Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs


Abstract
:
1. Introduction
2. The APD Database Features Useful for Peptide Design
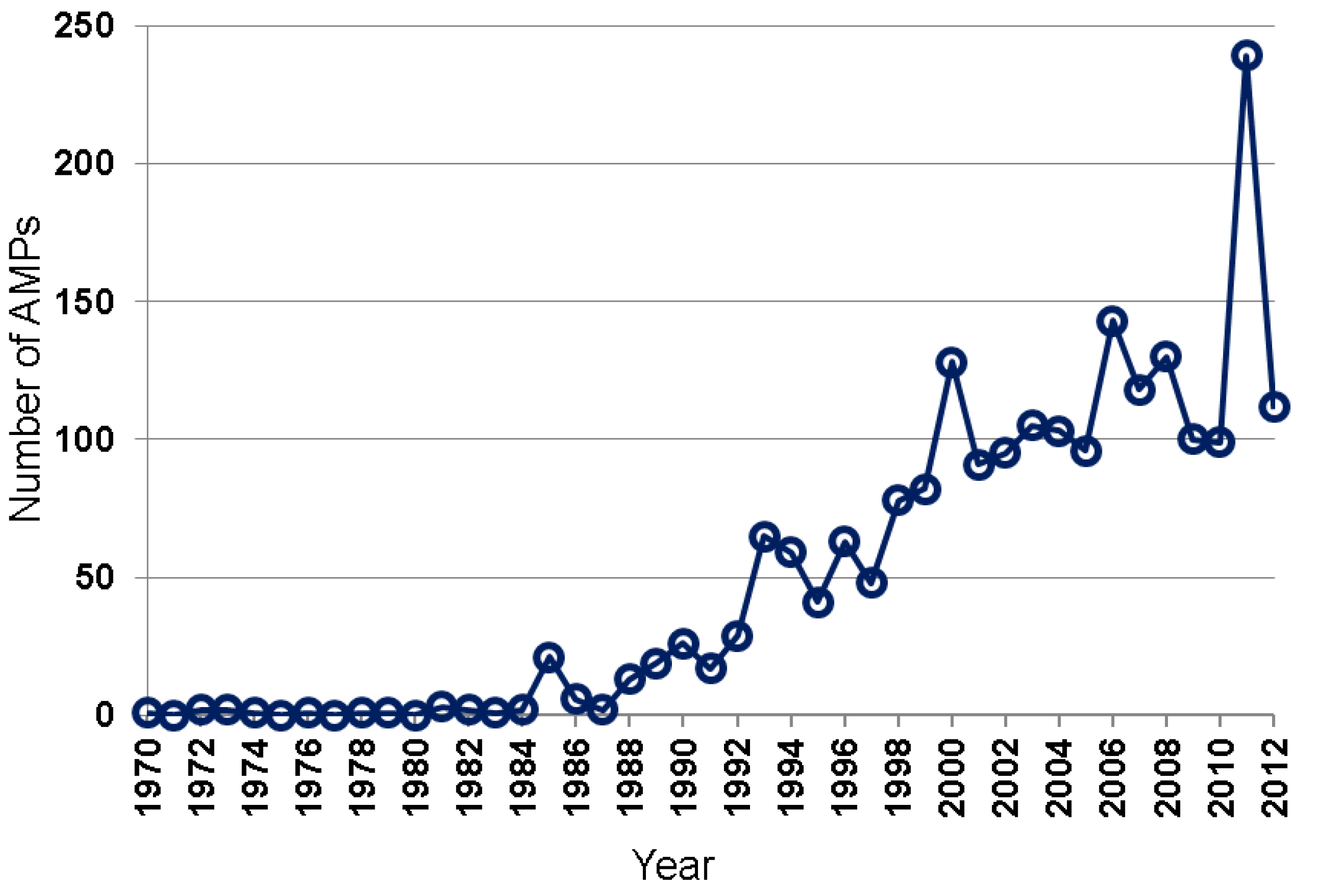
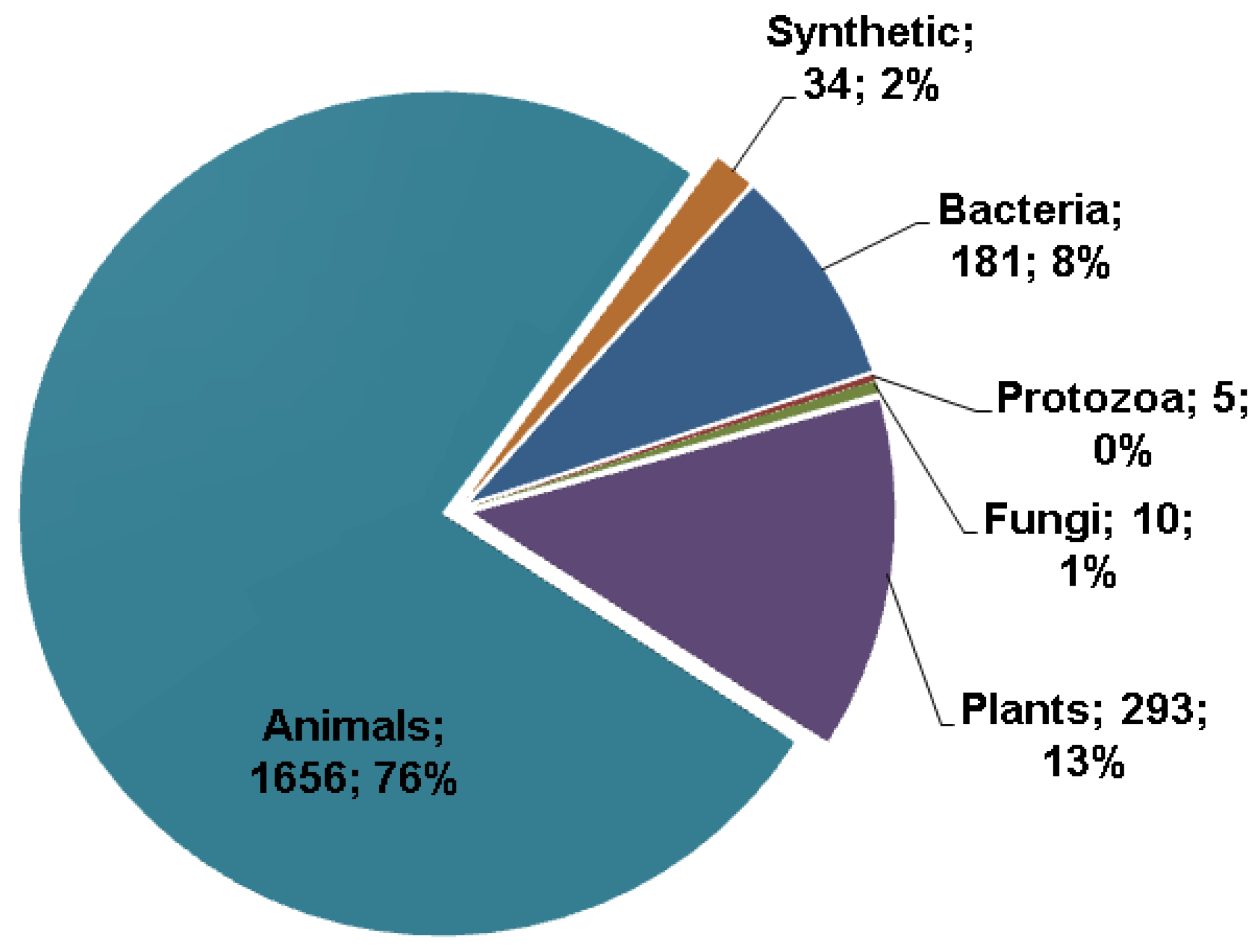
2.1. Peptide Sources

2.2. AMP Activity Annotated in the APD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide activity | Peptide number | Abundant amino acids |
|---|---|---|
| Antibacterial | 1768 | L, G, S, K |
| Antiviral | 158 | C, G, S, R |
| Antifungal | 777 | C, G, S, K |
| Antiparasitic | 48 | C, G, S, K |
| Insecticidal | 22 | L, G, T, K |
| Spermicidal | 9 | A, G, T, K |
| Anticancer | 145 | C, G, S, K |
| Hemolytic | 255 | L, G, S, K |
| Chemotactic | 41 | L, G/P, S, K |
2.3. Calculations of AMP Parameters and Amino Acid Composition
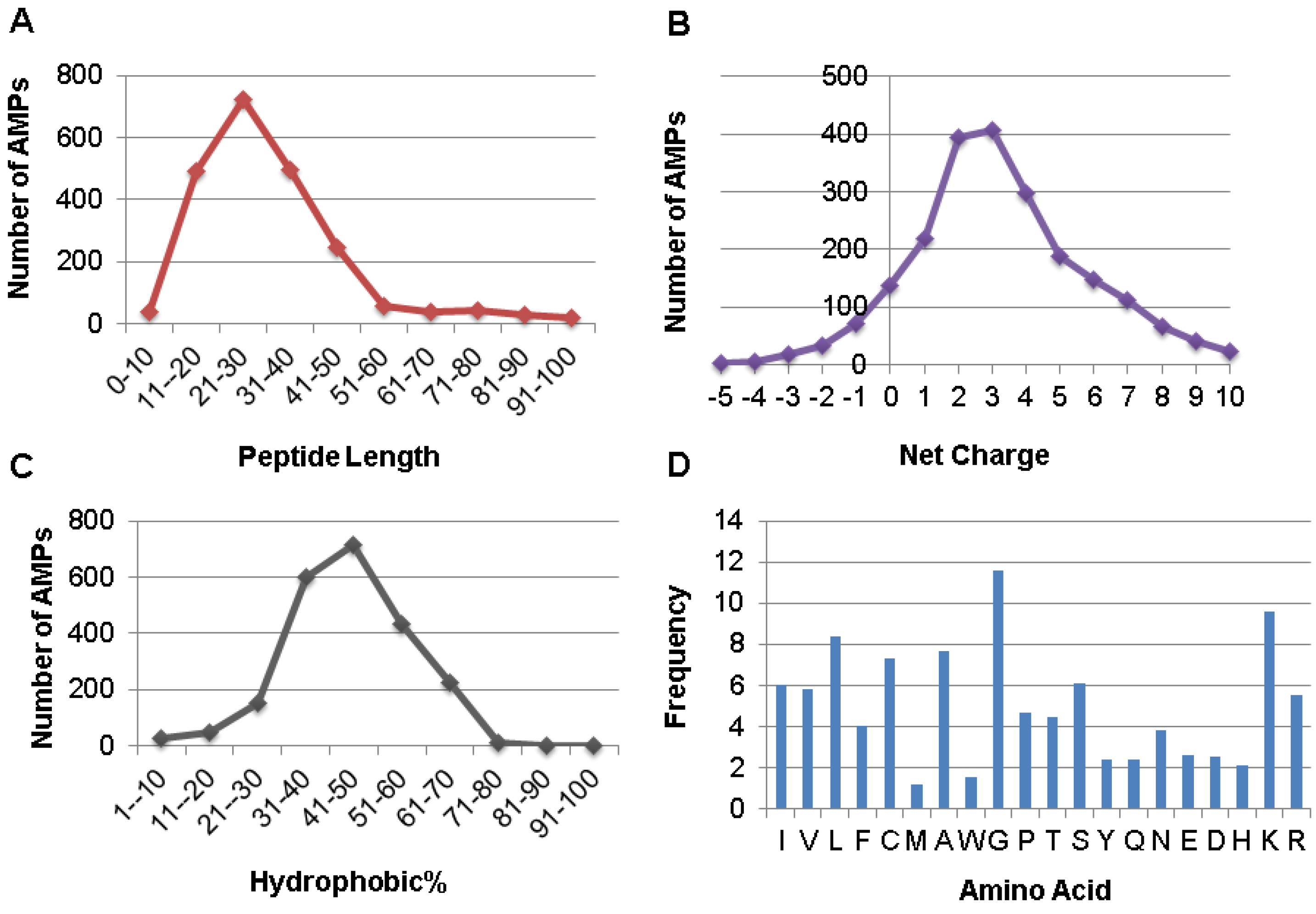
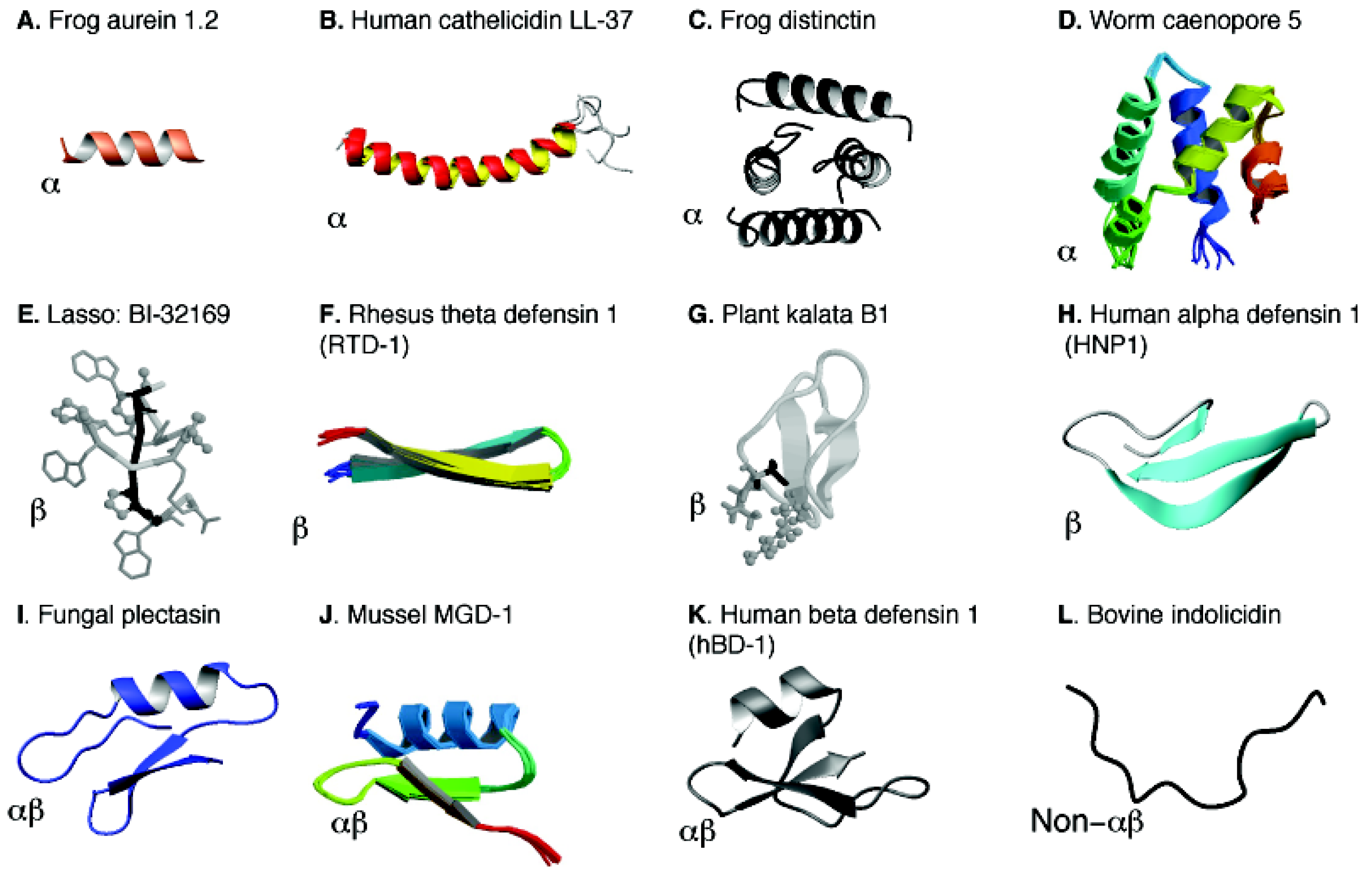
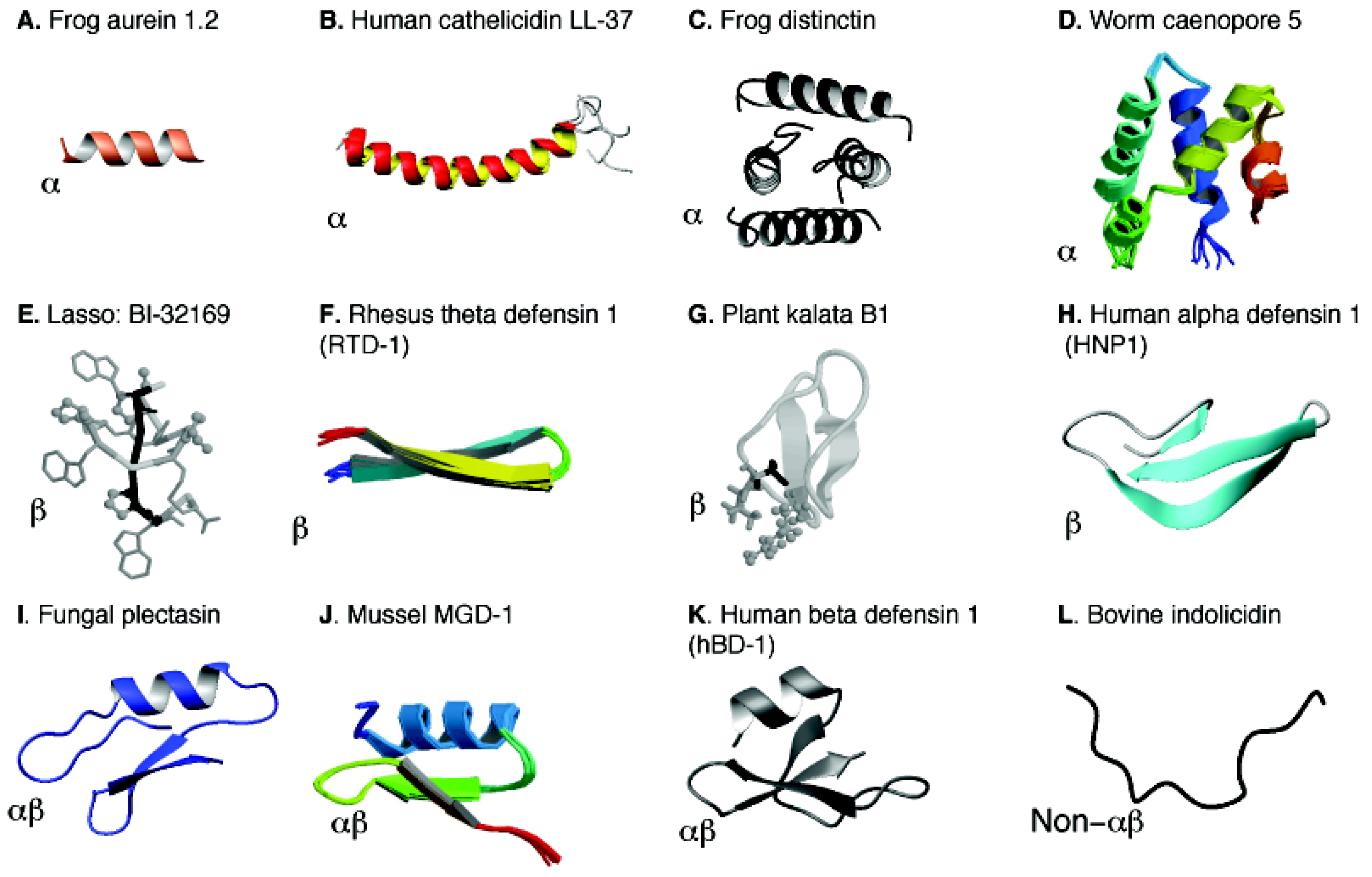
3. Peptide Discovery Based on the APD Database
3.1. The Linguistic Model
3.2. Database Screening
3.2.1. Identification of HIV-1 Inhibitory Peptides
| Peptide Name | S. aureus USA300 (MIC, μM) | Peptide Name | HIV-1 (EC50, μM) |
|---|---|---|---|
| Ascaphin-8 | 3.1 | Ascaphin-8 | 1.2 |
| DASamP1 | 3.1 | DASamP1 | 0.63 |
| DASamP2 | 6.2 | DASamP5 | 0.83 |
| Lycotoxin I | 3.1 | Ponericin L2 | 1.4 |
| Maculatin 1.3 | 6.2 | Brevinin-2 related | 1.65 |
| Piscidin 1 | 3.1 | Piscidin 1 | 2.1 |
3.2.2. Identification of Anti-Staphylococcal Peptides
3.3. Database-Guided Design of Antimicrobial Agents
3.3.1. De Novo Design of Anti-HIV Peptides
| Peptide name | Amino acid sequence | Design strategy | EC50 (μM) | MIC (μM) | Stability (% left) | Ref. |
|---|---|---|---|---|---|---|
| GLK-19 | GLKKLLGKLLKKLGKLLLK | G, L, K | >47.5 | 10 | NE | [9] |
| GLR-19 | GLRRLLGRLLRRLGRLLLR | K to R | 4.4 | >120 | 14.5 | [52] |
| GLRC-1 | GCRRLLGRLLRRLGRLLCR | C2-C18 | >43.8 | 30 | 20.8 | [54] |
| GLRC-2 | GLRCRLGRLLRRLGRCLLR | C4-C16 | 0.79 | 30 | 63.3 | [54] |
| GLRC-3 | GLRRLCGRLGRRLCRLLLR | C6-C14 | 2.8 | 7.5 | 1.4 | [54] |
| GLRC-4 | GCRRLCGRLGRRLCRLLCR | C2-C18; C6-C14 | 30.7 | 60 | 9.7 | [54] |
3.3.2. Ab Initio Design of Anti-MRSA Peptides
3.4. Template-Based Design and Optimization
4. Differences in Binding Targets and Mechanisms of Action of AMPs
5. Structural Annotation, Classification and Determination of AMPs
5.1. Structural Annotation and Classification
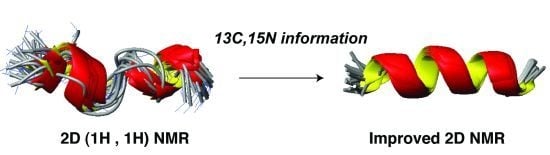
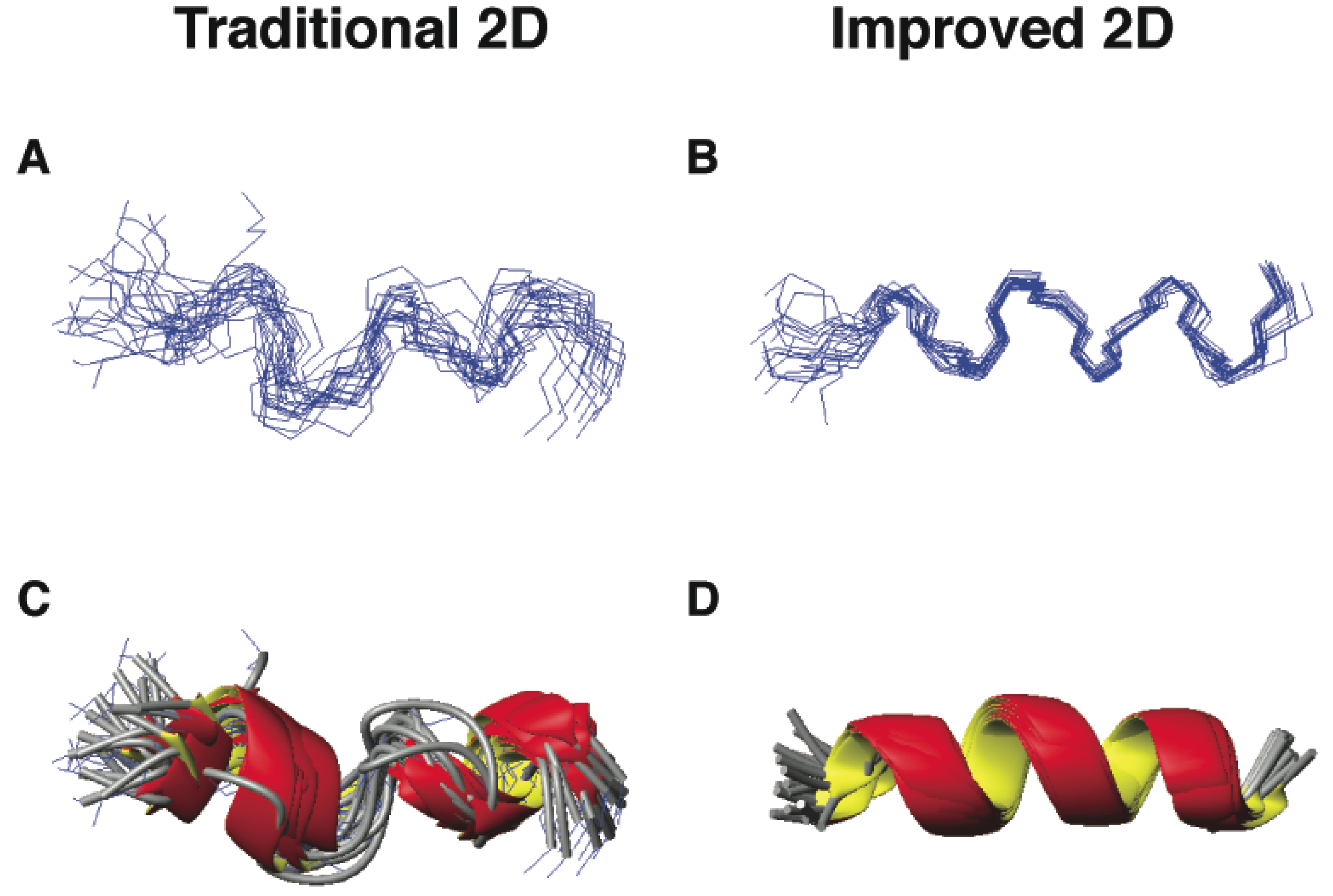
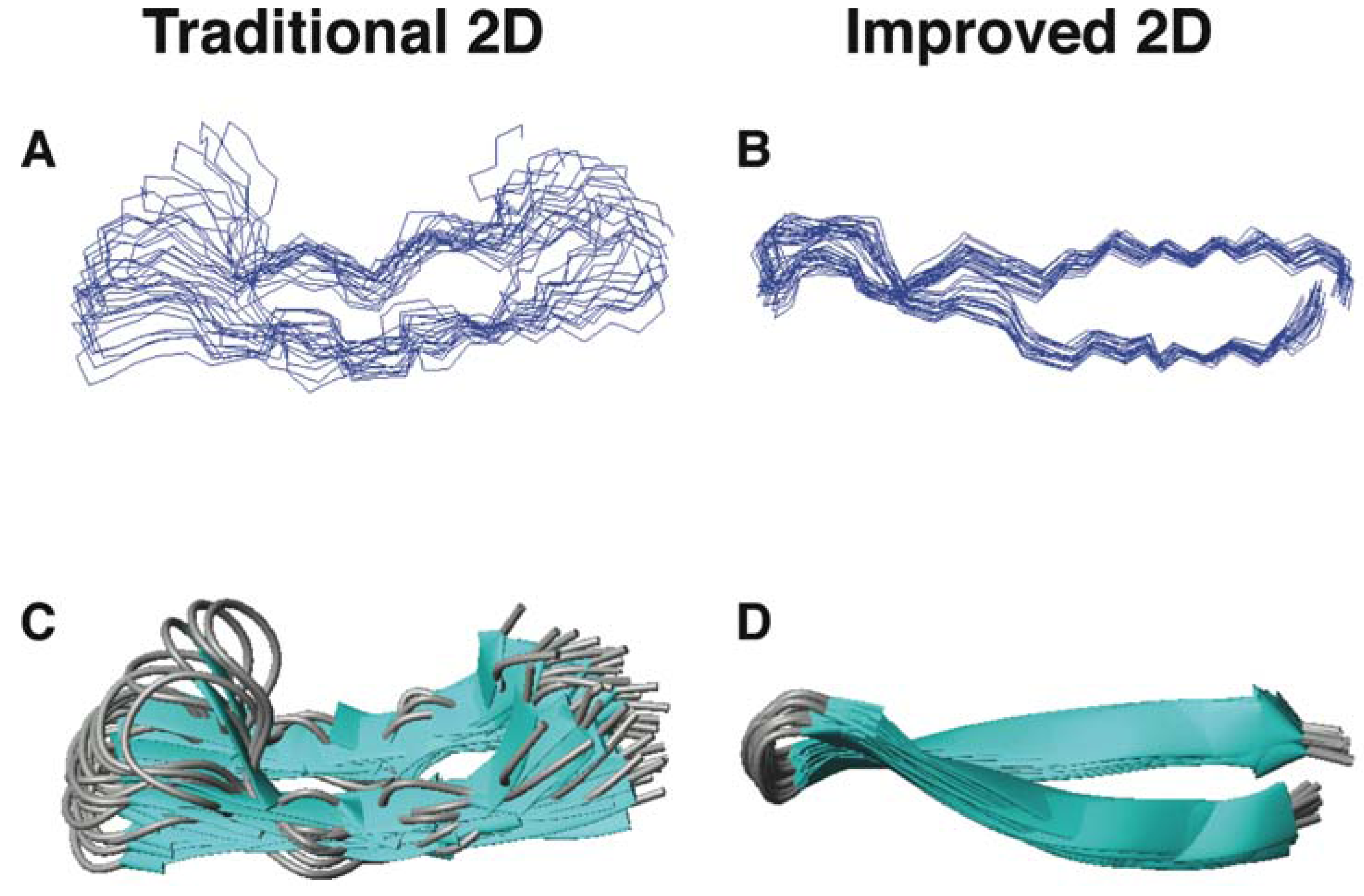
5.2. Structural Determination of Database-Designed Peptides by the Improved 2D NMR Method
| Nucleus | Traditional 2D NMR method [103] | Improved 2D NMR method [50] |
|---|---|---|
| 1H | TOCSY, DQF-COSY, and NOESY | TOCSY, DQF-COSY, and NOESY |
| 13C | Natural abundance (1H, 13C) HSQC | |
| 15N | Natural abundance (1H, 15N) HSQC |

5.3. Beyond 2D NMR
6. Concluding Remarks
Acknowledgements
Conflict of Interest
References
- Alexander Fleming (1881-1955): A Noble Life in Science. Available online: http://www.bl.uk/onlinegallery/features/beautifulminds/fleming.html/ (accessed on 2 May 2013).
- Dubos, R.J. Studies on a bactericidal agent extracted from a soil bacillus: I. Preparation of the agent. Its activity in vitro. J. Exp. Med. 1939, 70, 1–10. [Google Scholar] [CrossRef]
- Gran, L. On the effect of a polypeptide isolated from “Kalata-Kalata” (Oldenlandia affinis DC) on the oestrogen dominated uterus. Acta Pharmacol. Toxicol. (Copenh) 1973, 33, 400–408. [Google Scholar] [CrossRef]
- Tam, J.P.; Lu, Y.A.; Yang, J.L.; Chiu, K.W. An unusual structural motif of antimicrobial peptides containing end-to-end macrocycle and cystine-knot disulfides. Proc. Natl. Acad. Sci. USA. 1999, 96, 8913–8918. [Google Scholar]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef]
- Selsted, M.E.; Harwig, S.S.; Ganz, T.; Schilling, J.W.; Lehrer, R.I. Primary structures of three human neutrophil defensins. J. Clin. Invest. 1985, 76, 1436–1439. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, G. APD: The antimicrobial peptide database. Nucleic Acids Res. 2004, 32, D590–D592. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef]
- Seshadri Sundararajan, V.; Gabere, M.N.; Pretorius, A.; Adam, S.; Christoffels, A.; Lehväslaiho, M.; Archer, J.A.; Bajic, V.B. DAMPD: A manually curated antimicrobial peptide database. Nucleic Acids Res. 2012, 40, D1108–D1112. [Google Scholar] [CrossRef]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents. 2012, 39, 346–351. [Google Scholar] [CrossRef]
- Antiinfective peptides laboratory Tossi group. Available online: http://www.bbcm.univ.trieste.it/~tossi/amsdb.html (accessed on 3 May 2013).
- Whitmore, L.; Wallace, B.A. The Peptaibol Database: A database for sequences and structures of naturally occurring peptaibols. Nucleic Acids Res. 2004, 32, D593–D594. [Google Scholar] [CrossRef]
- Hammami, R.; Ben Hamida, J.; Vergoten, G.; Fliss, I. PhytAMP: A database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2009, 37, D963–D968. [Google Scholar] [CrossRef]
- Gueguen, Y.; Garnier, J.; Robert, L.; Lefranc, M.P.; Mougenot, I.; de Lorgeril, J.; Janech, M.; Gross, P.S.; Warr, G.W.; Cuthbertson, B.; et al. PenBase, the shrimp antimicrobial peptide penaeidin database: Sequence-based classification and recommended nomenclature. Dev. Comp. Immunol. 2006, 30, 283–288. [Google Scholar] [CrossRef]
- Novković, M.; Simunić, J.; Bojović, V.; Tossi, A.; Juretić, D. DADP: The database of anuran defense peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef]
- De Jong, A.; van Heel, A.J.; Kok, J.; Kuipers, O.P. BAGEL2: Mining for bacteriocins in genomic data. Nucleic Acids Res. 2010, 38, W647–W651. [Google Scholar] [CrossRef]
- Hammami, R.; Zouhir, A.; Le Lay, C.; Ben Hamida, J.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; He, X.; Duan, L.; Wu, G.; Bi, D.; Deng, Z.; Liu, W.; Ou, H.Y. ThioFinder: A web-based tool for the identification of thiopeptide gene clusters in DNA sequences. PLoS One 2012, 7, e45878. [Google Scholar]
- Wang, C.K.; Kaas, Q.; Chiche, L.; Craik, D.J. CyBase: A database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res. 2008, 36, D206–D210. [Google Scholar]
- Seebah, S.; Suresh, A.; Zhuo, S.; Choong, Y.H.; Chua, H.; Chuon, D.; Beuerman, R.; Verma, C. Defensins knowledgebase: A manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 2007, 35, D265–D268. [Google Scholar] [CrossRef]
- Wu, H.; Lu, H.; Huang, J.; Li, G.; Huang, Q. EnzyBase: A novel database for enzybiotic studies. BMC Microbiol. 2012, 12, 54. [Google Scholar] [CrossRef]
- Wade, D.; Englund, J. Synthetic antibiotic peptides database. Protein Pept. Lett. 2002, 9, 53–57. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z. RAPD: A database of recombinantly-produced antimicrobial peptides. FEMS Microbiol. Lett. 2008, 289, 126–129. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Zasloff, M. A database view of natural antimicrobial peptides: Nomenclature, classification and amino acid sequence analysis. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies; Wang, G., Ed.; CABI: Wallingford, England, 2010; pp. 1–21. [Google Scholar]
- Ma, Y.; Liu, C.; Liu, X.; Wu, J.; Yang, H.; Wang, Y.; Li, J.; Yu, H.; Lai, R. Peptidomics and genomics analysis of novel antimicrobial peptides from the frog, Rana nigrovittata. Genomics 2010, 95, 66–71. [Google Scholar]
- Yang, X.; Lee, W.H.; Zhang, Y. Extremely Abundant Antimicrobial Peptides Existed in the Skins of Nine Kinds of Chinese Odorous Frogs. J. Proteome Res. 2012, 11, 306–319. [Google Scholar] [CrossRef]
- Conlon, J.M. Structural diversity and species distribution of host-defense peptides in frog skin secretions. Cell. Mol. Life Sci. 2011, 68, 2303–2315. [Google Scholar] [CrossRef]
- Simmaco, M.; Kreil, G.; Barra, D. Bombinins, antimicrobial peptides from Bombina species. Biochim. Biophys. Acta 2009, 1788, 1551–1555. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellullar organisms. Nature 2002, 415, 359–365. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yuan, J.; Osapay, G.; Osapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef]
- Sørensen, O.E.; Gram, L.; Johnsen, A.H.; Andersson, E.; Bangsbøll, S.; Tjabringa, G.S.; Hiemstra, P.S.; Malm, J.; Egesten, A.; Borregaard, N. Processing of seminal plasma hCAP-18 to ALL-38 by gastricsin: A novel mechanism of generating antimicrobial peptides in vagina. J. Biol. Chem. 2003, 278, 28540–28546. [Google Scholar] [CrossRef]
- Silkin, L.; Hamza, S.; Kaufman, S.; Cobb, S.L.; Vederas, J.C. Spermicidal bacteriocins: Lacticin 3147 and subtilosin A. Bioorg. Med. Chem. Lett. 2008, 18, 3103–3106. [Google Scholar] [CrossRef]
- Ganz, T.; Lehrer, R.I. Defensins. Curr. Opin. Immunol. 1994, 6, 584–589. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Inter. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Mayer, M.L.; Easton, D.M.; Hancock, R.E.W. Fine tuning host responses in the face of infection: Emerging roles and clinical applications of host defense peptides. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies; Wang, G., Ed.; CABI: Wallingford, England, 2010; pp. 195–220. [Google Scholar]
- Lemaitre, B.; Hoffmann, J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Li, H.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Wang, G. Natural antimicrobial peptides as promising anti-HIV candidates. Curr. Topics Pept. Protein Res. 2012, 13, 93–110. [Google Scholar]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef]
- Mishra, B.; Wang, G. The importance of amino acid composition in natural AMPs: An evolutional, structural, and functional perspective. Frontier Immunol. 2012, 3, 221. [Google Scholar]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar] [CrossRef]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS One 2011, 6, e18476. [Google Scholar]
- Henriques, S.T.; Huang, Y.H.; Castanho, M.A.; Bagatolli, L.A.; Sonza, S.; Tachedjian, G.; Daly, N.L.; Craik, D.J. Phosphatidylethanolamine binding is a conserved feature of cyclotide-membrane interactions. J. Biol. Chem. 2012, 287, 33629–33643. [Google Scholar] [CrossRef]
- Iwamoto, K.; Hayakawa, T.; Murate, M.; Makino, A.; Ito, K.; Fujisawa, T.; Kobayashi, T. Curvature-dependent recognition of ethanolamine phospholipids by duramycin and cinnamycin. Biophys. J. 2007, 93, 1608–1619. [Google Scholar] [CrossRef]
- Zhao, M. Lantibiotics as probes for phosphatidylethanolamine. Amino Acids 2011, 41, 1071–1079. [Google Scholar] [CrossRef]
- Wang, G.; Li, Y.; Li, X. Correlation of three-dimensional structures with the antibacterial activity of a group of peptides designed based on a non-toxic bacterial membrane anchor. J. Biol. Chem. 2005, 280, 5803–5811. [Google Scholar] [CrossRef]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]
- Wang, G.; Watson, K.M.; Peterkofsky, A.; Buckheit, R.W., Jr. Identification of novel human immunodeficiency virus type 1 inhibitory peptides based on the antimicrobial peptide database. Antimicrob. Agents Chemother. 2010, 54, 1343–1346. [Google Scholar] [CrossRef]
- Menousek, J.; Mishra, B.; Hanke, M.L.; Heim, C.E.; Kielian, T.; Wang, G. Database screening and in vivo efficacy of antimicrobial peptides against methicillin-resistant Staphylococcus aureus USA300. Int. J. Antimicrob. Agents 2012, 39, 402–406. [Google Scholar] [CrossRef]
- Wang, G.; Buckheit, K.W.; Mishra, B.; Lushnikova, T.; Buckheit, R.W., Jr. De Novo Design of Antiviral and Antibacterial Peptides with Varying Loop structures. J. AIDS Clin. Res. 2011, S2:003. [Google Scholar] [CrossRef]
- Mishra, B.; Wang, G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef]
- Wang, G.; Watson, K.M.; Buckheit, R.W., Jr. Anti-human immunodeficiency virus type 1 activities of antimicrobial peptides derived from human and bovine cathelicidins. Antimicrob. Agents Chemother. 2008, 52, 3438–3440. [Google Scholar] [CrossRef]
- Liu, Y.; Xia, X.; Xu, L.; Wang, Y. Design of hybrid β-hairpin peptides with enhanced cell specificity and potent anti-inflammatory activity. Biomaterials 2013, 34, 237–250. [Google Scholar] [CrossRef]
- Fox, M.A.; Thwaite, J.E.; Ulaeto, D.O.; Atkins, T.P.; Atkins, H.S. Design and characterization of novel hybrid antimicrobial peptides based on cecropin A, LL-37 and magainin II. Peptides 2012, 33, 197–205. [Google Scholar] [CrossRef]
- Merrifield, R.B.; Merrifield, E.L.; Juvvadi, P.; Andreu, D.; Boman, H.G. Design and synthesis of antimicrobial peptides. Ciba Found Symp. 1994, 186, 5–20; discussion 20–26. [Google Scholar]
- Turpin, J.A. The next generation of HIV/AIDS drugs: Novel and developmental anti-HIV drugs and targets. Expert Rev. Anti Infect. Ther. 2003, 1, 97–128. [Google Scholar] [CrossRef]
- Buckheit, K.W.; Buckheit, R.W., Jr. Factors Important to the Prioritization and Development of Successful Topical Microbicides for HIV-1. Mol. Biol. Int. 2012, 2012, 781305. [Google Scholar]
- Conlon, J.M.; Kolodziejek, J.; Nowotny, N.; Leprince, J.; Vaudry, H.; Coquet, L.; Jouenne, T.; King, J.D. Characterization of antimicrobial peptides from the skin secretions of the Malaysian frogs, Odorrana hosii and Hylarana picturata (Anura:Ranidae). Toxicon 2008, 52, 465–473. [Google Scholar] [CrossRef]
- Wang, H.; Lu, Y.; Zhang, X.; Hu, Y.; Yu, H.; Liu, J.; Sun, J. The novel antimicrobial peptides from skin of Chinese broad-folded frog, Hylarana latouchii (Anura:Ranidae). Peptides 2009, 30, 273–282. [Google Scholar] [CrossRef]
- Souza, B.M.; Mendes, M.A.; Santos, L.D.; Marques, M.R.; Cesar, L.M.; Almeida, R.N.; Pagnocca, F.C.; Konno, K.; Palma, M.S. Structural and functional characterization of two novel peptide toxins isolated from the venom of the social wasp Polybia paulista. Peptides 2005, 26, 2157–2164. [Google Scholar] [CrossRef]
- Otvos, L., Jr; Insug, O.; Rogers, M.E.; Consolvo, P.J.; Condie, B.A.; Lovas, S.; Bulet, P.; Blaszczyk-Thurin, M. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar] [CrossRef]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-D-magainin: Chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 1990, 274, 151–155. [Google Scholar] [CrossRef]
- Merrifield, E.L.; Mitchell, S.A.; Ubach, J.; Boman, H.G.; Andreu, D.; Merrifield, R.B. D-enantiomers of 15-residue cecropin A-melittin hybrids. Int. J. Pept. Protein Res. 1995, 46, 214–220. [Google Scholar]
- Wang, G.; Keifer, P.A.; Peterkofsky, A. Solution structure of the N-terminal amphitropic domain of Escherichia coli glucose-specific enzyme IIA in membrane-mimetic micelles. Protein Sci. 2003, 12, 1087–1096. [Google Scholar] [CrossRef]
- Wang, G.; Peterkofsky, A.; Clore, G.M. A novel membrane anchor function for the N-terminal amphipathic sequence of the signal-transducing protein IIAGlucose of the Escherichia coli phosphotransferase system. J. Biol. Chem. 2000, 275, 39811–39814. [Google Scholar]
- Rozek, T.; Wegener, K.L.; Bowie, J.H.; Olver, I.N.; Carver, J.A.; Wallace, J.C.; Tyler, M.J. The antibiotic and anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria raniformis the solution structure of aurein 1.2. Eur. J. Biochem. 2000, 267, 5330–5341. [Google Scholar] [CrossRef]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Han, H.; Miller, D.W.; Wang, G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 2006, 128, 5776–5785. [Google Scholar]
- Li, X.; Li, Y.; Peterkofsky, A.; Wang, G. NMR studies of aurein 1.2 analogs. Biochim. Biophys. Acta 2006, 1758, 1203–1214. [Google Scholar] [CrossRef]
- Wang, G.; Epand, R.F.; Mishra, B.; Lushnikova, T.; Thomas, V.C.; Bayles, K.W.; Epand, R.M. Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob. Agents Chemother. 2012, 56, 845–856. [Google Scholar] [CrossRef]
- Wang, G. NMR studies of a model antimicrobial peptide in the micelles of SDS, dodecylphosphocholine, or dioctanoyl phosphatidylglycerol. Open Magn. Reson. J. 2008, 1, 9–15. [Google Scholar]
- Wong, J.H.; Legowska, A.; Rolka, K.; Ng, T.B.; Hui, M.; Cho, C.H.; Lam, W.W.; Au, S.W.; Gu, O.W.; Wan, D.C. Effects of cathelicidin and its fragments on three key enzymes of HIV-1. Peptides 2011, 32, 1117–1122. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F. Biophysical analysis of membrane targeting antimicrobial peptides: Membrane properties and design of peptides specifically targeting Gram-negative bacteria. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies; Wang, G., Ed.; CABI: Wallingford, England, 2010; pp. 116–127. [Google Scholar]
- Lohner, K. New strategies for novel antibiotics: Peptides targeting bacterial cell membranes. Gen. Physiol. Biophys. 2009, 28, 105–116. [Google Scholar] [CrossRef]
- Islam, M.R.; Nagao, J.; Zendo, T.; Sonomoto, K. Antimicrobial mechanism of lantibiotics. Biochem. Soc. Trans. 2012, 40, 1528–1533. [Google Scholar] [CrossRef]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Yaver, D.; Elvig-Jørgensen, S.G.; Sørensen, M.V.; Christensen, B.E.; Kjaerulff, S.; Frimodt-Moller, N.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef]
- De Kruijff, B.; van Dam, V.; Breukink, E. Lipid II: A central component in bacterial cell wall synthesis and a target for antibiotics. Prostaglandins Leukot. Essent. Fatty Acids 2008, 79, 117–121. [Google Scholar] [CrossRef]
- De Leeuw, E.; Li, C.; Zeng, P.; Li, C.; Diepeveen-de Buin, M.; Lu, W.Y.; Breukink, E.; Lu, W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010, 584, 1543–1548. [Google Scholar] [CrossRef]
- Demirkhanyan, L.H.; Marin, M.; Padilla-Parra, S.; Zhan, C.; Miyauchi, K.; Jean-Baptiste, M.; Novitskiy, G.; Lu, W.; Melikyan, G.B. Multifaceted mechanisms of HIV-1 entry inhibition by human α-defensin. J. Biol. Chem. 2012, 287, 28821–28838. [Google Scholar] [CrossRef]
- Wang, W.; Owen, S.M.; Rudolph, D.L.; Cole, A.M.; Hong, T.; Waring, A.J.; Lal, R.B.; Lehrer, R.I. Activity of alpha- and theta-defensins against primary isolates of HIV-1. J. Immunol. 2004, 173, 515–520. [Google Scholar]
- Ilić, N.; Novković, M.; Guida, F.; Xhindoli, D.; Benincasa, M.; Tossi, A.; Juretić, D. Selective antimicrobial activity and mode of action of adepantins, glycine-rich peptide antibiotics based on anuran antimicrobial peptide sequences. Biochim. Biophys. Acta 2013, 1828, 1004–1012. [Google Scholar] [CrossRef]
- Gonçalves, S.; Abade, J.; Teixeira, A.; Santos, N.C. Lipid composition is a determinant for human defensin HNP1 selectivity. Biopolymers 2012, 98, 313–321. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. New lytic peptides based on the d,L-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry 2003, 42, 9346–9354. [Google Scholar] [CrossRef]
- Oren, Z.; Shai, Y. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: Structure-function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar] [CrossRef]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef]
- Liang, J.F.; Kim, S.C. Not only the nature of peptide but also the characteristics of cell membrane determine the antimicrobial mechanism of a peptide. J. Pept. Res. 1999, 53, 518–522. [Google Scholar] [CrossRef]
- Teuber, M.; Bader, J. Action of polymyxin B on bacterial membranes: Phosphatidylglycerol- and cardiolipin-induced susceptibility to polymyxin B in Acholeplasma laidlawii B. Antimicrob. Agents Chemother. 1976, 9, 26–35. [Google Scholar] [CrossRef]
- Kobayashi, S.; Takeshima, K.; Park, C.B.; Kim, S.C.; Matsuzaki, K. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: Proline as a translocation promoting factor. Biochemistry 2000, 39, 8648–8654. [Google Scholar]
- Park, C.B.; Yi, K.S.; Matsuzaki, K.; Kim, M.S.; Kim, S.C. Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin II. Proc. Natl. Acad. Sci. USA 2000, 97, 8245–8250. [Google Scholar]
- Bystricky, V.; Ladzianska, K.; Halasa, M. Electron microscopy of the action of polymyxin on leptospirae. J. Bacteriol. 1962, 84, 864–865. [Google Scholar]
- Wada, A.; Wong, P.F.; Hojo, H.; Hasegawa, M.; Ichinose, A.; Llanes, R.; Kubo, Y.; Senba, M.; Ichinose, Y. Alarin but not its alternative-splicing form, GALP (Galanin-like peptide) has antimicrobial activity. Biochem. Biophys. Res. Commun. 2013. [Google Scholar] [CrossRef]
- Shapiro, H.M.; Natale, P.J.; Kamentsky, L.A. Estimation of membrane potentials of individual lymphocytes by flow cytometry. Proc. Natl. Acad. Sci. USA 1979, 76, 5728–5730. [Google Scholar] [CrossRef]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999, 341((Pt. 3)), 501–513. [Google Scholar]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar]
- Lee, C.C.; Sun, Y.; Qian, S.; Huang, H.W. Transmembrane pores formed by human antimicrobial peptide LL-37. Biophys J. 2011, 100, 1688–1696. [Google Scholar]
- Wang, G. Structural studies of antimicrobial peptides provide insight into their mechanisms of action. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic strategies; Wang, G., Ed.; CABI: Oxfordshire, England, 2010; pp. 141–168. [Google Scholar]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L. Molecular diversity in gene-coded, cationic antimicrobial polypeptides. Curr. Pharm. Des. 2002, 8, 743–761. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; John Wiley & Sons: New York, NY, USA, 1986. [Google Scholar]
- Franklin, J.C.; Ellena, J.F.; Jayasinghe, S.; Kelsh, L.P.; Cafiso, D.S. Structure of micelle-associated alamethicin from 1H-NMR. Evidence for conformational heterogeneity in a voltage-gated peptide. Biochemistry 1994, 33, 4036–4045. [Google Scholar]
- Abdul-Manan, N.; Hinton, J.F. Conformation states of gramicidin A along the pathway to the formation of channels in model membranes determined by 2D NMR and circular dichroism spectroscopy. Biochemistry 1994, 33, 6773–6783. [Google Scholar] [CrossRef]
- Schibli, D.J.; Hwang, P.M.; Vogel, H.J. The structure of the antimicrobial active center of lactoferricin B bound to sodium dodecyl sulfate micelles. FEBS Lett. 1999, 446, 213–217. [Google Scholar] [CrossRef]
- Wang, G.; Elliott, M.; Cogen, A.L.; Ezell, E.L.; Gallo, R.L.; Hancock, R.E.W. Structure, dynamics, antimicrobial and immune modulatory activities of human LL-23 and its single residue variants mutated based on homologous primate cathelicidins. Biochemistry 2012, 51, 653–664. [Google Scholar] [CrossRef]
- Greathouse, D.V.; Hinton, J.F.; Kim, K.S.; Koeppe, R.E., 2nd. Gramicidin A/short-chain phospholipid dispersions: Chain length dependence of gramicidin conformation and lipid organization. Biochemistry 1994, 33, 4291–4299. [Google Scholar] [CrossRef]
- Subasinghage, A.P.; O'Flynn, D.; Conlon, J.M.; Hewage, C.M. Conformational and membrane interaction studies of the antimicrobial peptide alyteserin-1c and its analogue [E4K] alyteserin-1c. Biochim. Biophys. Acta 2011, 1808, 1975–1984. [Google Scholar] [CrossRef]
- Keifer, P.A.; Peterkofsky, A.; Wang, G. Effects of detergent alkyl chain length and chemical structure on the properties of a micelle-bound bacterial membrane targeting peptide. Anal. Biochem. 2004, 331, 33–39. [Google Scholar]
- Wang, G.; Keifer, P.A.; Peterkofsky, A. Short-chain diacyl phosphatidylglycerol: Which one to use for the NMR structural determination of a membrane-associated peptide from Escherichia coli? Spectroscopy 2004, 18, 257–264. [Google Scholar] [CrossRef]
- Wang, G. Determination of solution structure and lipid micelle location of an engineered membrane peptide by using one NMR experiment and one sample. Biochim. Biophys. Acta 2007, 1768, 3271–3281. [Google Scholar] [CrossRef]
- Wang, G. Structural biology of antimicrobial peptides by NMR. Curr. Org. Chem. 2006, 10, 569–581. [Google Scholar] [CrossRef]
- Raimondo, D.; Andreotti, G.; Saint, N.; Amodeo, P.; Renzone, G.; Sanseverino, M.; Zocchi, I.; Molle, G.; Motta, A.; Scaloni, A. A folding-dependent mechanism of antimicrobial peptide resistance to degradation unveiled by solution structure of distinctin. Proc. Natl. Acad. Sci. USA 2005, 102, 6309–6314. [Google Scholar] [CrossRef]
- Mysliwy, J.; Dingley, A.J.; Stanisak, M.; Jung, S.; Lorenzen, I.; Roeder, T.; Leippe, M.; Grötzinger, J. Caenopore-5: The three-dimensional structure of an antimicrobial protein from Caenorhabditis elegans. Dev. Comp. Immunol. 2010, 34, 323–330. [Google Scholar]
- Nar, H.; Schmid, A.; Puder, C.; Potterat, O. High-resolution crystal structure of a lasso peptide. ChemMedChem 2010, 5, 1689–1692. [Google Scholar]
- Conibear, A.C.; Rosengren, K.J.; Harvery, P.J.; Craik, D.J. Structural characterization of the cyclic cystine ladder motif of θ-defensins. Biochemistry 2012, 51, 9718–9726. [Google Scholar] [CrossRef]
- Trabi, M.; Schirra, H.J.; Craik, D.J. Three-dimensional structure of RTD-1, a cyclic antimicrobial defensin from Rhesus macaque leukocytes. Biochemistry 2001, 40, 4211–4221. [Google Scholar]
- Saether, O.; Craik, D.J.; Campbell, I.D.; Sletten, K.; Juul, J.; Norman, D.G. Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1. Biochemistry 1995, 34, 4147–4158. [Google Scholar]
- Wei, G.; de Leeuw, E.; Pazgier, M.; Yuan, W.; Zou, G.; Wang, J.; Ericksen, B.; Lu, W.Y.; Lehrer, R.I.; Lu, W. Through the looking glass, mechanistic insights from enantiomeric human defensins. J. Biol. Chem. 2009, 284, 29180–29192. [Google Scholar] [CrossRef]
- Yang, Y.S.; Mitta, G.; Chavanieu, A.; Calas, B.; Sanchez, J.F.; Roch, P.; Aumelas, A. Solution structure and activity of the synthetic four-disulfide bond Mediterranean mussel defensin (MGD-1). Biochemistry 2000, 39, 14436–14447. [Google Scholar] [CrossRef]
- Bauer, F.; Schweimer, K.; Klüver, E.; Conejo-Garcia, J.R.; Forssmann, W.G.; Rösch, P.; Adermann, K.; Sticht, H. Structure determination of human and murine beta-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci. 2001, 10, 2470–2479. [Google Scholar] [CrossRef]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef]
- Rose, P.W.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dimitropoulos, D.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Prlic, A.; Quesada, M.; et al. The RCSB Protein Data Bank: New resources for research and education. Nucleic Acids Res. 2013, 41, D475–D482. [Google Scholar] [CrossRef]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef]
- Wang, G. Structure, dynamics and mapping of membrane-binding residues of micelle-bound antimicrobial peptides by natural abundance 13C NMR spectroscopy. Biochim. Biophys. Acta 2010, 1798, 114–121. [Google Scholar]
- Li, Y.; Li, X.; Li, H.; Lockridge, O.; Wang, G. A novel method for purifying recombinant human host defense cathelicidin LL-37 by utilizing its inherent property of aggregation. Protein Expr. Purif. 2007, 54, 157–165. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Wang, G. Cloning, expression, isotope labeling, and purification of human antimicrobial peptide LL-37 in Escherichia coli for NMR studies. Protein Expr. Purif. 2006, 47, 498–505. [Google Scholar] [CrossRef]
- Ikura, M.; Kay, L.E.; Bax, A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: Heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry 1990, 29, 4659–4667. [Google Scholar] [CrossRef]
- Porcelli, F.; Verardi, R.; Shi, L.; Henzler-Wildman, K.A.; Ramamoorthy, A.; Veglia, G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 2008, 47, 5565–5572. [Google Scholar]
- Bhunia, A.; Jana, J.; Ghosh, A.; Kar, R.K.; Biswas, A.; Ghosh, S.; Chatterjee, S. Human cathelicidin peptide LL37 binds telomeric G-Quadruplex. Mol. Biosys. 2013. [Google Scholar] [CrossRef]
- Turner, J.; Cho, Y.; Dinh, N.-N.; Waring, A.J.; Lehrer, R.I. Activities of LL-37, a cathelin-associated antimicrobial peptides of human neutrophils. Antimicrob. Agents Chemother 1998, 42, 2206–2214. [Google Scholar]
- Knappe, D.; Zahn, M.; Sauer, U.; Schiffer, G.; Sträter, N.; Hoffmann, R. Rational design of oncocin derivatives with superior protease stabilities and antibacterial activities based on the high-resolution structure of the oncocin-DnaK complex. ChemBioChem 2011, 12, 874–876. [Google Scholar]
- Koehn, F.E.; Carter, G.T. Rediscovering natural products as a source of new drugs. Discov. Med. 2005, 5, 159–164. [Google Scholar]
- Nisbet, L.J.; Moore, M. Will natural products remain an important source of drug research for the future? Curr. Opin. Biotechnol. 1997, 8, 708–712. [Google Scholar] [CrossRef]
- Wang, G. Database-aided prediction and design of novel antimicrobial peptides. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies; Wang, G., Ed.; CABI: Wallingford, England, 2010; pp. 72–86. [Google Scholar]
- Houghten, R.A.; Pinilla, C.; Blondelle, S.E.; Appel, J.R.; Dooley, C.T.; Cuervo, J.H. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991, 354, 84–86. [Google Scholar] [CrossRef]
- Krauson, A.J.; He, J.; Wimley, A.W.; Hoffmann, A.R.; Wimley, W.C. Synthetic Molecular Evolution of Pore-Forming Peptides by Iterative Combinatorial Library Screening. ACS Chem. Biol. 2013, 8, 823–831. [Google Scholar] [CrossRef]
- Blondelle, S.E.; Lohner, K. Optimization and high-throughput screening of antimicrobial peptides. Curr. Pharm. Des. 2010, 16, 3204–3211. [Google Scholar] [CrossRef]
- Lata, S.; Sharma, B.K.; Raghava, G.P. Analysis and prediction of antibacterial peptides. BMC Bioinformatics 2007, 8, 263. [Google Scholar]
- Lata, S.; Mishra, N.K.; Raghava, G.P. AntiBP2: Improved version of antibacterial peptide prediction. BMC Bioinformatics 2010, 11, S19. [Google Scholar]
- Lira, F.; Perez, P.S.; Baranauskas, J.A.; Nozawa, S.R. Prediction of antimicrobial activity of synthetic peptides by a decision tree model. Appl. Environ. Microbiol. 2013, 79, 3156–3159. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef]
- Yan, L.; Yan, Y.; Liu, H.; Lv, Q. Stepwise identification of potent antimicrobial peptides from human genome. Biosystems 2013. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hancock, R.E.; Cherkasov, A. AMPer: A database and an automated discovery tool for antimicrobial peptides. Bioinformatics 2007, 23, 1148–1155. [Google Scholar]
- Siezen, R.J.; Kuipers, O.P.; de Vos, W.M. Comparison of lantibiotic gene clusters and encoded proteins. Antonie Van Leeuwenhoek 1996, 69, 171–184. [Google Scholar] [CrossRef]
- Torrent, M.; di Tommaso, P.; Pulido, D.; Nogués, M.V.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An automated web server for prediction of protein antimicrobial regions. Bioinformatics 2012, 28, 130–131. [Google Scholar]
- Wu, W.K.; Wang, G.; Coffelt, S.B.; Betancourt, A.M.; Lee, C.W.; Fan, D.; Wu, K.; Yu, J.; Sung, J.J.; Cho, C.H. Emerging roles of the host defense peptide LL-37 in human cancer and its potential therapeutic applications. Int. J. Cancer 2010, 127, 1741–1747. [Google Scholar] [CrossRef]
- Rico-Mata, R.; de Leon-Rodriguez, L.M.; Avila, E.E. Effect of antimicrobial peptides derived from human cathelicidin LL-37 on Entamoeba histolytica trophozoites. Exp. Parasitol. 2013, 133, 300–306. [Google Scholar] [CrossRef]
- Johnson, L.M.; Gellman, S.H. α-Helix Mimicry with α/β-Peptides. Methods Enzymol. 2013, 523, 407–429. [Google Scholar] [CrossRef]
- Sarig, H.; Livne, L.; Held-Kuznetsov, V.; Zaknoon, F.; Ivankin, A.; Gidalevitz, D.; Mor, A. A miniature mimic of host defense peptides with systemic antibacterial efficacy. FASEB J. 2010, 24, 1904–1913. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Shai, Y. Short native antimicrobial peptides and engineered ultrashort lipopeptides: Similarities and differences in cell specificities and modes of action. Cell. Mol. Life Sci. 2011, 68, 2267–2280. [Google Scholar] [CrossRef]
- Wang, G. Chemical modifications of natural antimicrobial peptides and strategies for peptide engineering. Curr. Biotechnol. 2012, 1, 72–79. [Google Scholar]
- Schroeder, B.O.; Wu, Z.; Nuding, S.; Groscurth, S.; Marcinowski, M.; Beisner, J.; Buchner, J.; Schaller, M.; Stange, E.F.; Wehkamp, J. Reduction of disulphide bonds unmasks potent antimicrobial activity of human β-defensin 1. Nature 2011, 469, 419–423. [Google Scholar] [CrossRef]
- Aboye, T.L.; Ha, H.; Majumder, S.; Christ, F.; Debyser, Z.; Shekhtman, A.; Neamati, N.; Camarero, J.A. Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity. J. Med. Chem. 2012, 55, 10729–10734. [Google Scholar] [CrossRef]
- Wong, C.T.; Rowlands, D.K.; Wong, C.H.; Lo, T.W.; Nguyen, G.K.; Li, H.Y.; Tam, J.P. Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. Engl. 2012, 51, 5620–5624. [Google Scholar]
- Craik, D.J.; Swedberg, J.E.; Mylne, J.S.; Cemazar, M. Cyclotides as a basis for drug design. Expert Opin. Drug Discov. 2012, 7, 179–194. [Google Scholar] [CrossRef]
- Tally, F.P.; DeBruin, M.F. Development of daptomycin for gram-positive infections. J. Antimicrob. Chemother. 2000, 46, 523–526. [Google Scholar] [CrossRef]
- Eliopoulos, G.M.; Willey, S.; Reiszner, E.; Spitzer, P.G.; Caputo, G.; Moellering, R.C., Jr. In vitro and in vivo activity of LY 146032, a new cyclic lipopeptide antibiotic. Antimicrob. Agents Chemother. 1986, 30, 532–535. [Google Scholar]
- Vilhena, C.; Bettencourt, A. Daptomycin: A review of properties, clinical use, drug delivery and resistance. Mini Rev. Med. Chem. 2012, 12, 202–209. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, G. Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs. Pharmaceuticals 2013, 6, 728-758. https://doi.org/10.3390/ph6060728
Wang G. Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs. Pharmaceuticals. 2013; 6(6):728-758. https://doi.org/10.3390/ph6060728
Chicago/Turabian StyleWang, Guangshun. 2013. "Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs" Pharmaceuticals 6, no. 6: 728-758. https://doi.org/10.3390/ph6060728
APA StyleWang, G. (2013). Database-Guided Discovery of Potent Peptides to Combat HIV-1 or Superbugs. Pharmaceuticals, 6(6), 728-758. https://doi.org/10.3390/ph6060728