Imaging Biomarkers or Biomarker Imaging?



Abstract
:1. Introduction
2. PET & MRI: Selected Examples
3. The Role of Radiopharmaceuticals in PET and PET/MRI
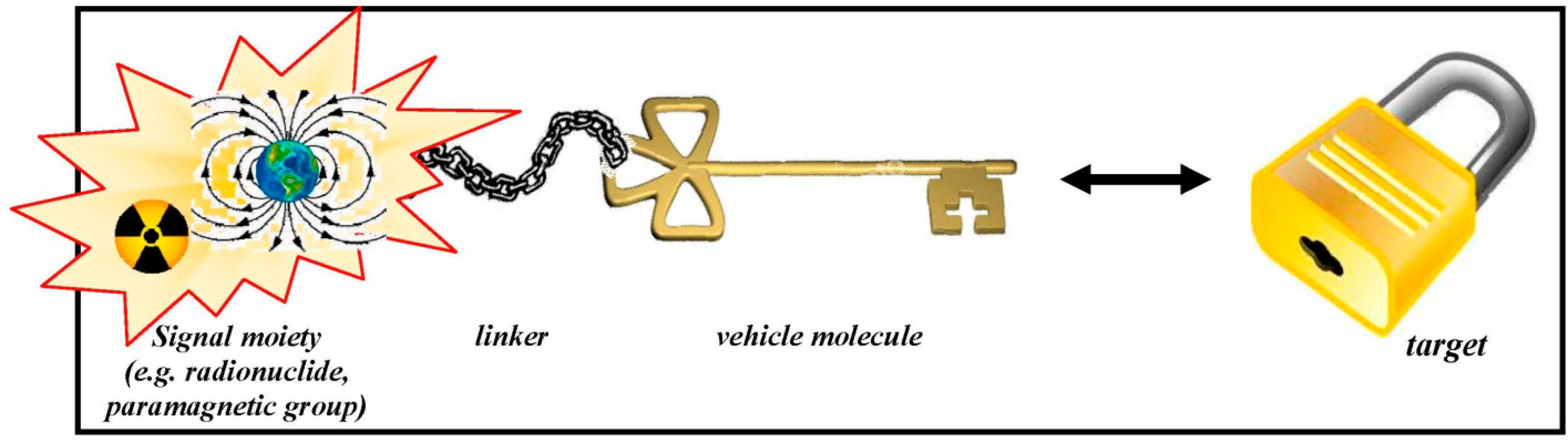

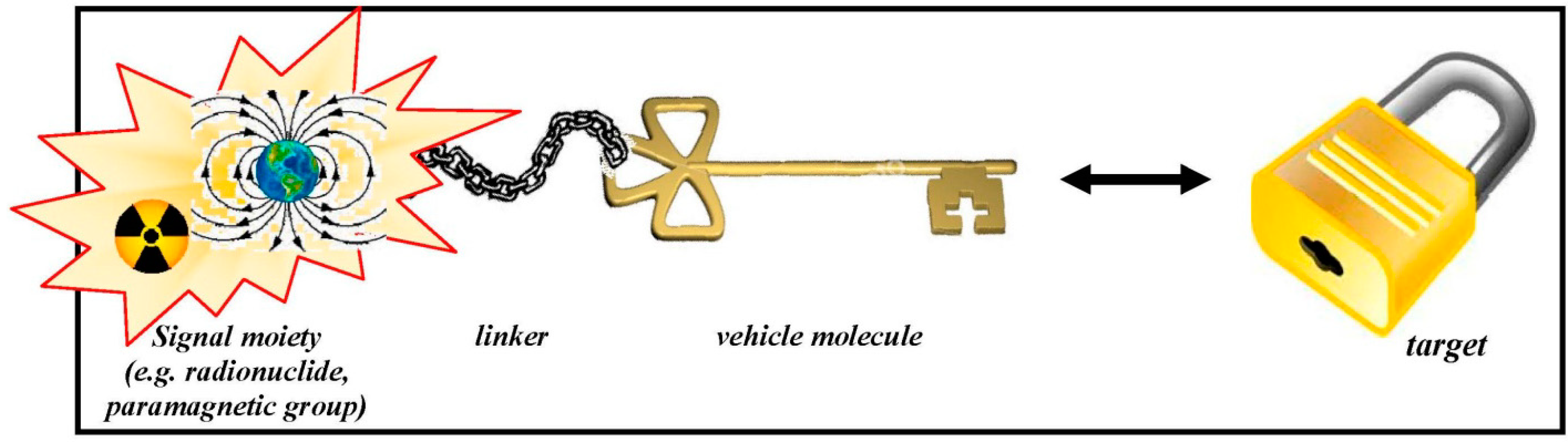{kind=link}
{kind=link}
{kind=link}
| Nuclide | Production | Half-Life |
|---|---|---|
| 18F (F−) | 18O(p,n)18F | 109.7 min |
| 18F (F2) | 20Ne(d,α)18F | 109.7 min |
| 11C | 14N(p,α)11C | 20.4 min |
| 13N | 16O(p,α)13N | 10.0 min |
| 15O | 14N(d,n)15O | 2.0 min |
| 64Cu | 64Ni(p,n)64Cu | 12.7 h |
| 86Y | 86Sr(p,n)86Y | 14.7 h |
| 76Br | 76Se(p,n)76Br | 16.0 h |
| 68Ga | 68Ge/68Ga generator | 67.6 min |
| 82Rb | 82Sr/82Rb-Generator | 1.3 min |
| 124I | 124Te(p,n)124I | 4.2 days |
- Availability of the radionuclide;
- Physical characteristics of the radionuclide;
- Radiochemical issues; and
- Radiopharmacological issues.
- Binding and/or uptake characteristics in terms of affinity, selectivity and unspecific binding;
- Pharmacokinetics yielding information on input function, elimination rate and principal fate of the compound;
- Metabolic issues with respect to formation of metabolites potentially interfering with the original signal and stability considerations;
- Involvement in different (patho-)physiological processes such as parallel representation of both vital tumour tissue and inflammation or combined information on perfusion and hypoxia; and
- Pharmaceutical issues referring to interactions of molecular probes and co-medication as well as influence of the pharmaceutical formulation of the molecular probe (pharmacokinetic and pharmacodynamic considerations; nota bene: pharmacodynamic effects are in general of no interest in molecular imaging using radiopharmaceuticals because of the minuscule mass of compounds that is administered. Therefore, pharmacodynamics are not considered herein).
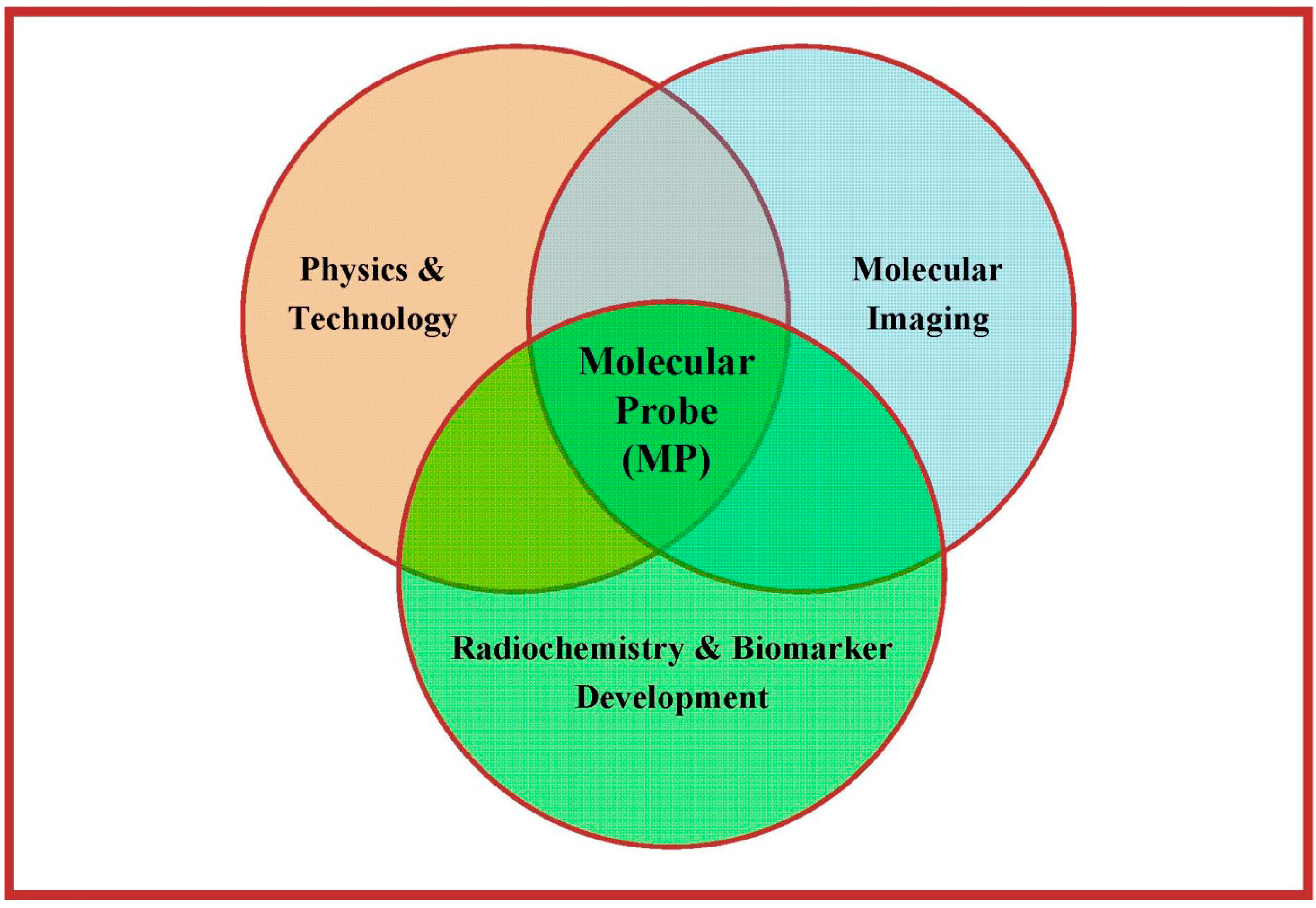
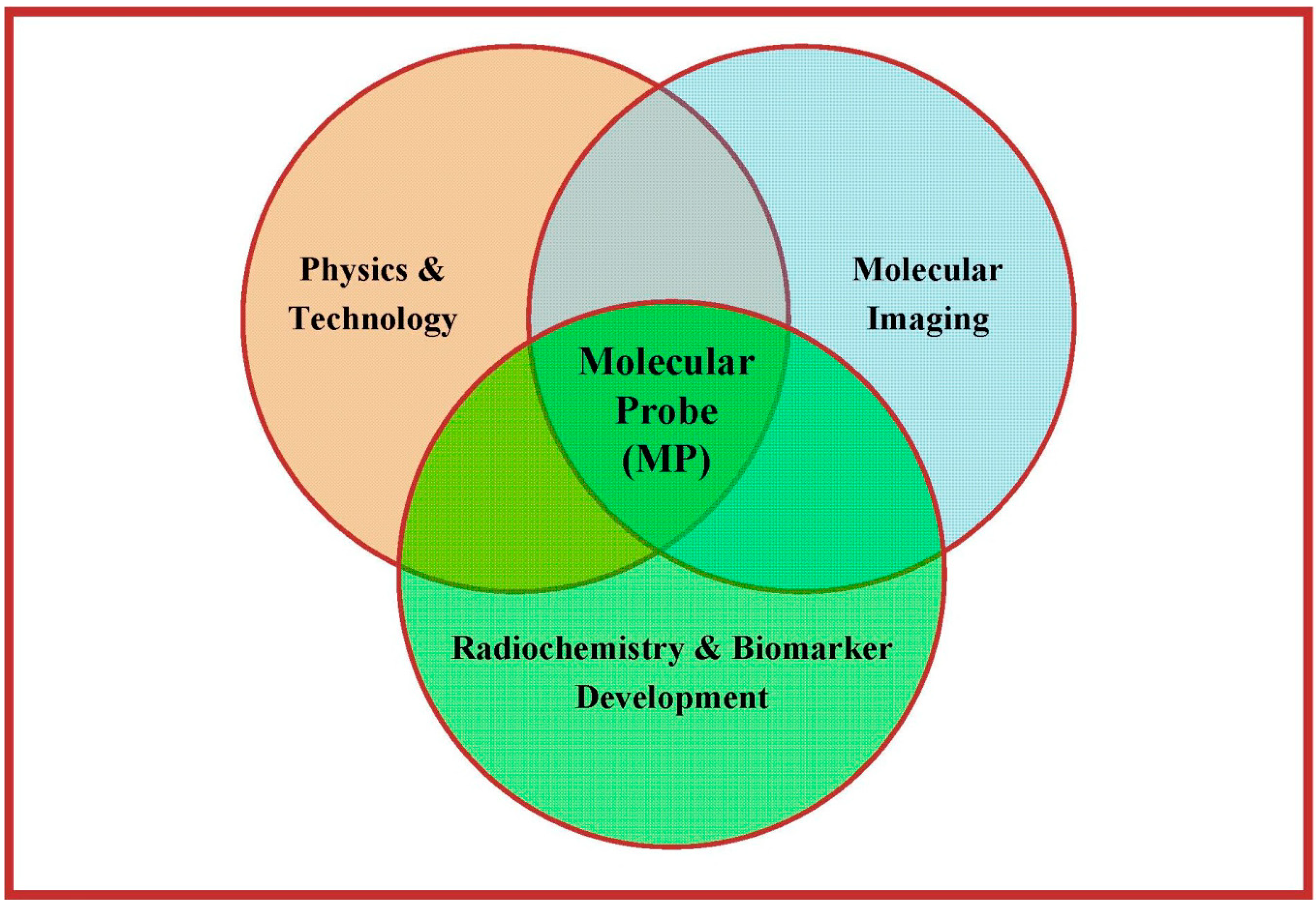
- Receptor systems and their subtypes;
- Transporter proteins;
- Antigens;
- Specific intra- or extracellular enzymes;
- Alterations in gene and protein expression;
- Changes in physiology and metabolism, such as hypoxia or differences in vascularisation and perfusion;
- Energy turnover.
4. Specific Radiochemical Considerations
- There is no stoichiometry between the reaction partners (i.e., the radionuclide and the precursor molecule)! A huge excess of the precursor is present in the reaction solution compared to the amount of radionuclide. An illustrative extrapolation reflecting this imbalance would be to react a whole shipload of precursor material with as little as one single sugar cube of radionuclide!
- Reactions that are unsuccessful under normal chemical circumstances are sometimes successful in radiochemistry and vice versa.
- Due to the very low mass of reaction partners (often 1 mg precursor or less), the use of special miniature equipment is demanded.
- Working with high amounts of radioactivity (several tens of GBq) requires powerful radiation protection devices. All radiolabelling procedures have to take place in a fully lead shielded environment (dedicated hot cells) under remote control using special equipment for manipulation like tele-tongs or robotic arms.
- For the routine delivery of radiopharmaceuticals with maximum quality, the radiosyntheses are performed in automated synthesis modules which can be remotely controlled from outside the shielded hot cells (quality assurance). The production sequences are programmed in advance based on optimization procedures and performed automatically with constant and reproducible quality.
- Any carrier (stable isotope of the radionuclide taking part in the reactions) influences the radiochemical conversion leading to a significant amount of indistinguishable, non-radioactive product that “dilutes” the final radiopharmaceutical. This is followed by a significant change in stoichiometry. This carrier also determines the maximum achievable specific radioactivity which is a measure for the radioactivity per mass unit (e.g., GBq/µmol or MBq/nmol) [35,36]. Such carrier is introduced into the radiosynthetic procedure either on purpose (due to production obligations) or by unwanted “contamination” of reaction partners or solvents. [36,37] In case of Carbon-11, even the 12C-CO2 always present in air may contribute significantly to a reduction in specific activity. In case of fluorine-18, the isotopically enriched target material (water, >98% 18O) also contains traces of non-radioactive fluorine-19 as dissolved fluoride anions. Typically, the content of 19F-fluoride is specified as <0.1 mg/L (=100 ppb) [38,39,40] and the total target volume usually is 2–5 mL. Hence, up to 0.2–0.5 ng of 19F-fluoride are present—that equals 10–26 pmol. However, 100 GBq of [18F]fluoride equal only 30.0 pg (=1.6 pmol) Therefore, up to a 16-fold excess of non-radioactive fluoride may be present in terms of amount of substance!
- When working with solutions with high radioactivity concentrations, one has to consider radiolysis as a major factor reducing product purity leading to formation of unwanted by-products. Especially when dealing with batches that are to be shipped (e.g., [18F]FDG and [18F]fluoride for labelling) concentrations up to 35 GBq/mL (700 GBq in 20 mL) can be reached [41]! In these cases, addition of stabilizers to the product matrix may be indicated.
5. Specific Radiopharmacological Considerations
- As already mentioned earlier in this review, the specific radioactivity is an essential parameter for the quantification and visualisation of the MP’s signal—the imaging biomarker. In many (patho-)physiological imaging procedures the actual number of target sites is limited and the process of binding between the MP and the specific target (e.g., receptor subtype, transporter, antigen binding pocket) is saturable. Then, the maximum available binding sites (Bmax) determine the maximum achievable signal. Specific radioactivity (as the measure for the ratio of non-radioactive to radioactive molecules with the identical structure and binding characteristics) therefore allows predicting the amount of detectable signal, reflecting the ratio of signal-evincing to signal-erasing binding. Consequently, the required specific radioactivity has to be higher when the density of the target sites is lower. Nota bene, the specific radioactivity should be kept constant throughout a (quantified) clinical study and should always be included as a co-variable in statistical analysis of the data (e.g., binding potential).
- The radionuclide as the signalling function of the MP only allows for the allocation and quantification of the annihilation events. But it does not reflect the integrity of the MP. Thus, it might occur that the radionuclide (or a small part of the molecule which includes the signalling radionuclide) is cleaved from the intact parent molecule through active metabolism. Subsequently, the measured signal would be wrongly assigned and quantification would be systematically biased. Therefore, knowledge on and quantification of the metabolic stability of the MP (both in target tissue and blood stream) are pivotal.
- The availability of the desired radionuclide and the potential alterations in the in vivo behaviour of the MP due to the insertion of this radionuclide unfortunately point into opposite directions. (cf. Figure 3) For example, gallium-68 is readily available through a radionuclide-generator system but, since it is a radiometal, has to be integrated into the MP’s backbone by addition of a bulky chelating group and complexation. This leads to a drastic change in the molecular structure and therefore the MP’s in vivo behaviour becomes unpredictable. This is true not only for gallium-68 but for all radiometals and leads to probable changes in pharmacokinetics of the so-labelled radiopharmaceuticals. As a consequence, there is—despite significant efforts throughout many years—only one MP labelled with a radiometal targeting a receptor system in the brain that found its way into clinical application, namely 99mTc-TRODAT [44,45]. Moreover, interactions between radiometal and chelator within a complex are usually much weaker than covalent binding. On the other hand, carbon-11 labelled radiotracers represent the authentic (unchanged) molecule. carbon-11 is always bound covalently within the vehicle molecule, most often as a [11C]methyl group attached to an amine, hydroxyl or carboxyl moiety. No isotopic effect can be observed and, consequently, organisms are not able to distinguish between the original compound and its 11C-labelled analogue. Finally, fluorine-18 is also attached covalently to the parent molecule. Since normally the target molecules do not bear a fluorine atom in their chemical structure the label has to be added (and not substituted). Although the structural change may appear small it can not be excluded that this will lead to pronounced changes in the MP’s characteristics—both, in vitro and in vivo. This could be in both directions: it might lead to reduced affinity, stability or selectivity but could also lead to ameliorated in vivo behaviour [24].

6. Regulatory Aspects and Quality Assessment
Author Contributions
Conflicts of Interest
References
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Lucignani, G. Imaging biomarkers: From research to patient care—A shift in view. Eur. J. Nucl. Med. Mol. Imaging 2001, 34, 1693–1697. [Google Scholar] [CrossRef]
- Peng, B.H.; Levin, C.S. Recent development in PET instrumentation. Curr. Pharm. Biotechnol. 2010, 11, 555–571. [Google Scholar] [CrossRef]
- Mittra, E.; Quon, A. Positron emission tomography/computed tomography: The current technology and applications. Radiol. Clin. N. Am. 2009, 47, 147–160. [Google Scholar] [CrossRef]
- Lecomte, R. Novel detector technology for clinical PET. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, S69–S85. [Google Scholar] [CrossRef]
- Lewellen, T.K. Recent developments in PET detector technology. Phys. Med. Biol. 2008, 53, 287–317. [Google Scholar] [CrossRef]
- Spanoudaki, V.C.; Ziegler, S.I. PET & SPECT instrumentation. Handb. Exp. Pharmacol. 2008, 185, 53–74. [Google Scholar] [CrossRef]
- Townsend, D.W. Positron emission tomography/computed tomography. Semin. Nucl. Med. 2008, 38, 152–166. [Google Scholar] [CrossRef]
- Forster, B.B.; MacKay, A.L.; Whittall, K.P.; Kiehl, K.A.; Smith, A.M.; Hare, R.D.; Liddle, P.F. Functional magnetic resonance imaging: The basics of blood-oxygen-level dependent (BOLD) imaging. Can. Assoc. Radiol. J. 1998, 49, 320–329. [Google Scholar]
- Hermann, P.; Kotek, J.; Kubícek, V.; Lukes, I. Gadolinium(III) complexes as MRI contrast agents: Ligand design and properties of the complexes. Dalton Trans. 2008, 21, 3027–3047. [Google Scholar]
- Lee, S.T.; Scott, A.M. Hypoxia positron emission tomography imaging with 18f-fluoromisonidazole. Semin. Nucl. Med. 2007, 37, 451–461. [Google Scholar] [CrossRef]
- Padhani, A. PET imaging of tumour hypoxia. Cancer Imaging 2006, 31, S117–S121. [Google Scholar]
- Smith, T.A. FDG uptake, tumour characteristics and response to therapy: A review. Nucl. Med. Commun. 1998, 19, 97–105. [Google Scholar] [CrossRef]
- Abouzied, M.M.; Crawford, E.S.; Nabi, H.A. 18F-FDG imaging: Pitfalls and artifacts. J. Nucl. Med. Technol. 2005, 33, 145–155. [Google Scholar]
- Pauwels, E.K.; Ribeiro, M.J.; Stoot, J.H.; McCready, V.R.; Bourguignon, M.; Mazière, B. FDG accumulation and tumor biology. Nucl. Med. Biol. 1998, 25, 317–322. [Google Scholar] [CrossRef]
- Breeman, W.A.; de Blois, E.; Sze Chan, H.; Konijnenberg, M.; Kwekkeboom, D.J.; Krenning, E.P. (68)Ga-labeled DOTA-peptides and (68)Ga-labeled radiopharmaceuticals for positron emission tomography: Current status of research, clinical applications, and future perspectives. Semin. Nucl. Med. 2011, 41, 314–321. [Google Scholar] [CrossRef]
- Lopci, E.; Nanni, C.; Rampin, L.; Rubello, D.; Fanti, S. Clinical applications of 68Ga-DOTANOC in neuroendocrine tumours. Minerva Endocrinol. 2008, 33, 277–281. [Google Scholar]
- Prata, M.I. Gallium-68: A new trend in PET radiopharmacy. Curr. Radiopharm. 2012, 5, 142–149. [Google Scholar] [CrossRef]
- Reubi, J.C.; Laissue, J.; Waser, B.; Horisberger, U.; Schaer, J.C. Expression of somatostatin receptors in normal, inflamed, and neoplastic human gastrointestinal tissues. Ann. N. Y. Acad. Sci. 1994, 733, 122–137. [Google Scholar] [CrossRef]
- Reubi, J.C.; Krenning, E.; Lamberts, S.W.; Kvols, L. Somatostatin receptors in malignant tissues. J. Steroid Biochem. Mol. Biol. 1990, 37, 1073–1077. [Google Scholar] [CrossRef]
- Zaidi, H.; Montandon, M.L.; Alavi, A. The clinical role of fusion imaging using PET, CT, and MR imaging. Magn. Reson. Imaging Clin. N. Am. 2010, 18, 133–149. [Google Scholar] [CrossRef]
- Beyer, T.; Townsend, D.W.; Brun, T.; Kinahan, P.E.; Charron, M.; Roddy, R.; Jerin, J.; Young, J.; Byars, L.; Nutt, R. A combined PET/CT scanner for clinical oncology. J. Nucl. Med. 2000, 41, 1369–1379. [Google Scholar]
- Moser, E.; Stadlbauer, A.; Windischberger, C.; Quick, H.H.; Ladd, M.E. Magnetic resonance imaging methodology. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, S30–S41. [Google Scholar] [CrossRef]
- Wadsak, W.; Mitterhauser, M. Basics and principles of radiopharmaceuticals for PET/CT. Eur. J. Radiol. 2010, 73, 461–469. [Google Scholar] [CrossRef]
- Ter-Pogossian, M.M.; Powers, W.E. Radioisotopes in Scientific Research; Pergamon Press: London, UK, 1958. [Google Scholar]
- Ido, T.; Wan, C.N.; Casella, V.; Fowler, J.S.; Wolf, A.P.; Reivich, M.; Kuhl, D.E. Labeled 2-deoxy-d-glucose analogs. 18F-labeled 2-deoxy-2-fluoro-d-glucose, 2-deoxy-2-fluoro-d-mannose and 14C-2-deoxy-2-fluoro-d-glucose. J. Label. Compd. Radiopharm. 1978, 14, 175–184. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.H.; Stoecklin, G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-d-glucose using aminopolyether supported nucleophilic substitution. J. Nucl. Med. 1986, 27, 235–238. [Google Scholar]
- Rowe, C.; Keefe, G.O.; Scott, A.M.; Tochon-Danguy, H.T. Positron emission tomography in neuroscience. Medicamundi 2005, 44, 9–16. [Google Scholar]
- Briner, W.H. Radiopharmaceuticals are drugs. Mod. Hosp. 1960, 95, 110–114. [Google Scholar]
- Del Vecchio, S.; Zannetti, A.; Fonti, R.; Pace, L.; Salvatore, M. Nuclear imaging in cancer theranostics. Q. J. Nucl. Med. Mol. Imaging 2007, 51, 152–163. [Google Scholar]
- Kallmerten, A.E.; Alexander, A.; Wager, K.M.; Jones, G.B. Microwave accelerated labeling methods in the synthesis of radioligands for positron emission tomography imaging. Curr. Radiopharm. 2011, 4, 343–354. [Google Scholar] [CrossRef]
- Velikyan, I.; Beyer, G.J.; Långström, B. Microwave-supported preparation of (68)Ga bioconjugates with high specific radioactivity. Bioconjug. Chem. 2004, 15, 554–560. [Google Scholar] [CrossRef]
- Wang, M.W.; Lin, W.Y.; Liu, K.; Masterman-Smith, M.; Kwang-Fu Shen, C. Microfluidics for positron emission tomography probe development. Mol. Imaging 2010, 9, 175–191. [Google Scholar]
- Elizarov, A.M. Microreactors for radiopharmaceutical synthesis. Lab Chip 2009, 9, 1326–1333. [Google Scholar] [CrossRef]
- Zeevaart, J.R.; Olsen, S. Recent trends in the concept of specific activity: Impact on radiochemical and radiopharmaceutical producers. Appl. Radiat. Isot. 2009, 64, 812–814. [Google Scholar] [CrossRef]
- De Goeij, J.J.M.; Bonardi, M.L. How to define the concepts specific activity, radioactive concentration, carrier, carrier-free and no-carrier added. J. Radioanal. Nucl. Chem. 2005, 263, 13–18. [Google Scholar] [CrossRef]
- Handbook of Radiopharmaceuticals: Radiochemistry and Applications, 1st ed.; Welch, M.J.; Redvanly, C.S. (Eds.) John Wiley & Sons: Chichester, UK, 2002; p. 43.
- Rotem Industries. Hyox 18 Enriched Water. Available online: http://www.rotem-medical.com/hyox18/ (accessed on 4 May 2012).
- Huayi Isotopes Co. Oxygen 18 Water 95 atom %. Specifications. Available online: http://www.huayi-isotopes.com/EnProductShow.asp?ID=136 (accessed on 4 May 2012).
- Isoflex. Oxygen-18 Enriched Water—98 atom %. Product Specifications. Available online: http://www.isoflex.com/imaging/O-18_wt-98.html (accessed on 4 May 2012).
- Eberl, S.; Eriksson, T.; Svedberg, O.; Norling, J.; Henderson, D.; Lam, P.; Fulham, M. High beam current operation of a PETtraceTM cyclotron for 18F− production. Appl. Radiat. Isot. 2012, 70, 922–930. [Google Scholar] [CrossRef]
- Fukumura, T.; Nakao, R.; Yamaguchi, M.; Suzuki, K. Stability of 11C-labeled PET radiopharmaceuticals. Appl. Radiat. Isot. 2004, 61, 1279–1287. [Google Scholar] [CrossRef]
- Lemaire, C.; Plenevaux, A.; Aerts, J.; del Fiore, G.; Brihaye, C.; le Bars, D.; Comar, D.; Luxen, A. Solid phase extraction—An alternative to the use of rotary evaporators for solvent removal in the rapid formulation of PET radiopharmaceuticals. J. Label. Compd. Radiopharm. 1999, 42, 63–75. [Google Scholar] [CrossRef]
- Meegalla, S.K.; Plössl, K.; Kung, M.-P.; Chumpradit, S.; Stevenson, D.A.; Kushner, S.A.; McElgin, W.T.; Mozley, P.D.; Kung, H.I.E. Synthesis and characterization of Tc-99m labeled tropanes as dopamine transporter imaging agents. J. Med. Chem. 1997, 40, 9–17. [Google Scholar] [CrossRef]
- Kung, H.F.; Kim, H.-J.; Kung, M.-R.; Meegalla, S.K.; Plössl, K.; Lee, H.-K. Imaging of dopamine transporters in humans with technetium-99m TRODAT-1. Eur. J. Nucl. Med. 1996, 23, 1527–1530. [Google Scholar] [CrossRef]
- Radiopharmaceutical Preparations. Radiopharmaceutica, 5.0/0125. In European Pharmacopoeia (Europaeisches Arzneibuch), 5th ed.; (5. Ausgabe Grundwerk); Official Austrian version; Verlag Oesterreich GmbH: Vienna, Germany, 2005; pp. 823–831. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mitterhauser, M.; Wadsak, W. Imaging Biomarkers or Biomarker Imaging? Pharmaceuticals 2014, 7, 765-778. https://doi.org/10.3390/ph7070765
Mitterhauser M, Wadsak W. Imaging Biomarkers or Biomarker Imaging? Pharmaceuticals. 2014; 7(7):765-778. https://doi.org/10.3390/ph7070765
Chicago/Turabian StyleMitterhauser, Markus, and Wolfgang Wadsak. 2014. "Imaging Biomarkers or Biomarker Imaging?" Pharmaceuticals 7, no. 7: 765-778. https://doi.org/10.3390/ph7070765
APA StyleMitterhauser, M., & Wadsak, W. (2014). Imaging Biomarkers or Biomarker Imaging? Pharmaceuticals, 7(7), 765-778. https://doi.org/10.3390/ph7070765