
Integrating Artificial Intelligence in Next-Generation Sequencing: Advances, Challenges, and Future Directions

 , , and
, , and 
Abstract

1. Introduction
2. AI in the Laboratory: Enhancing Research from Plan to Data Analysis
2.1. The Pre-Wet-Lab Phase
2.2. The Wet-Lab Phase
2.3. The Post-Wet-Lab Phase
3. AI in NGS Data Analysis
3.1. Machine Learning Approaches
3.2. Deep Learning Approaches
3.3. Hybrid and Ensemble Approaches
3.4. Deploying AI Within Clinical NGS Workflows
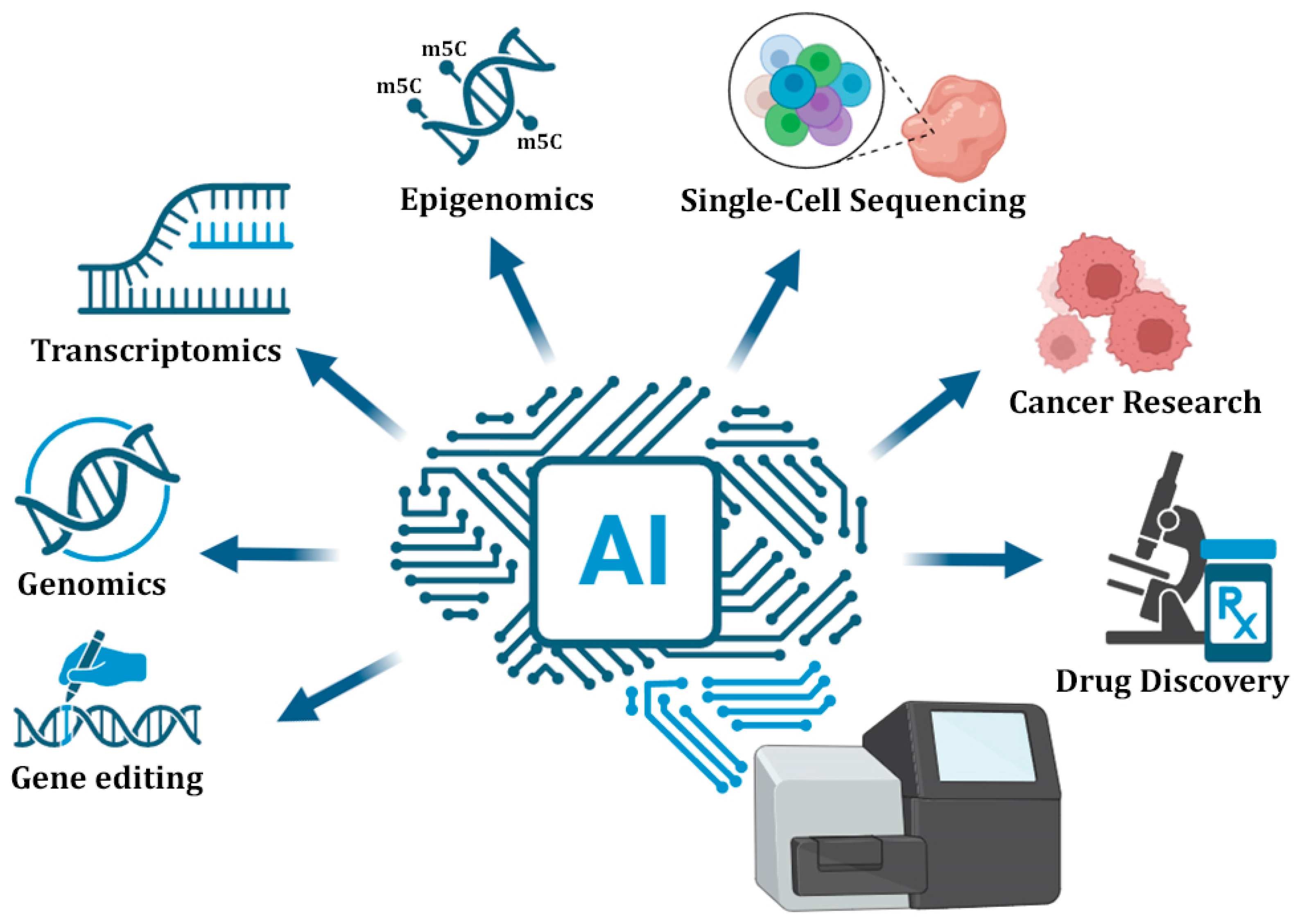
4. AI in NGS Applications
4.1. Genomics and Epigenomics
4.2. Transcriptomics
4.3. Single-Cell Sequencing
4.4. Cancer Research
4.5. Drug Discovery
5. AI-Driven Multi-Omics Integration and Clinical Translation
6. Challenges and Limitations
7. Future Perspectives—AI Integration into Third-Generation Sequencing
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AI | Artificial intelligence |
| ANN | Artificial neural network |
| ATAC-seq | Assay for transposase-accessible chromatin using sequencing |
| AUC | Area under the curve |
| CITE-seq | Cellular indexing of transcriptomes and epitopes by sequencing |
| CNN | Convolutional neural network |
| CNV | Copy number variation |
| CRF | Conditional random field |
| CTC | Connectionist temporal classification |
| ctDNA | Circulating tumor DNA |
| DL | Deep learning |
| DEA | Differential expression analysis |
| DMR | Differentially methylated region |
| EMBL-EBI | European Bioinformatics Institute |
| GATK | Genome Analysis Toolkit |
| GBM | Gradient boosting machine |
| GEO | Gene Expression Omnibus |
| GNN | Graph neural network |
| GRN | Gene regulatory network |
| HDP | Hierarchical Dirichlet process |
| HGP | Human Genome Project |
| HMM | Hidden Markov model |
| IGCN | Integrative graph convolutional network |
| IPD | Interpulse duration |
| LSTM | Long short-term memory |
| ML | Machine learning |
| m5C | 5-methylcytosine |
| m6A | N6-methyladenosine |
| hm5C | 5-hydroxymethylcytosine |
| ML | Machine learning |
| mRNA | Messenger RNA |
| NGS | Next-generation sequencing |
| ONT | Oxford Nanopore Technologies |
| PCA | Principal component analysis |
| PCR | Polymerase chain reaction |
| RF | Random forest |
| RNN | Recurrent neural network |
| scRNA-seq | Single-cell RNA sequencing |
| SdAs | Stacked denoising autoencoders |
| SNP | Single nucleotide polymorphism |
| SVM | Support vector machine |
| TCGA | The Cancer Genome Atlas |
| TGS | Third-generation sequencing |
| t-SNE | t-distributed stochastic neighbor embedding |
| UMAP | Uniform manifold approximation and projection |
| VAE | Variational autoencoder |
| WGBS | Whole-genome bisulfite sequencing |
References
- Watson, J.D.; Crick, F.H. The structure of DNA. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Morgan, M.; Patrinos, A. The human genome project: Lessons from large-scale biology. Science 2003, 300, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Green, E.D.; Watson, J.D.; Collins, F.S. Human genome project: Twenty-five years of big biology. Nature 2015, 526, 29–31. [Google Scholar] [CrossRef]
- Giani, A.M.; Gallo, G.R.; Gianfranceschi, L.; Formenti, G. Long walk to genomics: History and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J. 2020, 18, 9–19. [Google Scholar] [CrossRef]
- Caudai, C.; Galizia, A.; Geraci, F.; Le Pera, L.; Morea, V.; Salerno, E.; Via, A.; Colombo, T. Ai applications in functional genomics. Comput. Struct. Biotechnol. J. 2021, 19, 5762–5790. [Google Scholar] [CrossRef]
- Dixit, S.; Kumar, A.; Srinivasan, K.; Vincent, P.; Ramu Krishnan, N. Advancing genome editing with artificial intelligence: Opportunities, challenges, and future directions. Front. Bioeng. Biotechnol. 2023, 11, 1335901. [Google Scholar] [CrossRef]
- Koh, E.; Sunil, R.S.; Lam, H.Y.I.; Mutwil, M. Confronting the data deluge: How artificial intelligence can be used in the study of plant stress. Comput. Struct. Biotechnol. J. 2024, 23, 3454–3466. [Google Scholar] [CrossRef]
- Farhud, D.D.; Zokaei, S. Ethical issues of artificial intelligence in medicine and healthcare. Iran. J. Public Health 2021, 50, i–v. [Google Scholar] [CrossRef]
- Elendu, C.; Amaechi, D.C.; Elendu, T.C.; Jingwa, K.A.; Okoye, O.K.; John Okah, M.; Ladele, J.A.; Farah, A.H.; Alimi, H.A. Ethical implications of ai and robotics in healthcare: A review. Medicine 2023, 102, e36671. [Google Scholar] [CrossRef]
- Serrano, D.R.; Luciano, F.C.; Anaya, B.J.; Ongoren, B.; Kara, A.; Molina, G.; Ramirez, B.I.; Sanchez-Guirales, S.A.; Simon, J.A.; Tomietto, G.; et al. Artificial intelligence (ai) applications in drug discovery and drug delivery: Revolutionizing personalized medicine. Pharmaceutics 2024, 16, 1328. [Google Scholar] [CrossRef] [PubMed]
- Jamialahmadi, H.; Khalili-Tanha, G.; Nazari, E.; Rezaei-Tavirani, M. Artificial intelligence and bioinformatics: A journey from traditional techniques to smart approaches. Gastroenterol. Hepatol. Bed Bench 2024, 17, 241–252. [Google Scholar]
- Harrer, S.; Rane, R.V.; Speight, R.E. Generative ai agents are transforming biology research: High resolution functional genome annotation for multiscale understanding of life. EBioMedicine 2024, 109, 105446. [Google Scholar] [CrossRef]
- Ghose, A.K.; Abdullah, S.N.A.; Md Hatta, M.A.; Megat Wahab, P.E. DNA free crispr/dcas9 based transcriptional activation system for ugt76g1 gene in stevia rebaudiana bertoni protoplasts. Plants 2022, 11, 2393. [Google Scholar] [CrossRef]
- Yuan, Y.; Shi, Y.; Li, C.; Kim, J.; Cai, W.; Han, Z.; Feng, D.D. Deepgene: An advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinform. 2016, 17, 476. [Google Scholar] [CrossRef]
- Tegally, H.; San, J.E.; Giandhari, J.; de Oliveira, T. Unlocking the efficiency of genomics laboratories with robotic liquid-handling. BMC Genom. 2020, 21, 729. [Google Scholar] [CrossRef]
- Ng, N.; Gately, R.; Ooi, L. Automated liquid handling for microplate assays: A simplified user interface for the hamilton microlab star. J. Appl. Bioanal. 2020, 7, 11–18. [Google Scholar] [CrossRef]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. Deepcrispr: Optimized crispr guide rna design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Niu, R.; Peng, J.; Zhang, Z.; Shang, X. R-crispr: A deep learning network to predict off-target activities with mismatch, insertion and deletion in crispr-cas9 system. Genes 2021, 12, 1878. [Google Scholar] [CrossRef]
- Truong, V.; Viken, K.; Geng, Z.; Barkan, S.; Johnson, B.; Ebeling, M.C.; Montezuma, S.R.; Ferrington, D.A.; Dutton, J.R. Automating human induced pluripotent stem cell culture and differentiation of ipsc-derived retinal pigment epithelium for personalized drug testing. SLAS Technol. 2021, 26, 287–299. [Google Scholar] [CrossRef]
- Khan, S.; Møller, V.; Frandsen, R.; Mansourvar, M. Real-time ai-driven quality control for laboratory automation: A novel computer vision solution for the opentrons ot-2 liquid handling robot. Appl. Intell. 2025, 55, 524. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal snp and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. Crispresso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. Guide-seq enables genome-wide profiling of off-target cleavage by crispr-cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.S. Cas-offinder: A fast and versatile algorithm that searches for potential off-target sites of cas9 rna-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Cancellieri, S.; Canver, M.C.; Bombieri, N.; Giugno, R.; Pinello, L. Crispritz: Rapid, high-throughput and variant-aware in silico off-target site identification for crispr genome editing. Bioinformatics 2020, 36, 2001–2008. [Google Scholar] [CrossRef]
- Gao, L.; Yuan, J.; Hong, K.; Ma, N.L.; Liu, S.; Wu, X. Technological advancement spurs komagataella phaffii as a next-generation platform for sustainable biomanufacturing. Biotechnol. Adv. 2025, 82, 108593. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. Ncbi geo: Archive for gene expression and epigenomics data sets: 23-year update. Nucleic Acids Res. 2024, 52, D138–D144. [Google Scholar] [CrossRef]
- Thakur, M.; Brooksbank, C.; Finn, R.D.; Firth, H.V.; Foreman, J.; Freeberg, M.; Gurwitz, K.T.; Harrison, M.; Hulcoop, D.; Hunt, S.E.; et al. Embl’s european bioinformatics institute (embl-ebi) in 2024. Nucleic Acids Res. 2025, 53, D10–D19. [Google Scholar] [CrossRef]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. Encode data at the encode portal. Nucleic Acids Res. 2016, 44, D726–D732. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The cancer genome atlas (tcga): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Perrin, H.; Denorme, M.; Grosjean, J.; OMICtools Community; Dynomant, E.; Henry, V.; Pichon, F.; Darmoni, S.; Desfeux, A.; Gonzalez, B. Omictools: A community-driven search engine for biological data analysis. arXiv 2017, arXiv:1707.03659. [Google Scholar]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. Mageck enables robust identification of essential genes from genome-scale crispr/cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef]
- Hassani-Pak, K.; Singh, A.; Brandizi, M.; Hearnshaw, J.; Parsons, J.D.; Amberkar, S.; Phillips, A.L.; Doonan, J.H.; Rawlings, C. Knetminer: A comprehensive approach for supporting evidence-based gene discovery and complex trait analysis across species. Plant Biotechnol. J. 2021, 19, 1670–1678. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. Shinygo: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Grentner, A.; Ragueneau, E.; Gong, C.; Prinz, A.; Gansberger, S.; Oyarzun, I.; Hermjakob, H.; Griss, J. Reactomegsa: New features to simplify public data reuse. Bioinformatics 2024, 40, btae338. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Etcheverry, M.; Moulin-Frier, C.; Oudeyer, P.Y.; Levin, M. Ai-driven automated discovery tools reveal diverse behavioral competencies of biological networks. eLife 2025, 13, RP92683. [Google Scholar] [CrossRef]
- Yang, B.; Xu, Y.; Maxwell, A.; Koh, W.; Gong, P.; Zhang, C. Micrat: A novel algorithm for inferring gene regulatory networks using time series gene expression data. BMC Syst. Biol. 2018, 12, 115. [Google Scholar] [CrossRef]
- Pop, M.; Attwood, T.K.; Blake, J.A.; Bourne, P.E.; Conesa, A.; Gaasterland, T.; Hunter, L.; Kingsford, C.; Kohlbacher, O.; Lengauer, T.; et al. Biological databases in the age of generative artificial intelligence. Bioinform. Adv. 2025, 5, vbaf044. [Google Scholar] [CrossRef]
- Peng, Y.; Malin, B.A.; Rousseau, J.F.; Wang, Y.; Xu, Z.; Xu, X.; Weng, C.; Bian, J. From gpt to deepseek: Significant gaps remain in realizing ai in healthcare. J. Biomed. Inform. 2025, 163, 104791. [Google Scholar] [CrossRef]
- Luo, R.; Sun, L.; Xia, Y.; Qin, T.; Zhang, S.; Poon, H.; Liu, T.Y. Biogpt: Generative pre-trained transformer for biomedical text generation and mining. Brief. Bioinform. 2022, 23, bbac409. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, Q.; Shih, G. Deepseek is open-access and the next ai disrupter for radiology. Radiol. Adv. 2025, 2, umaf009. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Digby, B.; Finn, S.P.; Broin, P.Ó. Nf-core/circrna: A portable workflow for the quantification, mirna target prediction and differential expression analysis of circular rnas. BMC Bioinform. 2023, 24, 27. [Google Scholar] [CrossRef]
- Mpangase, P.T.; Frost, J.; Tikly, M.; Ramsay, M.; Hazelhurst, S. Nf-rnaseqcount: A nextflow pipeline for obtaining raw read counts from rna-seq data. S. Afr. Comput. J. 2021, 33, 2. [Google Scholar] [CrossRef]
- Liu, X.; Bienkowska, J.R.; Zhong, W. Geneteflow: A nextflow-based pipeline for analysing gene and transposable elements expression from rna-seq data. PLoS ONE 2020, 15, e0232994. [Google Scholar] [CrossRef]
- Hu, K.; Liu, H.; Lawson, N.D.; Zhu, L.J. Scatacpipe: A nextflow pipeline for comprehensive and reproducible analyses of single cell atac-seq data. Front. Cell Dev. Biol. 2022, 10, 981859. [Google Scholar] [CrossRef]
- Engelberg, A.B.; Avorn, J.; Kesselheim, A.S. A new way to contain unaffordable medication costs—Exercising the government’s existing rights. N. Engl. J. Med. 2022, 386, 1104–1106. [Google Scholar] [CrossRef]
- Song, Z.; Gurinovich, A.; Federico, A.; Monti, S.; Sebastiani, P. Nf-gwas-pipeline: A nextflow genome-wide association study pipeline. J. Open Source Softw. 2021, 6, 2957. [Google Scholar] [CrossRef]
- Twesigomwe, D.; Drogemoller, B.I.; Wright, G.E.B.; Siddiqui, A.; da Rocha, J.; Lombard, Z.; Hazelhurst, S. Stellarpgx: A nextflow pipeline for calling star alleles in cytochrome p450 genes. Clin. Pharmacol. Ther. 2021, 110, 741–749. [Google Scholar] [CrossRef]
- Patel, Y.; Zhu, C.; Yamaguchi, T.N.; Bugh, Y.Z.; Tian, M.; Holmes, A.; Fitz-Gibbon, S.T.; Boutros, P.C. Nftest: Automated testing of nextflow pipelines. Bioinformatics 2024, 40, btae081. [Google Scholar] [CrossRef]
- Holzer, M.; Marz, M. Poseidon: A nextflow pipeline for the detection of evolutionary recombination events and positive selection. Bioinformatics 2021, 37, 1018–1020. [Google Scholar] [CrossRef]
- Federico, A.; Karagiannis, T.; Karri, K.; Kishore, D.; Koga, Y.; Campbell, J.D.; Monti, S. Pipeliner: A nextflow-based framework for the definition of sequencing data processing pipelines. Front. Genet. 2019, 10, 614. [Google Scholar] [CrossRef]
- Vlasova, A.; Hermoso Pulido, T.; Camara, F.; Ponomarenko, J.; Guigo, R. Fa-nf: A functional annotation pipeline for proteins from non-model organisms implemented in nextflow. Genes 2021, 12, 1645. [Google Scholar] [CrossRef]
- Allain, F.; Romejon, J.; La Rosa, P.; Jarlier, F.; Servant, N.; Hupe, P. Geniac: Automatic configuration generator and installer for nextflow pipelines. Open Res. Eur. 2021, 1, 76. [Google Scholar] [CrossRef]
- Yukselen, O.; Turkyilmaz, O.; Ozturk, A.R.; Garber, M.; Kucukural, A. Dolphinnext: A distributed data processing platform for high throughput genomics. BMC Genom. 2020, 21, 310. [Google Scholar] [CrossRef]
- Babadi, M.; Fu, J.M.; Lee, S.K.; Smirnov, A.N.; Gauthier, L.D.; Walker, M.; Benjamin, D.I.; Zhao, X.; Karczewski, K.J.; Wong, I.; et al. Gatk-gcnv enables the discovery of rare copy number variants from exome sequencing data. Nat. Genet. 2023, 55, 1589–1597. [Google Scholar] [CrossRef]
- Brouard, J.S.; Bissonnette, N. Variant calling from rna-seq data using the gatk joint genotyping workflow. Methods Mol. Biol. 2022, 2493, 205–233. [Google Scholar]
- Tong, S.Y.; Fan, K.; Zhou, Z.W.; Liu, L.Y.; Zhang, S.Q.; Fu, Y.; Wang, G.Z.; Zhu, Y.; Yu, Y.C. Mvppt: A highly efficient and sensitive pathogenicity prediction tool for missense variants. Genom. Proteom. Bioinform. 2023, 21, 414–426. [Google Scholar] [CrossRef]
- Ng, S.; Masarone, S.; Watson, D.; Barnes, M.R. The benefits and pitfalls of machine learning for biomarker discovery. Cell Tissue Res. 2023, 394, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.I.; Song, B.; Taylor, C.; Vaske, C.J.; Benz, S.C.; Rabizadeh, S.; Soon-Shiong, P.; Szeto, C.W. A deep learning image-based intrinsic molecular subtype classifier of breast tumors reveals tumor heterogeneity that may affect survival. Breast Cancer Res. 2020, 22, 12. [Google Scholar] [CrossRef]
- Tang, Y.; Li, S.; Zhu, L.; Yao, L.; Li, J.; Sun, X.; Liu, Y.; Zhang, Y.; Fu, X. Improve clinical feature-based bladder cancer survival prediction models through integration with gene expression profiles and machine learning techniques. Heliyon 2024, 10, e38242. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Xu, L.; Wang, J.; Yue, Z.; Jing, Y.; Tai, S.; Yang, J.; Fang, X. Vcf2pcacluster: A simple, fast and memory-efficient tool for principal component analysis of tens of millions of snps. BMC Bioinform. 2024, 25, 173. [Google Scholar] [CrossRef] [PubMed]
- Cieslak, M.C.; Castelfranco, A.M.; Roncalli, V.; Lenz, P.H.; Hartline, D.K. T-distributed stochastic neighbor embedding (t-sne): A tool for eco-physiological transcriptomic analysis. Mar. Genom. 2020, 51, 100723. [Google Scholar] [CrossRef]
- Li, T.; Zou, Y.; Li, X.; Wong, T.K.F.; Rodrigo, A.G. Mugen-umap: Umap visualization and clustering of mutated genes in single-cell DNA sequencing data. BMC Bioinform. 2024, 25, 308. [Google Scholar] [CrossRef]
- Schmidt, B.; Hildebrandt, A. Deep learning in next-generation sequencing. Drug Discov. Today 2021, 26, 173–180. [Google Scholar] [CrossRef]
- Hwang, H.; Jeon, H.; Yeo, N.; Baek, D. Big data and deep learning for rna biology. Exp. Mol. Med. 2024, 56, 1293–1321. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, L.; Zhang, Y.; Han, X.; Deveci, M.; Parmar, M. A review of convolutional neural networks in computer vision. Artif. Intell. Rev. 2024, 57, 99. [Google Scholar] [CrossRef]
- Alharbi, W.S.; Rashid, M. A review of deep learning applications in human genomics using next-generation sequencing data. Hum. Genom. 2022, 16, 26. [Google Scholar] [CrossRef]
- Vaz, J.M.; Balaji, S. Convolutional neural networks (cnns): Concepts and applications in pharmacogenomics. Mol. Divers. 2021, 25, 1569–1584. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachari, K.; Lu, D.; Swift-Scott, A.; Yeraliyev, A.; Lee, K.; Huang, W.; Leng, S.; Jacobsen Skanderup, A. Accurate somatic variant detection using weakly supervised deep learning. Nat. Commun. 2022, 13, 4248. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.; Qian, S.; Zhang, H.; Zhang, S.; Kozlova, A.; Duan, J.; Xu, J.; He, X. Annotating functional effects of non-coding variants in neuropsychiatric cell types by deep transfer learning. PLoS Comput. Biol. 2022, 18, e1010011. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Beknazarov, N.; Poptsova, M. Deepz: A deep learning approach for z-DNA prediction. Methods Mol. Biol. 2023, 2651, 217–226. [Google Scholar]
- Mienye, D.; Swart, T.; Obaido, G. Recurrent neural networks: A comprehensive review of architectures, variants, and applications. Information 2024, 15, 517. [Google Scholar] [CrossRef]
- Liu, F.; Miao, Y.; Liu, Y.; Hou, T. Rnn-virseeker: A deep learning method for identification of short viral sequences from metagenomes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1840–1849. [Google Scholar] [CrossRef]
- Paggi, J.M.; Bejerano, G. A sequence-based, deep learning model accurately predicts rna splicing branchpoints. RNA 2018, 24, 1647–1658. [Google Scholar] [CrossRef]
- Gupta, G.; Saini, S. Davi:Deep learning based tool for alignment and single nucleotide variant identification. bioRxiv 2019, 778647. [Google Scholar]
- Chen, Y.; Xie, M.; Wen, J. Predicting gene expression from histone modifications with self-attention based neural networks and transfer learning. Front. Genet. 2022, 13, 1081842. [Google Scholar] [CrossRef] [PubMed]
- Aburass, S.; Dorgham, O.; Al Shaqsi, J. A hybrid machine learning model for classifying gene mutations in cancer using lstm, bilstm, cnn, gru, and glove. Syst. Soft Comput. 2024, 6, 200110. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, S.; Su, J.; Leung, A.W.; Lam, T.W.; Luo, R. Symphonizing pileup and full-alignment for deep learning-based long-read variant calling. Nat. Comput. Sci. 2022, 2, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Shafin, K.; Pesout, T.; Chang, P.-C.; Nattestad, M.; Kolesnikov, A.; Goel, S.; Baid, G.; Kolmogorov, M.; Eizenga, J.M.; Miga, K.H.; et al. Haplotype-aware variant calling with pepper-margin-deepvariant enables high accuracy in nanopore long-reads. Nat. Methods 2021, 18, 1322–1332. [Google Scholar] [CrossRef]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. Clinpred: Prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef]
- Choon, Y.W.; Choon, Y.F.; Nasarudin, N.A.; Al Jasmi, F.; Remli, M.A.; Alkayali, M.H.; Mohamad, M.S. Artificial intelligence and database for ngs-based diagnosis in rare disease. Front. Genet. 2023, 14, 1258083. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, Z.; Wei, Y.; Schmidt, B.; Liu, W. Deepfilter: A deep learning based variant filter for vardict. Tsinghua Sci. Technol. 2023, 28, 665–672. [Google Scholar] [CrossRef]
- Freed, D.; Pan, R.; Chen, H.; Li, Z.; Hu, J.; Aldana, R. Dnascope: High accuracy small variant calling using machine learning. bioRxiv 2022. [Google Scholar]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. Revel: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R.V. Dnabert: Pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with chromhmm. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Luo, R.; Pinello, L. Epinformer: A scalable deep learning framework for gene expression prediction by integrating promoter-enhancer sequences with multimodal epigenomic data. bioRxiv 2024. [Google Scholar]
- Singh, R.; Lanchantin, J.; Robins, G.; Qi, Y. Deepchrome: Deep-learning for predicting gene expression from histone modifications. Bioinformatics 2016, 32, i639–i648. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, A.; Singh, R.; Qi, Y. Deepdiff: Deep-learning for predicting differential gene expression from histone modifications. Bioinformatics 2018, 34, i891–i900. [Google Scholar] [CrossRef]
- Yan, F.; Telonis, A.G.; Yang, Q.; Jiang, L.; Garrett-Bakelman, F.E.; Sekeres, M.A.; Santini, V.; Ceccarelli, M.; Goel, N.; Garcia-Martinez, L.; et al. Genome-wide methylome modeling via generative ai incorporating long- and short-range interactions. Sci. Adv. 2025, 11, eadt4152. [Google Scholar] [CrossRef]
- Ying, K.; Song, J.; Cui, H.; Zhang, Y.; Li, S.; Chen, X.; Liu, H.; Eames, A.; McCartney, D.L.; Marioni, R.E.; et al. Methylgpt: A foundation model for the DNA methylome. bioRxiv 2024. [Google Scholar]
- Wang, Y.; Liu, T.; Xu, D.; Shi, H.; Zhang, C.; Mo, Y.Y.; Wang, Z. Predicting DNA methylation state of cpg dinucleotide using genome topological features and deep networks. Sci. Rep. 2016, 6, 19598. [Google Scholar] [CrossRef]
- Levy, J.J.; Titus, A.J.; Petersen, C.L.; Chen, Y.; Salas, L.A.; Christensen, B.C. Methylnet: An automated and modular deep learning approach for DNA methylation analysis. BMC Bioinform. 2020, 21, 108. [Google Scholar] [CrossRef]
- Levy, J.J.; Chen, Y.; Azizgolshani, N.; Petersen, C.L.; Titus, A.J.; Moen, E.L.; Vaickus, L.J.; Salas, L.A.; Christensen, B.C. Methylspwnet and methylcapsnet: Biologically motivated organization of dnam neural networks, inspired by capsule networks. NPJ Syst. Biol. Appl. 2021, 7, 33. [Google Scholar] [CrossRef]
- Kelley, D.R.; Snoek, J.; Rinn, J.L. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016, 26, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Avsec, Z.; Agarwal, V.; Visentin, D.; Ledsam, J.R.; Grabska-Barwinska, A.; Taylor, K.R.; Assael, Y.; Jumper, J.; Kohli, P.; Kelley, D.R. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 2021, 18, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Uzun, Y.; Zhu, Q.; Chen, C.; Tan, K. Scatac-pro: A comprehensive workbench for single-cell chromatin accessibility sequencing data. Genome Biol. 2020, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Liu, J.; Yang, Y.; Gu, C.; Han, Y.; Wu, B.; Jiang, Y.; Chen, G.; Heng, P.A. Scgraphformer: Unveiling cellular heterogeneity and interactions in scrna-seq data using a scalable graph transformer network. Commun. Biol. 2024, 7, 1463. [Google Scholar] [CrossRef]
- Yin, Q.; Wu, M.; Liu, Q.; Lv, H.; Jiang, R. Deephistone: A deep learning approach to predicting histone modifications. BMC Genom. 2019, 20, 193. [Google Scholar] [CrossRef]
- Wen, W.; Zhong, J.; Zhang, Z.; Jia, L.; Chu, T.; Wang, N.; Danko, C.G.; Wang, Z. Dhica: A deep transformer-based model enables accurate histone imputation from chromatin accessibility. Brief. Bioinform. 2024, 25, bbae459. [Google Scholar] [CrossRef]
- Schuette, G.; Lao, Z.; Zhang, B. Chromogen: Diffusion model predicts single-cell chromatin conformations. Sci. Adv. 2025, 11, eadr8265. [Google Scholar] [CrossRef]
- Wang, Y.; Kong, S.; Zhou, C.; Wang, Y.; Zhang, Y.; Fang, Y.; Li, G. A review of deep learning models for the prediction of chromatin interactions with DNA and epigenomic profiles. Brief. Bioinform. 2024, 26, bbae651. [Google Scholar] [CrossRef]
- Brozek, A.; Theodoris, C.V. Ai learns from chromatin data to uncover gene interactions. Nature 2025, 637, 799–800. [Google Scholar] [CrossRef]
- Saadh, M.J.; Ahmed, H.H.; Kareem, R.A.; Yadav, A.; Ganesan, S.; Shankhyan, A.; Sharma, G.C.; Naidu, K.S.; Rakhmatullaev, A.; Sameer, H.N.; et al. Advanced machine learning framework for enhancing breast cancer diagnostics through transcriptomic profiling. Discov. Oncol. 2025, 16, 334. [Google Scholar] [CrossRef]
- Eraslan, G.; Simon, L.M.; Mircea, M.; Mueller, N.S.; Theis, F.J. Single-cell rna-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef] [PubMed]
- Svensson, V.; Gayoso, A.; Yosef, N.; Pachter, L. Interpretable factor models of single-cell rna-seq via variational autoencoders. Bioinformatics 2020, 36, 3418–3421. [Google Scholar] [CrossRef] [PubMed]
- Gronbech, C.H.; Vording, M.F.; Timshel, P.N.; Sonderby, C.K.; Pers, T.H.; Winther, O. Scvae: Variational auto-encoders for single-cell gene expression data. Bioinformatics 2020, 36, 4415–4422. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Bendl, J.; Girdhar, K.; Schadt, E.E.; Roussos, P. Functional interpretation of genetic variants using deep learning predicts impact on chromatin accessibility and histone modification. Nucleic Acids Res. 2019, 47, 10597–10611. [Google Scholar] [CrossRef]
- Chen, Y.; Sim, A.; Wan, Y.K.; Yeo, K.; Lee, J.J.X.; Ling, M.H.; Love, M.I.; Goke, J. Context-aware transcript quantification from long-read rna-seq data with bambu. Nat. Methods 2023, 20, 1187–1195. [Google Scholar] [CrossRef]
- de Sainte Agathe, J.M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. Spliceai-visual: A free online tool to improve spliceai splicing variant interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef]
- Barbosa, P.; Savisaar, R.; Carmo-Fonseca, M.; Fonseca, A. Computational prediction of human deep intronic variation. Gigascience 2022, 12, giad085. [Google Scholar] [CrossRef]
- Strauch, Y.; Lord, J.; Niranjan, M.; Baralle, D. Ci-spliceai-improving machine learning predictions of disease causing splicing variants using curated alternative splice sites. PLoS ONE 2022, 17, e0269159. [Google Scholar] [CrossRef]
- Chao, K.H.; Mao, A.; Salzberg, S.L.; Pertea, M. Splam: A deep-learning-based splice site predictor that improves spliced alignments. Genome Biol. 2024, 25, 243. [Google Scholar] [CrossRef]
- Erfanian, N.; Heydari, A.A.; Feriz, A.M.; Ianez, P.; Derakhshani, A.; Ghasemigol, M.; Farahpour, M.; Razavi, S.M.; Nasseri, S.; Safarpour, H.; et al. Deep learning applications in single-cell genomics and transcriptomics data analysis. Biomed. Pharmacother. 2023, 165, 115077. [Google Scholar] [CrossRef] [PubMed]
- Gayoso, A.; Steier, Z.; Lopez, R.; Regier, J.; Nazor, K.L.; Streets, A.; Yosef, N. Joint probabilistic modeling of single-cell multi-omic data with totalvi. Nat. Methods 2021, 18, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Qu, H.; Qu, D.; Zhao, M. Trajectory inference with cell-cell interactions (ticci): Intercellular communication improves the accuracy of trajectory inference methods. Bioinformatics 2025, 41, btaf027. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Gorin, G.; Pachter, L. Trajectory inference from single-cell genomics data with a process time model. PLoS Comput. Biol. 2025, 21, e1012752. [Google Scholar] [CrossRef]
- Huguet, G.; Magruder, D.S.; Tong, A.; Fasina, O.; Kuchroo, M.; Wolf, G.; Krishnaswamy, S. Manifold interpolating optimal-transport flows for trajectory inference. Adv. Neural Inf. Process Syst. 2022, 35, 29705–29718. [Google Scholar]
- Smolander, J.; Junttila, S.; Venalainen, M.S.; Elo, L.L. Scshaper: An ensemble method for fast and accurate linear trajectory inference from single-cell rna-seq data. Bioinformatics 2022, 38, 1328–1335. [Google Scholar] [CrossRef]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef]
- Alum, E.U. Ai-driven biomarker discovery: Enhancing precision in cancer diagnosis and prognosis. Discov. Oncol. 2025, 16, 313. [Google Scholar] [CrossRef]
- Unger, M.; Kather, J.N. Deep learning in cancer genomics and histopathology. Genome Med. 2024, 16, 44. [Google Scholar] [CrossRef]
- Tellez-Gabriel, M.; Ory, B.; Lamoureux, F.; Heymann, M.F.; Heymann, D. Tumour heterogeneity: The key advantages of single-cell analysis. Int. J. Mol. Sci. 2016, 17, 2142. [Google Scholar] [CrossRef]
- Gillis, S.; Roth, A. Pyclone-vi: Scalable inference of clonal population structures using whole genome data. BMC Bioinform. 2020, 21, 571. [Google Scholar] [CrossRef] [PubMed]
- Lu, B. Cancer phylogenetic inference using copy number alterations detected from DNA sequencing data. Cancer Pathog. Ther. 2025, 3, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Lee, H.O.; Lee, S.; Ryu, D.E.; Lee, S.; Xue, C.; Kim, S.J.; Kim, K.; Barkas, N.; Park, P.J.; et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell rna-seq data. Genome Res. 2018, 28, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of t cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Nguyen, L.; Van Hoeck, A.; Cuppen, E. Machine learning-based tissue of origin classification for cancer of unknown primary diagnostics using genome-wide mutation features. Nat. Commun. 2022, 13, 4013. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, Z.; Namburi, S.; Pattison, A.; Posner, A.; Balachander, S.; Paisie, C.A.; Reddi, H.V.; Rueter, J.; Gill, A.J.; et al. Cup-ai-dx: A tool for inferring cancer tissue of origin and molecular subtype using rna gene-expression data and artificial intelligence. EBioMedicine 2020, 61, 103030. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, L. Ai-driven multi-omics integration for multi-scale predictive modeling of genotype-environment-phenotype relationships. Comput. Struct. Biotechnol. J. 2025, 27, 265–277. [Google Scholar] [CrossRef]
- Taroni, J.N.; Grayson, P.C.; Hu, Q.; Eddy, S.; Kretzler, M.; Merkel, P.A.; Greene, C.S. Multiplier: A transfer learning framework for transcriptomics reveals systemic features of rare disease. Cell Syst. 2019, 8, 380–394.e4. [Google Scholar] [CrossRef]
- Wu, K.E.; Yost, K.E.; Chang, H.Y.; Zou, J. Babel enables cross-modality translation between multiomic profiles at single-cell resolution. Proc. Natl. Acad. Sci. USA 2021, 118, e2023070118. [Google Scholar] [CrossRef]
- Truesdell, P.; Chang, J.; Coto Villa, D.; Dai, M.; Zhao, Y.; McIlwain, R.; Young, S.; Hiley, S.; Craig, A.W.; Babak, T. Pharmacogenomic discovery of genetically targeted cancer therapies optimized against clinical outcomes. NPJ Precis. Oncol. 2024, 8, 186. [Google Scholar] [CrossRef]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-nnet: An artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar] [CrossRef] [PubMed]
- Mhandire, D.Z.; Goey, A.K.L. The value of pharmacogenetics to reduce drug-related toxicity in cancer patients. Mol. Diagn. Ther. 2022, 26, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Guo, H.; Zhao, Y.; Liu, Z.; Wang, C.; Bu, J.; Sun, T.; Wei, J. Liquid biopsy in cancer current: Status, challenges and future prospects. Signal Transduct. Target. Ther. 2024, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Norouzkhani, N.; Mobaraki, H.; Varmazyar, S.; Zaboli, H.; Mohamadi, Z.; Nikeghbali, G.; Bagheri, K.; Marivany, N.; Najafi, M.; Nozad Varjovi, M.; et al. Artificial intelligence networks for assessing the prognosis of gastrointestinal cancer to immunotherapy based on genetic mutation features: A systematic review and meta-analysis. BMC Gastroenterol. 2025, 25, 310. [Google Scholar] [CrossRef]
- Kong, J.; Zhao, X.; Singhal, A.; Park, S.; Bachelder, R.; Shen, J.; Zhang, H.; Moon, J.; Ahn, C.; Ock, C.Y.; et al. Prediction of immunotherapy response using mutations to cancer protein assemblies. Sci. Adv. 2024, 10, eado9746. [Google Scholar] [CrossRef]
- You, Y.; Lai, X.; Pan, Y.; Zheng, H.; Vera, J.; Liu, S.; Deng, S.; Zhang, L. Artificial intelligence in cancer target identification and drug discovery. Signal Transduct. Target. Ther. 2022, 7, 156. [Google Scholar] [CrossRef]
- Huang, K.; Xiao, C.; Glass, L.M.; Sun, J. Moltrans: Molecular interaction transformer for drug-target interaction prediction. Bioinformatics 2021, 37, 830–836. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. Deepdr: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Kuru, H.I.; Tastan, O.; Cicek, A.E. Matchmaker: A deep learning framework for drug synergy prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2334–2344. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. Deeppurpose: A deep learning library for drug-target interaction prediction. Bioinformatics 2021, 36, 5545–5547. [Google Scholar] [CrossRef]
- Duan, X.P.; Qin, B.D.; Jiao, X.D.; Liu, K.; Wang, Z.; Zang, Y.S. New clinical trial design in precision medicine: Discovery, development and direction. Signal Transduct. Target. Ther. 2024, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xing, Y.; Sun, K.; Guo, Y. Omiembed: A unified multi-task deep learning framework for multi-omics data. Cancers 2021, 13, 3047. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Li, X.; Coleman, K.; Schroeder, A.; Ma, N.; Irwin, D.J.; Lee, E.B.; Shinohara, R.T.; Li, M. Spagcn: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 2021, 18, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Jiang, S.; Zhang, S.; Xu, M.; Jia, X. Graph-in-graph convolutional network for hyperspectral image classification. IEEE Trans. Neural Netw. Learn. Syst. 2022, 35, 1157–1171. [Google Scholar] [CrossRef]
- Wang, T.; Shao, W.; Huang, Z.; Tang, H.; Zhang, J.; Ding, Z.; Huang, K. Mogonet integrates multi-omics data using graph convolutional networks allowing patient classification and biomarker identification. Nat. Commun. 2021, 12, 3445. [Google Scholar] [CrossRef]
- Zhang, L.; Song, R.; Tan, W.; Ma, L.; Zhang, W. Igcn: A provably informative gcn embedding for semi-supervised learning with extremely limited labels. IEEE Trans. Pattern Anal. Mach. Intell. 2024, 46, 8396–8409. [Google Scholar] [CrossRef]
- Tran, A.T.; Zeevi, T.; Payabvash, S. Strategies to improve the robustness and generalizability of deep learning segmentation and classification in neuroimaging. BioMedInformatics 2025, 5, 20. [Google Scholar] [CrossRef]
- Soliman, A.; Li, Z.; Parwani, A.V. Artificial intelligence’s impact on breast cancer pathology: A literature review. Diagn. Pathol. 2024, 19, 38. [Google Scholar] [CrossRef]
- Minton, K. Predicting variant pathogenicity with alphamissense. Nat. Rev. Genet. 2023, 24, 804. [Google Scholar] [CrossRef]
- Caswell, R.C.; Gunning, A.C.; Owens, M.M.; Ellard, S.; Wright, C.F. Assessing the clinical utility of protein structural analysis in genomic variant classification: Experiences from a diagnostic laboratory. Genome Med. 2022, 14, 77. [Google Scholar] [CrossRef]
- Quazi, S. Artificial intelligence and machine learning in precision and genomic medicine. Med. Oncol. 2022, 39, 120. [Google Scholar] [CrossRef] [PubMed]
- Wadden, J.J. Defining the undefinable: The black box problem in healthcare artificial intelligence. J. Med. Ethics 2021, 48, 107529. [Google Scholar] [CrossRef] [PubMed]
- Linardatos, P.; Papastefanopoulos, V.; Kotsiantis, S. Explainable ai: A review of machine learning interpretability methods. Entropy 2020, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Vadapalli, S.; Abdelhalim, H.; Zeeshan, S.; Ahmed, Z. Artificial intelligence and machine learning approaches using gene expression and variant data for personalized medicine. Brief. Bioinform. 2022, 23, bbac191. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein data bank (pdb): The single global macromolecular structure archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar]
- Karuppasamy, M.P.; Venkateswaran, S.; Subbiah, P. Pdb-2-pbv3.0: An updated protein block database. J. Bioinform. Comput. Biol. 2020, 18, 2050009. [Google Scholar] [CrossRef]
- Reiff, S.B.; Schroeder, A.J.; Kirli, K.; Cosolo, A.; Bakker, C.; Mercado, L.; Lee, S.; Veit, A.D.; Balashov, A.K.; Vitzthum, C.; et al. The 4d nucleome data portal as a resource for searching and visualizing curated nucleomics data. Nat. Commun. 2022, 13, 2365. [Google Scholar] [CrossRef]
- Oluwadare, O.; Highsmith, M.; Turner, D.; Lieberman Aiden, E.; Cheng, J. Gsdb: A database of 3D chromosome and genome structures reconstructed from hi-c data. BMC Mol. Cell Biol. 2020, 21, 60. [Google Scholar] [CrossRef]
- Li, C.; Dong, X.; Fan, H.; Wang, C.; Ding, G.; Li, Y. The 3DGD: A database of genome 3D structure. Bioinformatics 2014, 30, 1640–1642. [Google Scholar] [CrossRef]
- Haripriya, R.; Khare, N.; Pandey, M. Privacy-preserving federated learning for collaborative medical data mining in multi-institutional settings. Sci. Rep. 2025, 15, 12482. [Google Scholar] [CrossRef]
- Cesaro, A.; Hoffman, S.C.; Das, P.; de la Fuente-Nunez, C. Challenges and applications of artificial intelligence in infectious diseases and antimicrobial resistance. NPJ Antimicrob. Resist. 2025, 3, 2. [Google Scholar] [CrossRef]
- Acosta, J.N.; Falcone, G.J.; Rajpurkar, P.; Topol, E.J. Multimodal biomedical ai. Nat. Med. 2022, 28, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Gangwal, A.; Ansari, A.; Ahmad, I.; Azad, A.K.; Kumarasamy, V.; Subramaniyan, V.; Wong, L.S. Generative artificial intelligence in drug discovery: Basic framework, recent advances, challenges, and opportunities. Front. Pharmacol. 2024, 15, 1331062. [Google Scholar] [CrossRef] [PubMed]
- Goktas, P.; Grzybowski, A. Shaping the future of healthcare: Ethical clinical challenges and pathways to trustworthy ai. J. Clin. Med. 2025, 14, 1605. [Google Scholar] [CrossRef]
- Scarano, C.; Veneruso, I.; De Simone, R.R.; Di Bonito, G.; Secondino, A.; D’Argenio, V. The third-generation sequencing challenge: Novel insights for the omic sciences. Biomolecules 2024, 14, 568. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Boza, V.; Brejova, B.; Vinar, T. Deepnano: Deep recurrent neural networks for base calling in minion nanopore reads. PLoS ONE 2017, 12, e0178751. [Google Scholar] [CrossRef]
- Wan, Y.K.; Hendra, C.; Pratanwanich, P.N.; Goke, J. Beyond sequencing: Machine learning algorithms extract biology hidden in nanopore signal data. Trends Genet. 2022, 38, 246–257. [Google Scholar] [CrossRef]
- Pages-Gallego, M.; de Ridder, J. Comprehensive benchmark and architectural analysis of deep learning models for nanopore sequencing basecalling. Genome Biol. 2023, 24, 71. [Google Scholar] [CrossRef] [PubMed]
- Baid, G.; Cook, D.E.; Shafin, K.; Yun, T.; Llinares-López, F.; Berthet, Q.; Belyaeva, A.; Töpfer, A.; Wenger, A.M.; Rowell, W.J.; et al. Deepconsensus improves the accuracy of sequences with a gap-aware sequence transformer. Nat. Biotechnol. 2023, 41, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.U.; Liu, Q.; Perdomo, J.E.; Fang, L.; Wang, K. A survey of algorithms for the detection of genomic structural variants from long-read sequencing data. Nat. Methods 2023, 20, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Yu, X.; Chen, L.; Lee, Y.L.; Xin, C.; Wong, A.O.K.; Jain, M.; Kesharwani, R.K.; Sedlazeck, F.J.; Luo, R. Clair3-rna: A deep learning-based small variant caller for long-read rna sequencing data. bioRxiv 2025. [Google Scholar]
- Senol Cali, D.; Kim, J.S.; Ghose, S.; Alkan, C.; Mutlu, O. Nanopore sequencing technology and tools for genome assembly: Computational analysis of the current state, bottlenecks and future directions. Brief. Bioinform. 2019, 20, 1542–1559. [Google Scholar] [CrossRef]
- Ni, P.; Huang, N.; Zhang, Z.; Wang, D.P.; Liang, F.; Miao, Y.; Xiao, C.L.; Luo, F.; Wang, J. Deepsignal: Detecting DNA methylation state from nanopore sequencing reads using deep-learning. Bioinformatics 2019, 35, 4586–4595. [Google Scholar] [CrossRef]
- McIntyre, A.B.R.; Alexander, N.; Grigorev, K.; Bezdan, D.; Sichtig, H.; Chiu, C.Y.; Mason, C.E. Single-molecule sequencing detection of n6-methyladenine in microbial reference materials. Nat. Commun. 2019, 10, 579. [Google Scholar] [CrossRef]
- Rand, A.C.; Jain, M.; Eizenga, J.M.; Musselman-Brown, A.; Olsen, H.E.; Akeson, M.; Paten, B. Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods 2017, 14, 411–413. [Google Scholar] [CrossRef]
- Lorenz, D.A.; Sathe, S.; Einstein, J.M.; Yeo, G.W. Direct rna sequencing enables m(6)a detection in endogenous transcript isoforms at base-specific resolution. RNA 2020, 26, 19–28. [Google Scholar] [CrossRef]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m(6)a rna modifications in native rna sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, X.; Wu, B.; Wang, H.; Xi, F.; Kohnen, M.V.; Reddy, A.S.N.; Gu, L. Quantitative profiling of n(6)-methyladenosine at single-base resolution in stem-differentiating xylem of populus trichocarpa using nanopore direct rna sequencing. Genome Biol. 2021, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. Rna modifications detection by comparative nanopore direct rna sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H. Nanodoc: Rna modification detection using nanopore raw reads with deep one-class classification. bioRxiv 2021. [Google Scholar]
- Pratanwanich, P.N.; Yao, F.; Chen, Y.; Koh, C.W.Q.; Wan, Y.K.; Hendra, C.; Poon, P.; Goh, Y.T.; Yap, P.M.L.; Chooi, J.Y.; et al. Identification of differential rna modifications from nanopore direct rna sequencing with xpore. Nat. Biotechnol. 2021, 39, 1394–1402. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Pipeline | Application | Features | Reference |
|---|---|---|---|
| nf-core/circrna | circRNA and miRNA analysis | circRNA quantification, miRNA target prediction, differential analysis | [45] |
| nf-rnaSeqCount | RNA-seq quantification | QC, alignment, count quantification, MultiQC reporting | [46] |
| GeneTEFlow | Gene + TE expression | STAR/RSEM + SQuIRE quantification, DE analysis, Docker containerization | [47] |
| scATACpipe | scATAC-seq processing | Preprocessing, fragment/BED/BAM outputs, ArchR clustering, interactive HTML report | [48] |
| polishCLR | Genome assembly polishing | PacBio CLR polishing with Illumina data, haplotig purging, outputs quality-checked assemblies and logs | [49] |
| nf-gwas-pipeline | GWAS analysis | Automates genotype QC, population structure correction, association, and visualization | [50] |
| StellarPGx | Pharmacogenomics | CYP star allele calling, variant annotation, phasing and clinical interpretation | [51] |
| NFTest | Pipeline testing | Automated functional testing of Nextflow workflows using synthetic test cases | [52] |
| PoSeiDon | Positive selection and recombination analysis | Runs PAML CODEML for detecting positive selection and recombination in nucleotide alignments; configurable for HPC/Singularity deployments | [53] |
| Pipeliner | Sequencing data preprocessing | Modular processing for bulk and single-cell RNA-Seq; leverages Nextflow + Conda for reproducible workflows | [54] |
| FA-nf | Functional annotation | Nextflow-based functional annotation of novel genomes using Pfam, GO terms, database scaffolding | [55] |
| Geniac | Nextflow add-on | Auto-generates config files + containers; linter enforces standardized outputs | [56] |
| DolphinNext | Workflow management | GUI platform built on Nextflow; drag-and-drop pipeline design, monitoring, containerized reproducibility | [57] |
| NGS/TGS Application | Tool/Platform | Functionality | Algorithm Type |
|---|---|---|---|
| Variant Calling | DeepVariant | SNP and indel calling | CNN |
| DNAscope | High-accuracy genotyping | ML | |
| Clair3, Clairvoyante | Long-read variant calling | CNN | |
| Clair3-RNA | Small variant caller for long-read RNA sequencing data | DL | |
| PEPPER-Margin-DeepVariant | Alignment and consensus-based variant calling | DL | |
| ClinPred | Pathogenicity prediction for missense variants | Ensemble (XGBoost + RF) | |
| REVEL | Aggregated predictions for the pathogenicity of variant effects | Ensemble | |
| DeepFilter | Variant call filtering | DL | |
| Splicing Prediction | SpliceAI | Splice-disruptive variants prediction | CNN |
| Splam | Splice junctions in DNA prediction | DL/CNN | |
| Transcriptomics | Bambu | Transcript discovery and quantification from long-read RNA-Seq data | ML |
| Deep Count Autoencoder (DCA) | Gene expression denoising | DL | |
| scVI/scVAE | Batch correction, embedding, differential expression | DL | |
| MultiPLIER | Interpretable feature extraction across large transcriptomic cohorts | ML | |
| Cox-nnet | Patient prognosis prediction from high-throughput RNA-Seq data | ANN | |
| BABEL | Cross-modality translation between multi-omic profiles | DL | |
| Genomics | DNABERT | Genome-wide prediction of promoters, splice sites, and transcription factor binding sites | Bidirectional encoder representation from transformers |
| Methylation Analysis | MethylNet | DNA methylation prediction | DL |
| MethylSPWNet | Classification of CpGs into biologically relevant capsules | DL | |
| DeepMethyl | CpG methylation prediction | DL | |
| DiffuCpG | Methylation imputation | DL | |
| MethylGPT | Methylation value prediction | DL | |
| Chromatin Accessibility | Basset | Prediction of accessible chromatin regions | CNN |
| Enformer | Prediction of variant effects on gene expression | DL | |
| scATAC-pro | Quality assessment, analysis, and visualization of single-cell chromatin accessibility sequencing data | VAE | |
| Histone Modifications | DeepHistone | Histone modification patterns prediction | NN/DL |
| dHICA | Histone mark imputation and prediction of their modifications | DL | |
| Single-Cell Analysis | CopyKAT | CNV inference from scRNA-seq | Integrative Bayesian segmentation approach |
| scGraphformer | Unveiling cellular heterogeneity and interactions in scRNA-seq data | Transformer-based GNN | |
| TotalVI | Multi-modal data analysis/joint analysis of CITE-seq data | VAE | |
| scANVI | Cell state/transcriptomics data annotation | DL | |
| scShaper | Accurate linear trajectory inference | Ensemble | |
| 3D Genome Structure | ChromoGen | Single-cell chromatin conformation modeling | Generative model |
| Cancer Research | PyClone-VI | Inference of clonal population structures using whole genome data | Bayesian statistical method |
| HoneyBADGER | Identification of CNVs and heterozygosity loss in individual cells from single-cell RNA-Seq | HMM-integrated Bayesian hierarchical model | |
| CUPLR | Tissue of origin classification for cancer of unknown primary diagnostics | ML | |
| CUP-AI-DX | Inference of cancer tissue of origin and molecular subtyping using gene expression data | CNN | |
| Drug Discovery | DeepDR | Drug repurposing | DL |
| DeepPurpose | Drug–target interaction prediction | DL | |
| MolTrans | Drug–target interaction prediction | DL | |
| MatchMaker | Drug synergy prediction | DL | |
| Clinical Diagnostics | AlphaMissense | Variant pathogenicity prediction | ML |
| Multi-Omics Integration | OmiEmbed | Multi-omics data analysis | DL |
| MOGONET | Patient classification and biomarker identification | GNN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athanasopoulou, K.; Michalopoulou, V.-I.; Scorilas, A.; Adamopoulos, P.G. Integrating Artificial Intelligence in Next-Generation Sequencing: Advances, Challenges, and Future Directions. Curr. Issues Mol. Biol. 2025, 47, 470. https://doi.org/10.3390/cimb47060470
Athanasopoulou K, Michalopoulou V-I, Scorilas A, Adamopoulos PG. Integrating Artificial Intelligence in Next-Generation Sequencing: Advances, Challenges, and Future Directions. Current Issues in Molecular Biology. 2025; 47(6):470. https://doi.org/10.3390/cimb47060470
Chicago/Turabian StyleAthanasopoulou, Konstantina, Vasiliki-Ioanna Michalopoulou, Andreas Scorilas, and Panagiotis G. Adamopoulos. 2025. "Integrating Artificial Intelligence in Next-Generation Sequencing: Advances, Challenges, and Future Directions" Current Issues in Molecular Biology 47, no. 6: 470. https://doi.org/10.3390/cimb47060470
APA StyleAthanasopoulou, K., Michalopoulou, V.-I., Scorilas, A., & Adamopoulos, P. G. (2025). Integrating Artificial Intelligence in Next-Generation Sequencing: Advances, Challenges, and Future Directions. Current Issues in Molecular Biology, 47(6), 470. https://doi.org/10.3390/cimb47060470