Dysregulation of the Mammalian Target of Rapamycin and p27Kip1 Promotes Intimal Hyperplasia in Diabetes Mellitus

{kind=link}
Abstract
:1. Introduction
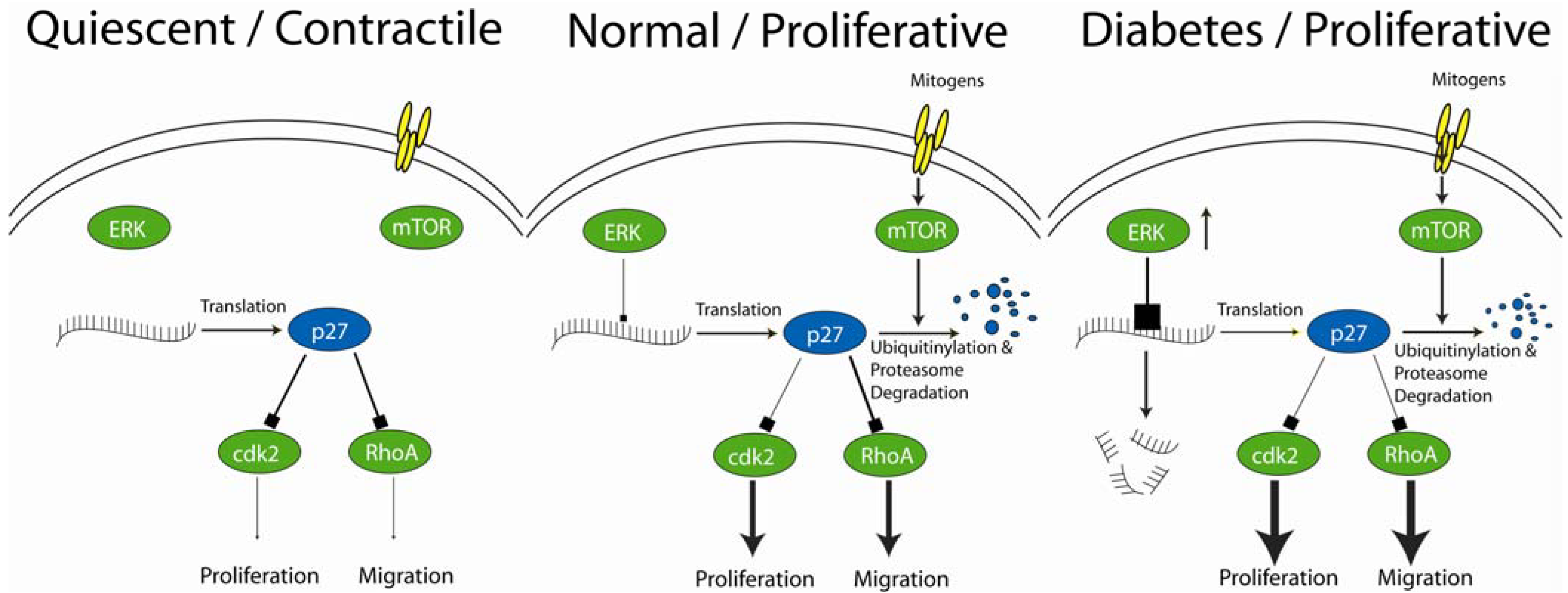
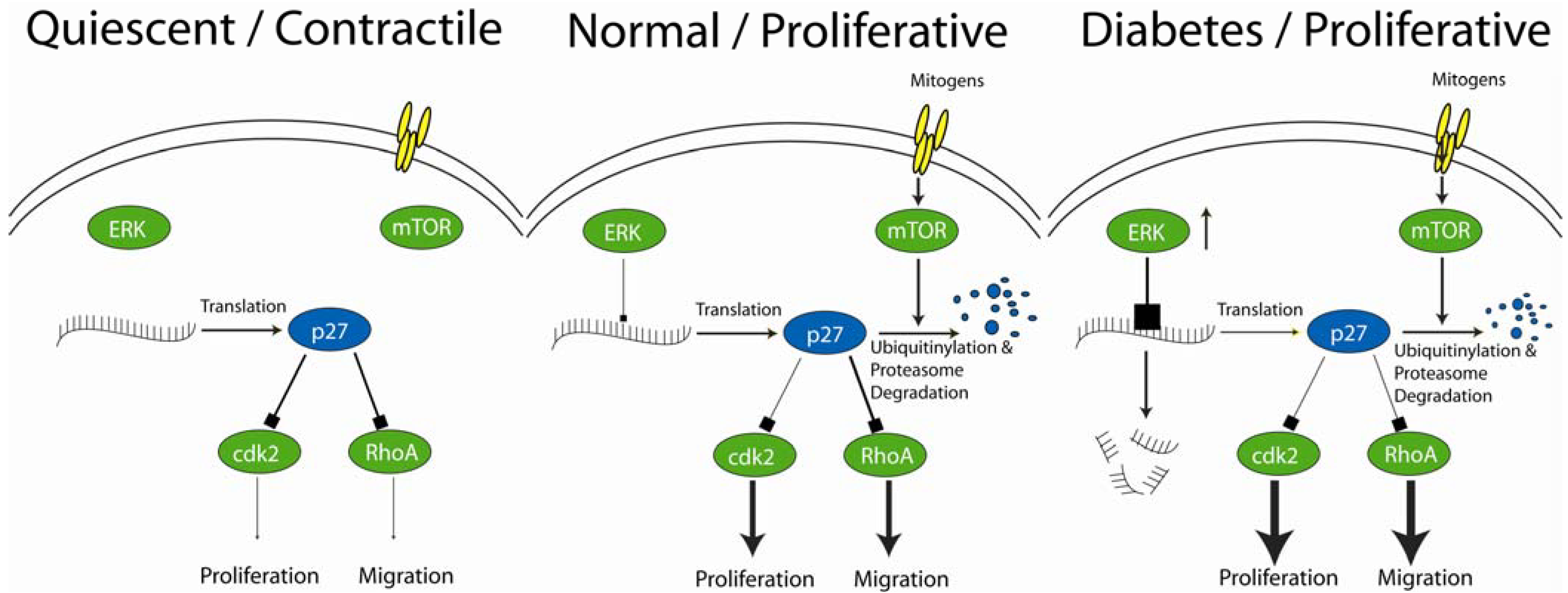
2. Role of the Cyclin-Dependent Kinase Inhibitor, p27Kip1, in Cardiovascular Disease
2.1. The Vascular Response to Injury
2.2. Intimal Thickening is Blocked by Elevated Levels of the Cyclin Dependent Kinase Inhibitor, p27Kip1
3. Clinical Use of mTOR Inhibitors in the Treatment of Cardiovascular Disease
4. Changes in the Molecular Mechanisms Regulating Cell Proliferation and Migration under Diabetic Conditions
4.1. Role of Ang II in VSMC Insulin Resistance
4.2. Hyperglycemia and IGF-1 Activation of ERK
4.3. Changes in Insulin Signaling in Response to Changes in IGFR Expression
5. Effects of Diabetes Mellitus on Efficacy of mTOR Inhibitors in Preventing In-Stent Restenosis
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Ross, R. The pathogenesis of atherosclerosis—An update. N. Engl. J. Med. 1986, 314, 488–500. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.A. The pathogenesis of atherosclerosis (second of two parts). N. Engl. J. Med. 1976, 295, 420–425. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.A. The pathogenesis of atherosclerosis (first of two parts). N. Engl. J. Med. 1976, 295, 369–377. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar]
- Ross, R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis: Current understanding of mechanisms and future strategies in therapy. Transplant Proc. 1993, 25, 2041–2043. [Google Scholar]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012, 110, 875–88. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Van der Wal, A.C.; Das, P.K.; Tigges, A.J.; Becker, A.E. Adhesion molecules on the endothelium and mononuclear cells in human atherosclerotic lesions. Am. J. Pathol. 1992, 141, 1427–1433. [Google Scholar]
- Wood, K.M.; Cadogan, M.D.; Ramshaw, A.L.; Parums, D.V. The distribution of adhesion molecules in human atherosclerosis. Histopathology 1993, 22, 437–444. [Google Scholar] [CrossRef]
- Geng, J.G.; Bevilacqua, M.P.; Moore, K.L.; McIntyre, T.M.; Prescott, S.M.; Kim, J.M.; Bliss, G.A.; Zimmerman, G.A.; McEver, R.P. Rapid neutrophil adhesion to activated endothelium mediated by GMP-140. Nature 1990, 343, 757–760. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Stengelin, S.; Gimbrone, M.A., Jr; Seed, B. Endothelial leukocyte adhesion molecule 1: An inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science 1989, 243, 1160–1165. [Google Scholar]
- Charo, I.F.; Taubman, M.B. Chemokines in the pathogenesis of vascular disease. Circ. Res. 2004, 95, 858–866. [Google Scholar] [CrossRef]
- Heeneman, S.; Sluimer, J.C.; Daemen, M.J. Angiotensin-converting enzyme and vascular remodeling. Circ. Res. 2007, 101, 441–454. [Google Scholar] [CrossRef]
- Boehm, M.; Yoshimoto, T.; Crook, M.F.; Nallamshetty, S.; True, A.; Nabel, G.J.; Nabel, E.G. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002, 21, 3390–3401. [Google Scholar] [CrossRef]
- Hara, T.; Kamura, T.; Nakayama, K.; Oshikawa, K.; Hatakeyama, S. Degradation of p27(Kip1) at the G(0)-G(1) transition mediated by a Skp2-independent ubiquitination pathway. J. Biol. Chem. 2001, 276, 48937–48943. [Google Scholar]
- Kamura, T.; Hara, T.; Matsumoto, M.; Ishida, N.; Okumura, F.; Hatakeyama, S.; Yoshida, M.; Nakayama, K.; Nakayama, K.I. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat. Cell Biol. 2004, 6, 1229–1235. [Google Scholar] [CrossRef]
- Kotoshiba, S.; Kamura, T.; Hara, T.; Ishida, N.; Nakayama, K.I. Molecular dissection of the interaction between p27 and Kip1 ubiquitylation-promoting complex, the ubiquitin ligase that regulates proteolysis of p27 in G1 phase. J. Biol. Chem. 2005, 280, 17694–17700. [Google Scholar]
- Carrano, A.C.; Eytan, E.; Hershko, A.; Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999, 1, 193–199. [Google Scholar] [CrossRef]
- Montagnoli, A.; Fiore, F.; Eytan, E.; Carrano, A.C.; Draetta, G.F.; Hershko, A.; Pagano, M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999, 13, 1181–1189. [Google Scholar] [CrossRef]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Miyake, S.; Ishida, N.; Hatakeyama, S.; Kitagawa, M.; Iemura, S.; Natsume, T.; Nakayama, K.I. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 2004, 6, 661–672. [Google Scholar] [CrossRef]
- Koff, A.; Polyak, K. p27KIP1, an inhibitor of cyclin-dependent kinases. Prog. Cell Cycle Res. 1995, 1, 141–147. [Google Scholar] [CrossRef]
- Bresnahan, W.A.; Boldogh, I.; Ma, T.; Albrecht, T.; Thompson, E.A. Cyclin E/Cdk2 activity is controlled by different mechanisms in the G0 and G1 phases of the cell cycle. Cell Growth Differ. 1996, 7, 1283–1290. [Google Scholar]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Nakayama, M.; Amano, M.; Katsumi, A.; Kaneko, T.; Kawabata, S.; Takefuji, M.; Kaibuchi, K. Rho-kinase and myosin II activities are required for cell type and environment specific migration. Genes Cells 2005, 10, 107–117. [Google Scholar] [CrossRef]
- Moss, S.C.; Lightell, D.J., Jr; Marx, S.O.; Marks, A.R.; Woods, T.C. Rapamycin regulates endothelial cell migration through regulation of the cyclin-dependent kinase inhibitor p27Kip1. J. Biol. Chem. 2010, 285, 11991–11997. [Google Scholar]
- Besson, A.; Gurian-West, M.; Schmidt, A.; Hall, A.; Roberts, J.M. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004, 18, 862–876. [Google Scholar]
- Gallo, R.; Padurean, A.; Jayaraman, T.; Marx, S.; Roque, M.; Adelman, S.; Chesebro, J.; Fallon, J.; Fuster, V.; Marks, A.; Badimon, J.J. Inhibition of intimal thickening after balloon angioplasty in porcine coronary arteries by targeting regulators of the cell cycle. Circulation 1999, 99, 2164–2170. [Google Scholar] [CrossRef]
- Luo, Y.; Marx, S.O.; Kiyokawa, H.; Koff, A.; Massague, J.; Marks, A.R. Rapamycin resistance tied to defective regulation of p27Kip1. Mol. Cell. Biol. 1996, 16, 6744–6751. [Google Scholar]
- Marx, S.O.; Jayaraman, T.; Go, L.O.; Marks, A.R. Rapamycin-FKBP inhibits cell cycle regulators of proliferation in vascular smooth muscle cells. Circ. Res. 1995, 76, 412–417. [Google Scholar] [CrossRef]
- Sun, J.; Marx, S.O.; Chen, H.J.; Poon, M.; Marks, A.R.; Rabbani, L.E. Role for p27(Kip1) in Vascular Smooth Muscle Cell Migration. Circulation 2001, 103, 2967–2972. [Google Scholar] [CrossRef]
- Chen, D.; Krasinski, K.; Sylvester, A.; Chen, J.; Nisen, P.D.; Andres, V. Downregulation of cyclin-dependent kinase 2 activity and cyclin A promoter activity in vascular smooth muscle cells by p27(KIP1), an inhibitor of neointima formation in the rat carotid artery. J. Clin. Invest. 1997, 99, 2334–2341. [Google Scholar] [CrossRef]
- Lightell, D.J., Jr.; Moss, S.C.; Woods, T.C. Loss of Canonical Insulin Signaling Accelerates Vascular Smooth Muscle Cell Proliferation and Migration Through Changes in p27Kip1 Regulation. Endocrinology 2011, 152, 651–658. [Google Scholar] [CrossRef]
- Roque, M.; Reis, E.D.; Cordon-Cardo, C.; Taubman, M.B.; Fallon, J.T.; Fuster, V.; Badimon, J.J. Effect of p27 deficiency and rapamycin on intimal hyperplasia: In vivo and in vitro studies using a p27 knockout mouse model. Lab Invest. 2001, 81, 895–903. [Google Scholar] [CrossRef]
- Wu, Y.J.; Sala-Newby, G.B.; Shu, K.T.; Yeh, H.I.; Nakayama, K.I.; Nakayama, K.; Newby, A.C.; Bond, M. S-phase kinase-associated protein-2 (Skp2) promotes vascular smooth muscle cell proliferation and neointima formation in vivo. J. Vasc. Surg. 2009, 50, 1135–1142. [Google Scholar]
- Song, P.; Wang, S.; He, C.; Liang, B.; Viollet, B.; Zou, M.H. AMPKalpha2 Deletion Exacerbates Neointima Formation by Upregulating Skp2 in Vascular Smooth Muscle Cells. Circ. Res. 2011, 109, 1230–1239. [Google Scholar] [CrossRef]
- Martin, K.A.; Rzucidlo, E.M.; Merenick, B.L.; Fingar, D.C.; Brown, D.J.; Wagner, R.J.; Powell, R.J. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am. J. Physiol. Cell Physiol. 2004, 286, C507–C517. [Google Scholar] [CrossRef]
- Song, G.J.; Barrick, S.; Leslie, K.L.; Bauer, P.M.; Alonso, V.; Friedman, P.A.; Fiaschi-Taesch, N.M.; Bisello, A. The scaffolding protein EBP50 promotes vascular smooth muscle cell proliferation and neointima formation by regulating Skp2 and p21(cip1). Arterioscler. Thromb. Vasc. Biol. 2012, 32, 33–41. [Google Scholar] [CrossRef]
- Bond, M.; Sala-Newby, G.B.; Wu, Y.J.; Newby, A.C. Biphasic effect of p21Cip1 on smooth muscle cell proliferation: Role of PI 3-kinase and Skp2-mediated degradation. Cardiovasc. Res. 2006, 69, 198–206. [Google Scholar] [CrossRef]
- Diez-Juan, A.; Andres, V. The growth suppressor p27(Kip1) protects against diet-induced atherosclerosis. Faseb. J. 2001, 15, 1989–1995. [Google Scholar]
- Akyurek, L.M.; Boehm, M.; Olive, M.; Zhou, A.X.; San, H.; Nabel, E.G. Deficiency of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1 accelerates atherogenesis in apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2010, 396, 359–363. [Google Scholar] [CrossRef]
- Shahzad, K.; Thati, M.; Wang, H.; Kashif, M.; Wolter, J.; Ranjan, S.; He, T.; Zhou, Q.; Blessing, E.; Bierhaus, A.; Nawroth, P.P.; Isermann, B. Minocycline reduces plaque size in diet induced atherosclerosis via p27(Kip1). Atherosclerosis 2011, 219, 74–83. [Google Scholar] [CrossRef]
- Fattori, R.; Piva, T. Drug-eluting stents in vascular intervention. Lancet 2003, 361, 247–249. [Google Scholar] [CrossRef]
- Bhaskar, P.T.; Hay, N. The two TORCs and Akt. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Hresko, R.C.; Mueckler, M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 40406–40416. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell. 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar]
- Marso, S.P.; Gimple, L.W.; Philbrick, J.T.; DiMarco, J.P. Effectiveness of percutaneous coronary interventions to prevent recurrent coronary events in patients on chronic hemodialysis. Am. J. Cardiol. 1998, 82, 378–380. [Google Scholar]
- Koyanagi, T.; Nishida, H.; Kitamura, M.; Endo, M.; Koyanagi, H.; Kawaguchi, M.; Magosaki, N.; Sumiyoshi, T.; Hosoda, S. Comparison of clinical outcomes of coronary artery bypass grafting and percutaneous transluminal coronary angioplasty in renal dialysis patients. Ann. Thorac. Surg. 1996, 61, 1793–1796. [Google Scholar] [CrossRef]
- Azar, R.R.; Prpic, R.; Ho, K.K.; Kiernan, F.J.; Shubrooks, S.J., Jr; Baim, D.S.; Popma, J.J.; Kuntz, R.E.; Cohen, D.J. Impact of end-stage renal disease on clinical and angiographic outcomes after coronary stenting. Am. J. Cardiol. 2000, 86, 485–489. [Google Scholar] [CrossRef]
- Elezi, S.; Kastrati, A.; Pache, J.; Wehinger, A.; Hadamitzky, M.; Dirschinger, J.; Neumann, F.J.; Schomig, A. Diabetes mellitus and the clinical and angiographic outcome after coronary stent placement. J. Am. Coll. Cardiol. 1998, 32, 1866–1873. [Google Scholar]
- Woods, T.; Marks, A. Drug-Eluting Stents. Ann. Rev. Med. 2004, 55, 169–178. [Google Scholar] [CrossRef]
- Stettler, C.; Allemann, S.; Egger, M.; Windecker, S.; Meier, B.; Diem, P. Efficacy of drug eluting stents in patients with and without diabetes mellitus: Indirect comparison of controlled trials. Heart 2006, 92, 650–657. [Google Scholar] [CrossRef]
- Kastrati, A.; Massberg, S.; Ndrepepa, G. Is diabetes the achilles’ heel of limus-eluting stents? Circulation 2011, 124, 869–872. [Google Scholar] [CrossRef]
- Stone, G.W.; Kedhi, E.; Kereiakes, D.J.; Parise, H.; Fahy, M.; Serruys, P.W.; Smits, P.C. Differential clinical responses to everolimus-eluting and Paclitaxel-eluting coronary stents in patients with and without diabetes mellitus. Circulation 2011, 124, 893–900. [Google Scholar]
- Wendt, T.; Bucciarelli, L.; Qu, W.; Lu, Y.; Yan, S.F.; Stern, D.M.; Schmidt, A.M. Receptor for advanced glycation endproducts (RAGE) and vascular inflammation: Insights into the pathogenesis of macrovascular complications in diabetes. Curr. Atheroscler. Rep. 2002, 4, 228–237. [Google Scholar] [CrossRef]
- Jones, J.I.; Prevette, T.; Gockerman, A.; Clemmons, D.R. Ligand occupancy of the alpha-V-beta3 integrin is necessary for smooth muscle cells to migrate in response to insulin-like growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 2482–2487. [Google Scholar]
- Maile, L.A.; Capps, B.E.; Ling, Y.; Xi, G.; Clemmons, D.R. Hyperglycemia alters the responsiveness of smooth muscle cells to insulin-like growth factor-I. Endocrinology 2007, 148, 2435–2443. [Google Scholar] [CrossRef]
- Folli, F.; Kahn, C.R.; Hansen, H.; Bouchie, J.L.; Feener, E.P. Angiotensin II inhibits insulin signaling in aortic smooth muscle cells at multiple levels. A potential role for serine phosphorylation in insulin/angiotensin II crosstalk. J. Clin. Invest. 1997, 100, 2158–2169. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—Dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Taniyama, Y.; Hitomi, H.; Shah, A.; Alexander, R.W.; Griendling, K.K. Mechanisms of reactive oxygen species-dependent downregulation of insulin receptor substrate-1 by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1142–1147. [Google Scholar]
- Nakanishi, H.; Brewer, K.A.; Exton, J.H. Activation of the zeta isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1993, 268, 13–16. [Google Scholar]
- Kim, T.N.; Kim, S.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Vascular Inflammation in Patients with Impaired Glucose Tolerance and Type 2 Diabetes: Analysis with 18F-Fluorodeoxyglucose Positron Emission Tomography. Circ. Cardiovasc. Imaging 2010, 3, 142–148. [Google Scholar]
- Preis, S.R.; Hwang, S.J.; Coady, S.; Pencina, M.J.; D'Agostino, R.B., Sr; Savage, P.J.; Levy, D.; Fox, C.S. Trends in all-cause and cardiovascular disease mortality among women and men with and without diabetes mellitus in the Framingham Heart Study, 1950 to 2005. Circulation 2009, 119, 1728–1735. [Google Scholar]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H., Jr; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998, 352, 854–865. [CrossRef]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef]
- Jonas, M.; Edelman, E.R.; Groothuis, A.; Baker, A.B.; Seifert, P.; Rogers, C. Vascular neointimal formation and signaling pathway activation in response to stent injury in insulin-resistant and diabetic animals. Circ. Res. 2005, 97, 725–733. [Google Scholar] [CrossRef]
- Gogg, S.; Smith, U.; Jansson, P.A. Increased MAPK activation and impaired insulin signaling in subcutaneous microvascular endothelial cells in type 2 diabetes: The role of endothelin-1. Diabetes 2009, 58, 2238–2245. [Google Scholar] [CrossRef]
- Lightell, D.J., Jr.; Woods, T.C. Relative Resistance to mTOR Inhibition in Vascular Smooth Muscle Cells of Diabetic Donors. Ochsner. J. 2013, 13, 56–60. [Google Scholar]
- Henriksen, E.J.; Jacob, S.; Kinnick, T.R.; Teachey, M.K.; Krekler, M. Selective angiotensin II receptor receptor antagonism reduces insulin resistance in obese Zucker rats. Hypertension 2001, 38, 884–890. [Google Scholar] [CrossRef]
- Igarashi, M.; Hirata, A.; Yamaguchi, H.; Tsuchiya, H.; Ohnuma, H.; Tominaga, M.; Daimon, M.; Kato, T. Candesartan inhibits carotid intimal thickening and ameliorates insulin resistance in balloon-injured diabetic rats. Hypertension 2001, 38, 1255–1259. [Google Scholar] [CrossRef]
- Arnqvist, H.J.; Bornfeldt, K.E.; Chen, Y.; Lindstrom, T. The insulin-like growth factor system in vascular smooth muscle: Interaction with insulin and growth factors. Metabolism 1995, 44, 58–66. [Google Scholar] [CrossRef]
- Johansson, G.S.; Arnqvist, H.J. Insulin and IGF-I action on insulin receptors, IGF-I receptors, and hybrid insulin/IGF-I receptors in vascular smooth muscle cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1124–E1130. [Google Scholar] [CrossRef]
- Chisalita, S.I.; Arnqvist, H.J. Expression and function of receptors for insulin-like growth factor-I and insulin in human coronary artery smooth muscle cells. Diabetologia 2005, 48, 2155–2161. [Google Scholar] [CrossRef]
- Moxham, C.P.; Duronio, V.; Jacobs, S. Insulin-like growth factor I receptor beta-subunit heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I and insulin receptor heterodimers. J. Biol. Chem. 1989, 264, 13238–13244. [Google Scholar]
- Bailyes, E.M.; Nave, B.T.; Soos, M.A.; Orr, S.R.; Hayward, A.C.; Siddle, K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: Quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem. J. 1997, 327, 209–215. [Google Scholar]
- Soos, M.A.; Siddle, K. Immunological relationships between receptors for insulin and insulin-like growth factor I. Evidence for structural heterogeneity of insulin-like growth factor I receptors involving hybrids with insulin receptors. Biochem. J. 1989, 263, 553–563. [Google Scholar]
- Seely, B.L.; Reichart, D.R.; Takata, Y.; Yip, C.; Olefsky, J.M. A functional assessment of insulin/insulin-like growth factor-I hybrid receptors. Endocrinology 1995, 136, 1635–1641. [Google Scholar] [CrossRef]
- Soos, M.A.; Field, C.E.; Siddle, K. Purified hybrid insulin/insulin-like growth factor-I receptors bind insulin-like growth factor-I, but not insulin, with high affinity. Biochem. J. 1993, 290, 419–426. [Google Scholar]
- Engberding, N.; San Martin, A.; Martin-Garrido, A.; Koga, M.; Pounkova, L.; Lyons, E.; Lassegue, B.; Griendling, K.K. Insulin-Like Growth Factor-1 Receptor Expression Masks the Antiinflammatory and Glucose Uptake Capacity of Insulin in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 408–415. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Woods, T.C. Dysregulation of the Mammalian Target of Rapamycin and p27Kip1 Promotes Intimal Hyperplasia in Diabetes Mellitus. Pharmaceuticals 2013, 6, 716-727. https://doi.org/10.3390/ph6060716
Woods TC. Dysregulation of the Mammalian Target of Rapamycin and p27Kip1 Promotes Intimal Hyperplasia in Diabetes Mellitus. Pharmaceuticals. 2013; 6(6):716-727. https://doi.org/10.3390/ph6060716
Chicago/Turabian StyleWoods, Thomas Cooper. 2013. "Dysregulation of the Mammalian Target of Rapamycin and p27Kip1 Promotes Intimal Hyperplasia in Diabetes Mellitus" Pharmaceuticals 6, no. 6: 716-727. https://doi.org/10.3390/ph6060716
APA StyleWoods, T. C. (2013). Dysregulation of the Mammalian Target of Rapamycin and p27Kip1 Promotes Intimal Hyperplasia in Diabetes Mellitus. Pharmaceuticals, 6(6), 716-727. https://doi.org/10.3390/ph6060716