
Controlling Persister and Biofilm Cells of Gram-Negative Bacteria with a New 1,3,5-Triazine Derivative

Abstract
:1. Introduction
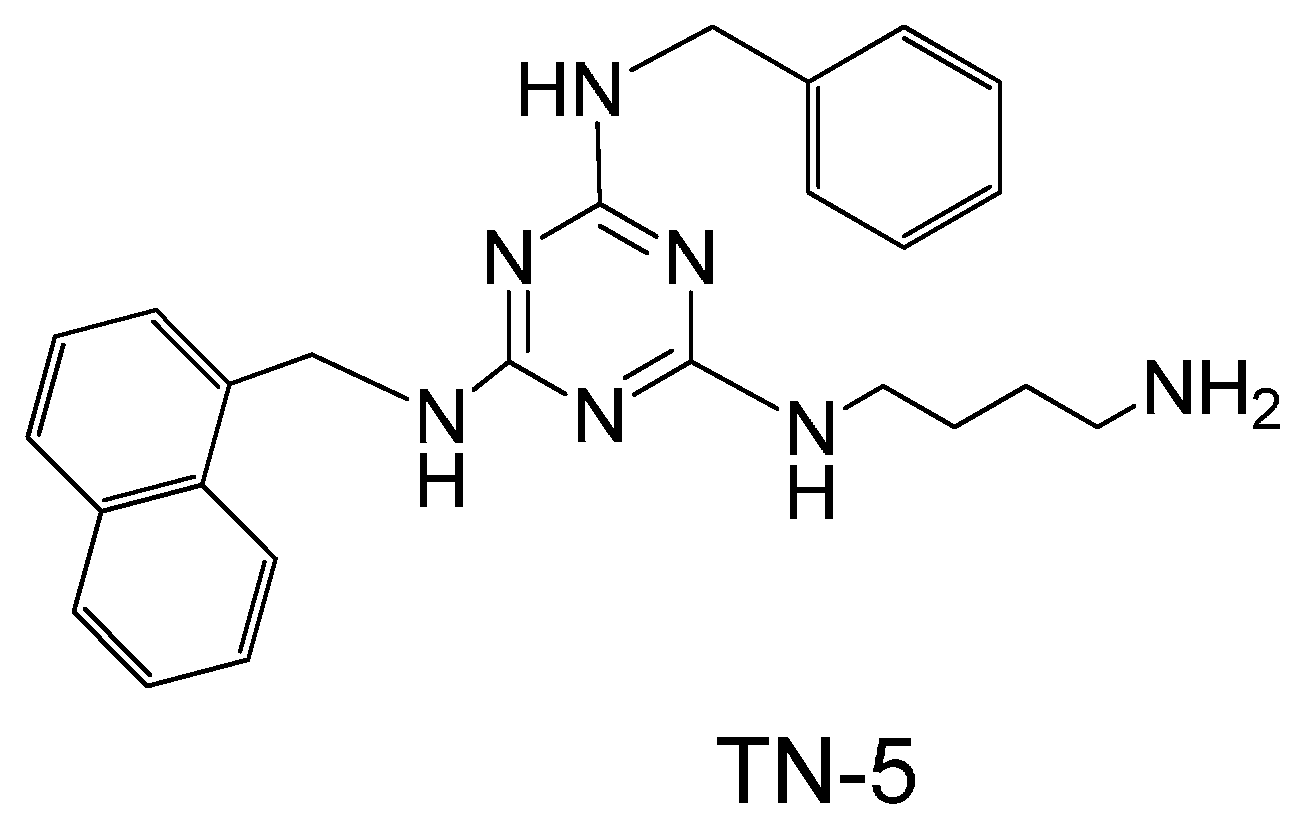
2. Results
2.1. Minimum Inhibitory Concentration (MIC) and Minimal Bactericidal Concentration (MBC) of TN-5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Test | Bacterial Strains | ||
|---|---|---|---|
| E. coli RP437 | P. aeruginosa PAO1 | P. aeruginosa PDO300 | |
| MIC (µM) | 12.8 | 12.8 | 12.8 |
| MBC (µM) | >96 | >96 | >96 |
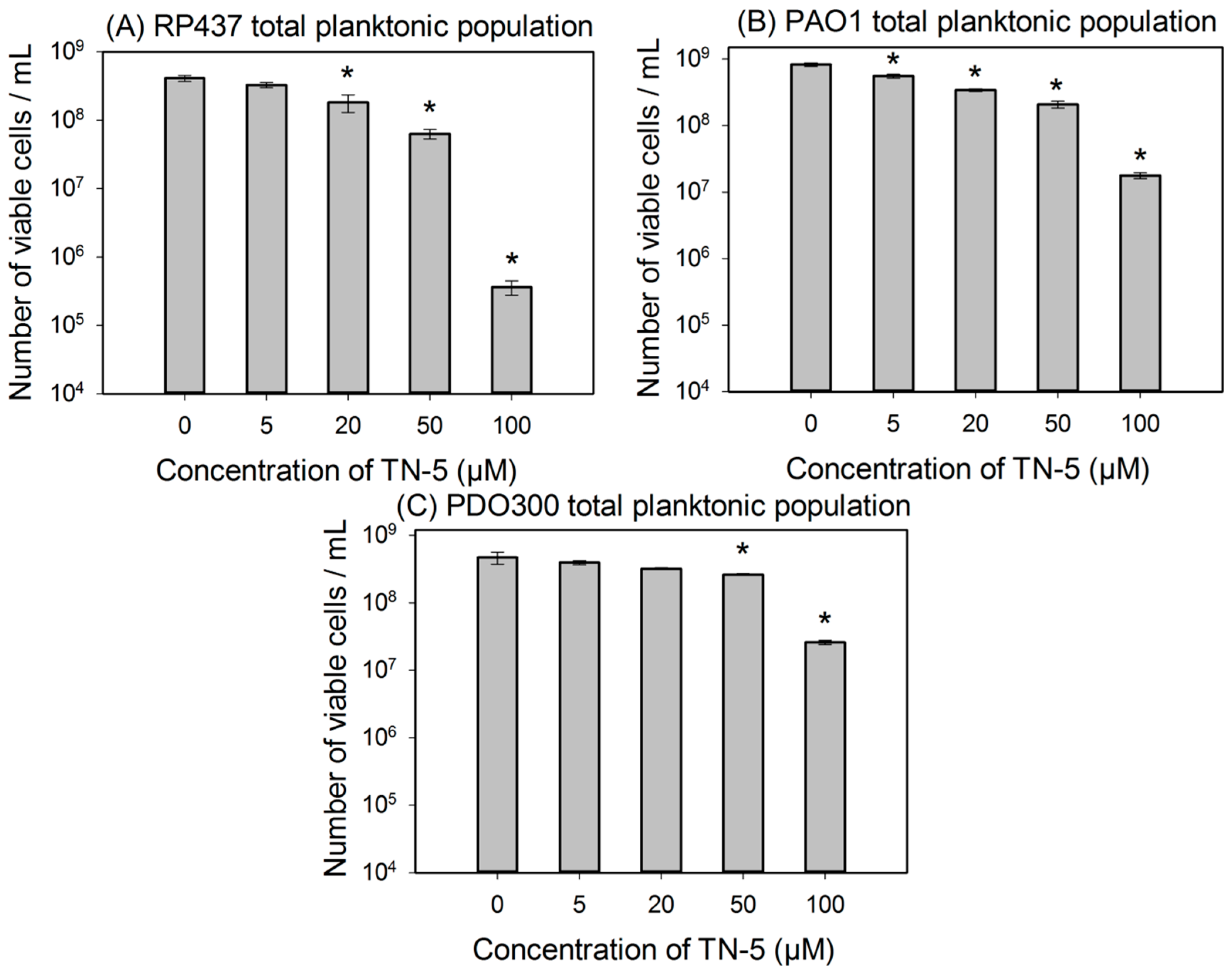
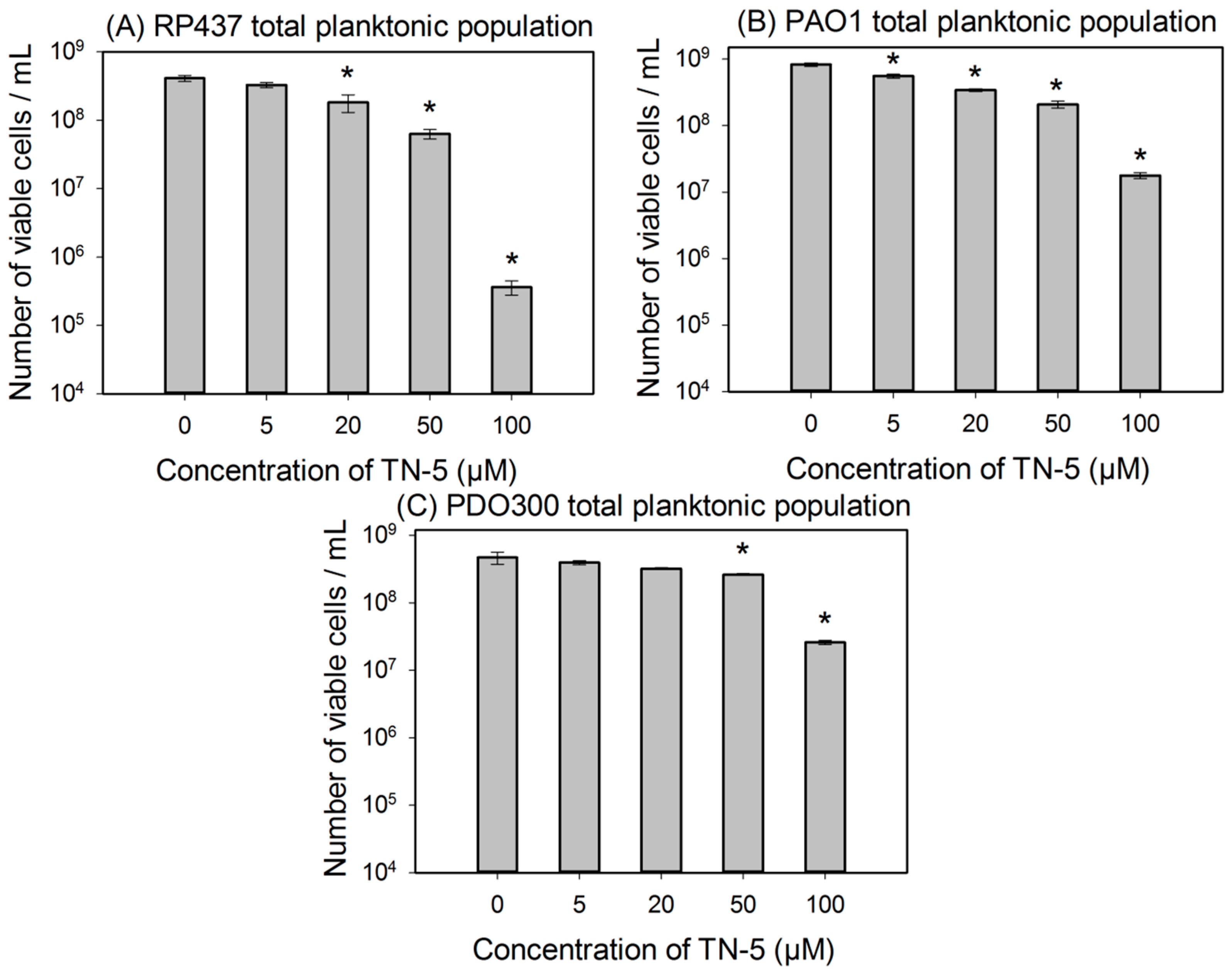
2.2. Antimicrobial Effects of TN-5 on Planktonic Cells
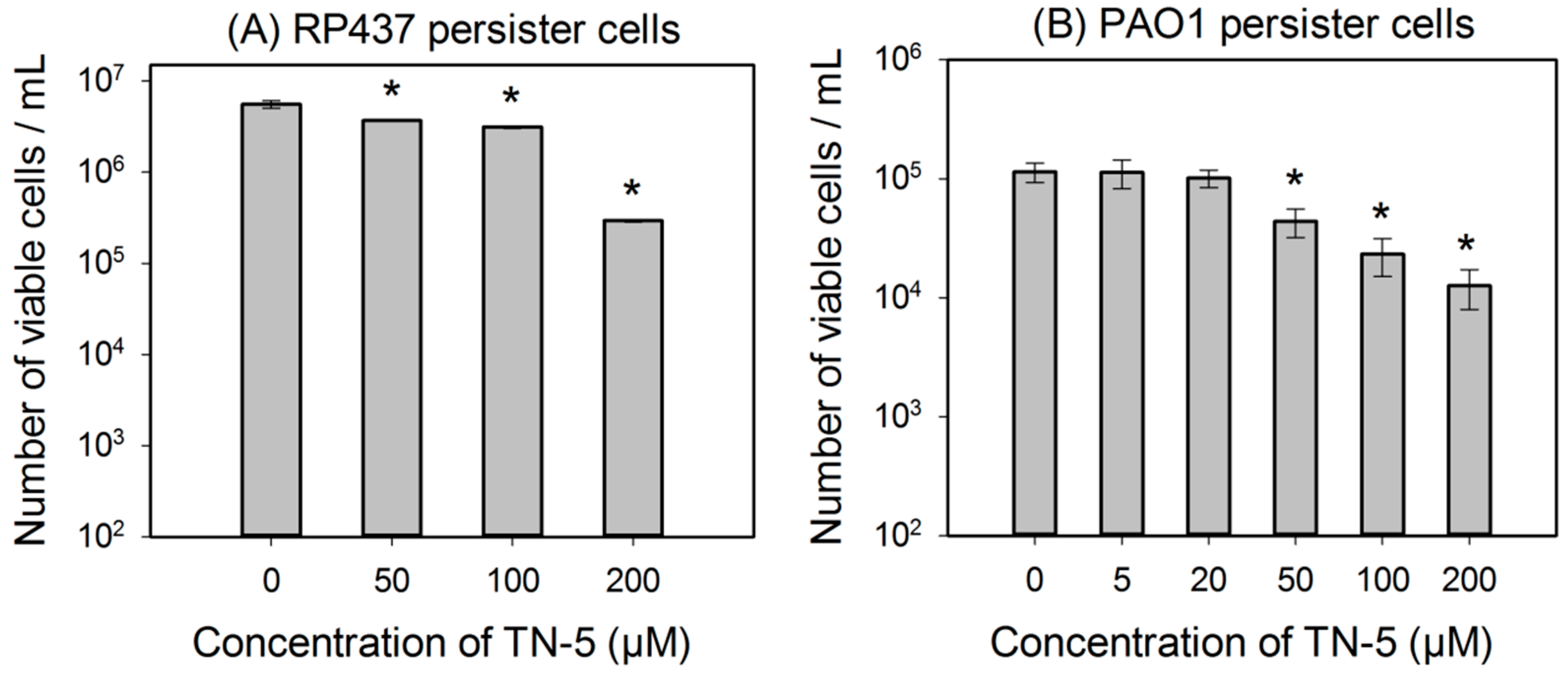
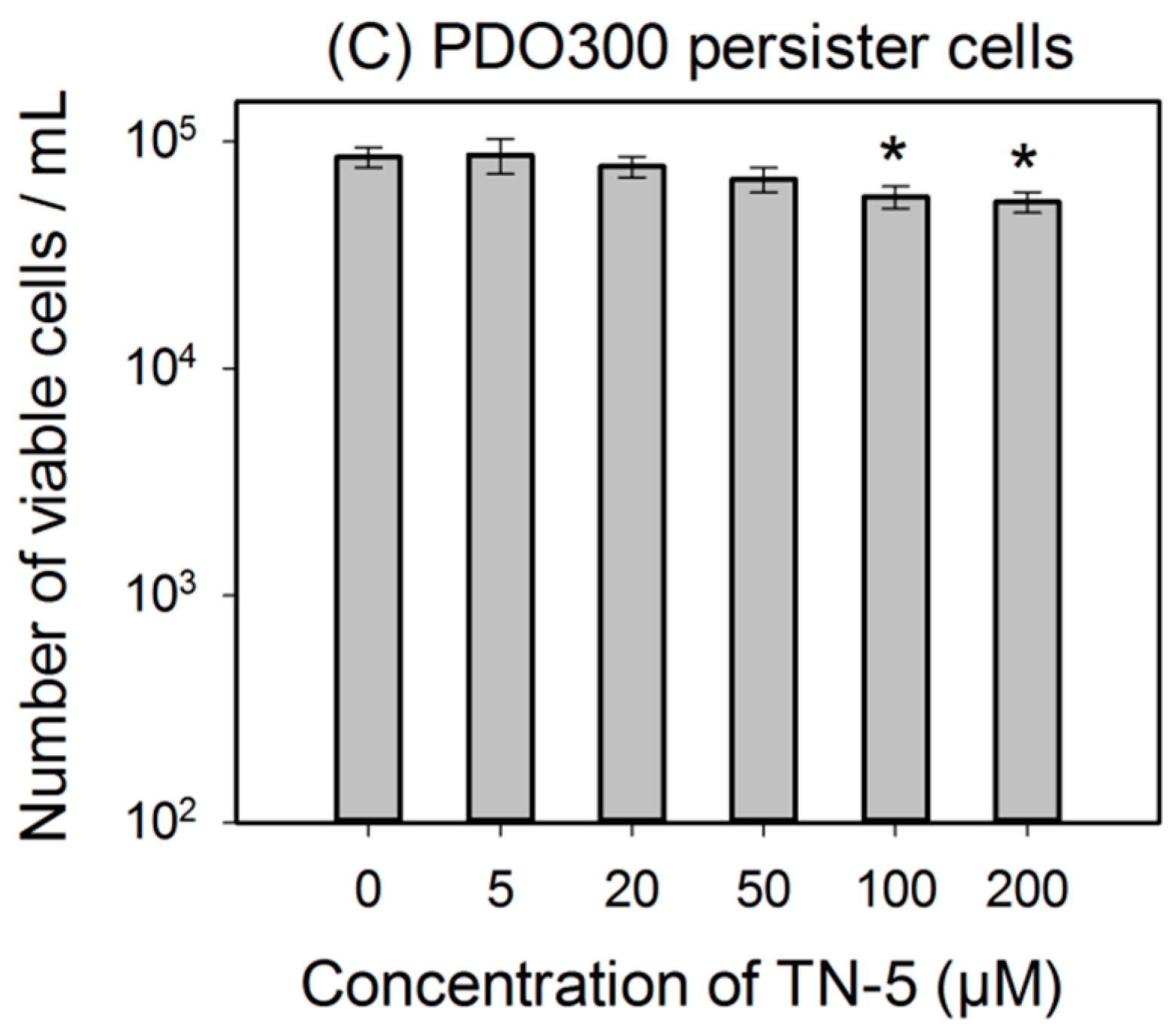
2.3. Antimicrobial Effects of TN-5 on Persister Cells
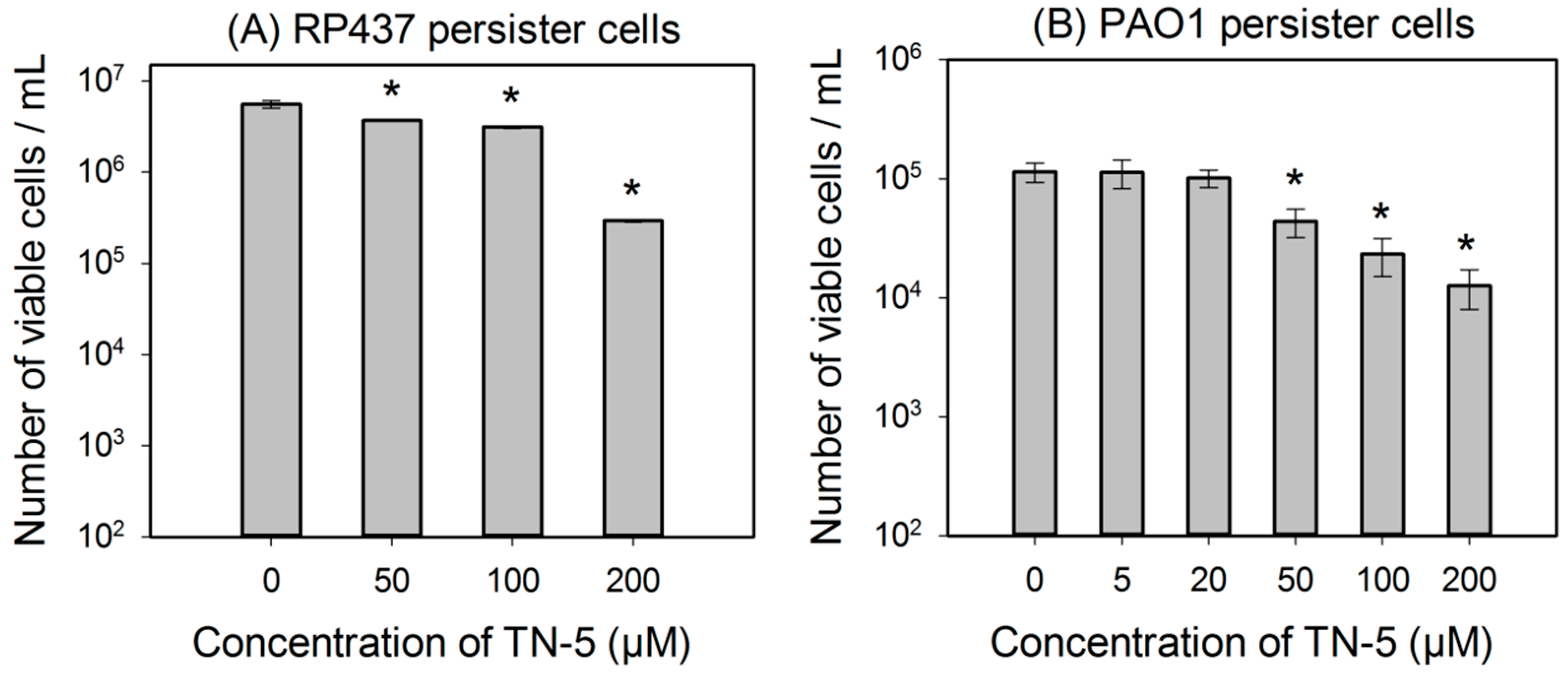
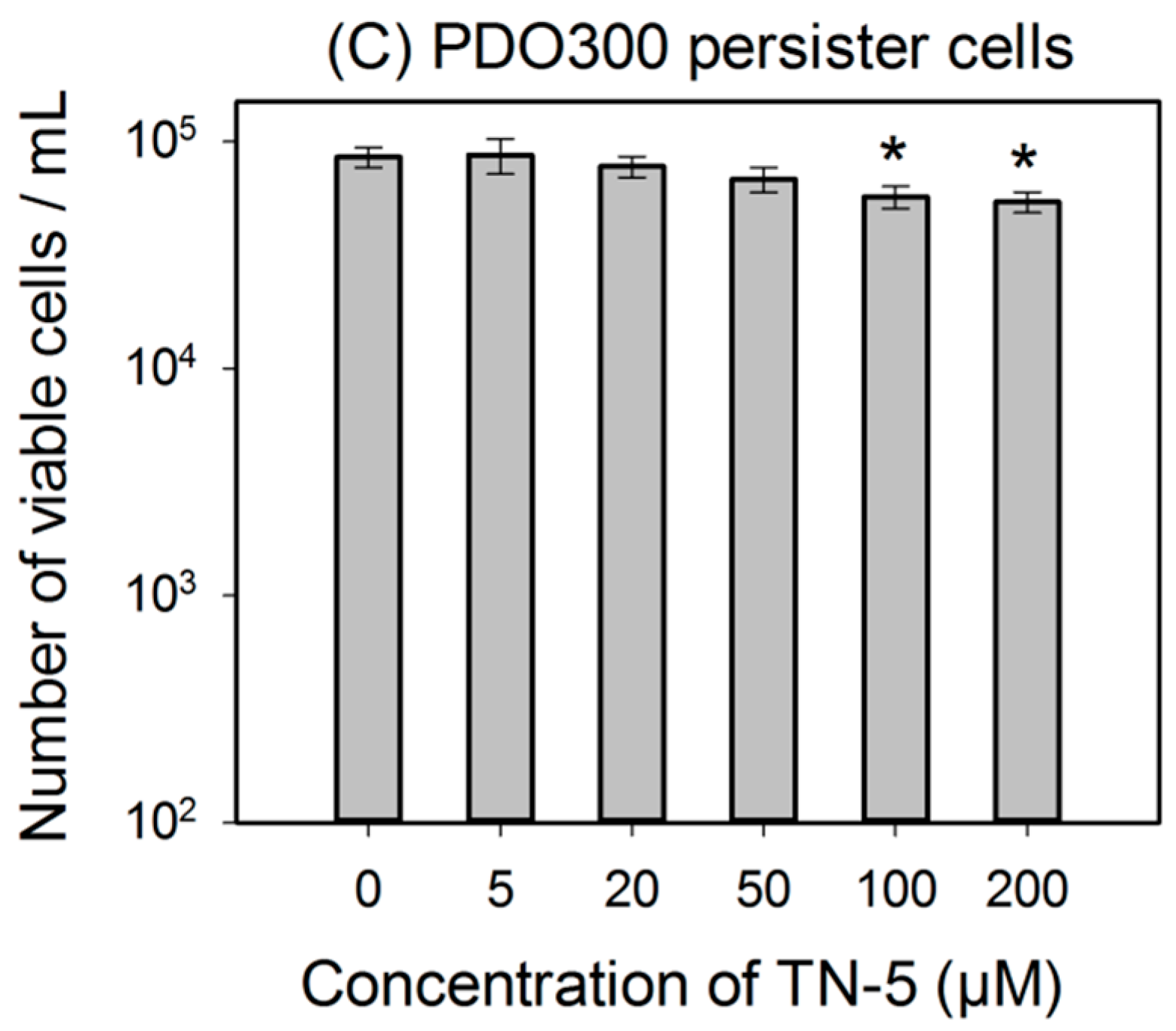
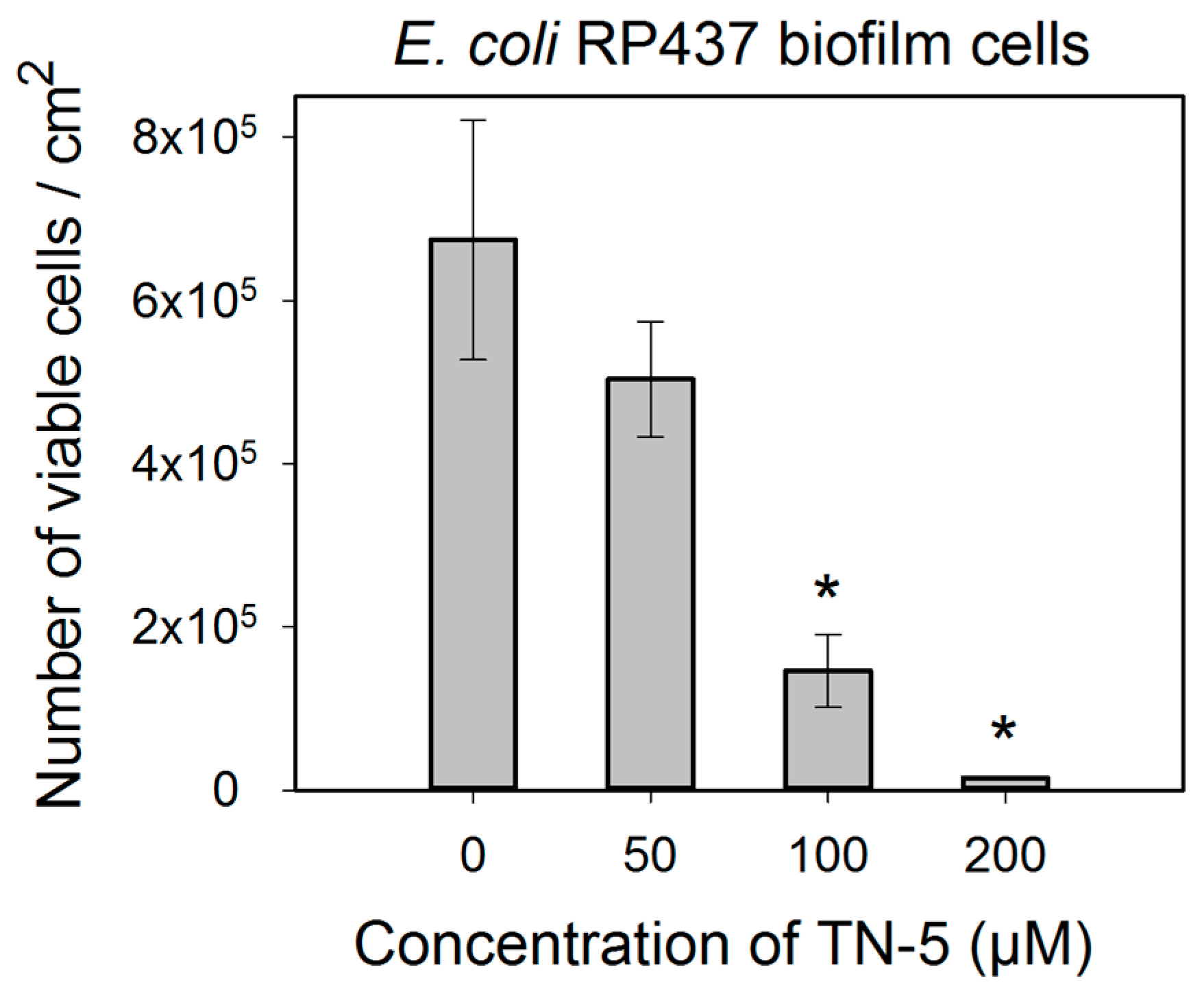
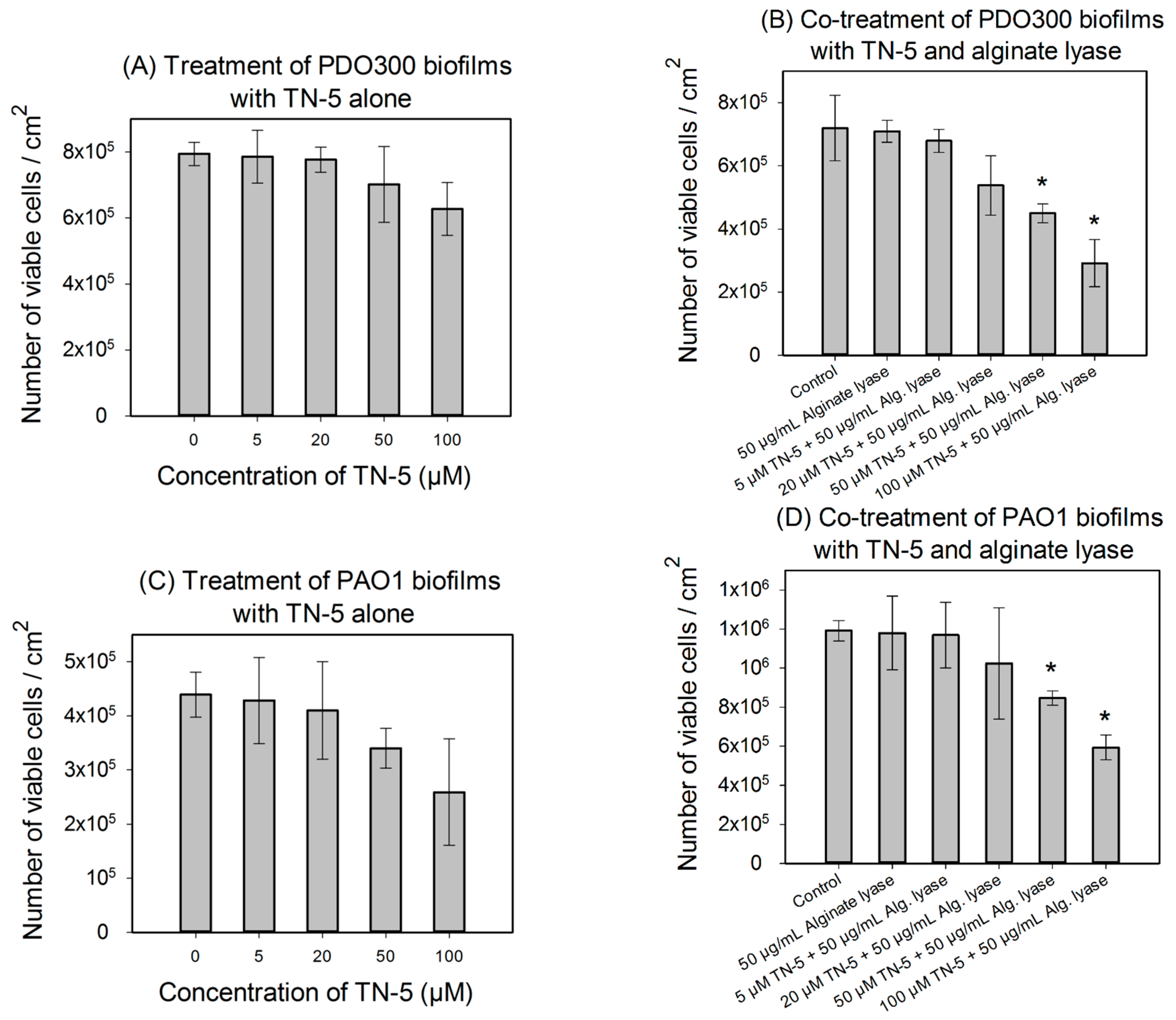
2.4. Antimicrobial Effects of TN-5 on Biofilm Cells
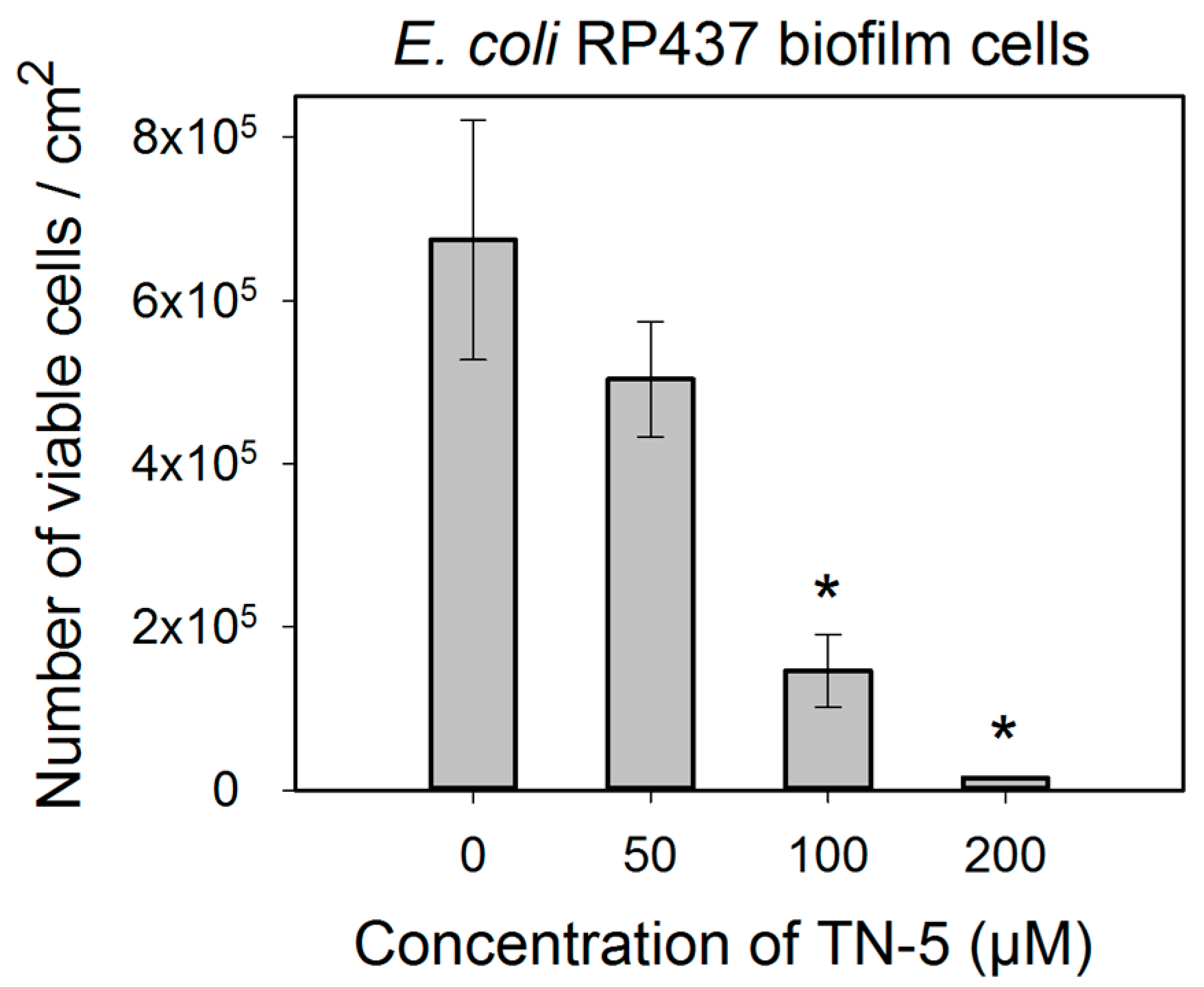
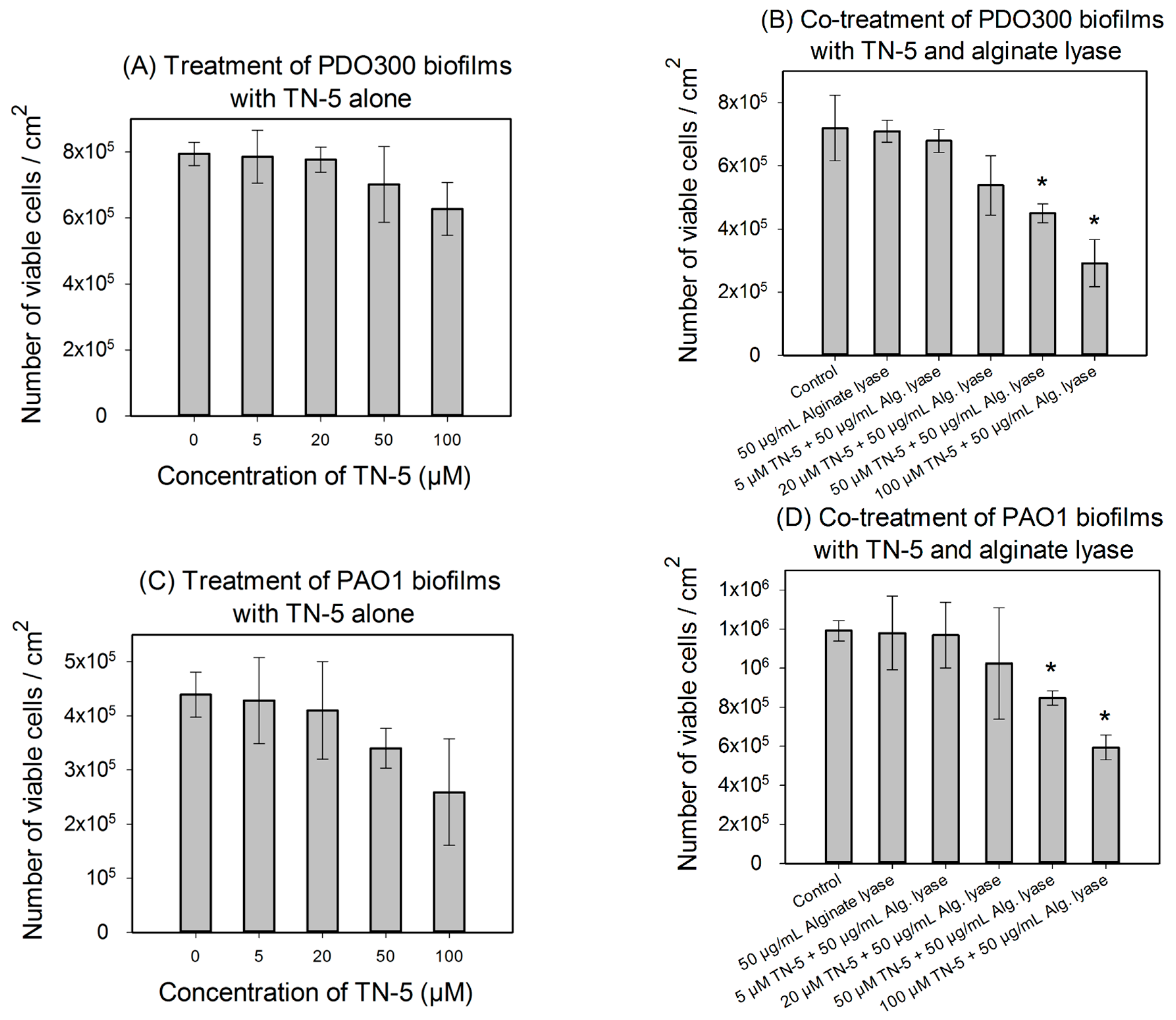
3. Discussion
4. Experimental Section
4.1. Chemical Synthesis of TN-5
4.2. Bacterial Strains and Growth Media
4.3. MIC and MBC Values of TN-5
4.4. Effects on Planktonic Cells
4.5. Persister Isolation and Treatment
4.6. Biofilm Experiments
4.7. Alginate Lyase Enhanced the Effects of TN-5 on P. aeruginosa Biofilm Cells
4.8 Statistical Analysis
Supplementary Files
Supplementary File 1Acknowledgements
Author Contributions
Conflicts of Interest
References
- Fleming, A. On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. Brit. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Myers, R.S.; Aldrich, R.H.; Howard, R.W.; Walsh, R.A. The use of gauze inoculated with Penicillium notatum or impregnated with crude penicillin in the treatment of surface infections. New Engl. J. Med. 1944, 231, 761–764. [Google Scholar] [CrossRef]
- Kardos, N.; Demain, A.L. Penicillin: The medicine with the greatest impact on therapeutic outcomes. Appl. Microbiol. Biotechnol. 2011, 92, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Witte, W.; Cuny, C.; Klare, I.; Nubel, U.; Strommenger, B.; Werner, G. Emergence and spread of antibiotic-resistant Gram-positive bacterial pathogens. Int. J. Med. Microbiol. 2008, 298, 365–377. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.M.; Hodgkinson, J.T.; Sore, H.F.; Welch, M.; Salmond, G.P.; Spring, D.R. Combating multidrug-resistant bacteria: Current strategies for the discovery of novel antibacterials. Angew. Chem. Int. Ed. 2013, 52, 10706–10733. [Google Scholar] [CrossRef] [PubMed]
- Davies, J. Inactivation of antibiotics and the dissemination of resistance genes. Science 1994, 264, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells: Molecular mechanisms related to antibiotic tolerance. In Antibiotic Resistance; Springer Berlin Heidelberg: Berlin, Germany, 2012; Volume 211, pp. 121–133. [Google Scholar]
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Barlow, M. What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol. Biol. 2009, 532, 397–411. [Google Scholar] [PubMed]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Mathee, K.; Ciofu, O.; Sternberg, C.; Lindum, P.W.; Campbell, J.I.; Jensen, P.; Johnsen, A.H.; Givskov, M.; Ohman, D.E.; Molin, S.; et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: A mechanism for virulence activation in the cystic fibrosis lung. Microbiology 1999, 145, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Pritt, B.; O’Brien, L.; Winn, W. Mucoid Pseudomonas in cystic fibrosis. Am. J. Clin. Pathol. 2007, 128, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Cabral, D.A.; Loh, B.A.; Speert, D.P. Mucoid Pseudomonas aeruginosa resists nonopsonic phagocytosis by human neutrophils and macrophages. Pediatr. Res. 1987, 22, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Govan, J.R.; Harris, G.S. Pseudomonas aeruginosa and cystic fibrosis: Unusual bacterial adaptation and pathogenesis. Microbiol. Sci. 1986, 3, 302–308. [Google Scholar] [PubMed]
- Torcato, I.M.; Huang, Y.H.; Franquelim, H.G.; Gaspar, D.; Craik, D.J.; Castanho, M.A.; Henriques, S.T. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biomembranes 2013, 1828, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Wetzler, M.; Barron, A.E. Functional synergy between antimicrobial peptoids and peptides against Gram-negative bacteria. Antimicrob. Agents Ch. 2011, 55, 5399–5402. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides the ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef] [PubMed]
- Izadpanah, A.; Gallo, R.L. Antimicrobial peptides. J. Am. Acad. Dermatol. 2005, 52, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Graslund, A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef] [PubMed]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.P.; Bellm, L.A.; Epstein, J.B.; Sonis, S.T.; Symonds, R.P. Antimicrobial therapy to prevent or treat oral mucositis. Lancet Infect. Dis. 2003, 3, 405–412. [Google Scholar] [CrossRef]
- Zhang, L.; Parente, J.; Harris, S.M.; Woods, D.E.; Hancock, R.E.; Falla, T.J. Antimicrobial peptide therapeutics for cystic fibrosis. Antimicrob. Agents Chemother. 2005, 49, 2921–2927. [Google Scholar] [CrossRef] [PubMed]
- Yedery, R.D.; Reddy, K.V. Antimicrobial peptides as microbicidal contraceptives: Prophecies for prophylactics—a mini review. Eur. J. Contracep. Repr. 2005, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Laverty, G.; Gorman, S.P.; Gilmore, B.F. The potential of antimicrobial peptides as biocides. Int. J. Mol. Sci. 2011, 12, 6566–6596. [Google Scholar] [CrossRef] [PubMed]
- Noto, P.B.; Abbadessa, G.; Cassone, M.; Mateo, G.D.; Agelan, A.; Wade, J.D.; Szabo, D.; Kocsis, B.; Nagy, K.; Rozgonyi, F.; et al. Alternative stabilities of a proline-rich antibacterial peptide in vitro and in vivo. Protein Sci. 2008, 17, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, O.; Grotzinger, J.; Cascorbi, I.; Jung, S. Antimicrobial peptides and proteins of the horse-insights into a well-armed organism. Vet. Res. 2011, 42, 98. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Lichtenstein, A.K.; Ganz, T. Defensins: Antimicrobial and cytotoxic peptides of mammalian cells. Annu. Rev. Immunol. 1993, 11, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Min, J.K.; Liu, Z.G.; Young, A.; Deshazer, H.; Gao, T.; Chang, Y.T.; Kallenbach, N.R. Synthesis and biological evaluation of novel 1,3,5-triazine derivatives as antimicrobial agents. Bioorg. Med. Chem. Lett. 2008, 18, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Hay, I.D.; Remminghorst, U.; Rehm, B.H. MucR, a novel membrane-associated regulator of alginate biosynthesis in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2009, 75, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Som, A.; Vemparala, S.; Ivanov, I.; Tew, G.N. Synthetic mimics of antimicrobial peptides. Biopolymers 2008, 90, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Thaker, H.D.; Som, A.; Ayaz, F.; Lui, D.; Pan, W.; Scott, R.W.; Anguita, J.; Tew, G.N. Synthetic mimics of antimicrobial peptides with immunomodulatory responses. J. Am. Chem. Soc. 2012, 134, 11088–11091. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Eckert, R.; Pharm, T.; Simanian, M.D.; Hu, C.; Yarbrough, D.K.; Qi, F.; Anderson, M.H.; Shi, W. Novel synthetic antimicrobial peptides against Streptococcus mutans. Antimicrob. Agents Chemother. 2007, 51, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Diehnelt, C.W. Peptide array based discovery of synthetic antimicrobial peptides. Front. Microbiol. 2013, 4, 402. [Google Scholar] [CrossRef] [PubMed]
- Bahar, A.A.; Liu, Z.; Totsingan, F.; Buitrago, C.; Kallenbach, N.; Ren, D. Synthetic dendrimeric peptide active against biofilm and persister cells of Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 2015, 99, 8125–8135. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, D.J.; Wyckoff, T.J.; Starkey, M.; Keyser, R.; Azadi, P.; O’Toole, G.A.; Parsek, M.R. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. USA 2003, 100, 7907–7912. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.Y.; Liu, Z.G.; Young, A.W.; Mark, S.L.; Kallenbach, N.R.; Ren, D.C. Effects of Trp- and Arg-containing antimicrobial peptide structure on inhibition of Escherichia coli planktonic growth and biofilm formation. Appl. Environ. Microbiol. 2010, 76, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Lu, Y.A.; Yang, J.L. Antimicrobial dendrimeric peptides. Eur. J. Biochem. 2002, 269, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J.S.; Houts, S.E. Isolation and behavior of Escherichia coli deletion mutants lacking chemotaxis functions. J. Bacteriol. 1982, 151, 106–113. [Google Scholar] [PubMed]
- DeVries, C.A.; Ohman, D.E. Mucoid-to-nonmucoid conversion in alginate-producing Pseudomonas aeruginosa often results from spontaneous mutations in algt, encoding a putative alternate sigma factor, and shows evidence for autoregulation. J. Bacteriol. 1994, 176, 6677–6687. [Google Scholar] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Niepa, T.H.R.; Gilbert, J.L.; Ren, D.C. Controlling Pseudomonas aeruginosa persister cells by weak electrochemical currents and synergistic effects with tobramycin. Biomaterials 2012, 33, 7356–7365. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.C.; Bahar, A.A.; Syed, H.; Ren, D.C. Reverting antibiotic tolerance of Pseudomonas aeruginosa PAO1 persister cells by (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. PloS ONE 2012, 7, e45778. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, M.; Zhou, C.H.; Kallenbach, N.R.; Ren, D.C. Control of bacterial persister cells by Trp/Arg-containing antimicrobial peptides. Appl. Environ. Microbiol. 2011, 77, 4878–4885. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahar, A.A.; Liu, Z.; Garafalo, M.; Kallenbach, N.; Ren, D. Controlling Persister and Biofilm Cells of Gram-Negative Bacteria with a New 1,3,5-Triazine Derivative. Pharmaceuticals 2015, 8, 696-710. https://doi.org/10.3390/ph8040696
Bahar AA, Liu Z, Garafalo M, Kallenbach N, Ren D. Controlling Persister and Biofilm Cells of Gram-Negative Bacteria with a New 1,3,5-Triazine Derivative. Pharmaceuticals. 2015; 8(4):696-710. https://doi.org/10.3390/ph8040696
Chicago/Turabian StyleBahar, Ali Adem, Zhigang Liu, Meagan Garafalo, Neville Kallenbach, and Dacheng Ren. 2015. "Controlling Persister and Biofilm Cells of Gram-Negative Bacteria with a New 1,3,5-Triazine Derivative" Pharmaceuticals 8, no. 4: 696-710. https://doi.org/10.3390/ph8040696
APA StyleBahar, A. A., Liu, Z., Garafalo, M., Kallenbach, N., & Ren, D. (2015). Controlling Persister and Biofilm Cells of Gram-Negative Bacteria with a New 1,3,5-Triazine Derivative. Pharmaceuticals, 8(4), 696-710. https://doi.org/10.3390/ph8040696