J.A.; Nephew, K.P.; Burow, M.E.; et al. Effects of the Endocrine-Disrupting Chemical DDT on Self-Renewal and Differentiation of Human Mesenchymal Stem Cells. Environ. Health Perspect. 2015, 123, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Mangum, L.H.; Howell, G.E.; Chambers, J.E. Exposure to p,p′-DDE Enhances Differentiation of 3T3-L1 Preadipocytes in a Model of Sub-Optimal Differentiation. Toxicol. Lett. 2015, 238, 65–71. [Google Scholar] [CrossRef]
- King, S.E.; Nilsson, E.; Beck, D.; Skinner, M.K. Adipocyte Epigenetic Alterations and Potential Therapeutic Targets in Transgenerationally Inherited Lean and Obese Phenotypes Following Ancestral Exposures. Adipocyte 2019, 8, 362–378. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.; Mangum, L. Exposure to Bioaccumulative Organochlorine Compounds Alters Adipogenesis, Fatty Acid Uptake, and Adipokine Production in NIH3T3-L1 Cells. Toxicol. In Vitro 2011, 25, 394–402. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.; Karey, E.; Moshier, E.; Lindtner, C.; La Frano, M.R.; Newman, J.W.; Buettner, C. Perinatal Exposure of Mice to the Pesticide DDT Impairs Energy Expenditure and Metabolism in Adult Female Offspring. PLoS ONE 2014, 9, 103337. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Murinova, L.; Trnovec, T.; Loffredo, C.; Washington, K.; Mitra, P.; Dutta, S. Biomarkers Linking PCB Exposure and Obesity. Curr. Pharm. Biotechnol. 2014, 15, 1058–1068. [Google Scholar] [CrossRef]
- Buha Djordjevic, A.; Antonijevic, E.; Curcic, M.; Milovanovic, V.; Antonijevic, B. Endocrine-Disrupting Mechanisms of Polychlorinated Biphenyls. Curr. Opin. Toxicol. 2020, 19, 42–49. [Google Scholar] [CrossRef]
- Buha, A.; Antonijević, B.; Bulat, Z.; Jaćević, V.; Milovanović, V.; Matović, V. The Impact of Prolonged Cadmium Exposure and Co-Exposure with Polychlorinated Biphenyls on Thyroid Function in Rats. Toxicol. Lett. 2013, 221, 83–90. [Google Scholar] [CrossRef]
- Domazet, S.L.; Grøntved, A.; Jensen, T.K.; Wedderkopp, N.; Andersen, L.B. Higher Circulating Plasma Polychlorinated Biphenyls (PCBs) in Fit and Lean Children: The European Youth Heart Study. Environ. Int. 2020, 136, 105481. [Google Scholar] [CrossRef]
- Warner, M.; Rauch, S.; Coker, E.S.; Harley, K.; Kogut, K.; Sjödin, A.; Eskenazi, B. Obesity in Relation to Serum Persistent Organic Pollutant Concentrations in CHAMACOS Women. Environ. Epidemiol. 2018, 2, e032. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, I.K.; Porta, M.; Steffes, M.; Jacobs, D.R. Relationship between Serum Concentrations of Persistent Organic Pollutants and the Prevalence of Metabolic Syndrome among Non-Diabetic Adults: Results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 2007, 50, 1841–1851. [Google Scholar] [CrossRef]
- Donat-Vargas, C.; Gea, A.; Sayon-Orea, C.; Carlos, S.; Martinez-Gonzalez, M.A.; Bes-Rastrollo, M. Association between Dietary Intakes of PCBs and the Risk of Obesity: The SUN Project. J. Epidemiol. Community Health 2014, 68, 834–841. [Google Scholar] [CrossRef]
- Lee, D.H.; Lind, L.; Jacobs, D.R.; Salihovic, S.; van Bavel, B.; Lind, P.M. Associations of Persistent Organic Pollutants with Abdominal Obesity in the Elderly: The Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) Study. Environ. Int. 2012, 40, 170–178. [Google Scholar] [CrossRef]
- Valvi, D.; Mendez, M.A.; Martinez, D.; Grimalt, J.O.; Torrent, M.; Sunyer, J.; Vrijheid, M. Prenatal Concentrations of Polychlorinated Biphenyls, DDE, and DDT and Overweight in Children: A Prospective Birth Cohort Study. Environ. Health Perspect. 2012, 120, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Steffes, M.W.; Sjödin, A.; Jones, R.S.; Needham, L.L.; Jacobs, D.R. Low Dose Organochlorine Pesticides and Polychlorinated Biphenyls Predict Obesity, Dyslipidemia, and Insulin Resistance among People Free of Diabetes. PLoS ONE 2011, 6, e15977. [Google Scholar] [CrossRef] [PubMed]
- Hertz-Picciotto, I.; Charles, M.J.; James, R.A.; Keller, J.A.; Willman, E.; Teplin, S. In Utero Polychlorinated Biphenyl Exposures in Relation to Fetal and Early Childhood Growth. Epidemiology 2005, 16, 648–656. [Google Scholar] [CrossRef]
- Hassan, W.; Ahmed, H.; Murtaza, G.; Umar, M.I.; Iqbal, F.M. Role of Polychlorinated Biphenyls as EDCs in Metabolic Disorders; Springer: Cham, Switzerland, 2021; pp. 161–174. [Google Scholar]
- Bourez, S.; Le Lay, S.; Van den Daelen, C.; Louis, C.; Larondelle, Y.; Thomé, J.-P.; Schneider, Y.-J.; Dugail, I.; Debier, C. Accumulation of Polychlorinated Biphenyls in Adipocytes: Selective Targeting to Lipid Droplets and Role of Caveolin-1. PLoS ONE 2012, 7, e31834. [Google Scholar] [CrossRef]
- Bourez, S.; Van den Daelen, C.; Le Lay, S.; Poupaert, J.; Larondelle, Y.; Thomé, J.-P.; Schneider, Y.-J.; Dugail, I.; Debier, C. The Dynamics of Accumulation of PCBs in Cultured Adipocytes Vary with the Cell Lipid Content and the Lipophilicity of the Congener. Toxicol. Lett. 2013, 216, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Arsenescu, V.; Arsenescu, R.I.; King, V.; Swanson, H.; Cassis, L.A. Polychlorinated Biphenyl-77 Induces Adipocyte Differentiation and Proinflammatory Adipokines and Promotes Obesity and Atherosclerosis. Environ. Health Perspect. 2008, 116, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Taxvig, C.; Dreisig, K.; Boberg, J.; Nellemann, C.; Schelde, A.B.; Pedersen, D.; Boergesen, M.; Mandrup, S.; Vinggaard, A.M. Differential Effects of Environmental Chemicals and Food Contaminants on Adipogenesis, Biomarker Release and PPARγ Activation. Mol. Cell. Endocrinol. 2012, 361, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wu, G.; Cheng, X.; Fan, J.; Peng, J.; Su, H.; Xu, Z.; Cao, M.; Long, Z.; Hao, Y.; et al. Oleanolic Acid Attenuates PCBs-Induced Adiposity and Insulin Resistance via HNF1b-Mediated Regulation of Redox and PPARγ Signaling. Free Radic. Biol. Med. 2018, 124, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Wen, Q.; Ren, Q.; Du, Y.; Xie, X. Polychlorinated Biphenyl Congener 180 (PCB 180) Regulates Mitotic Clonal Expansion and Enhances Adipogenesis through Modulation of C/EBPβ SUMOylation in Preadipocytes. Food Chem. Toxicol. 2021, 152, 112205. [Google Scholar] [CrossRef]
- Baker, N.A.; Karounos, M.; English, V.; Fang, J.; Wei, Y.; Stromberg, A.; Sunkara, M.; Morris, A.J.; Swanson, H.I.; Cassis, L.A. Coplanar Polychlorinated Biphenyls Impair Glucose Homeostasis in Lean C57BL/6 Mice and Mitigate Beneficial Effects of Weight Loss on Glucose Homeostasis in Obese Mice. Environ. Health Perspect. 2013, 121, 105–110. [Google Scholar] [CrossRef]
- Gourronc, F.A.; Robertson, L.W.; Klingelhutz, A.J. A Delayed Proinflammatory Response of Human Preadipocytes to PCB126 Is Dependent on the Aryl Hydrocarbon Receptor. Environ. Sci. Pollut. Res. 2018, 25, 16481–16492. [Google Scholar] [CrossRef]
- Wu, H.; Yu, W.; Meng, F.; Mi, J.; Peng, J.; Liu, J.; Zhang, X.; Hai, C.; Wang, X. Polychlorinated Biphenyls-153 Induces Metabolic Dysfunction through Activation of ROS/NF-ΚB Signaling via Downregulation of HNF1b. Redox Biol. 2017, 12, 300–310. [Google Scholar] [CrossRef]
- Kim, Y.A.; Kim, H.Y.; Oh, Y.J.; Kwon, W.Y.; Lee, M.H.; Bae, J.Y.; Woo, M.S.; Kim, J.M.; Yoo, Y.H. Polychlorinated Biphenyl 138 Exposure-Mediated Lipid Droplet Enlargement Endows Adipocytes with Resistance to TNF-α-Induced Cell Death. Toxicol. Lett. 2018, 292, 55–62. [Google Scholar] [CrossRef]
- Baker, N.A.; English, V.; Sunkara, M.; Morris, A.J.; Pearson, K.J.; Cassis, L.A. Resveratrol Protects against Polychlorinated Biphenyl-Mediated Impairment of Glucose Homeostasis in Adipocytes. J. Nutr. Biochem. 2013, 24, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Gourronc, F.A.; Perdew, G.H.; Robertson, L.W.; Klingelhutz, A.J. PCB126 Blocks the Thermogenic Beiging Response of Adipocytes. Environ. Sci. Pollut. Res. 2020, 27, 8897–8904. [Google Scholar] [CrossRef]
- Kamata, R.; Nakajima, D.; Shiraishi, F. Measurement of the Agonistic Activities of Monohydroxylated Polychlorinated Biphenyls at the Retinoid X and Retinoic Acid Receptors Using Recombinant Yeast Cells. Toxicol. In Vitro 2019, 57, 9–17. [Google Scholar] [CrossRef]
- Wahlang, B.; Falkner, K.C.; Gregory, B.; Ansert, D.; Young, D.; Conklin, D.J.; Bhatnagar, A.; McClain, C.J.; Cave, M. Polychlorinated Biphenyl 153 Is a Diet-Dependent Obesogen That Worsens Nonalcoholic Fatty Liver Disease in Male C57BL6/J Mice. J. Nutr. Biochem. 2013, 24, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Huang, Y.J.; Gao, S.; Chen, L.Q.; Zhang, M.L.; Du, Z.Y. Sex-Specific Alterations of Lipid Metabolism in Zebrafish Exposed to Polychlorinated Biphenyls. Chemosphere 2019, 221, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Scinicariello, F.; Buser, M.C. Urinary Polycyclic Aromatic Hydrocarbons and Childhood Obesity: NHANES (2001–2006). Environ. Health Perspect. 2014, 122, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Poursafa, P.; Dadvand, P.; Amin, M.M.; Hajizadeh, Y.; Ebrahimpour, K.; Mansourian, M.; Pourzamani, H.; Sunyer, J.; Kelishadi, R. Association of Polycyclic Aromatic Hydrocarbons with Cardiometabolic Risk Factors and Obesity in Children. Environ. Int. 2018, 118, 203–210. [Google Scholar] [CrossRef]
- Kim, H.W.; Kam, S.; Lee, D.H. Synergistic Interaction between Polycyclic Aromatic Hydrocarbons and Environmental Tobacco Smoke on the Risk of Obesity in Children and Adolescents: The U.S. National Health and Nutrition Examination Survey 2003-2008. Environ. Res. 2014, 135, 354–360. [Google Scholar] [CrossRef]
- Yin, W.; Hou, J.; Xu, T.; Cheng, J.; Li, P.; Wang, L.; Zhang, Y.; Wang, X.; Hu, C.; Huang, C.; et al. Obesity Mediated the Association of Exposure to Polycyclic Aromatic Hydrocarbon with Risk of Cardiovascular Events. Sci. Total Environ. 2018, 616–617, 841–854. [Google Scholar] [CrossRef]
- Hou, J.; Sun, H.; Huang, X.; Zhou, Y.; Zhang, Y.; Yin, W.; Xu, T.; Cheng, J.; Chen, W.; Yuan, J. Exposure to Polycyclic Aromatic Hydrocarbons and Central Obesity Enhanced Risk for Diabetes among Individuals with Poor Lung Function. Chemosphere 2017, 185, 1136–1143. [Google Scholar] [CrossRef]
- Li, K.; Yin, R.; Wang, Y.; Zhao, D. Associations between Exposure to Polycyclic Aromatic Hydrocarbons and Metabolic Syndrome in U.S. Adolescents: Cross-Sectional Results from the National Health and Nutrition Examination Survey (2003–2016) Data. Environ. Res. 2021, 202, 111747. [Google Scholar] [CrossRef]
- May, P.; Bremond, P.; Sauzet, C.; Piccerelle, P.; Grimaldi, F.; Champion, S.; Villard, P.-H. In Vitro Cocktail Effects of PCB-DL (PCB118) and Bulky PCB (PCB153) with BaP on Adipogenesis and on Expression of Genes Involved in the Establishment of a Pro-Inflammatory State. Int. J. Mol. Sci. 2018, 19, 841. [Google Scholar] [CrossRef]
- Podechard, N.; Fardel, O.; Corolleur, M.; Bernard, M.; Lecureur, V. Inhibition of Human Mesenchymal Stem Cell-Derived Adipogenesis by the Environmental Contaminant Benzo(a)Pyrene. Toxicol. In Vitro 2009, 23, 1139–1144. [Google Scholar] [CrossRef]
- Rathore, K.; Cekanova, M. Effects of Environmental Carcinogen Benzo(a)Pyrene on Canine Adipose-Derived Mesenchymal Stem Cells. Res. Vet. Sci. 2015, 103, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhang, H.; Maher, C.; Arteaga-Solis, E.; Champagne, F.A.; Wu, L.; McDonald, J.D.; Yan, B.; Schwartz, G.J.; Miller, R.L. Prenatal Polycyclic Aromatic Hydrocarbon, Adiposity, Peroxisome Proliferator-Activated Receptor (PPAR) γ Methylation in Offspring, Grand-Offspring Mice. PLoS ONE 2014, 9, e110706. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, L.; Nakamura, B.; Li, X.; Blumberg, B.; Luderer, U. In Utero Exposure to Benzo[a]Pyrene Increases Adiposity and Causes Hepatic Steatosis in Female Mice, and Glutathione Deficiency Is Protective. Toxicol. Lett. 2013, 223, 260–267. [Google Scholar] [CrossRef]
- Gato, W.E.; Hunter, D.A.; Whitby, S.L.; Mays, C.A.; Yau, W. Investigating Susceptibility to Diabetes Using Features of the Adipose Tissue in Response to In Utero Polycyclic Aromatic Hydrocarbons Exposure. Diabetes Metab. J. 2016, 40, 494. [Google Scholar] [CrossRef]
- Irigaray, P.; Lacomme, S.; Mejean, L.; Belpomme, D. Ex Vivo Study of Incorporation into Adipocytes and Lipolysis-Inhibition Effect of Polycyclic Aromatic Hydrocarbons. Toxicol. Lett. 2009, 187, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Irigaray, P.; Ogier, V.; Jacquenet, S.; Notet, V.; Sibille, P.; Mejean, L.; Bihain, B.E.; Yen, F.T. Benzo[a]Pyrene Impairs Beta-Adrenergic Stimulation of Adipose Tissue Lipolysis and Causes Weight Gain in Mice. A Novel Molecular Mechanism of Toxicity for a Common Food Pollutant. FEBS J. 2006, 273, 1362–1372. [Google Scholar] [CrossRef]
- Guo, J.; Huang, J.; Wang, Q.; Fang, L.; Zhang, S.; Li, B.; Lv, L.; Chen, M.; Wang, C. Maternal Exposure to Phenanthrene during Gestation Disturbs Glucose Homeostasis in Adult Mouse Offspring. Chemosphere 2021, 270, 128635. [Google Scholar] [CrossRef]
- Kim, Y.H.; Lee, Y.S.; Lee, D.H.; Kim, D.S. Polycyclic Aromatic Hydrocarbons Are Associated with Insulin Receptor Substrate 2 Methylation in Adipose Tissues of Korean Women. Environ. Res. 2016, 150, 47–51. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Peng, S.H.; Chou, M.J. Obesity Enhances Carcinogen 7, 12-Dimethylbenz [a] Anthracene -Induced Tumorigenesis in Vitro and in Vivo. Food Chem. Toxicol. 2017, 110, 156–164. [Google Scholar] [CrossRef]
- Warner, M.; Mocarelli, P.; Brambilla, P.; Wesselink, A.; Samuels, S.; Signorini, S.; Eskenazi, B. Diabetes, Metabolic Syndrome, and Obesity in Relation to Serum Dioxin Concentrations: The Seveso Women’s Health Study. Environ. Health Perspect. 2013, 121, 906–911. [Google Scholar] [CrossRef]
- Chang, J.-W.; Chen, H.-L.; Su, H.-J.; Lee, C.-C. Abdominal Obesity and Insulin Resistance in People Exposed to Moderate-to-High Levels of Dioxin. PLoS ONE 2016, 11, e0145818. [Google Scholar] [CrossRef] [PubMed]
- Uemura, H.; Arisawa, K.; Hiyoshi, M.; Kitayama, A.; Takami, H.; Sewachika, F.; Dakeshita, S.; Nii, K.; Satoh, H.; Sumiyoshi, Y.; et al. Prevalence of Metabolic Syndrome Associated with Body Burden Levels of Dioxin and Related Compounds among Japan’s General Population. Environ. Health Perspect. 2009, 117, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.; Chen, H.L.; Su, H.J.; Liao, P.C.; Guo, H.R.; Lee, C.C. Dioxin Exposure and Insulin Resistance in Taiwanese Living near a Highly Contaminated Area. Epidemiology 2010, 21, 56–61. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.; Birnbaum, L.S. Childhood Obesity and Environmental Chemicals. Mt. Sinai J. Med. J. Transl. Personal. Med. 2011, 78, 22–48. [Google Scholar] [CrossRef] [PubMed]
- Girer, N.G.; Tomlinson, C.R.; Elferink, C.J. The Aryl Hydrocarbon Receptor in Energy Balance: The Road from Dioxin-Induced Wasting Syndrome to Combating Obesity with AHR Ligands. Int. J. Mol. Sci. 2021, 22, 49. [Google Scholar] [CrossRef]
- Hsu, H.F.; Tsou, T.C.; Chao, H.R.; Kuo, Y.T.; Tsai, F.Y.; Yeh, S.C. Effects of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on Adipogenic Differentiation and Insulin-Induced Glucose Uptake in 3T3-L1 Cells. J. Hazard. Mater. 2010, 182, 649–655. [Google Scholar] [CrossRef]
- Hanlon, P.R.; Cimafranca, M.A.; Liu, X.; Cho, Y.C.; Jefcoate, C.R. Microarray Analysis of Early Adipogenesis in C3H10T1/2 Cells: Cooperative Inhibitory Effects of Growth Factors and 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Toxicol. Appl. Pharmacol. 2005, 207, 39–58. [Google Scholar] [CrossRef]
- Choi, E.M.; Suh, K.S.; Jung, W.W.; Park, S.Y.; Chin, S.O.; Rhee, S.Y.; Kim Pak, Y.; Chon, S. Glabridin Attenuates Antiadipogenic Activity Induced by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Murine 3T3-L1 Adipocytes. J. Appl. Toxicol. 2018, 38, 1426–1436. [Google Scholar] [CrossRef]
- Hanlon, P.R.; Ganem, L.G.; Cho, Y.C.; Yamamoto, M.; Jefcoate, C.R. AhR- and ERK-Dependent Pathways Function Synergistically to Mediate 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Suppression of Peroxisome Proliferator-Activated Receptor-Γ1 Expression and Subsequent Adipocyte Differentiation. Toxicol. Appl. Pharmacol. 2003, 189, 11–27. [Google Scholar] [CrossRef]
- Liu, P.C.C.; Phillips, M.A.; Matsumura, F. Alteration by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin of CCAAT/Enhancer Binding Protein Correlates with Suppression of Adipocyte Differentiation in 3T3-L1 Cells. Mol. Pharmacol. 1996, 49, 989–997. [Google Scholar]
- Nishiumi, S.; Yabushita, Y.; Furuyashiki, T.; Fukuda, I.; Ashida, H. Involvement of SREBPs in 2,3,7,8-Tetrachlorodibenzo-p-Dioxin-Induced Disruption of Lipid Metabolism in Male Guinea Pig. Toxicol. Appl. Pharmacol. 2008, 229, 281–289. [Google Scholar] [CrossRef]
- Brewster, D.W. Fumio Matsumura Reduction of Adipose Tissue Lipoprotein Lipase Activity as a Result of in Vivo Administration of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin to the Guinea Pig. Biochem. Pharmacol. 1988, 37, 2247–2253. [Google Scholar] [CrossRef]
- Brulport, A.; Le Corre, L.; Chagnon, M.C. Chronic Exposure of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Induces an Obesogenic Effect in C57BL/6J Mice Fed a High Fat Diet. Toxicology 2017, 390, 43–52. [Google Scholar] [CrossRef]
- Duval, C.; Teixeira-Clerc, F.; Leblanc, A.F.; Touch, S.; Emond, C.; Guerre-Millo, M.; Lotersztajn, S.; Barouki, R.; Aggerbeck, M.; Coumoul, X. Chronic Exposure to Low Doses of Dioxin Promotes Liver Fibrosis Development in the C57BL/6J Diet-Induced Obesity Mouse Model. Environ. Health Perspect. 2017, 125, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Hoyeck, M.P.; Merhi, R.C.; Blair, H.L.; Spencer, C.D.; Payant, M.A.; Martin Alfonso, D.I.; Zhang, M.; Matteo, G.; Chee, M.J.; Bruin, J.E. Female Mice Exposed to Low Doses of Dioxin during Pregnancy and Lactation Have Increased Susceptibility to Diet-Induced Obesity and Diabetes. Mol. Metab. 2020, 42, 101104. [Google Scholar] [CrossRef]
- Matteo, G.; Hoyeck, M.P.; Blair, H.L.; Zebarth, J.; Rick, K.R.C.; Williams, A.; Gagné, R.; Buick, J.K.; Yauk, C.L.; Bruin, J.E. Prolonged Low-Dose Dioxin Exposure Impairs Metabolic Adaptability to High-Fat Diet Feeding in Female but Not Male Mice. Endocrinology 2021, 162, bqab050. [Google Scholar] [CrossRef]
- Baralić, K.; Živancevic, K.; Bozic, D.; Jennen, D.; Buha Djordjevic, A.; Antonijevic Miljakovic, E.; Đukic-Cosic, D. Potential Genomic Biomarkers of Obesity and Its Comorbidities for Phthalates and Bisphenol A Mixture: In Silico Toxicogenomic Approach. Biocell 2022, 46, 519–533. [Google Scholar] [CrossRef]
- Hong, S.H.; Sung, Y.A.; Hong, Y.S.; Ha, E.; Jeong, K.; Chung, H.; Lee, H. Urinary Bisphenol A Is Associated with Insulin Resistance and Obesity in Reproductive-Aged Women. Clin. Endocrinol. 2017, 86, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Park, Y.J.; Kim, M.J.; Kim, S.; Choi, S.; Park, J.; Cho, Y.H.; Hong, S.; Yoo, J.; Park, H.; et al. Associations of Urinary Concentrations of Phthalate Metabolites, Bisphenol A, and Parabens with Obesity and Diabetes Mellitus in a Korean Adult Population: Korean National Environmental Health Survey (KoNEHS) 2015–2017. Environ. Int. 2021, 146, 106227. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-e.; Choi, B.K.; Jee, S.H. Urinary Bisphenol A, Phthalate Metabolites, and Obesity: Do Gender and Menopausal Status Matter? Environ. Sci. Pollut. Res. 2020, 27, 34300–34310. [Google Scholar] [CrossRef]
- Carwile, J.L.; Michels, K.B. Urinary Bisphenol A and Obesity: NHANES 2003–2006. Environ. Res. 2011, 111, 825–830. [Google Scholar] [CrossRef]
- Amin, M.M.; Ebrahim, K.; Hashemi, M.; Shoshtari-Yeganeh, B.; Rafiei, N.; Mansourian, M.; Kelishadi, R. Association of Exposure to Bisphenol A with Obesity and Cardiometabolic Risk Factors in Children and Adolescents. Int. J. Environ. Health Res. 2019, 29, 94–106. [Google Scholar] [CrossRef]
- Li, D.K.; Miao, M.; Zhou, Z.J.; Wu, C.; Shi, H.; Liu, X.; Wang, S.; Yuan, W. Urine Bisphenol-A Level in Relation to Obesity and Overweight in School-Age Children. PLoS ONE 2013, 8, 11–13. [Google Scholar] [CrossRef]
- Wang, H.X.; Zhou, Y.; Tang, C.X.; Wu, J.G.; Chen, Y.; Jiang, Q.W. Association between Bisphenol A Exposure and Body Mass Index in Chinese School Children: A Cross-Sectional Study. Environ. Health Glob. Access Sci. Source 2012, 11, 79. [Google Scholar] [CrossRef]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol a Enhances Adipogenic Differentiation of Human Adipose Stromal/Stem Cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, B.; Hou, M.; Pan, X.; Li, X. The Environmental Obesogen Bisphenol A Promotes Adipogenesis by Increasing the Amount of 11β-Hydroxysteroid Dehydrogenase Type 1 in the Adipose Tissue of Children. Int. J. Obes. 2013, 37, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Atlas, E.; Pope, L.; Wade, M.G.; Kawata, A.; Boudreau, A.; Boucher, J.G. Bisphenol A Increases AP2 Expression in 3T3L1 by Enhancing the Transcriptional Activity of Nuclear Receptors at the Promoter. Adipocyte 2014, 3, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.G.; Boudreau, A.; Atlas, E. Bisphenol a Induces Differentiation of Human Preadipocytes in the Absence of Glucocorticoid and Is Inhibited by an Estrogen-Receptor Antagonist. Nutr. Diabetes 2014, 4, e102. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Nigro, C.; Oriente, F.; Formisano, P.; Miele, C.; Beguinot, F. Low-Dose Bisphenol-a Promotes Epigenetic Changes at Pparγ Promoter in Adipose Precursor Cells. Nutrients 2020, 12, 3498. [Google Scholar] [CrossRef]
- Masuno, H.; Iwanami, J.; Kidani, T.; Sakayama, K.; Honda, K. Bisphenol A Accelerates Terminal Differentiation of 3T3-L1 Cells into Adipocytes through the Phosphatidylinositol 3-Kinase Pathway. Toxicol. Sci. 2005, 84, 319–327. [Google Scholar] [CrossRef]
- Ariemma, F.; D’Esposito, V.; Liguoro, D.; Oriente, F.; Cabaro, S.; Liotti, A.; Cimmino, I.; Longo, M.; Beguinot, F.; Formisano, P.; et al. Low-Dose Bisphenol-A Impairs Adipogenesis and Generates Dysfunctional 3T3-L1 Adipocytes. PLoS ONE 2016, 11, e0150762. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, E.; Li, T.; Rosen, E.D. Exposure of Adipocytes to Bisphenol-A in Vitro Interferes with Insulin Action without Enhancing Adipogenesis. PLoS ONE 2018, 13, e0201122. [Google Scholar] [CrossRef]
- Valentino, R.; D’Esposito, V.; Passaretti, F.; Liotti, A.; Cabaro, S.; Longo, M.; Perruolo, G.; Oriente, F.; Beguinot, F.; Formisano, P. Bisphenol-A Impairs Insulin Action and up-Regulates Inflammatory Pathways in Human Subcutaneous Adipocytes and 3T3-L1 Cells. PLoS ONE 2013, 8, e82099. [Google Scholar] [CrossRef]
- Hugo, E.R.; Brandebourg, T.D.; Woo, J.G.; Loftus, J.; Alexander, J.W.; Ben-Jonathan, N. Bisphenol A at Environmentally Relevant Doses Inhibits Adiponectin Release from Human Adipose Tissue Explants and Adipocytes. Environ. Health Perspect. 2008, 116, 1642–1647. [Google Scholar] [CrossRef] [PubMed]
- Angle, B.M.; Do, R.P.; Ponzi, D.; Stahlhut, R.W.; Drury, B.E.; Nagel, S.C.; Welshons, W.V.; Besch-Williford, C.L.; Palanza, P.; Parmigiani, S.; et al. Metabolic Disruption in Male Mice Due to Fetal Exposure to Low but Not High Doses of Bisphenol A (BPA): Evidence for Effects on Body Weight, Food Intake, Adipocytes, Leptin, Adiponectin, Insulin and Glucose Regulation. Reprod. Toxicol. 2013, 42, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Somm, E.; Schwitzgebel, V.M.; Toulotte, A.; Cederroth, C.R.; Combescure, C.; Nef, S.; Aubert, M.L.; Hüppi, P.S. Perinatal Exposure to Bisphenol A Alters Early Adipogenesis in the Rat. Environ. Health Perspect. 2009, 117, 1549–1555. [Google Scholar] [CrossRef]
- Lejonklou, M.H.; Dunder, L.; Bladin, E.; Pettersson, V.; Rönn, M.; Lind, L.; Waldén, T.B.; Lind, P.M. Effects of Low-Dose Developmental Bisphenol a Exposure on Metabolic Parameters and Gene Expression in Male and Female Fischer 344 Rat Offspring. Environ. Health Perspect. 2017, 125, 6. [Google Scholar] [CrossRef]
- Yang, M.; Chen, M.; Wang, J.; Xu, M.; Sun, J.; Ding, L.; Lv, X.; Ma, Q.; Bi, Y.; Liu, R.; et al. Bisphenol a Promotes Adiposity and Inflammation in a Nonmonotonic Dose-Response Way in 5-Week-Old Male and Female C57BL/6J Mice Fed a Low-Calorie Diet. Endocrinology 2016, 157, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Ferrini, M.G.; Jellyman, J.K.; Han, G.; Michael, G.; Angeles, L.; Drew, C.R.; Angeles, L.; Angeles, L. In Vivo and In Vitro Bisphenol A Exposure Effects on Adiposity. J. Dev. Orig. Health Dis. 2019, 9, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Van Esterik, J.C.J.; Dollé, M.E.T.; Lamoree, M.H.; van Leeuwen, S.P.J.; Hamers, T.; Legler, J.; Van der Ven, L.T.M. Programming of Metabolic Effects in C57BL/6JxFVB Mice by Exposure to Bisphenol A during Gestation and Lactation. Toxicology 2014, 321, 40–52. [Google Scholar] [CrossRef]
- Boucher, J.G.; Boudreau, A.; Ahmed, S.; Atlas, E. In Vitro Effects of Bisphenol A β-D-Glucuronide (BPA-G) on Adipogenesis in Human and Murine Preadipocytes. Environ. Health Perspect. 2015, 123, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Chamorro-García, R.; Kirchner, S.; Li, X.; Janesick, A.; Casey, S.C.; Chow, C.; Blumberg, B. Bisphenol A Diglycidyl Ether Induces Adipogenic Differentiation of Multipotent Stromal Stem Cells through a Peroxisome Proliferator-Activated Receptor Gamma-Independent Mechanism. Environ. Health Perspect. 2012, 120, 984–989. [Google Scholar] [CrossRef]
- Martínez, M.; Blanco, J.; Rovira, J.; Kumar, V.; Domingo, J.L.; Schuhmacher, M. Bisphenol A Analogues (BPS and BPF) Present a Greater Obesogenic Capacity in 3T3-L1 Cell Line. Food Chem. Toxicol. 2020, 140, 111298. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.G.; Ahmed, S.; Atlas, E. Bisphenol S Induces Adipogenesis in Primary Human Preadipocytes from Female Donors. Endocrinology 2016, 157, 1397–1407. [Google Scholar] [CrossRef]
- Ahmed, S.; Atlas, E. Bisphenol S- and Bisphenol A-Induced Adipogenesis of Murine Preadipocytes Occurs through Direct Peroxisome Proliferator-Activated Receptor Gamma Activation. Int. J. Obes. 2016, 40, 1566–1573. [Google Scholar] [CrossRef]
- Boucher, J.G.; Gagné, R.; Rowan-Carroll, A.; Boudreau, A.; Yauk, C.L.; Atlas, E. Bisphenol A and Bisphenol S Induce Distinct Transcriptional Profiles in Differentiating Human Primary Preadipocytes. PLoS ONE 2016, 11, e0163318. [Google Scholar] [CrossRef]
- Ahn, Y.A.; Baek, H.; Choi, M.; Park, J.; Son, S.J.; Seo, H.J.; Jung, J.; Seong, J.K.; Lee, J.; Kim, S. Adipogenic Effects of Prenatal Exposure to Bisphenol S (BPS) in Adult F1 Male Mice. Sci. Total Environ. 2020, 728, 138759. [Google Scholar] [CrossRef]
- Skledar, D.G.; Carino, A.; Trontelj, J.; Troberg, J.; Distrutti, E.; Marchianò, S.; Tomašič, T.; Zega, A.; Finel, M.; Fiorucci, S.; et al. Endocrine Activities and Adipogenic Effects of Bisphenol AF and Its Main Metabolite. Chemosphere 2019, 215, 870–880. [Google Scholar] [CrossRef]
- Stojanoska, M.M.; Milosevic, N.; Milic, N.; Abenavoli, L. The Influence of Phthalates and Bisphenol A on the Obesity Development and Glucose Metabolism Disorders. Endocrine 2017, 55, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.M.; Ebrahimpour, K.; Parastar, S.; Shoshtari-Yeganeh, B.; Hashemi, M.; Mansourian, M.; Poursafa, P.; Fallah, Z.; Rafiei, N.; Kelishadi, R. Association of Urinary Concentrations of Phthalate Metabolites with Cardiometabolic Risk Factors and Obesity in Children and Adolescents. Chemosphere 2018, 211, 547–556. [Google Scholar] [CrossRef]
- Hatch, E.E.; Nelson, J.W.; Qureshi, M.M.; Weinberg, J.; Moore, L.L.; Singer, M.; Webster, T.F. Association of Urinary Phthalate Metabolite Concentrations with Body Mass Index and Waist Circumference: A Cross-Sectional Study of NHANES Data, 1999-2002. Environ. Health Glob. Access Sci. Source 2008, 7, 27. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, H.; Lee, J.; Cho, G.; Choi, S.; Choi, G.; Kim, S.Y.; Eun, S.H.; Suh, E.; Kim, S.K.; et al. Association of Diethylhexyl Phthalate with Obesityrelated Markers and Body Mass Change from Birth to 3 Months of Age. J. Epidemiol. Community Health 2016, 70, 466–472. [Google Scholar] [CrossRef]
- Mansouri, V.; Ebrahimpour, K.; Poursafa, P.; Riahi, R.; Shoshtari-Yeganeh, B.; Hystad, P.; Kelishadi, R. Exposure to Phthalates and Bisphenol A Is Associated with Higher Risk of Cardiometabolic Impairment in Normal Weight Children. Environ. Sci. Pollut. Res. 2019, 26, 18604–18614. [Google Scholar] [CrossRef]
- Teitelbaum, S.L.; Mervish, N.; Moshier, L.E.; Vangeepuram, N.; Galvez, M.P.; Calafat, A.M.; Silva, M.J.; Brenner, L.B.; Wolff, M.S. Associations between Phthalate Metabolite Urinary Concentrations and Body Size Measures in New York City Children. Environ. Res. 2012, 112, 186–193. [Google Scholar] [CrossRef]
- Desvergne, B.; Feige, J.N.; Casals-Casas, C. PPAR-Mediated Activity of Phthalates: A Link to the Obesity Epidemic? Mol. Cell. Endocrinol. 2009, 304, 43–48. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Rossi, D.; Zoete, V.; Métivier, R.; Tudor, C.; Anghel, S.I.; Grosdidier, A.; Lathion, C.; Engelborghs, Y.; et al. The Endocrine Disruptor Monoethyl-Hexyl-Phthalate Is a Selective Peroxisome Proliferator-Activated Receptor γ Modulator That Promotes Adipogenesis. J. Biol. Chem. 2007, 282, 19152–19166. [Google Scholar] [CrossRef] [PubMed]
- Hurst, C.H.; Waxman, D.J. Activation of PPARalpha and PPARgamma by Environmental Phthalate Monoesters. Toxicol. Sci. 2003, 74, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Yu, K.S.; Lu, K.; Yu, X. Benzyl Butyl Phthalate Promotes Adipogenesis in 3T3-L1 Preadipocytes: A High Content Cellomics and Metabolomic Analysis. Toxicol. In Vitro 2016, 32, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, S.; Sumida, M.; Endoh, Y.; Kurita, A.; Yamaguchi, A.; Watanabe, T.; Kohda, T.; Tsukiyama, Y.; Fujimoto, Y. Curcumin Inhibits Adipogenesis Induced by Benzyl Butyl Phthalate in 3T3-L1 Cells. Toxicol. Appl. Pharmacol. 2017, 329, 158–164. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, W.; Duan, X.; Duan, Y.; Sun, H. Promoting Differentiation and Lipid Metabolism Are the Primary Effects for DINP Exposure on 3T3-L1 Preadipocytes. Environ. Pollut. 2019, 255, 113154. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Sun, S.C.; Chiang, C.K.; Wang, C.C.; Chan, D.C.; Chen, H.J.; Liu, S.H.; Yang, R. Sen Plasticizer Di(2-Ethylhexyl)Phthalate Interferes with Osteoblastogenesis and Adipogenesis in a Mouse Model. J. Orthop. Res. 2018, 36, 1124–1134. [Google Scholar] [CrossRef]
- Hao, X.; Guan, X.; Zhao, X.; Ji, M.; Wen, X.; Chen, P.; Chen, F.; Yang, J.; Lian, Q.; Ye, L.; et al. Phthalate Inhibits Leydig Cell Differentiation and Promotes Adipocyte Differentiation. Chemosphere 2021, 262, 127855. [Google Scholar] [CrossRef] [PubMed]
- Sargis, R.M.; Johnson, D.N.; Choudhury, R.A.; Brady, M.J. Environmental Endocrine Disruptors Promote Adipogenesis in the 3T3-L1 Cell Line through Glucocorticoid Receptor Activation. Obesity 2010, 18, 1283–1288. [Google Scholar] [PubMed]
- Singh, N.; Dalal, V.; Kumar, P. Molecular Docking and Simulation Analysis for Elucidation of Toxic Effects of Dicyclohexyl Phthalate (DCHP) in Glucocorticoid Receptor-Mediated Adipogenesis. Mol. Simul. 2020, 46, 9–21. [Google Scholar] [CrossRef]
- Schmidt, J.-S.; Schaedlich, K.; Fiandanese, N.; Pocar, P.; Fischer, B. Effects of Di(2-Ethylhexyl) Phthalate (DEHP) on Female Fertility and Adipogenesis in C3H/N Mice. Environ. Health Perspect. 2012, 120, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.C.; Wang, C.H.; Yeh, S.C.; Lin, Y.H.; Kuo, Y.T.; Liao, C.W.; Tsai, F.Y.; Lin, W.Y.; Chuang, W.H.; Tsou, T.C. Comparative Microarray Analyses of Mono(2-Ethylhexyl)Phthalate Impacts on Fat Cell Bioenergetics and Adipokine Network. Cell Biol. Toxicol. 2017, 33, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Ellero-Simatos, S.; Claus, S.P.; Benelli, C.; Forest, C.; Letourneur, F.; Cagnard, N.; Beaune, P.H.; de Waziers, I. Combined Transcriptomic-1H NMR Metabonomic Study Reveals That Monoethylhexyl Phthalate Stimulates Adipogenesis and Glyceroneogenesis in Human Adipocytes. J. Proteome Res. 2011, 10, 5493–5502. [Google Scholar]
- Qi, W.; Xu, Q.; Xu, Y.; Wang, Z.; Yang, L.; Guo, S.; Shi, Y.; Zhao, T.; Zhou, L.; Ye, L. Effect of Notch Pathway on Lipid Accumulation Induced by Mono-2-Ethylhexyl Phthalate on 3T3-L1 Cells. Ecotoxicol. Environ. Saf. 2021, 208, 111472. [Google Scholar] [CrossRef]
- Qi, W.; Zhou, L.; Zhao, T.; Ding, S.; Xu, Q.; Han, X.; Zhao, Y.; Song, X.; Zhao, T.; Zhang, X.; et al. Effect of the TYK-2/STAT-3 Pathway on Lipid Accumulation Induced by Mono-2-Ethylhexyl Phthalate. Mol. Cell. Endocrinol. 2019, 484, 52–58. [Google Scholar] [CrossRef]
- Hsu, J.W.; Nien, C.Y.; Yeh, S.C.; Tsai, F.Y.; Chen, H.W.; Lee, T.S.; Chen, S.L.; Kao, Y.H.; Tsou, T.C. Phthalate Exposure Causes Browning-like Effects on Adipocytes in Vitro and in Vivo. Food Chem. Toxicol. 2020, 142, 111487. [Google Scholar] [CrossRef]
- Chiang, H.c.; Kuo, Y.T.; Shen, C.C.; Lin, Y.H.; Wang, S.L.; Tsou, T.C. Mono(2-Ethylhexyl)Phthalate Accumulation Disturbs Energy Metabolism of Fat Cells. Arch. Toxicol. 2016, 90, 589–601. [Google Scholar] [CrossRef]
- Hao, C. Perinatal Exposure to Diethyl-Hexyl-Phthalate Induces Obesity in Mice. Front. Biosci. 2013, E5, E653. [Google Scholar] [CrossRef]
- Hao, C.; Cheng, X.; Xia, H.; Ma, X. The Endocrine Disruptor Mono-(2-Ethylhexyl) Phthalate Promotes Adipocyte Differentiation and Induces Obesity in Mice. Biosci. Rep. 2012, 32, 619–629. [Google Scholar] [CrossRef]
- Gu, H.; Liu, Y.; Wang, W.; Ding, L.; Teng, W.; Liu, L. In Utero Exposure to Di-(2-Ethylhexyl) Phthalate Induces Metabolic Disorder and Increases Fat Accumulation in Visceral Depots of C57BL/6J Mice Offspring. Exp. Ther. Med. 2016, 12, 3806–3812. [Google Scholar] [CrossRef]
- Lee, K.I.; Chiang, C.W.; Lin, H.C.; Zhao, J.F.; Li, C.T.; Shyue, S.K.; Lee, T.S. Maternal Exposure to Di-(2-Ethylhexyl) Phthalate Exposure Deregulates Blood Pressure, Adiposity, Cholesterol Metabolism and Social Interaction in Mouse Offspring. Arch. Toxicol. 2016, 90, 1211–1224. [Google Scholar] [CrossRef]
- Rajesh, P.; Sathish, S.; Srinivasan, C.; Selvaraj, J.; Balasubramanian, K. Phthalate Is Associated with Insulin Resistance in Adipose Tissue of Male Rat: Role of Antioxidant Vitamins. J. Cell Biochem. 2013, 114, 558–569. [Google Scholar] [CrossRef]
- Zhang, J.; Powell, C.A.; Kay, M.K.; Park, M.H.; Meruvu, S.; Sonkar, R.; Choudhury, M. A Moderate Physiological Dose of Benzyl Butyl Phthalate Exacerbates the High Fat Diet-Induced Diabesity in Male Mice. Toxicol. Res. 2020, 9, 353–370. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, H.; Xu, Q.; Han, X.; Zhao, Y.; Song, X.; Zhao, T.; Ye, L. The Effect of Di-2-Ethylhexyl Phthalate on Inflammation and Lipid Metabolic Disorder in Rats. Ecotoxicol. Environ. Saf. 2019, 170, 391–398. [Google Scholar] [CrossRef]
- Campioli, E.; Martinez-Arguelles, D.B.; Papadopoulos, V. In Utero Exposure to the Endocrine Disruptor Di-(2-Ethylhexyl) Phthalate Promotes Local Adipose and Systemic Inflammation in Adult Male Offspring. Nutr. Diabetes 2014, 4, e115. [Google Scholar] [CrossRef]
- Manteiga, S.; Lee, K. Monoethylhexyl Phthalate Elicits an Inflammatory Response in Adipocytes Characterized by Alterations in Lipid and Cytokine Pathways. Environ. Health Perspect. 2017, 125, 615–622. [Google Scholar] [CrossRef]
- Sonkar, R.; Powell, C.A.; Choudhury, M. Benzyl Butyl Phthalate Induces Epigenetic Stress to Enhance Adipogenesis in Mesenchymal Stem Cells. Mol. Cell. Endocrinol. 2016, 431, 109–122. [Google Scholar] [CrossRef]
- Xu, Q.; Qi, W.; Zhang, Y.; Wang, Q.; Ding, S.; Han, X.; Zhao, Y.; Song, X.; Zhao, T.; Zhou, L.; et al. DNA Methylation of JAK3/STAT5/PPARγ Regulated the Changes of Lipid Levels Induced by Di (2-Ethylhexyl) Phthalate and High-Fat Diet in Adolescent Rats. Environ. Sci. Pollut. Res. 2020, 27, 30232–30242. [Google Scholar] [CrossRef] [PubMed]
- Moody, L.; Kougias, D.; Jung, P.M.; Digan, I.; Hong, A.; Gorski, A.; Chen, H.; Juraska, J.; Pan, Y.X. Perinatal Phthalate and High-Fat Diet Exposure Induce Sex-Specific Changes in Adipocyte Size and DNA Methylation. J. Nutr. Biochem. 2019, 65, 15–25. [Google Scholar] [CrossRef]
- Su, H.; Yuan, P.; Lei, H.; Zhang, L.; Deng, D.; Zhang, L.; Chen, X. Long-Term Chronic Exposure to Di-(2-Ethylhexyl)-Phthalate Induces Obesity via Disruption of Host Lipid Metabolism and Gut Microbiota in Mice. Chemosphere 2022, 287, 132414. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.J.; Cheng, X.J.; Xia, H.F.; Ma, X. The Endocrine Disruptor Diethylstilbestrol Induces Adipocyte Differentiation and Promotes Obesity in Mice. Toxicol. Appl. Pharmacol. 2012, 263, 102–110. [Google Scholar] [CrossRef]
- Newbold, R.R.; Padilla-Banks, E.; Jefferson, W.N. Environmental Estrogens and Obesity. Mol. Cell. Endocrinol. 2009, 304, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.E.; Troisi, R.; Palmer, J.R.; Wise, L.A.; Titus, L.; Strohsnitter, W.C.; Ricker, W.; Hyer, M.; Hoover, R.N. Prenatal Diethylstilbestrol Exposure and Risk of Obesity in Adult Women. J. Dev. Orig. Health Dis. 2014, 6, 201–207. [Google Scholar] [CrossRef]
- Jensen, E.T.; Longnecker, M.P. Pharmacologic Sex Hormones in Pregnancy in Relation to Offspring Obesity. Obesity 2014, 22, 2406–2412. [Google Scholar] [CrossRef][Green Version]
- Biasiotto, G.; Zanella, I.; Masserdotti, A.; Pedrazzani, R.; Papa, M.; Caimi, L.; Di Lorenzo, D. Municipal Wastewater Affects Adipose Deposition in Male Mice and Increases 3T3-L1 Cell Differentiation. Toxicol. Appl. Pharmacol. 2016, 297, 32–40. [Google Scholar] [CrossRef]
- Tsou, T.C.; Yeh, S.C.; Hsu, J.W.; Tsai, F.Y. Estrogenic Chemicals at Body Burden Levels Attenuate Energy Metabolism in 3T3-L1 Adipocytes. J. Appl. Toxicol. 2017, 37, 1537–1546. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Ikeda, T.; Zhou, X.K.; Wang, H.; Zheng, X.E.; Giri, D.D.; Elemento, O.; Verma, A.; Miyazawa, M.; Mukherjee, S.; et al. Supplemental Estrogen and Caloric Restriction Reduce Obesity-Induced Periprostatic White Adipose Inflammation in Mice. Carcinogenesis 2019, 40, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Escher, B.I.; Stapleton, H.M.; Schymanski, E.L. Tracking Complex Mixtures of Chemicals in Our Changing Environment. Science 2020, 367, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, T.; Hu, W.; Wang, X.; Xu, B.; Lin, Z.; Hofer, T.; Stefanoff, P.; Chen, Y.; Wang, X.; et al. Association between Exposure to a Mixture of Phenols, Pesticides, and Phthalates and Obesity: Comparison of Three Statistical Models. Environ. Int. 2019, 123, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Lyche, J.L.; Nourizadeh-Lillabadi, R.; Almaas, C.; Stavik, B.; Berg, V.; Skåre, J.U.; Alestrøm, P.; Ropstad, E. Natural Mixtures of Persistent Organic Pollutants (POP) Increase Weight Gain, Advance Puberty, and Induce Changes in Gene Expression Associated with Steroid Hormones and Obesity in Female Zebrafish. J. Toxicol. Environ. Health Part A 2010, 73, 1032–1057. [Google Scholar] [CrossRef] [PubMed]
- Berntsen, H.F.; Berg, V.; Thomsen, C.; Ropstad, E.; Zimmer, K.E. The Design of an Environmentally Relevant Mixture of Persistent Organic Pollutants for Use in In Vivo and In Vitro Studies. J. Toxicol. Environ. Health Part A 2017, 80, 1002–1016. [Google Scholar] [CrossRef]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the Roles of PPARγ in Adipocytes via Dynamic Change of Transcription Complex. Front. Endocrinol. 2018, 9, 473. [Google Scholar] [CrossRef]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ Is Required for the Differentiation of Adipose Tissue In Vivo and In Vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Wang, Q.A.; Zhang, F.; Jiang, L.; Ye, R.; An, Y.; Shao, M.; Tao, C.; Gupta, R.K.; Scherer, P.E. Peroxisome Proliferator-Activated Receptor γ and Its Role in Adipocyte Homeostasis and Thiazolidinedione-Mediated Insulin Sensitization. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef]
- Ros Pérez, M.; Medina-Gómez, G. Obesity, Adipogenesis and Insulin Resistance. Endocrinol. Nutr. 2011, 58, 360–369. [Google Scholar] [CrossRef]
- Farmer, S.R. Regulation of PPARγ Activity during Adipogenesis. Int. J. Obes. 2005, 29, S13–S16. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the Global Map of Adipogenesis and Beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Hsu, C.-H.; Wang, X.; Sakai, S.; Freeman, M.W.; Gonzalez, F.J.; Spiegelman, B.M. C/EBPα Induces Adipogenesis through PPARγ: A Unified Pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-E.; Ge, K. Transcriptional and Epigenetic Regulation of PPARγ Expression during Adipogenesis. Cell Biosci. 2014, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, K.; Kano, F.; Shiota, K.; Murata, M. Expression of the Peroxisome Proliferator Activated Receptor γ Gene Is Repressed by DNA Methylation in Visceral Adipose Tissue of Mouse Models of Diabetes. BMC Biol. 2009, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ Signaling and Metabolism: The Good, the Bad and the Future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Janesick, A.; Blumberg, B. Minireview: PPARγ as the Target of Obesogens. J. Steroid Biochem. Mol. Biol. 2011, 127, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Pallottini, V.; Bulzomi, P.; Galluzzo, P.; Martini, C.; Marino, M. Estrogen Regulation of Adipose Tissue Functions: Involvement of Estrogen Receptor Isoforms. IDDT 2008, 8, 52–60. [Google Scholar] [CrossRef]
- Foryst-Ludwig, A.; Clemenz, M.; Hohmann, S.; Hartge, M.; Sprang, C.; Frost, N.; Krikov, M.; Bhanot, S.; Barros, R.; Morani, A.; et al. Metabolic Actions of Estrogen Receptor Beta (ERβ) Are Mediated by a Negative Cross-Talk with PPARγ. PLoS Genet. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Jeong, S.; Yoon, M. 17β-Estradiol Inhibition of PPARγ-Induced Adipogenesis and Adipocyte-Specific Gene Expression. Acta Pharm. Sin. 2011, 32, 230–238. [Google Scholar] [CrossRef]
- Yepuru, M.; Eswaraka, J.; Kearbey, J.D.; Barrett, C.M.; Raghow, S.; Veverka, K.A.; Miller, D.D.; Dalton, J.T.; Narayanan, R. Estrogen Receptor-β-Selective Ligands Alleviate High-Fat Diet- and Ovariectomy-Induced Obesity in Mice. J. Biol. Chem. 2010, 285, 31292–31303. [Google Scholar] [CrossRef]
- González-Granillo, M.; Savva, C.; Li, X.; Fitch, M.; Pedrelli, M.; Hellerstein, M.; Parini, P.; Korach-André, M.; Gustafsson, J.-Å. ERβ Activation in Obesity Improves Whole Body Metabolism via Adipose Tissue Function and Enhanced Mitochondria Biogenesis. Mol. Cell. Endocrinol. 2019, 479, 147–158. [Google Scholar] [CrossRef]
- Bian, X.; Liu, T.; Zhou, M.; He, G.; Ma, Y.; Shi, Y.; Wang, Y.; Tang, H.; Kang, X.; Yang, M.; et al. Absence of Estrogen Receptor Beta Leads to Abnormal Adipogenesis during Early Tendon Healing by an Up-regulation of PPARγ Signalling. J. Cell Mol. Med. 2019, 23, 7406–7416. [Google Scholar] [CrossRef] [PubMed]
- Dieudonné, M.N.; Leneveu, M.C.; Giudicelli, Y.; Pecquery, R. Evidence for Functional Estrogen Receptors α and β in Human Adipose Cells: Regional Specificities and Regulation by Estrogens. Am. J. Physiol.-Cell Physiol. 2004, 286, C655–C661. [Google Scholar] [CrossRef]
- Davis, K.E.; Neinast, D.M.; Sun, K.; Skiles, M.W.; Bills, D.J.; Zehr, A.J.; Zeve, D.; Hahner, D.L.; Cox, W.D.; Gent, M.L.; et al. The Sexually Dimorphic Role of Adipose and Adipocyte Estrogen Receptors in Modulating Adipose Tissue Expansion, Inflammation, and Fibrosis. Mol. Metab. 2013, 2, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Moore, T.M.; Drew, B.G.; Ribas, V.; Wanagat, J.; Civelek, M.; Segawa, M.; Wolf, D.M.; Norheim, F.; Seldin, M.M.; et al. Estrogen Receptor α Controls Metabolism in White and Brown Adipocytes by Regulating Polg1 and Mitochondrial Remodeling. Sci. Transl. Med. 2020, 12, eaax8096. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.S.; Frank, A.P.; Fátima, L.A.; Palmer, B.F.; Öz, O.K.; Clegg, D.J. Activation of Estrogen Receptor Alpha Induces Beiging of Adipocytes. Mol. Metab. 2018, 18, 51–59. [Google Scholar] [CrossRef]
- Blüher, M. Importance of Estrogen Receptors in Adipose Tissue Function. Mol. Metab. 2013, 2, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Hoa, N.; Ge, L.; Blumberg, B.; Levin, E.R. Mechanisms by Which Membrane and Nuclear ER Alpha Inhibit Adipogenesis in Cells Isolated From Female Mice. Endocrinology 2020, 161, bqaa175. [Google Scholar] [CrossRef]
- Bitirim, C.V.; Ozer, Z.B.; Akcali, K.C. Estrogen Receptor Alpha Regulates the Expression of Adipogenic Genes Genetically and Epigenetically in Rat Bone Marrow-Derived Mesenchymal Stem Cells. PeerJ 2021, 9, e12071. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Hur, J.-Y.; Seo, H.S.; Jeong, Y.-A.; Lee, J.K.; Oh, M.-J.; Kim, T.; Saw, H.S.; Kim, S.H. The Ratio of Estrogen Receptor α to Estrogen Receptor β in Adipose Tissue Is Associated with Leptin Production and Obesity. Steroids 2007, 72, 592–599. [Google Scholar] [CrossRef]
- Le Magueresse-Battistoni, B. Adipose Tissue and Endocrine-Disrupting Chemicals: Does Sex Matter? Int. J. Environ. Res. Public Health 2020, 17, 9403. [Google Scholar] [CrossRef]
- Lee, R.A.; Harris, C.A.; Wang, J.-C. Glucocorticoid Receptor and Adipocyte Biology. Nucl. Recept. Res. 2018, 5. [Google Scholar] [CrossRef]
- Abdou, H.-S.; Atlas, E.; Haché, R.J.G. A Positive Regulatory Domain in CCAAT/Enhancer Binding Protein β (C/EBPβ) Is Required for the Glucocorticoid-Mediated Displacement of Histone Deacetylase 1 (HDAC1) from the C/Ebpα Promoter and Maximum Adipogenesis. Endocrinology 2013, 154, 1454–1464. [Google Scholar] [CrossRef]
- Steger, D.J.; Grant, G.R.; Schupp, M.; Tomaru, T.; Lefterova, M.I.; Schug, J.; Manduchi, E.; Stoeckert, C.J.; Lazar, M.A. Propagation of Adipogenic Signals through an Epigenomic Transition State. Genes Dev. 2010, 24, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Merrett, J.E.; Bo, T.; Psaltis, P.J.; Proud, C.G. Identification of DNA Response Elements Regulating Expression of CCAAT/Enhancer-Binding Protein (C/EBP) β and δ and MAP Kinase-Interacting Kinases during Early Adipogenesis. Adipocyte 2020, 9, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Asada, M.; Rauch, A.; Shimizu, H.; Maruyama, H.; Miyaki, S.; Shibamori, M.; Kawasome, H.; Ishiyama, H.; Tuckermann, J.; Asahara, H. DNA Binding-Dependent Glucocorticoid Receptor Activity Promotes Adipogenesis via Krüppel-like Factor 15 Gene Expression. Lab. Investig. 2011, 91, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Bauerle, K.T.; Hutson, I.; Scheller, E.L.; Harris, C.A. Glucocorticoid Receptor Signaling Is Not Required for In Vivo Adipogenesis. Endocrinology 2018, 159, 2050–2061. [Google Scholar] [CrossRef]
- Park, Y.-K.; Ge, K. Glucocorticoid Receptor Accelerates, but Is Dispensable for, Adipogenesis. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef]
- Desarzens, S.; Faresse, N. Adipocyte Glucocorticoid Receptor Has a Minor Contribution in Adipose Tissue Growth. J. Endocrinol. 2016, 230, 1–11. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aaseth, J.; Javorac, D.; Djordjevic, A.B.; Bulat, Z.; Skalny, A.V.; Zaitseva, I.P.; Aschner, M.; Tinkov, A.A. The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies. Toxics 2022, 10, 65. https://doi.org/10.3390/toxics10020065
Aaseth J, Javorac D, Djordjevic AB, Bulat Z, Skalny AV, Zaitseva IP, Aschner M, Tinkov AA. The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies. Toxics. 2022; 10(2):65. https://doi.org/10.3390/toxics10020065
Chicago/Turabian StyleAaseth, Jan, Dragana Javorac, Aleksandra Buha Djordjevic, Zorica Bulat, Anatoly V. Skalny, Irina P. Zaitseva, Michael Aschner, and Alexey A. Tinkov. 2022. "The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies" Toxics 10, no. 2: 65. https://doi.org/10.3390/toxics10020065
APA StyleAaseth, J., Javorac, D., Djordjevic, A. B., Bulat, Z., Skalny, A. V., Zaitseva, I. P., Aschner, M., & Tinkov, A. A. (2022). The Role of Persistent Organic Pollutants in Obesity: A Review of Laboratory and Epidemiological Studies. Toxics, 10(2), 65. https://doi.org/10.3390/toxics10020065