
Bioinformatic RNA-Seq Functional Profiling of the Tumor Suppressor Gene OPCML in Ovarian Cancers: The Multifunctional, Pleiotropic Impacts of Having Three Ig Domains



Abstract

1. Introduction
2. Materials and Methods
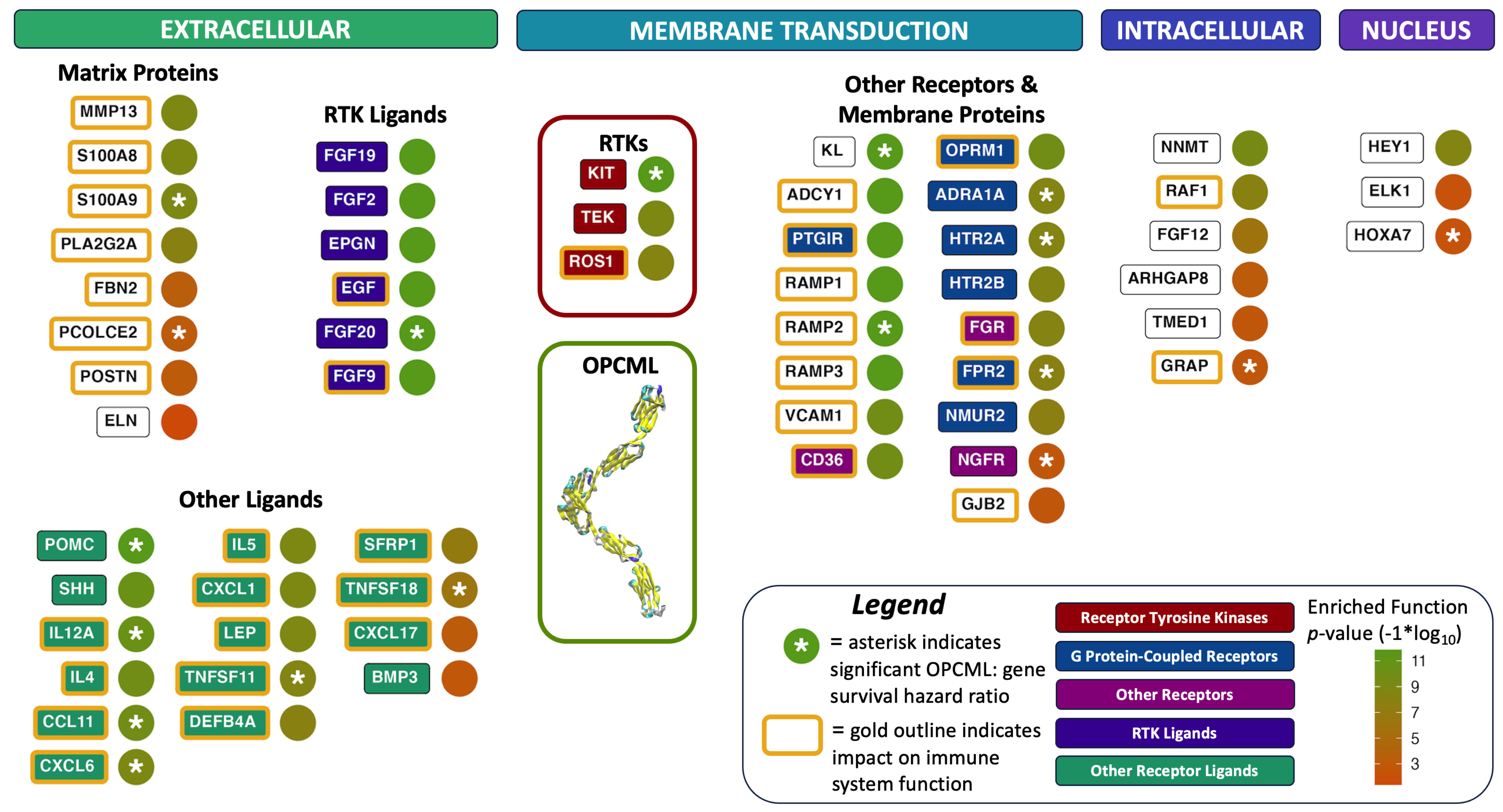
3. Results
3.1. Novel Computational Approach
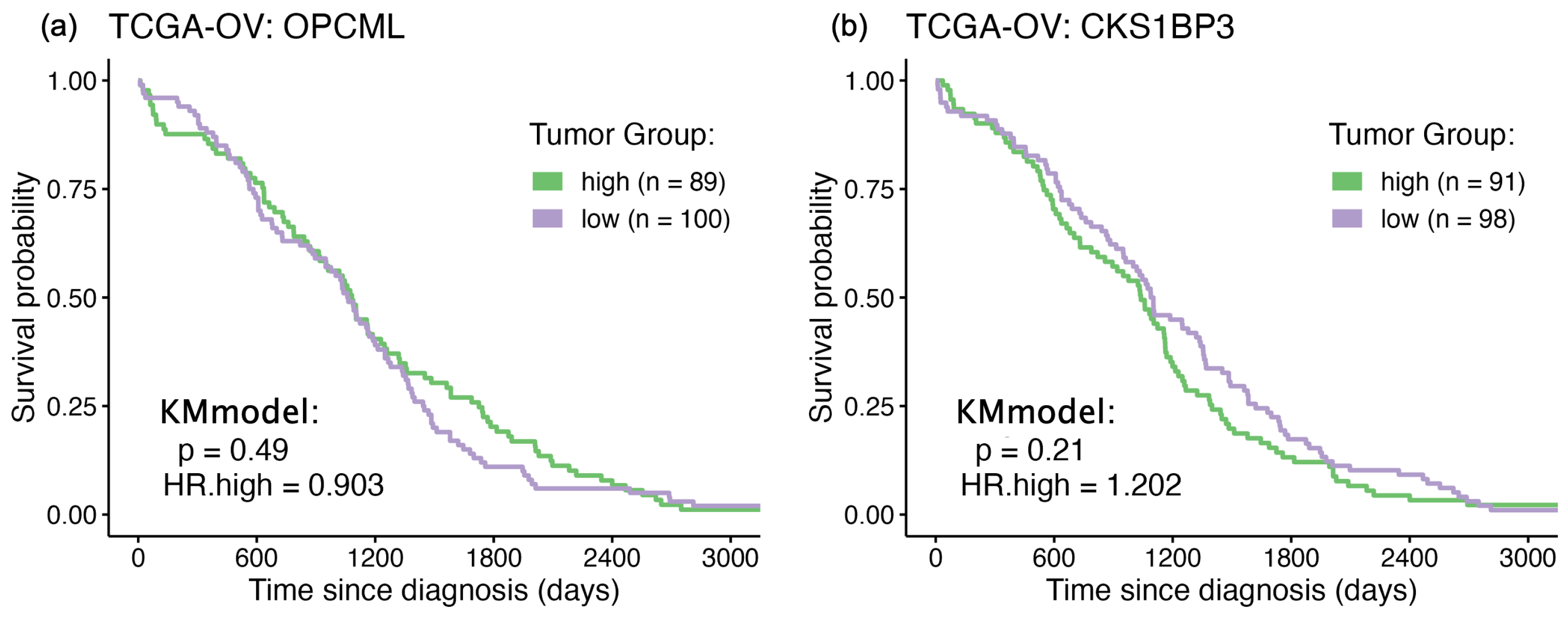
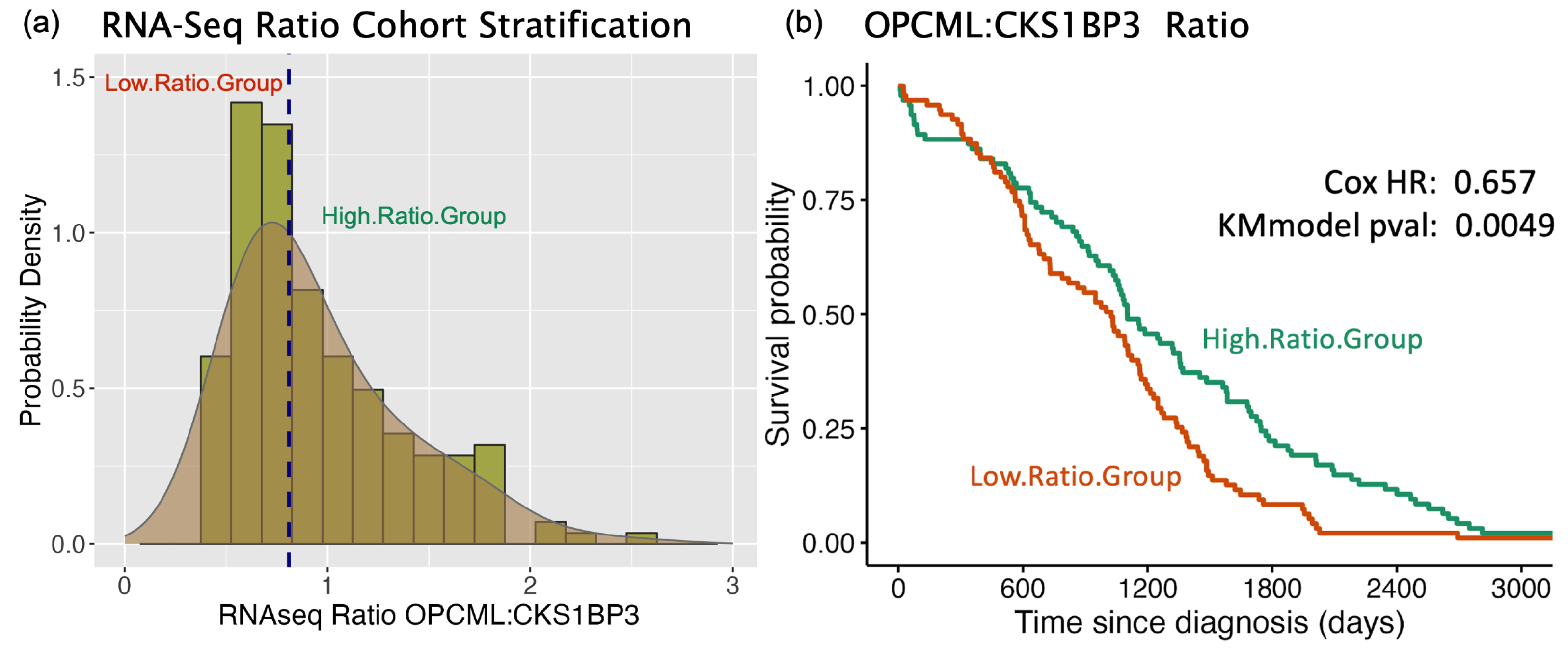
3.2. Survival Curve HR Analyses
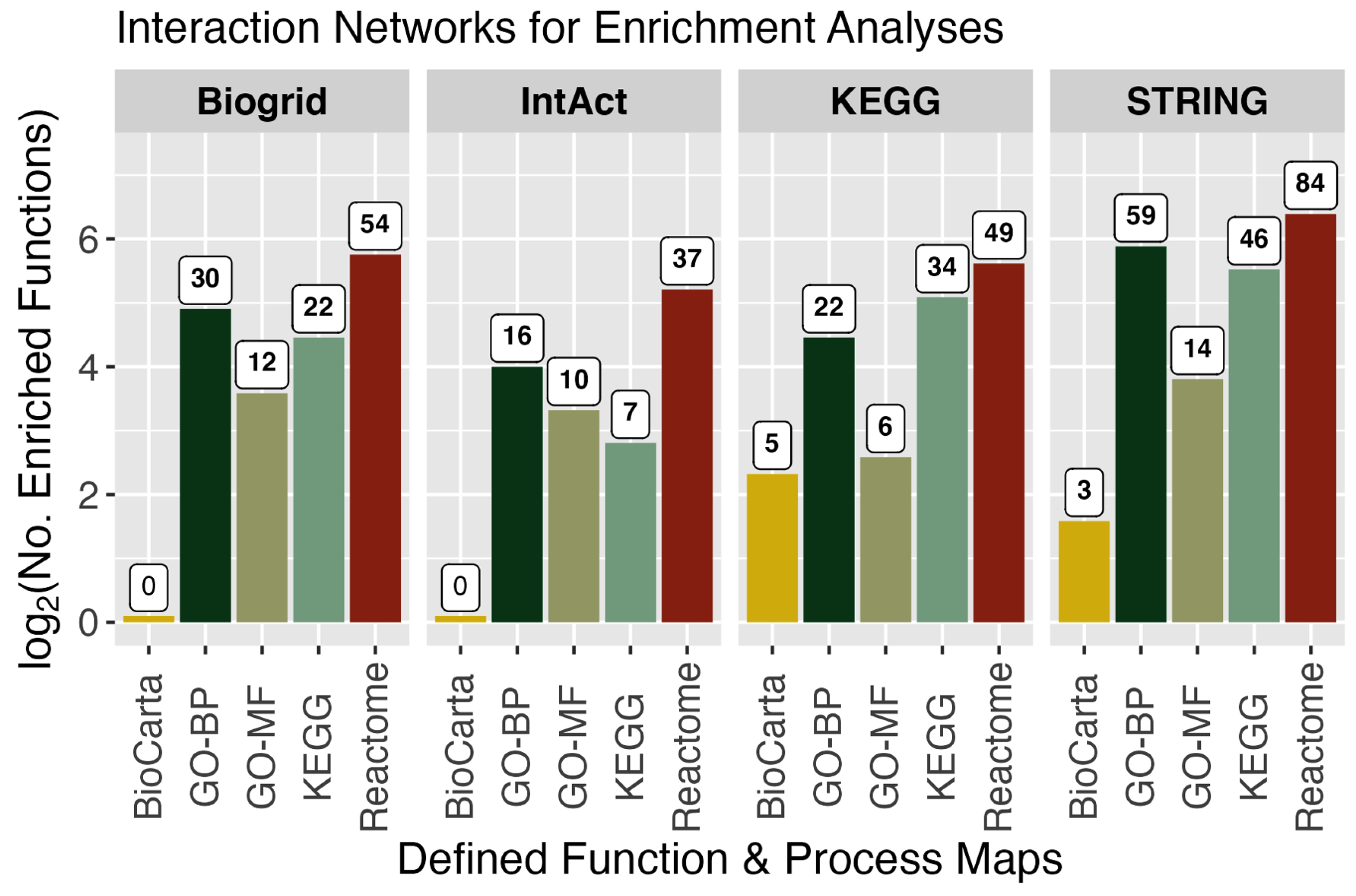
3.3. Functional Enrichment Analyses
3.4. Validation
4. Discussion
4.1. RTKs, Ligands, and Signaling
4.2. OPCML: Multifunctional Tumor Suppressor
4.3. Translational Application of Results
4.4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TSG | tumor suppressor gene |
| OPCML | opioid binding and cell adhesion molecule |
| OV | ovarian cancer |
| HR | Cox hazard ratios |
| HRz | Z-score transformed hazard ratios |
| Ig | immunoglobin binding domain |
| GO | Gene Ontology Database |
| GSEA | Gene Set Enrichment Analysis Database |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| FDR | false discovery rate |
| RTK | receptor tyrosine kinase signaling pathways |
Appendix A
Appendix A.1. Hierarchical Clustering Results
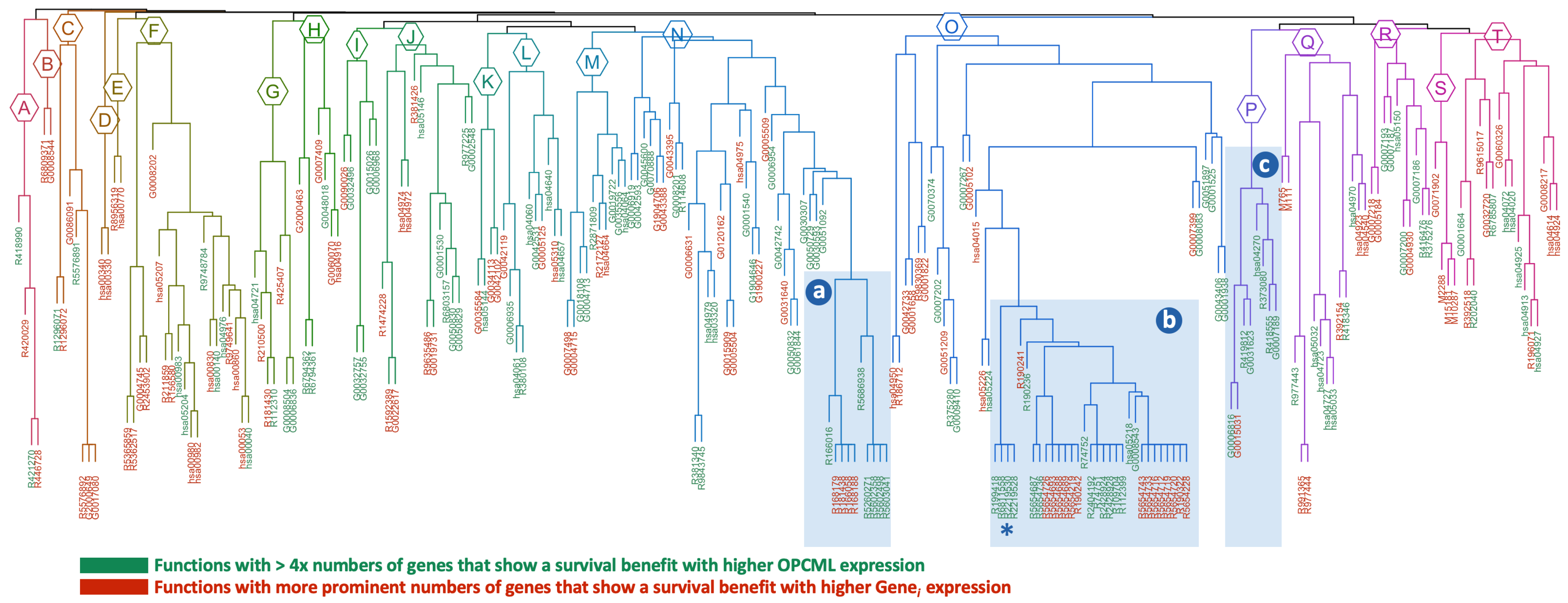
Appendix A.2. Validation Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA-OV | Validation | ||||
|---|---|---|---|---|---|
| Gene | Entrez ID | HR | p-Value | Source | Description |
| KIT | 3815 | 0.644 | 0.0154 | GSE26712 | KIT proto-oncogene, receptor tyrosine kinase |
| TEK | 7010 | 0.520 | 0.0247 | GSE26712 | TEK proto-oncogene, receptor tyrosine kinase |
| FGF2 | 2247 | 0.40 | 0.0153 | GSE19829 | Fibroblast growth factor 2 |
| POSTN | 10631 | 0.53 | 1.20 | GSE9891 | Periostin, extracellular matrix protein |
| MMP13 | 4322 | 0.82 | 0.0026 | GSE9891 | Matrix metallopeptidase 13 |
| PPARG | 5468 | 0.11 | 9.00 | GSE19829 | Peroxisome proliferator activated receptor |
| CKS1BP3 | 246715 | 0.51 | 0.0099 | GSE14764 | CDC28 Protein Kinase Regulatory Subunit BP3 |
| ANGPTL4 | 51129 | 0.42 | 0.0452 | GSE19829 | Angiopoietin like 4 |
| ALX4 | 60529 | 1.526 | 0.0184 | GSE26712 | ALX homeobox 4 |
| CRYBB1 | 1414 | 1.473 | 0.0305 | GSE26712 | Crystallin beta B1 |
| ADRA1A | 148 | 1.803 | 0.0013 | GSE26712 | Adrenoceptor alpha 1A |
| SSX1 | 6756 | 0.69 | 0.0389 | GSE26712 | SSX family member 1 |
| ASCL1 | 429 | 0.672 | 0.0258 | GSE26712 | Achaete-scute family bHLH transcription |
| PCOLCE2 | 26577 | 0.68 | 0.0309 | GSE26712 | Procollagen C-endopeptidase enhancer 2 |
| TNFRSF | 8794 | 1.683 | 0.0045 | GSE26712 | TNF receptor superfamily member 10c |
| CXCR2 | 3579 | 1.481 | 0.0296 | GSE26712 | C-X-C motif chemokine receptor 2 |
| GJA4 | 2701 | 0.629 | 0.0121 | GSE26712 | Gap junction protein alpha 4 |
| S100A11P1 | 10491 | 0.633 | 0.014 | GSE26712 | S100A11 pseudogene 1 |
References
- Wilson, D.; Kim, D.; Clarke, G.; MarshallClarke, S.; Moss, D. A family of glycoproteins (GP55), which inhibit neurite outgrowth, are members of the Ig superfamily and are related to OBCAM, neurotrimin, LAMP and CEPU-1. J. Cell Sci. 1996, 109, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Hancox, K.; Gooley, A.; Jeffrey, P. AvGp50, a predominantly axonally expressed glycoprotein, is a member of the IgLON’s subfamily of cell adhesion molecules (CAMs). Mol. Brain Res. 1997, 44, 273–285. [Google Scholar] [CrossRef]
- Sellar, G.; Watt, K.; Rabiasz, G.; Stronach, E.; Li, L.; Miller, E.; Massie, C.; Miller, J.; Contreras-Moreira, B.; Scott, D.; et al. OPCML at 11q25 is epigenetically inactivated and has tumor-suppressor function in epithelial ovarian cancer. Nat. Genet. 2003, 34, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rabiasz, G.; Scott, D.; Miller, E.; Stronach, E.; Taylor, K.; Ntougkos, E.; Gabra, H.; Smyth, J.; Sellar, G. Microarray analysis of OPCML tumour suppressor function in the SKOV-3 ovarian cancer cell line. Br. J. Cancer 2004, 91, S54. [Google Scholar]
- Ntougkos, E.; Rush, R.; Scott, D.; Frankenberg, T.; Gabra, H.; Smyth, J.; Sellar, G. The IgLON family in epithelial ovarian cancer: Expression profiles and clinicopathologic correlates. Clin. Cancer Res. 2005, 11, 5764–5768. [Google Scholar] [CrossRef]
- Teodoridis, J.; Hall, J.; Marsh, S.; Kannall, H.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005, 65, 8961–8967. [Google Scholar] [CrossRef]
- Wu, S.Y.; Sood, A.K. New Roles Opined for OPCML. Cancer Discov. 2012, 2, 115–116. [Google Scholar] [CrossRef]
- Zanini, E.; Louis, L.S.; Antony, J.; Karali, E.; Okon, I.S.; Mckie, A.B.; Vaughan, S.; El-Bahrawy, M.; Stebbing, J.; Recchi, C.; et al. The Tumor-Suppressor Protein OPCML Potentiates Anti-EGFR- and Anti-HER2-Targeted Therapy in HER2-Positive Ovarian and 594 Breast Cancer. Mol. Cancer Ther. 2017, 16, 2246–2256. [Google Scholar] [CrossRef]
- Antony, J.; Zanini, E.; Kelly, Z.; Tan, T.Z.; Karali, E.; Alomary, M.; Jung, Y.; Nixon, K.; Cunnea, P.; Fotopoulou, C.; et al. The tumour suppressor OPCML promotes AXL inactivation by the phosphatase PTPRG in ovarian cancer. EMBO Rep. 2018, 19, e45670. [Google Scholar] [CrossRef]
- McKie, A.B.; Vaughan, S.; Zanini, E.; Okon, I.S.; Louis, L.; de Sousa, C.; Greene, M.I.; Wang, Q.; Agarwal, R.; Shaposhnikov, D.; et al. The OPCML Tumor Suppressor Functions as a Cell Surface Repressor-Adaptor, Negatively Regulating Receptor Tyrosine Kinases in Epithelial Ovarian Cancer. Cancer Discov. 2012, 2, 156–171. [Google Scholar] [CrossRef]
- Birtley, J.R.; Alomary, M.; Zanini, E.; Antony, J.; Maben, Z.; Weaver, G.C.; Von Arx, C.; Mura, M.; Marinho, A.T.; Lu, H.; et al. Inactivating mutations and X-ray crystal structure of the tumor suppressor OPCML reveal cancer-associated functions. Nat. Commun. 2019, 10, 3134. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Zanini, E.; Birtley, J.R.; Gabra, H.; Recchi, C. Emerging roles for the GPI-anchored tumor suppressor OPCML in cancers. Cancer Gene Ther. 2021, 28, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef]
- Cao, X.; Wu, B.; Hertz, M.I. Empirical null distribution-based modeling of multi-class differential gene expression detection. J. Appl. Stat. 2013, 40, 347–357. [Google Scholar] [CrossRef]
- Gauran, I.I.M.; Park, J.; Lim, J.; Park, D.; Zylstra, J.; Peterson, T.; Kann, M.; Spouge, J.L. Empirical null estimation using zero-inflated discrete mixture distributions and its application to protein domain data. Biometrics 2018, 74, 458–471. [Google Scholar] [CrossRef]
- Ulgen, E.; Ozisik, O.; Sezerman, O.U. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data Through Active Subnetworks. Front. Genet. 2019, 10, 858. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Tanabe, S.; Quader, S.; Ono, R.; Cabral, H.; Aoyagi, K.; Hirose, A.; Yokozaki, H.; Sasaki, H. Molecular Network Profiling in Intestinal- and Diffuse-Type Gastric Cancer. Cancers 2020, 12, 3833. [Google Scholar] [CrossRef]
- Garelja, M.L.; Au, M.; Brimble, M.A.; Gingell, J.J.; Hendrikse, E.R.; Lovell, A.; Prodan, N.; Sexton, P.M.; Siow, A.; Walker, C.S.; et al. Molecular Mechanisms of Class B GPCR Activation: Insights from Adrenomedullin Receptors. ACS Pharmacol. Transl. Sci. 2020, 3, 246–262. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Jarvia, N.L.; Balu-Iyera, S.V. Immunogenicity of Therapeutic Biological Modalities- Lessons from Hemophilia A Therapies. J. Pharm. Sci. 2023, 112, 2347–2370. [Google Scholar] [CrossRef] [PubMed]
- Schaible, P. Modifying enzyme replacement therapy—A perspective. J. Cell. Mol. Med. 2023, 27, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Faggiano, S.; Mozzarelli, A. Enzyme Replacement Therapy for Genetic Disorders Associated with Enzyme Deficiency. Curr. Med. Chem. 2022, 29, 489–525. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Wade, J.D. Current challenges in peptide-based drug discovery. Front. Chem. 2014, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Khamko, R.; Wasenang, W.; Daduang, J.; Settasatian, C.; Limpaiboon, T. Combined OPCML and AXL Expression as a Prognostic Marker and OPCML Enhances AXL Inhibitor in Cholangiocarcinoma. In Vivo 2022, 36, 1168–1177. [Google Scholar] [CrossRef]
- Tonti, N.; Golia D’Auge, T.; Cuccu, I.; De Angelis, E.; D’Oria, O.; Perniola, G.; Lagana, A.S.; Etrusco, A.; Ferrari, F.; Saponara, S.; et al. The Role of Tumor Biomarkers in Tailoring the Approach to Advanced Ovarian Cancer. Int. J. Mol. Sci. 2024, 25, 11239. [Google Scholar] [CrossRef]
- D’Auge, T.G.; Giannini, A.; Bogani, G.; Di Dio, C.; Lagana, A.S.; Di Donato, V.; Salerno, M.G.; Caserta, D.; Chiantera, V.; Vizza, E.; et al. Prevention, Screening, Treatment and Follow-Up of Gynecological Cancers: State of Art and Future Perspectives. Clin. Exp. Obstet. Gynecol. 2023, 50, 160. [Google Scholar] [CrossRef]


| ID | p-Value | ×Fold | Survival Group | Description |
|---|---|---|---|---|
| a: cluster 16 (n = 10) | ||||
| R5602498 | 1.51 | 6.48 | OPCML+ | MyD88 deficiency (TLR2/4) |
| R5603041 | 1.51 | 6.07 | OPCML+ | IRAK4 deficiency (TLR2/4) |
| R5686938 | 3.58 | 4.86 | OPCML+ | Regulation of TLR by endogenous ligand |
| R166016 | 1.18 | 1.71 | OPCML+ | Toll-like receptor 4 (TLR4) Cascade |
| R5602358 | 1.64 | 3.24 | OPCML+ | Diseases associated with the TLR signaling cascade |
| R5260271 | 1.64 | 3.24 | OPCML+ | Diseases of immune system |
| R181438 | 9.19 | 1.74 | GENE+ | Toll-like receptor 2 (TLR2) Cascade |
| R168179 | 9.19 | 1.74 | GENE+ | Toll-like receptor TLR1:TLR2 Cascade |
| R166058 | 9.19 | 1.78 | GENE+ | MyD88:MAL(TIRAP) cascade initiated on plasma membrane |
| R168188 | 9.19 | 1.78 | GENE+ | Toll-like receptor TLR6:TLR2 cascade |
| b: cluster 1 (n = 31; top 10 shown) | ||||
| R2219530 | 1.41 | 2.52 | OPCML+ | Constitutive signaling by aberrant PI3K in cancer |
| R2219528 | 4.14 | 1.87 | OPCML+ | PI3K/AKT signaling in cancer |
| R6811558 | 4.14 | 1.87 | OPCML+ | PI5P, PP2A, and IER3 regulate PI3K/AKT signaling |
| R199418 | 4.96 | 1.75 | OPCML+ | Negative regulation of the PI3K/AKT network |
| R109704 | 5.83 | 2.76 | OPCML+ | PI3K cascade |
| R112399 | 8.44 | 2.53 | OPCML+ | IRS-mediated signalling |
| R5654219 | 9.69 | 6.07 | GENE+ | Phospholipase C-mediated cascade: FGFR1 |
| R2428928 | 9.69 | 2.34 | OPCML+ | IRS-related events triggered by IGF1R |
| R2428924 | 9.69 | 2.29 | OPCML+ | IGF1R signaling cascade |
| R74751 | 9.69 | 2.25 | OPCML+ | Insulin receptor signalling cascade |
| c: cluster 6 (n = 8) | ||||
| R418555 | 1.69 | 2.35 | OPCML+ | G alpha (s) signalling events |
| R419812 | 1.84 | 16.2 | OPCML+ | Calcitonin-like ligand receptors |
| R373080 | 2.96 | 4.0 | OPCML+ | Class B/2 (Secretin family receptors) |
| hsa04270 | 8.68 | 3.04 | OPCML+ | Vascular smooth muscle contraction |
| G0007189 | 1.49 | 4.77 | OPCML+ | adenylate cyclase-activating G protein-coupled receptor |
| G0031623 | 8.13 | 3.74 | OPCML+ | receptor internalization |
| G0015031 | 0.0007 | 5.28 | GENE+ | external membrane protein transport |
| G0006816 | 0.0106 | 2.31 | OPCML+ | calcium ion transport |
| OPCML Pathway | ||||||
|---|---|---|---|---|---|---|
| Dependent | Independent | |||||
| ID | p-Value | ×Fold | OVca | Top 3 OPCML+ | Top 3 | Description |
| R2219530 | 1.41 | 2.52 | 323.3 | KIT, EGF, FGF2 | FGF9 | Constitutive signaling by aberrant PI3K in cancer |
| R2219528 | 4.14 | 1.87 | 323.3 | KIT, EGF, FGF2 | FGF9 | PI3K/AKT signaling in Cancer |
| R6811558 | 4.14 | 1.87 | 323.3 | KIT, EGF, FGF2 | FGF9 | PI5P, PP2A, and IER3 regulate PI3K/AKT signaling |
| R199418 | 4.96 | 1.75 | 323.3 | KIT, EGF, FGF2 | FGF9 | Negative regulation of the PI3K/AKT network |
| hsa04640 | 1.69 | 1.91 | 271.0 | KIT, IL5, CD36 | IL4 | Hematopoietic cell lineage |
| R6785807 | 1.42 | 3.01 | 257.5 | FGF2, POMC, NOS2 | IL4 | Interleukin-4 and Interleukin-13 signaling |
| hsa04072 | 0.0029 | 1.76 | 255.9 | KIT, EGF, CXCR2 | DGKK, GRM6 | Phospholipase D signaling pathway |
| G0051897 | 1.74 | 2.31 | 221.8 | KIT, EGF, FGF2 | LEP, ERFE | Positive regulation of phosphatidylinositol 3-kinase/protein kinase B signal transduction |
| hsa04913 | 0.0001 | 1.98 | 201.9 | STAR, CYP1A1, HSD3B2 | Ovarian steroidogenesis | |
| G0006954 | 1.49 | 1.87 | 197.3 | KIT, CXCR2, ADM | Inflammatory response | |
| R375276 | 1.86 | 2.69 | 173.1 | GAL, POMC, CXCL1 | GRP, GPR37L1, PENK | Peptide ligand-binding receptors |
| hsa04927 | 1.10 | 2.25 | 150.8 | STAR, POMC, HSD3B2 | Cortisol synthesis and secretion | |
| hsa04657 | 1.07 | 1.99 | 150.7 | CXCL1, IL5, MMP13 | IL4 | IL-17 signaling pathway |
| G0001938 | 1.74 | 2.64 | 127.9 | EGF, FGF2, CCL11 | Positive regulation of endothelial cell proliferation | |
| hsa04020 | 2.58 | 2.44 | 122.9 | EGF, FGF2, SLN | FGF9, GDNF, SMIM6 | Calcium signaling pathway |
| G0042531 | 0.0002 | 3.28 | 122.6 | KIT, HES1, IL12A | IL4 | Positive regulation of tyrosine phosphorylation of STAT protein |
| hsa04060 | 0.0016 | 1.77 | 109.1 | CXCL1, CXCR2, IL5 | IL4, LEP | Cytokine-cytokine receptor interaction |
| G0043406 | 0.0017 | 2.92 | 96.7 | EGF, FGF2, TNFSF11 | Positive regulation of MAP kinase activity | |
| hsa04979 | 0.0002 | 1.98 | 95.6 | STAR, CD36, ANGPTL4 | Cholesterol metabolism | |
| R114608 | 0.0077 | 1.74 | 93.8 | EGF, CD36, SELP | HRG | Platelet degranulation |
| hsa04064 | 0.0001 | 2.0 | 91.9 | CXCL1, VCAM1, LBP | LAT | NF-kappa B signaling pathway |
| G0048018 | 0.0133 | 2.92 | 88.6 | EGF, NTS, CCL11 | WNT7A | Receptor ligand activity |
| G0006935 | 1.01 | 2.74 | 85.2 | FGF2, CXCL1, CXCR2 | Chemotaxis | |
| R381340 | 1.24 | 2.13 | 84.0 | CD36, ANGPTL4, PPARG | LEP | Transcriptional regulation of white adipocyte differentiation |
| R9843745 | 3.30 | 1.85 | 84.0 | CD36, ANGPTL4, PPARG | LEP | Adipogenesis |
| G0051092 | 6.92 | 2.28 | 83.6 | ROR1, SPHK1, NTS | CRNN | Positive regulation of NF-kappaB transcription factor activity |
| G0001525 | 0.0058 | 2.43 | 77.5 | EGF, HEY1, HOXA7 | LEP | Angiogenesis |
| G0008201 | 0.0009 | 3.99 | 76.2 | POSTN, SFRP1, LTF | HRG, AOC1 | Heparin binding |
| G0004713 | 5.10 | 2.81 | 75.3 | KIT, FGR, BTK | ROS1 | Protein tyrosine kinase activity |
| G0070374 | 0.0008 | 2.54 | 59.0 | FGF2, CD36, TNFSF11 | ARHGAP8 | Positive regulation of ERK1 and ERK2 cascade |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marsh, A.G.; Görtler, F.; Hafizi, S.; Gabra, H. Bioinformatic RNA-Seq Functional Profiling of the Tumor Suppressor Gene OPCML in Ovarian Cancers: The Multifunctional, Pleiotropic Impacts of Having Three Ig Domains. Curr. Issues Mol. Biol. 2025, 47, 405. https://doi.org/10.3390/cimb47060405
Marsh AG, Görtler F, Hafizi S, Gabra H. Bioinformatic RNA-Seq Functional Profiling of the Tumor Suppressor Gene OPCML in Ovarian Cancers: The Multifunctional, Pleiotropic Impacts of Having Three Ig Domains. Current Issues in Molecular Biology. 2025; 47(6):405. https://doi.org/10.3390/cimb47060405
Chicago/Turabian StyleMarsh, Adam G., Franziska Görtler, Sassan Hafizi, and Hani Gabra. 2025. "Bioinformatic RNA-Seq Functional Profiling of the Tumor Suppressor Gene OPCML in Ovarian Cancers: The Multifunctional, Pleiotropic Impacts of Having Three Ig Domains" Current Issues in Molecular Biology 47, no. 6: 405. https://doi.org/10.3390/cimb47060405
APA StyleMarsh, A. G., Görtler, F., Hafizi, S., & Gabra, H. (2025). Bioinformatic RNA-Seq Functional Profiling of the Tumor Suppressor Gene OPCML in Ovarian Cancers: The Multifunctional, Pleiotropic Impacts of Having Three Ig Domains. Current Issues in Molecular Biology, 47(6), 405. https://doi.org/10.3390/cimb47060405