
Assembly and Comparative Analysis of Complete Mitochondrial Genome Sequence of Endangered Medicinal Plant Trichopus zeylanicus

 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Plant Collection and Genomic DNA Extraction
2.2. Library Preparation, Sequencing, and Mitochondrial Genome Assembly
2.3. Mitochondrial Genome Annotation
2.4. Identification of Repetitive Elements
2.5. Identification of RNA Editing Sites
2.6. Chloroplast and Mitochondrial Genome Migration
2.7. Phylogenetic Analysis
3. Results and Discussion
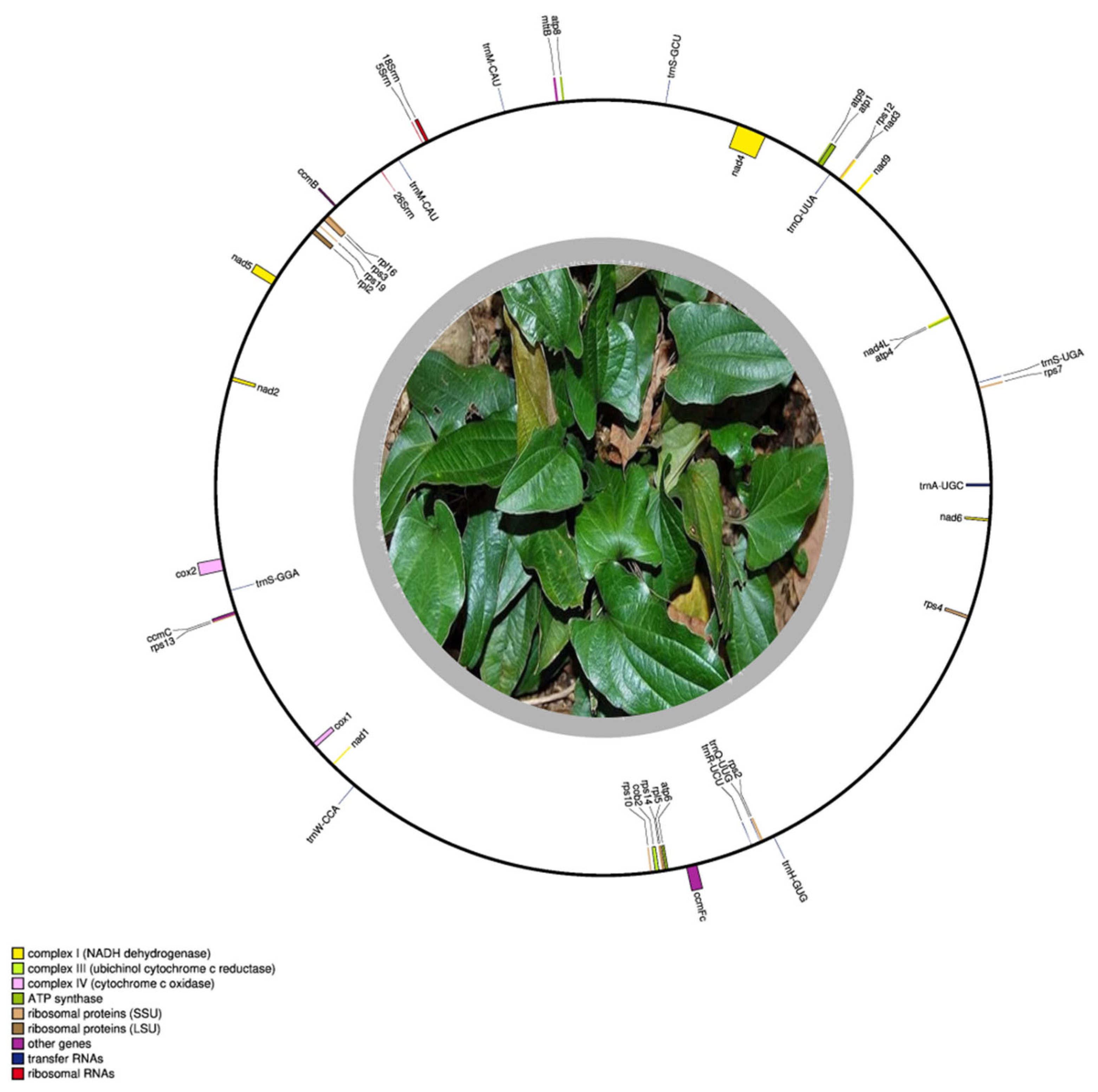
3.1. The Structure, Organization, and Composition of the T. zeylanicus Mitogenome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | GenBank Accession Number | Genome Size (bp) | GC Content (%) | Repetitive Sequence % | Chloroplast-Derived Sequences (%) | Genes | RNA-Editing Sites in PCGs * | ||
|---|---|---|---|---|---|---|---|---|---|
| PCGs * | tRNAs | rRNAs | |||||||
| Trichopus zeylanicus | OR830326 | 709,127 | 45.9 | 0.69 | 6.7 | 32 | 17 | 324 | 324 |
| Cocos nucifera | NC_031696.1 | 678,653 | 45.5 | 17.26 | 5.07 | 72 | 23 | 734 | 734 |
| Phoenix dactylifera | NC_016740.1 | 715,001 | 45.1 | 1.6 | 10.3 | 38 | 30 | 491 | 491 |
| Asparagus officinalis | NC_053642.1 | 492,062 | 45.9 | 5.7 | 4.11 | 36 | 17 | 810 | 810 |
| Pandanus odorifer | NC_080521.1 | 330,962 | 45.7 | 1.3 | 5.2 | 32 | 6 | 325 | 325 |
3.2. Gene Features of the Mitochondrial Genome of T. zeylanicus
| Group of Genes | Gene Names |
|---|---|
| Complex I (NADH dehydrogenase) | nad1, nad2(2), nad3, nad4(3), nad4l, nad5, nad6, nad9 |
| Complex III (ubiquinol cytochrome c reductase) | cob2|cob |
| Complex IV (cytochrome c oxidase) | cox1, cox2(2) |
| Complex V (ATP synthase) | atp1, atp4, atp6, atp8, atp9 |
| Cytochrome c biogenesis | ccmB, ccmc, ccmfc(5) |
| Ribosomal proteins (SSU) | rps2(1), rps3, rps4, rps7, rps10, rps12, rps13, rps14, rps19 |
| Ribosomal proteins (LSU) | rpl2, rpl5, rpl16 |
| Transport membrane protein | mttB(1) |
| Ribosomal RNAs | rrn26, rrn5, rrn18 |
| Transfer RNAs | trnA-UGC(2), trnC-GCA, trnH-GUG, trnM-CAU, trnN-GUU, trnQ-UUA(1), trnQ-UUG, trnR-UCU(1), trnS-GCU, trnS-GGA, trnS-UGA(1), trnW-CCA, trnY-GUA |
3.3. Repetitive Sequence Analysis
3.4. RNA-Editing Site Prediction
3.5. Codon Usage Analysis
3.6. Analysis of Genes Under Selective Pressure
3.7. Chloroplast-Derived Mitochondrial Genome Sequences
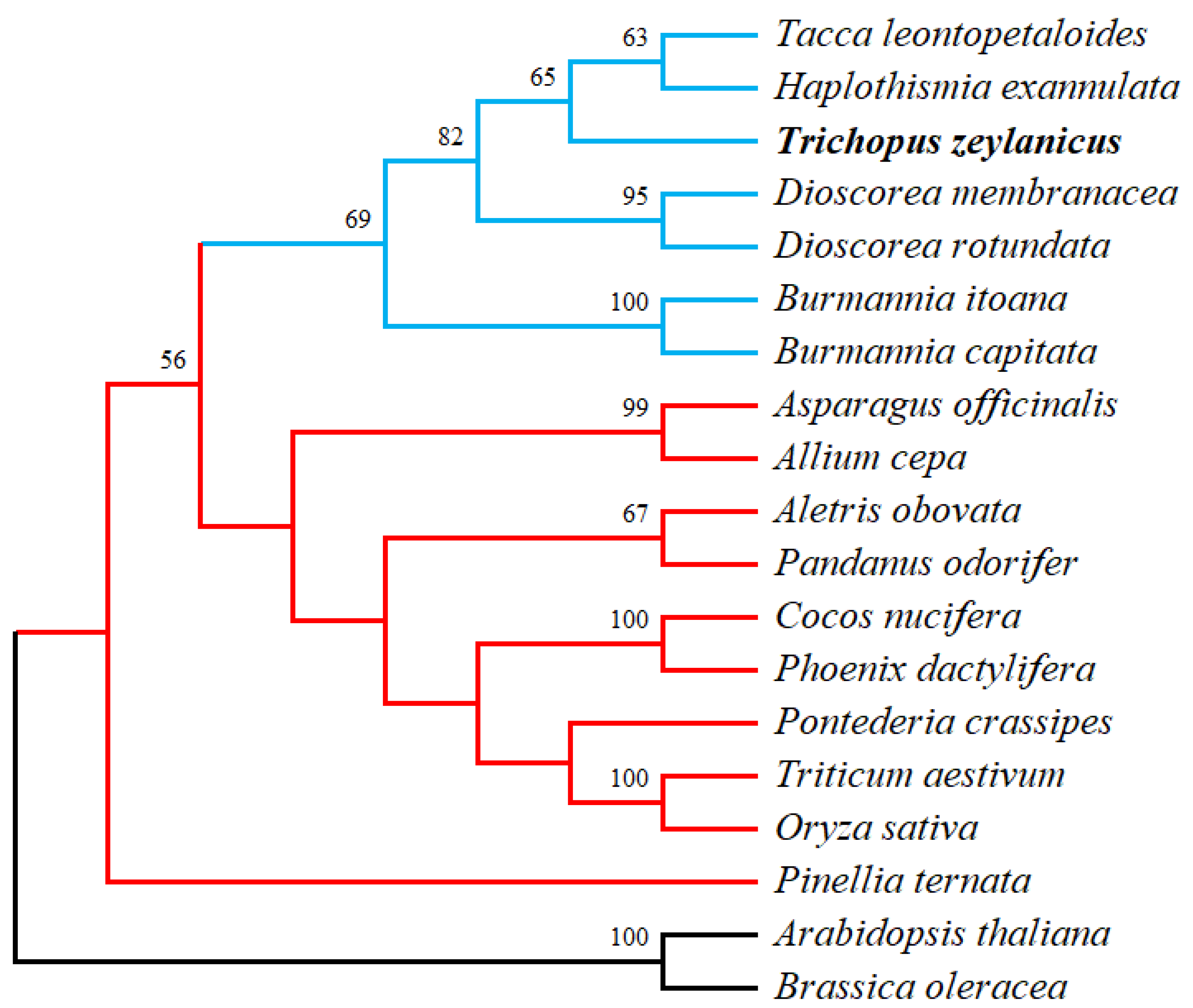
3.8. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nielsen, B.L. Plant mitochondrial DNA. Front. Biosci. 2017, 22, 4531. [Google Scholar] [CrossRef]
- Handa, H. Linear plasmids in plant mitochondria: Peaceful coexistences or malicious invasions? Mitochondrion 2008, 8, 15–25. [Google Scholar] [CrossRef]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant Mitochondrial Genome Diversity: The Genomics Revolution. In Plant Genome Diversity; Springer: Vienna, Austria, 2012; Volume 1, pp. 123–144. [Google Scholar]
- Wu, Z.; Liao, X.; Zhang, X.; Tembrock, L.R.; Broz, A. Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 2022, 60, 160–168. [Google Scholar] [CrossRef]
- Wynn, E.L.; Christensen, A.C. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence and Evolution. G3 Genes|Genomes|Genet. 2019, 9, 549–559. [Google Scholar] [CrossRef]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and Variable Rates of Repeat-Mediated Mitochondrial Genome Rearrangement in a Genus of Plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef] [PubMed]
- Duminil, J. Mitochondrial Genome and Plant Taxonomy. Methods Mol. Biol. 2014, 1115, 121–140. [Google Scholar] [PubMed]
- Takenaka, M.; Verbitskiy, D.; van der Merwe, J.A.; Zehrmann, A.; Brennicke, A. The process of RNA editing in plant mitochondria. Mitochondrion 2008, 8, 35–46. [Google Scholar] [CrossRef]
- Hiesel, R.; Wissinger, B.; Schuster, W.; Brennicke, A. RNA Editing in Plant Mitochondria. Science 1989, 246, 1632–1634. [Google Scholar] [CrossRef]
- Mulligan, R.M. RNA editing site recognition in higher plant mitochondria. J. Hered. 1999, 90, 338–344. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Salucci, S.; Faenza, I.; Blalock, W.L. Alternative Splicing, RNA Editing, and the Current Limits of Next Generation Sequencing. Genes 2023, 14, 1386. [Google Scholar] [CrossRef]
- Araya, A. RNA editing in plant mitochondria, cytoplasmic male sterility and plant breeding. Electron. J. Biotechnol. 1998, 1, 31–39. [Google Scholar] [CrossRef]
- Pushpangadan, P. ‘Arogyappacha’ (trichopus zeylanicus gaerin), the ‘ginseng’ of kani tribes of agashyar hills (kerala) for ever green healh and vitality. Anc. Sci. Life 1988, 8, 13–16. [Google Scholar] [PubMed]
- Manza, M.M.; Saj, O.P. Cytotoxic and Antimicrobial Studies on Arogyapacha or Kerala Ginseng Leaf Extracts. Int. J. Pharm. Chem. Biol. Sci. 2013, 3, 315–319. [Google Scholar]
- Pushpangadan, P.; Rajasekharan, S.; Subramaniam, A.; Latha, P.G.; Evans, D.A.; Raj, R.V. Further on the pharmacology of Trichopus zeylanicus. Anc. Sci. Life 1995, 14, 127–135. [Google Scholar]
- Chacko, S.; Sethuraman, M.G.; George, V.P.P. Phytochemical constituents of Trichopus zeylanicus ssp. travancoricus. J. Medicnal Aromat. Plant Sci. 2002, 24, 703–706. [Google Scholar]
- Subramoniam, A.; Madhavachandran, V.; Rajasekharan, S.; Pushpangadan, P. Aphrodisiac property of Trichopus zeylanicus extract in male mice. J. Ethnopharmacol. 1997, 57, 21–27. [Google Scholar] [CrossRef]
- Tharakan, B.; Dhanasekaran, M.; Brown-Borg, H.M.; Manyam, B.V. Trichopus zeylanicus combats fatigue without amphetamine-mimetic activity. Phytother. Res. 2006, 20, 165–168. [Google Scholar] [CrossRef]
- Avinash, K.; Pushpangadan, P.S.; Chopra, C.L. Adaptogenic activity of seeds of Trichopus zeylanicus gaertn, the ginseng of kerala. Anc. Sci. Life 1989, 8, 212–219. [Google Scholar]
- Tharakan, B.; Dhanasekaran, M.; Manyam, B.V. Antioxidant and DNA protecting properties of anti-fatigue herb Trichopus zeylanicus. Phytother. Res. 2005, 19, 669–673. [Google Scholar] [CrossRef]
- Chase, M.W.; Christenhusz, M.J.M.; Fay, M.F.; Byng, J.W.; Judd, W.S.; Soltis, D.E.; Mabberley, D.J.; Sennikov, A.N.; Soltis, P.S.; Stevens, P.F. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Lindley, J. Aristolochiaceae. In Edwards’s Botanical Register; James Ridgway & Sons: London, UK, 1832; p. 18. [Google Scholar]
- Thwaites, G.H.K. Enumeratio Plantarum Zeylaniae; Part 4; Dulau & Co.: London, UK, 1861. [Google Scholar]
- Brenan, J.P.M.; Hutchinson, J. The Families of Flowering PlantsFamilies of Flowering Plants. Kew Bull. 2007, 14, 477. [Google Scholar] [CrossRef]
- Chellappan, B.V.; Shidhi, P.R.; Vijayan, S.; Rajan, V.S.; Sasi, A.; Nair, A.S. High quality draft genome of arogyapacha (Trichopus zeylanicus), an important medicinal plant endemic to Western Ghats of India. G3 Genes Genomes Genet. 2019, 9, 2395–2404. [Google Scholar] [CrossRef]
- Biju, V.C.; Shidhi, P.R.; Vijayan, S.; Rajan, V.S.; Sasi, A.; Janardhanan, A.; Nair, A.S. The Complete Chloroplast Genome of Trichopus zeylanicus, and Phylogenetic Analysis with Dioscoreales. Plant Genome 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Healey, A.; Furtado, A.; Cooper, T.; Henry, R.J. Protocol: A simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 2014, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef]
- Hackl, T.; Hedrich, R.; Schultz, J.; Förster, F. Proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- Walenz, B.P.; Koren, S.; Bergman, N.H.; Phillippy, A.M.; Miller, J.R.; Berlin, K. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Arakawa, K.; Tomita, M. The GC Skew Index: A Measure of Genomic Compositional Asymmetry and the Degree of Replicational Selection. Evol. Bioinform. 2007, 3, 159–168. [Google Scholar] [CrossRef]
- Kurtz, S. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Lenz, H.; Hein, A.; Knoop, V. Plant organelle RNA editing and its specificity factors: Enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinform. 2018, 19, 255. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Jin, G.; Nakhleh, L.; Snir, S.; Tuller, T. Maximum likelihood of phylogenetic networks. Bioinformatics 2006, 22, 2604–2611. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Caddick, L.R.; Wilkin, P.; Rudall, P.J.; Hedderson, T.A.J.; Chase, M.W. Yams Reclassified: A Recircumscription of Dioscoreaceae and Dioscoreales. Taxon 2002, 51, 103–114. [Google Scholar] [CrossRef]
- Sheng, W.; Deng, J.; Wang, C.; Kuang, Q. The garden asparagus (Asparagus officinalis L.) mitochondrial genome revealed rich sequence variation throughout whole sequencing data. Front. Plant Sci. 2023, 14, 1140043. [Google Scholar] [CrossRef]
- Aljohi, H.A.; Liu, W.; Lin, Q.; Zhao, Y.; Zeng, J.; Alamer, A.; Alanazi, I.O.; Alawad, A.O.; Al-Sadi, A.M.; Hu, S.; et al. Complete Sequence and Analysis of Coconut Palm (Cocos nucifera) Mitochondrial Genome. PLoS ONE 2016, 11, e0163990. [Google Scholar] [CrossRef]
- Mower, J.P. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 2020, 53, 203–213. [Google Scholar] [CrossRef]
- Sloan, D.B.; Wu, Z. History of Plastid DNA Insertions Reveals Weak Deletion and AT Mutation Biases in Angiosperm Mitochondrial Genomes. Genome Biol. Evol. 2014, 6, 3210–3221. [Google Scholar] [CrossRef]
- Fang, Y.; Wu, H.; Zhang, T.; Yang, M.; Yin, Y.; Pan, L.; Yu, X.; Zhang, X.; Hu, S.; Al-Mssallem, I.S.; et al. A Complete Sequence and Transcriptomic Analyses of Date Palm (Phoenix dactylifera L.) Mitochondrial Genome. PLoS ONE 2012, 7, e37164. [Google Scholar] [CrossRef]
- Sperisen, C.; Büchler, U.; Gugerli, F.; Mátyás, G.; Geburek, T.; Vendramin, G.G. Tandem repeats in plant mitochondrial genomes: Application to the analysis of population differentiation in the conifer Norway spruce. Mol. Ecol. 2001, 10, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, Z.; Li, T.; Zhao, J. Highly active repeat-mediated recombination in the mitogenome of the aquatic grass Hygroryza aristata. BMC Plant Biol. 2024, 24, 644. [Google Scholar] [CrossRef]
- Davila, J.I.; Arrieta-Montiel, M.P.; Wamboldt, Y.; Cao, J.; Hagmann, J.; Shedge, V.; Xu, Y.-Z.; Weigel, D.; Mackenzie, S.A. Double-Strand Break Repair Processes Drive Evolution of the Mitochondrial Genome in Arabidopsis. BMC Biol. 2011, 9, 64. [Google Scholar] [CrossRef]
- Bendich, A.J.; Rogers, S.O. The Biological and Evolutionary Consequences of Competition between DNA Sequences That Benefit the Cell and DNA Sequences That Benefit Themselves. Nucleic Acids Res. 2025, 53, gkaf589. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Sugita, M. RNA Editing and Its Molecular Mechanism in Plant Organelles. Genes 2016, 8, 5. [Google Scholar] [CrossRef]
- Maier, R.M.; Zeltz, P.; Kössel, H.; Bonnard, G.; Gualberto, J.M.; Grienenberger, J.M. RNA editing in plant mitochondria and chloroplasts. Plant Mol. Biol. 1996, 32, 343–365. [Google Scholar] [CrossRef]
- Hiesel, R.; Combettes, B.; Brennicke, A. Evidence for RNA editing in mitochondria of all major groups of land plants except the Bryophyta. Proc. Natl. Acad. Sci. USA 1994, 91, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, X. Analysis of synonymous codon usage patterns in different plant mitochondrial genomes. Mol. Biol. Rep. 2009, 36, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Gu, Y.; Zhou, J.; Lu, M.; Wang, J.; Lu, K.; Zeng, Y.; Tan, X. De novo assembly of the complete mitochondrial genomes of two Camellia-oil tree species reveals their multibranch conformation and evolutionary relationships. Sci. Rep. 2025, 15, 2899. [Google Scholar] [CrossRef]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Abrosimov, R.; Baeken, M.W.; Hauf, S.; Wittig, I.; Hajieva, P.; Perrone, C.E.; Moosmann, B. Mitochondrial complex I inhibition triggers NAD+-independent glucose oxidation via successive NADPH formation, “futile” fatty acid cycling, and FADH2 oxidation. Geroscience 2024, 46, 3635–3658. [Google Scholar] [CrossRef]
- Liu, Q.; Cai, Y.D.; Ma, L.; Liu, H.; Linghu, T.; Guo, S.; Wei, S.; Song, F.; Tian, L.; Cai, W. Relaxed purifying selection pressure drives accelerated and dynamic gene rearrangements in thrips (Insecta: Thysanoptera) mitochondrial genomes. Int. J. Biol. Macromol. 2023, 253, 126742. [Google Scholar] [CrossRef]
- Mower, J.P.; Jain, K.; Hepburn, N.J. The Role of Horizontal Transfer in Shaping the Plant Mitochondrial Genome. Adv. Bot. Res. 2012, 63, 41–69. [Google Scholar]
- Sasikala, N.; Ramasubbu, R. Population status and floral biology of Trichopus zeylanicus ssp. travancoricus Burkill ex K. Narayanan (Dioscoreaceae), an important ethnomedicinal plant of the southern Western Ghats, India. J. Threat. Taxa 2019, 11, 13156–13161. [Google Scholar] [CrossRef]
- Merckx, V.; Schols, P.; de Kamer, H.M.; Maas, P.; Huysmans, S.; Smets, E. Phylogeny and Evolution of Burmanniaceae (Dioscoreales) Based on Nuclear and Mitochondrial Data. Am. J. Bot. 2006, 93, 1684–1698. [Google Scholar] [CrossRef]





| No. | Size | Copy | Start | End | Sequence |
|---|---|---|---|---|---|
| 1 | 25 | 3 | 35,388 | 35,462 | GTCTCATAGGTTACATGGAATACCG |
| 2 | 17 | 2 | 127,171 | 127,204 | AGTGAATCAGATCGTAG |
| 3 | 27 | 2 | 127,199 | 127,252 | TGGTAGTTCGCGTGCTCAAGTGAAATG |
| 4 | 17 | 2 | 141,404 | 141,440 | CAAGGCAAGGTCAGGCT |
| 5 | 17 | 3 | 141,404 | 141,446 | CAAGGCAAGGTCAGGCT |
| 6 | 17 | 2 | 201,046 | 201,079 | AACCCTGATCGTCTTCC |
| 7 | 22 | 3 | 219,866 | 219,933 | CGTGCATGTCACCGTCTCCACC |
| 8 | 18 | 2 | 224,271 | 224,308 | TGTTGTTGCAATACCCGT |
| 9 | 13 | 2 | 254,962 | 254,986 | TCAAAGTGAGAAC |
| 10 | 13 | 2 | 381,379 | 381,403 | TTGTATATCCAAA |
| 11 | 19 | 2 | 385,131 | 385,169 | AATAGTAATAGTTCTATTC |
| 12 | 13 | 2 | 458,381 | 458,411 | TAGCCTCTAACTC |
| 13 | 25 | 2 | 472,717 | 472,764 | GTAGCATGAAGAAAGCAGAAGTGGA |
| 14 | 27 | 2 | 487,362 | 487,427 | ATACTTGCAGCGGGGATTCTACCTCTT |
| 15 | 21 | 2 | 536,618 | 536,665 | TCGGCGTCCGTCTATCTATTG |
| 16 | 21 | 2 | 570,837 | 570,876 | TTGATAATCCTACTCTTTTCC |
| 17 | 25 | 3 | 596,343 | 596,417 | CGAAGAAAGCACTACACCTGGCAGG |
| 18 | 14 | 2 | 691,088 | 691,117 | AGGGACTGCCTGGAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chellappan, B.V.; Shidhi, P.R.; Sasi, A.; Ibrahim, R.I.H.; Zahra, H.A. Assembly and Comparative Analysis of Complete Mitochondrial Genome Sequence of Endangered Medicinal Plant Trichopus zeylanicus. Curr. Issues Mol. Biol. 2025, 47, 553. https://doi.org/10.3390/cimb47070553
Chellappan BV, Shidhi PR, Sasi A, Ibrahim RIH, Zahra HA. Assembly and Comparative Analysis of Complete Mitochondrial Genome Sequence of Endangered Medicinal Plant Trichopus zeylanicus. Current Issues in Molecular Biology. 2025; 47(7):553. https://doi.org/10.3390/cimb47070553
Chicago/Turabian StyleChellappan, Biju Vadakkemukadiyil, P. R. Shidhi, Anu Sasi, Rashid Ismael Hag Ibrahim, and Hamad Abu Zahra. 2025. "Assembly and Comparative Analysis of Complete Mitochondrial Genome Sequence of Endangered Medicinal Plant Trichopus zeylanicus" Current Issues in Molecular Biology 47, no. 7: 553. https://doi.org/10.3390/cimb47070553
APA StyleChellappan, B. V., Shidhi, P. R., Sasi, A., Ibrahim, R. I. H., & Zahra, H. A. (2025). Assembly and Comparative Analysis of Complete Mitochondrial Genome Sequence of Endangered Medicinal Plant Trichopus zeylanicus. Current Issues in Molecular Biology, 47(7), 553. https://doi.org/10.3390/cimb47070553