Protein Kinase C Inhibitors as Modulators of Vascular Function and Their Application in Vascular Disease

Abstract
:1. Introduction
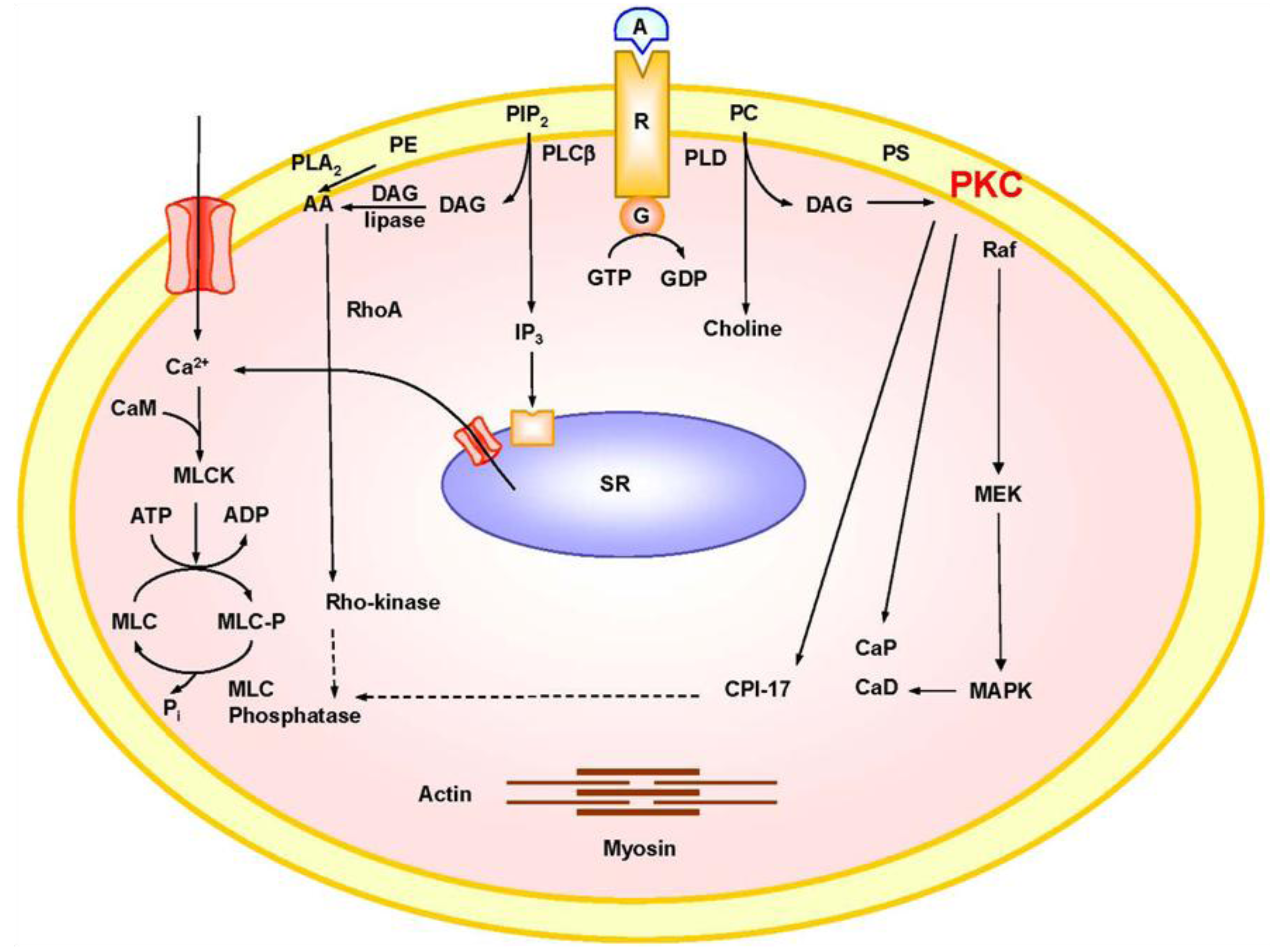
2. Mechanisms of VSM Contraction
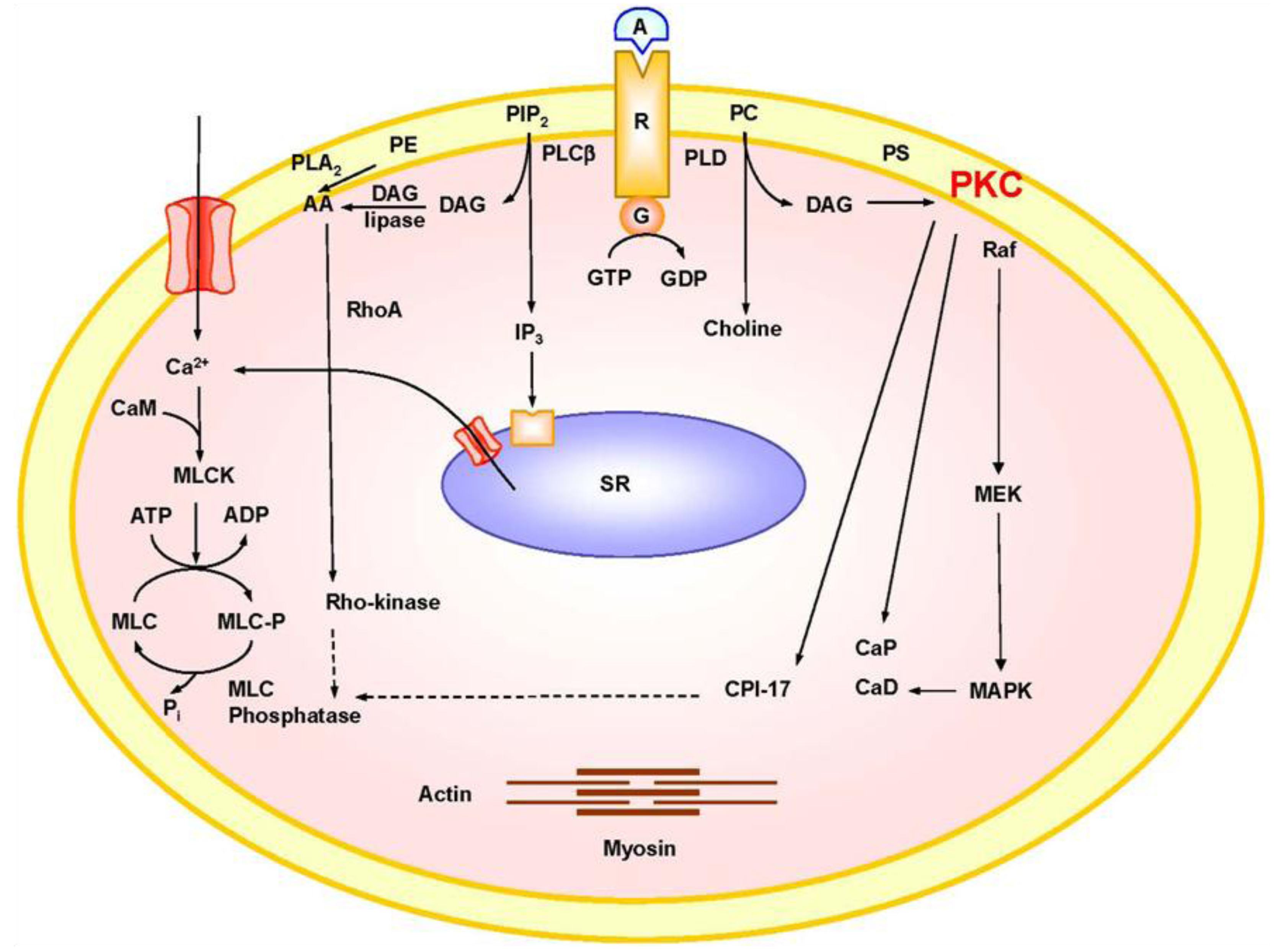
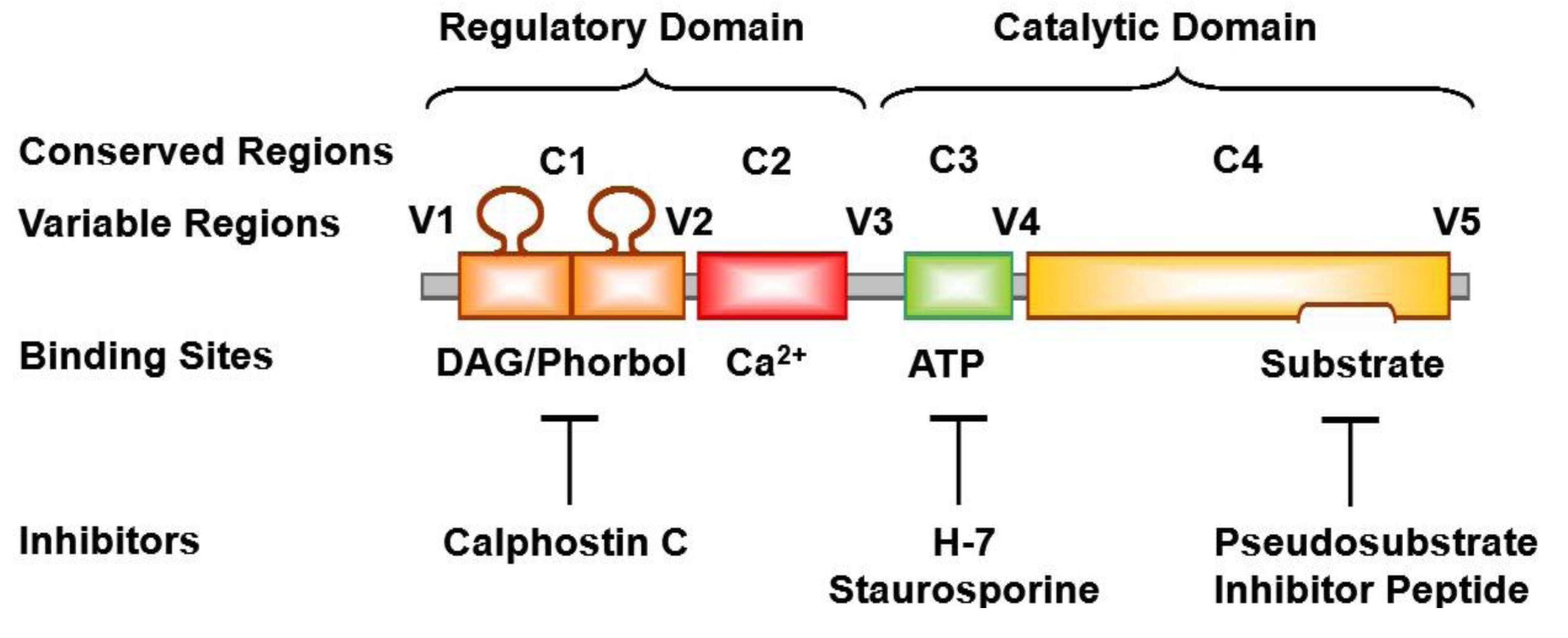
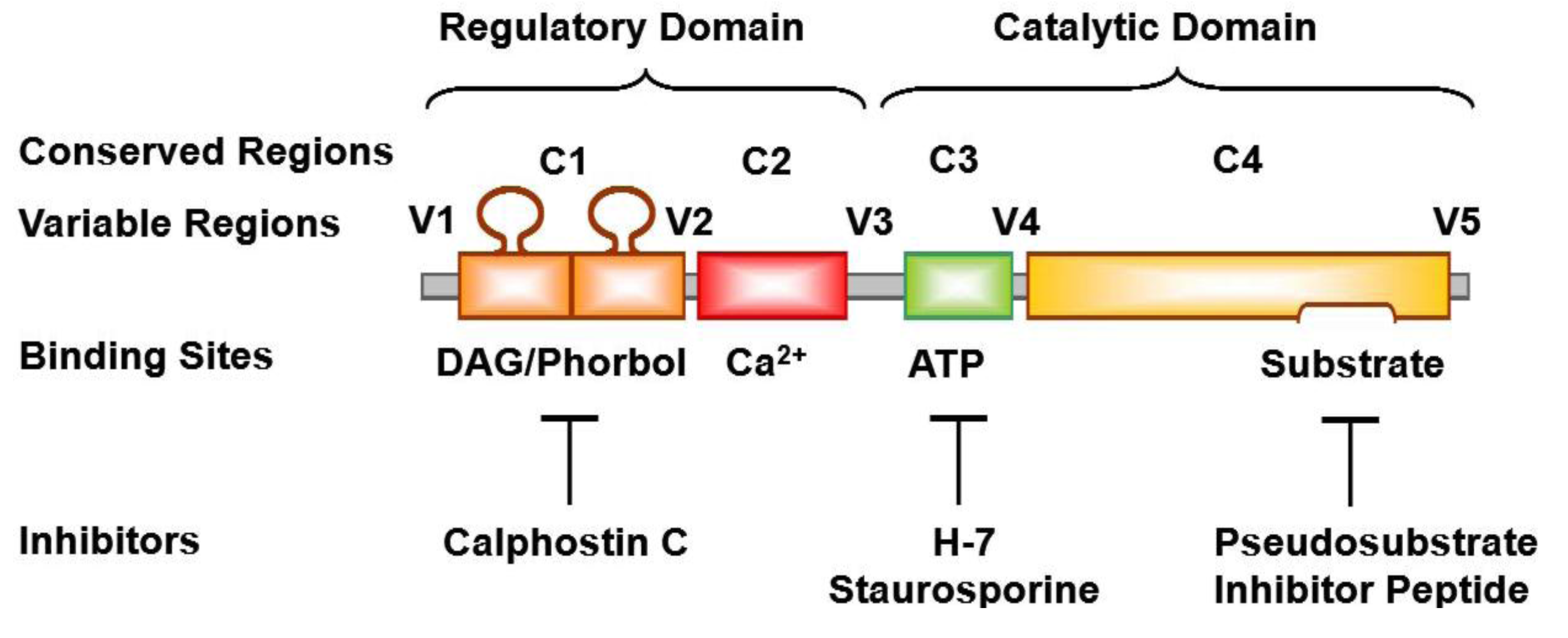
3. PKC Isoforms

{kind=link}
{kind=link}
{kind=link}
| PKC | MW (kDa) | Blood Vessel | Resting Cell | Activated Cell | Ref |
|---|---|---|---|---|---|
| Classic | |||||
| α | 74–82 | Rat aorta | Cytosolic | Nuclear | [35] |
| Rat Carotid artery | Cytosolic | Membrane | [36] | ||
| Rat mesenteric artery | Cytosolic/membrane | Cytosolic/Membrane | [37] | ||
| Porcine coronary artery | Cytosolic | Membrane | [38] | ||
| Bovine aorta | Cytosolic | Membrane | [39] | ||
| Ferret portal vein | Cytosolic | Surface membrane | [40] | ||
| β | 80–82 | Rat aorta | Cytosolic | Nuclear | [35] |
| Rat Carotid artery | Cytosolic | Membrane | [36] | ||
| γ | 70–82 | Rat mesenteric artery | Cytosolic | Cytosolic | [37] |
| Novel | |||||
| δ | 76–82 | Rat aorta | Cytoskeleton/organelle | Cytoskeleton/organelle | [41] |
| Rat mesenteric artery | Membrane | Membrane | [37] | ||
| ε | 90–97 | Rat mesenteric artery | Cytosolic/membrane | Cytosolic/membrane | [37] |
| Ferret aorta | Cytosol | Surface membrane | [42] | ||
| Porcine coronary artery | Cytosolic | Membrane | [38] | ||
| η | NIH 3T3 fibroblasts | Cytosolic/membrane | Membrane | [43] | |
| Atypical | |||||
| ζ | 64–82 | Rat aorta | Perinuclear | Intranuclear | [41] |
| Rat mesenteric artery | Cytosolic | Cytosolic | [37] | ||
| Ferret aorta, portal vein | Perinuclear | Intranuclear | [42] | ||
| λ/ι | 70 | Rabbit femoral artery | Cytosolic | Cytosolic | [44] |
| Rabbit portal vein | |||||
4. PKC Substrates
5. PKC Distribution
6. PKC Function
7. PKC Activators
8. PKC Inhibitors
| Chemical Group | Example | Site of Action | Specificity |
|---|---|---|---|
| 1-(5-isoquinolinesulfonyl)-2-methylpiperazines | H-7 | Catalytic domain Compete with ATP at the ATP binding site | Also, inhibits cyclic AMP and cyclic GMP-dependent protein kinases |
| Microbial Alkaloids, Products of Streptomyces | Staurosporine SCH47112 | Catalytic domain, ATP binding site | Also, inhibits MLC kinase and tyrosine kinase |
| Benzophenanthridine Alkaloids | Chelerythrine | Catalytic domain | Competitive inhibitor with histone IIIS |
| Indocarbazoles | Gö6976 | Catalytic domain | Ca2+-dependent α- and βI-PKC |
| Bisindolylmaleimide Staurosporine Analogs | GF109203X Ro-318220 Midostaurin (PKC412, CGP41251) Ruboxistaurin (LY333531) | Catalytic domain | PKC isozymes α, βI, βII, γ, δ and ε. Ruboxistaurin mesylate is a selective antagonist of PKC βI and PKC βII. |
| Perylenequinone Metabolites from Cladosporium cladosporioides | Calphostin C (UCN-1028A) | Regulatory domain | Binds to the regulatory domain at DAG/phorbol ester binding site |
| Membrane lipids | Sphingosine | Regulatory domain | Competitive inhibitor with phosphatidylserine |
| Other: | Adriamycin Aminoacridine Apigenin Cercosporin Chlorpromazine Dexniguldipine Polymixin B Sangivamycin Tamoxifen Trifluoperazine UCN-01, UCN-02 |
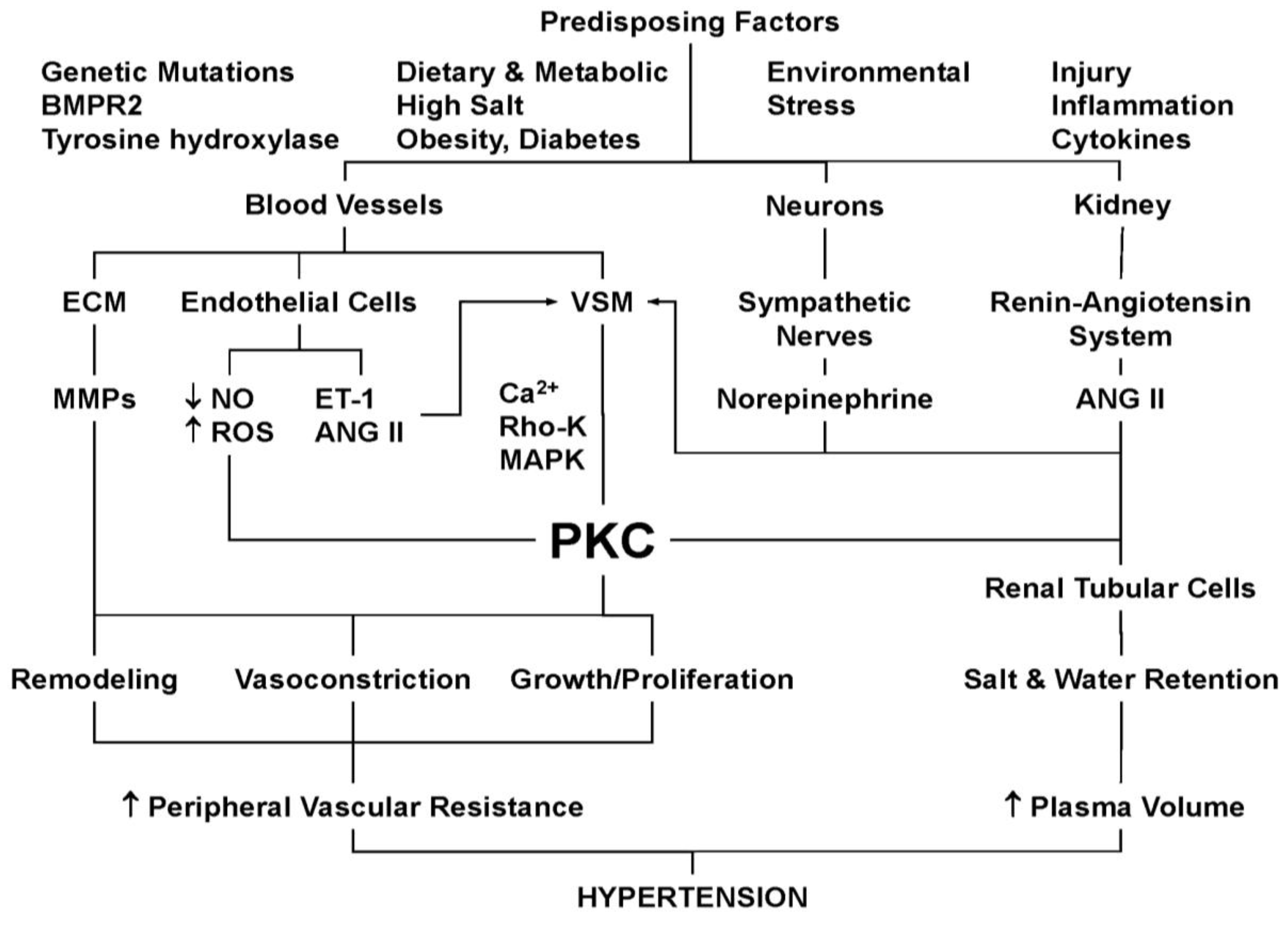
9. PKC and Hypertension
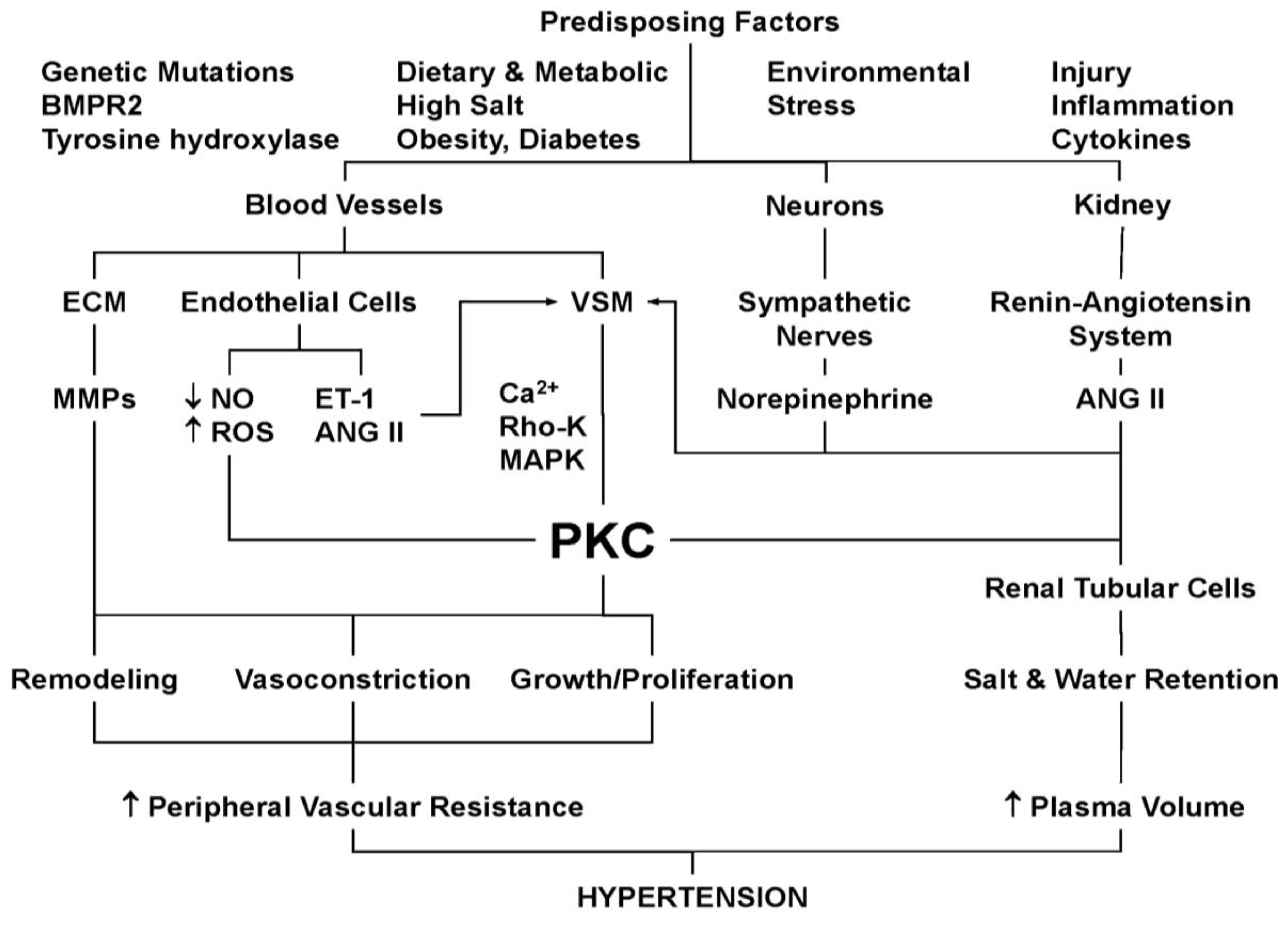
10. PKC and VSM Growth and Reactivity in Hypertension
11. PKC in Genetic Hypertension
12. PKC and Human Essential Hypertension
13. PKC and Aortic Constriction-Induced Hypertension
14. PKC, Endothelial Dysfunction and Hypertension
15. PKC, Oxidative Stress and Hypertension
16. PKC, MMPs and Vascular Remodeling in Hypertension
17. PKC in Salt-Sensitive Hypertension
18. PKC, Neuronal Dysfunction and Hypertension
19. PKC, Metabolic Dysfunction and Hypertension
20. PKC, Vascular Inflammation and Hypertension
21. PKC and Pulmonary Hypertension
22. PKC and Hypertension-in-Pregnancy and Preeclampsia
23. PKC Inhibitors as Modulators of Vascular Function in Hypertension
List of abbreviations
| ANG II | angiotensin II |
| ATP | adenosine triphosphate |
| BP | blood pressure |
| CPI-17 | PKC-potentiated phosphatase inhibitor protein-17 kDa |
| CAM | calmodulin |
| DAG | diacylglycerol |
| EC | endothelial cell |
| ET-1 | endothelin-1 |
| HTN | hypertension |
| IP3 | inositol 1,4,5-trisphosphate |
| MAPK | mitogen-activated protein kinase |
| MARCKs | myristoylated alanine-rich C-kinase substrate |
| MMP | matrix metalloproteinase |
| MEK | MAPK kinase |
| MLC | myosin light chain |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| O2−• | superoxide |
| PDBu | phorbol 12,13-dibutyrate |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PLC | phospholipase C |
| PKC | protein kinase C |
| PMA | phorbol myristate acetate |
| RACKs | receptors for activated C-kinase |
| RAS | renin-angiotensin system |
| Rho-kinase | Rho-associated kinase |
| ROS | reactive oxygen species |
| SHR | spontaneously hypertensive rat |
| TPA | 12-o-tetradecanoylphorbol-13-acetate |
| VSMC | vascular smooth muscle cell |
| WKY | Wistar-Kyoto |
Acknowledgments
References
- Cardillo, C.; Kilcoyne, C.M.; Quyyumi, A.A.; Cannon, R.O., III; Panza, J.A. Selective defect in nitric oxide synthesis may explain the impaired endothelium-dependent vasodilation in patients with essential hypertension. Circulation 1998, 97, 851–856. [Google Scholar] [CrossRef]
- Heitzer, T.; Wenzel, U.; Hink, U.; Krollner, D.; Skatchkov, M.; Stahl, R.A.; MacHarzina, R.; Brasen, J.H.; Meinertz, T.; Munzel, T. Increased NAD(P)H oxidase-mediated superoxide production in renovascular hypertension: evidence for an involvement of protein kinase C. Kidney Int. 1999, 55, 252–260. [Google Scholar] [CrossRef]
- Ungvari, Z.; Csiszar, A.; Huang, A.; Kaminski, P.M.; Wolin, M.S.; Koller, A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation 2003, 108, 1253–1258. [Google Scholar] [CrossRef]
- Libby, P. Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nutr. 2006, 83, 456S–460S. [Google Scholar]
- Nijm, J.; Wikby, A.; Tompa, A.; Olsson, A.G.; Jonasson, L. Circulating levels of proinflammatory cytokines and neutrophil-platelet aggregates in patients with coronary artery disease. Am. J. Cardiol. 2005, 95, 452–456. [Google Scholar] [CrossRef]
- McLachlan, C.S.; Chua, W.C.; Wong, P.T.; Kah, T.L.; Chen, C.; El Oakley, R.M. Homocysteine is positively associated with cytokine IL-18 plasma levels in coronary artery bypass surgery patients. Biofactors 2005, 23, 69–73. [Google Scholar]
- Laviades, C.; Varo, N.; Fernandez, J.; Mayor, G.; Gil, M.J.; Monreal, I.; Diez, J. Abnormalities of the extracellular degradation of collagen type I in essential hypertension. Circulation 1998, 98, 535–540. [Google Scholar] [CrossRef]
- Ergul, A.; Portik-Dobos, V.; Hutchinson, J.; Franco, J.; Anstadt, M.P. Downregulation of vascular matrix metalloproteinase inducer and activator proteins in hypertensive patients. Am. J. Hypertens. 2004, 17, 775–782. [Google Scholar] [CrossRef]
- Watts, S.W.; Rondelli, C.; Thakali, K.; Li, X.; Uhal, B.; Pervaiz, M.H.; Watson, R.E.; Fink, G.D. Morphological and biochemical characterization of remodeling in aorta and vena cava of DOCA-salt hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2438–H2448. [Google Scholar] [CrossRef]
- Hussain, S.; Assender, J.W.; Bond, M.; Wong, L.F.; Murphy, D.; Newby, A.C. Activation of protein kinase Czeta is essential for cytokine-induced metalloproteinase-1, -3, and -9 secretion from rabbit smooth muscle cells and inhibits proliferation. J. Biol. Chem. 2002, 277, 27345–27352. [Google Scholar]
- Park, M.J.; Park, I.C.; Lee, H.C.; Woo, S.H.; Lee, J.Y.; Hong, Y.J.; Rhee, C.H.; Lee, Y.S.; Lee, S.H.; Shim, B.S.; et al. Protein kinase C-alpha activation by phorbol ester induces secretion of gelatinase B/MMP-9 through ERK 1/2 pathway in capillary endothelial cells. Int. J. Oncol. 2003, 22, 137–143. [Google Scholar]
- Mountain, D.J.; Singh, M.; Menon, B.; Singh, K. Interleukin-1beta increases expression and activity of matrix metalloproteinase-2 in cardiac microvascular endothelial cells: role of PKCalpha/beta1 and MAPKs. Am. J. Physiol. Cell. Physiol. 2007, 292, C867–C875. [Google Scholar] [CrossRef]
- Tsai, B.M.; Wang, M.; Pitcher, J.M.; Meldrum, K.K.; Meldrum, D.R. Hypoxic pulmonary vasoconstriction and pulmonary artery tissue cytokine expression are mediated by protein kinase C. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1215–L1219. [Google Scholar] [CrossRef]
- Ramana, K.V.; Chandra, D.; Srivastava, S.; Bhatnagar, A.; Srivastava, S.K. Aldose reductase mediates the mitogenic signals of cytokines. Chem. Biol. Interact. 2003, 143–144, 587–596. [Google Scholar] [CrossRef]
- Ramana, K.V.; Tammali, R.; Reddy, A.B.; Bhatnagar, A.; Srivastava, S.K. Aldose reductase-regulated tumor necrosis factor-alpha production is essential for high glucose-induced vascular smooth muscle cell growth. Endocrinology 2007, 148, 4371–4384. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatas. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar]
- Khalil, R.A.; van Breemen, C. Sustained contraction of vascular smooth muscle: calcium influx or C-kinase activation? J. Pharmacol. Exp. Ther. 1988, 244, 537–542. [Google Scholar]
- Horowitz, A.; Menice, C.B.; Laporte, R.; Morgan, K.G. Mechanisms of smooth muscle contraction. Physiol. Rev. 1996, 76, 967–1003. [Google Scholar]
- Salamanca, D.A.; Khalil, R.A. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem. Pharmacol. 2005, 70, 1537–1547. [Google Scholar] [CrossRef]
- Berridge, M.J.; Irvine, R.F. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature 1984, 312, 315–321. [Google Scholar] [CrossRef]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar]
- Morgan, K.G.; Khalil, R.A.; Suematsu, E.; Katsuyama, H. Calcium-dependent and calcium-independent pathways of signal transduction in smooth muscle. Jpn. J. Pharmacol. 1992, 58 (Suppl. 2), 47P–53P. [Google Scholar]
- Nishimura, J.; Khalil, R.A.; van Breemen, C. Agonist-induced vascular tone. Hypertension 1989, 13, 835–844. [Google Scholar] [CrossRef]
- Jiang, M.J.; Morgan, K.G. Intracellular calcium levels in phorbol ester-induced contractions of vascular muscle. Am. J. Physiol. 1987, 253, H1365–H1371. [Google Scholar]
- Takai, Y.; Kishimoto, A.; Iwasa, Y.; Kawahara, Y.; Mori, T.; Nishizuka, Y. Calcium-dependent activation of a multifunctional protein kinase by membrane phospholipids. J. Biol. Chem. 1979, 254, 3692–3695. [Google Scholar]
- Newton, A.C. Protein kinase C: structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef]
- Klevit, R.E.; Herriott, J.R.; Horvath, S.J. Solution structure of a zinc finger domain of yeast ADR1. Proteins 1990, 7, 215–226. [Google Scholar] [CrossRef]
- Coussens, L.; Parker, P.J.; Rhee, L.; Yang-Feng, T.L.; Chen, E.; Waterfield, M.D.; Francke, U.; Ullrich, A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science 1986, 233, 859–866. [Google Scholar]
- Parker, C.A.; Takahashi, K.; Tao, T.; Morgan, K.G. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. Am. J. Physiol. 1994, 267, C1262–C1270. [Google Scholar]
- Ono, Y.; Fujii, T.; Ogita, K.; Kikkawa, U.; Igarashi, K.; Nishizuka, Y. Protein kinase C zeta subspecies from rat brain: its structure, expression, and properties. Proc. Natl. Acad. Sci. USA 1989, 86, 3099–3103. [Google Scholar]
- Ohno, S.; Konno, Y.; Akita, Y.; Yano, A.; Suzuki, K. A point mutation at the putative ATP-binding site of protein kinase C alpha abolishes the kinase activity and renders it down-regulation-insensitive. A molecular link between autophosphorylation and down-regulation. J. Biol. Chem. 1990, 265, 6296–6300. [Google Scholar]
- Schaap, D.; Parker, P.J.; Bristol, A.; Kriz, R.; Knopf, J. Unique substrate specificity and regulatory properties of PKC-epsilon: a rationale for diversity. FEBS Lett. 1989, 243, 351–357. [Google Scholar] [CrossRef]
- Osada, S.; Mizuno, K.; Saido, T.C.; Suzuki, K.; Kuroki, T.; Ohno, S. A new member of the protein kinase C family, nPKC theta, predominantly expressed in skeletal muscle. Mol. Cell. Biol. 1992, 12, 3930–3938. [Google Scholar]
- Bacher, N.; Zisman, Y.; Berent, E.; Livneh, E. Isolation and characterization of PKC-L, a new member of the protein kinase C-related gene family specifically expressed in lung, skin, and heart. Mol. Cell. Biol. 1991, 11, 126–133. [Google Scholar]
- Haller, H.; Quass, P.; Lindschau, C.; Luft, F.C.; Distler, A. Platelet-derived growth factor and angiotensin II induce different spatial distribution of protein kinase C-alpha and -beta in vascular smooth muscle cells. Hypertension 1994, 23, 848–852. [Google Scholar] [CrossRef]
- Singer, H.A. Phorbol ester-induced stress and myosin light chain phosphorylation in swine carotid medial smooth muscle. J. Pharmacol. Exp. Ther. 1990, 252, 1068–1074. [Google Scholar]
- Ohanian, V.; Ohanian, J.; Shaw, L.; Scarth, S.; Parker, P.J.; Heagerty, A.M. Identification of protein kinase C isoforms in rat mesenteric small arteries and their possible role in agonist-induced contraction. Circ. Res. 1996, 78, 806–812. [Google Scholar] [CrossRef]
- Kanashiro, C.A.; Altirkawi, K.A.; Khalil, R.A. Preconditioning of coronary artery against vasoconstriction by endothelin-1 and prostaglandin F2alpha during repeated downregulation of epsilon-protein kinase C. J. Cardiovasc. Pharmacol. 2000, 35, 491–501. [Google Scholar] [CrossRef]
- Watanabe, M.; Hachiya, T.; Hagiwara, M.; Hidaka, H. Identification of type III protein kinase C in bovine aortic tissue. Arch. Biochem. Biophys. 1989, 273, 165–169. [Google Scholar]
- Khalil, R.A.; Lajoie, C.; Morgan, K.G. In situ determination of [Ca2+]i threshold for translocation of the alpha-protein kinase C isoform. Am. J. Physiol. 1994, 266, C1544–C1551. [Google Scholar]
- Liou, Y.M.; Morgan, K.G. Redistribution of protein kinase C isoforms in association with vascular hypertrophy of rat aorta. Am. J. Physiol. 1994, 267, C980–C989. [Google Scholar]
- Khalil, R.A.; Lajoie, C.; Resnick, M.S.; Morgan, K.G. Ca(2+)-independent isoforms of protein kinase C differentially translocate in smooth muscle. Am. J. Physiol. 1992, 263, C714–C719. [Google Scholar]
- Goodnight, J.A.; Mischak, H.; Kolch, W.; Mushinski, J.F. Immunocytochemical localization of eight protein kinase C isozymes overexpressed in NIH 3T3 fibroblasts. Isoform-specific association with microfilaments, Golgi, endoplasmic reticulum, and nuclear and cell membranes. J. Biol. Chem. 1995, 270, 9991–10001. [Google Scholar]
- Gailly, P.; Gong, M.C.; Somlyo, A.V.; Somlyo, A.P. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. J. Physiol. 1997, 500, 95–109. [Google Scholar]
- Makowske, M.; Rosen, O.M. Complete activation of protein kinase C by an antipeptide antibody directed against the pseudosubstrate prototope. J. Biol. Chem. 1989, 264, 16155–16159. [Google Scholar]
- Orr, J.W.; Keranen, L.M.; Newton, A.C. Reversible exposure of the pseudosubstrate domain of protein kinase C by phosphatidylserine and diacylglycerol. J. Biol. Chem. 1992, 267, 15263–15266. [Google Scholar]
- House, C.; Kemp, B.E. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science 1987, 238, 1726–1728. [Google Scholar]
- Dekker, L.V.; McIntyre, P.; Parker, P.J. Mutagenesis of the regulatory domain of rat protein kinase C-eta. A molecular basis for restricted histone kinase activity. J. Biol. Chem. 1993, 268, 19498–19504. [Google Scholar]
- Kanashiro, C.A.; Khalil, R.A. Signal transduction by protein kinase C in mammalian cells. Clin. Exp. Pharmacol. Physiol. 1998, 25, 974–985. [Google Scholar] [CrossRef]
- Wang, J.K.; Walaas, S.I.; Sihra, T.S.; Aderem, A.; Greengard, P. Phosphorylation and associated translocation of the 87-kDa protein, a major protein kinase C substrate, in isolated nerve terminals. Proc. Natl. Acad. Sci. USA 1989, 86, 2253–2256. [Google Scholar] [CrossRef]
- Hartwig, J.H.; Thelen, M.; Rosen, A.; Janmey, P.A.; Nairn, A.C.; Aderem, A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature 1992, 356, 618–622. [Google Scholar] [CrossRef]
- Katada, T.; Gilman, A.G.; Watanabe, Y.; Bauer, S.; Jakobs, K.H. Protein kinase C phosphorylates the inhibitory guanine-nucleotide-binding regulatory component and apparently suppresses its function in hormonal inhibition of adenylate cyclase. Eur. J. Biochem. 1985, 151, 431–437. [Google Scholar] [CrossRef]
- Barman, S.A.; Zhu, S.; White, R.E. Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L149–L155. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Bosca, L.; Tamargo, J.; Perez-Vizcaino, F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ. Res. 2003, 93, 656–663. [Google Scholar] [CrossRef]
- Limas, C.J. Phosphorylation of cardiac sarcoplasmic reticulum by a calcium-activated, phospholipid-dependent protein kinase. Biochem. Biophys. Res. Commun. 1980, 96, 1378–1383. [Google Scholar] [CrossRef]
- Rosoff, P.M.; Stein, L.F.; Cantley, L.C. Phorbol esters induce differentiation in a pre-B-lymphocyte cell line by enhancing Na+/H+ exchange. J. Biol. Chem. 1984, 259, 7056–7060. [Google Scholar]
- Aviv, A. Cytosolic Ca2+, Na+/H+ antiport, protein kinase C trio in essential hypertension. Am. J. Hypertens. 1994, 7, 205–212. [Google Scholar]
- Schwienbacher, C.; Jockusch, B.M.; Rudiger, M. Intramolecular interactions regulate serine/threonine phosphorylation of vinculin. FEBS Lett. 1996, 384, 71–74. [Google Scholar] [CrossRef]
- Woodsome, T.P.; Eto, M.; Everett, A.; Brautigan, D.L.; Kitazawa, T. Expression of CPI-17 and myosin phosphatase correlates with Ca(2+) sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J. Physiol. 2001, 535, 553–564. [Google Scholar] [CrossRef]
- Inagaki, M.; Yokokura, H.; Itoh, T.; Kanmura, Y.; Kuriyama, H.; Hidaka, H. Purified rabbit brain protein kinase C relaxes skinned vascular smooth muscle and phosphorylates myosin light chain. Arch. Biochem. Biophys. 1987, 254, 136–141. [Google Scholar] [CrossRef]
- Newton, A.C. Regulation of protein kinase C. Curr. Opin. Cell. Biol. 1997, 9, 161–167. [Google Scholar] [CrossRef]
- Mochly-Rosen, D.; Gordon, A.S. Anchoring proteins for protein kinase C: A means for isozyme selectivity. FASEB J. 1998, 12, 35–42. [Google Scholar]
- Kraft, A.S.; Anderson, W.B. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature 1983, 301, 621–623. [Google Scholar] [CrossRef]
- Hyatt, S.L.; Klauck, T.; Jaken, S. Protein kinase C is localized in focal contacts of normal but not transformed fibroblasts. Mol. Carcinog. 1990, 3, 45–53. [Google Scholar] [CrossRef]
- Kose, A.; Saito, N.; Ito, H.; Kikkawa, U.; Nishizuka, Y.; Tanaka, C. Electron microscopic localization of type I protein kinase C in rat Purkinje cells. J. Neurosci. 1988, 8, 4262–4268. [Google Scholar]
- Cogolludo, A.; Moreno, L.; Lodi, F.; Tamargo, J.; Perez-Vizcaino, F. Postnatal maturational shift from PKCzeta and voltage-gated K+ channels to RhoA/Rho kinase in pulmonary vasoconstriction. Cardiovasc. Res. 2005, 66, 84–93. [Google Scholar] [CrossRef]
- Draeger, A.; Wray, S.; Babiychuk, E.B. Domain architecture of the smooth-muscle plasma membrane: regulation by annexins. Biochem. J. 2005, 387, 309–314. [Google Scholar] [CrossRef]
- Thelen, M.; Rosen, A.; Nairn, A.C.; Aderem, A. Regulation by phosphorylation of reversible association of a myristoylated protein kinase C substrate with the plasma membrane. Nature 1991, 351, 320–322. [Google Scholar] [CrossRef]
- Cazaubon, S.M.; Parker, P.J. Identification of the phosphorylated region responsible for the permissive activation of protein kinase C. J. Biol. Chem. 1993, 268, 17559–17563. [Google Scholar]
- Leventhal, P.S.; Bertics, P.J. Activation of protein kinase C by selective binding of arginine-rich polypeptides. J. Biol. Chem. 1993, 268, 13906–13913. [Google Scholar]
- Ron, D.; Mochly-Rosen, D. Agonists and antagonists of protein kinase C function, derived from its binding proteins. J. Biol. Chem. 1994, 269, 21395–21398. [Google Scholar]
- Housey, G.M.; Johnson, M.D.; Hsiao, W.L.; O'Brian, C.A.; Murphy, J.P.; Kirschmeier, P.; Weinstein, I.B. Overproduction of protein kinase C causes disordered growth control in rat fibroblasts. Cell 1988, 52, 343–354. [Google Scholar] [CrossRef]
- Dallas, A.; Khalil, R.A. Ca2+ antagonist-insensitive coronary smooth muscle contraction involves activation of epsilon-protein kinase C-dependent pathway. Am. J. Physiol. Cell. Physiol. 2003, 285, C1454–C1463. [Google Scholar]
- Khalil, R.A.; Menice, C.B.; Wang, C.L.; Morgan, K.G. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circ. Res. 1995, 76, 1101–1108. [Google Scholar] [CrossRef]
- Mii, S.; Khalil, R.A.; Morgan, K.G.; Ware, J.A.; Kent, K.C. Mitogen-activated protein kinase and proliferation of human vascular smooth muscle cells. Am. J. Physiol. 1996, 270, H142–H150. [Google Scholar]
- Adam, L.P.; Gapinski, C.J.; Hathaway, D.R. Phosphorylation sequences in h-caldesmon from phorbol ester-stimulated canine aortas. FEBS Lett. 1992, 302, 223–226. [Google Scholar] [CrossRef]
- D’Angelo, G.; Graceffa, P.; Wang, C.A.; Wrangle, J.; Adam, L.P. Mammal-specific, ERK-dependent, caldesmon phosphorylation in smooth muscle. Quantitation using novel anti-phosphopeptide antibodies. J. Biol. Chem. 1999, 274, 30115–30121. [Google Scholar]
- Hedges, J.C.; Oxhorn, B.C.; Carty, M.; Adam, L.P.; Yamboliev, I.A.; Gerthoffer, W.T. Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. Am. J. Physiol. Cell. Physiol. 2000, 278, C718–C726. [Google Scholar]
- Bazzi, M.D.; Nelsestuen, G.L. Protein kinase C interaction with calcium: A phospholipid-dependent process. Biochemistry 1990, 29, 7624–7630. [Google Scholar] [CrossRef]
- Nishizuka, Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995, 9, 484–496. [Google Scholar]
- Szallasi, Z.; Smith, C.B.; Pettit, G.R.; Blumberg, P.M. Differential regulation of protein kinase C isozymes by bryostatin 1 and phorbol 12-myristate 13-acetate in NIH 3T3 fibroblasts. J. Biol. Chem. 1994, 269, 2118–2124. [Google Scholar]
- Giardina, J.B.; Tanner, D.J.; Khalil, R.A. Oxidized-LDL enhances coronary vasoconstriction by increasing the activity of protein kinase C isoforms alpha and epsilon. Hypertension 2001, 37, 561–568. [Google Scholar] [CrossRef]
- Claro, S.; Kanashiro, C.A.; Oshiro, M.E.; Ferreira, A.T.; Khalil, R.A. alpha- and epsilon-protein kinase C activity during smooth muscle cell apoptosis in response to gamma-radiation. J. Pharmacol. Exp. Ther. 2007, 322, 964–972. [Google Scholar] [CrossRef]
- Li, W.; Zhang, J.; Bottaro, D.P.; Pierce, J.H. Identification of serine 643 of protein kinase C-delta as an important autophosphorylation site for its enzymatic activity. J. Biol. Chem. 1997, 272, 24550–24555. [Google Scholar]
- Keranen, L.M.; Dutil, E.M.; Newton, A.C. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol. 1995, 5, 1394–1403. [Google Scholar] [CrossRef]
- Edwards, A.S.; Newton, A.C. Phosphorylation at conserved carboxyl-terminal hydrophobic motif regulates the catalytic and regulatory domains of protein kinase C. J. Biol. Chem. 1997, 272, 18382–18390. [Google Scholar] [CrossRef]
- Eichholtz, T.; de Bont, D.B.; de Widt, J.; Liskamp, R.M.; Ploegh, H.L. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J. Biol. Chem. 1993, 268, 1982–1986. [Google Scholar]
- Clement, S.; Tasinato, A.; Boscoboinik, D.; Azzi, A. The effect of alpha-tocopherol on the synthesis, phosphorylation and activity of protein kinase C in smooth muscle cells after phorbol 12-myristate 13-acetate down-regulation. Eur. J. Biochem. 1997, 246, 745–749. [Google Scholar]
- Cain, A.E.; Khalil, R.A. Pathophysiology of essential hypertension: role of the pump, the vessel, and the kidney. Semin. Nephrol. 2002, 22, 3–16. [Google Scholar]
- Wang, S.; Desai, D.; Wright, G.; Niles, R.M.; Wright, G.L. Effects of protein kinase C alpha overexpression on A7r5 smooth muscle cell proliferation and differentiation. Exp. Cell. Res. 1997, 236, 117–126. [Google Scholar] [CrossRef]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; Hodge, S.E.; Knowles, J.A. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef]
- Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Lane, K.B.; Morgan, N.V.; Wheeler, L.; Phillips, J.A., III; Newman, J.; Williams, D.; Galie, N.; et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am. J. Hum. Genet. 2001, 68, 92–102. [Google Scholar] [CrossRef]
- Aldred, M.A.; Vijayakrishnan, J.; James, V.; Soubrier, F.; Gomez-Sanchez, M.A.; Martensson, G.; Galie, N.; Manes, A.; Corris, P.; Simonneau, G.; et al. MPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 212–213. [Google Scholar]
- Song, Y.; Jones, J.E.; Beppu, H.; Keaney, J.F., Jr.; Loscalzo, J.; Zhang, Y.Y. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 2005, 112, 553–562. [Google Scholar] [CrossRef]
- Hassel, S.; Eichner, A.; Yakymovych, M.; Hellman, U.; Knaus, P.; Souchelnytskyi, S. Proteins associated with type II bone morphogenetic protein receptor (BMPR-II) and identified by two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 1346–1358. [Google Scholar] [CrossRef]
- Shibata, R.; Morita, S.; Nagai, K.; Miyata, S.; Iwasaki, T. Effects of H-7 (protein kinase inhibitor) and phorbol ester on aortic strips from spontaneously hypertensive rats. Eur. J. Pharmacol. 1990, 175, 261–271. [Google Scholar] [CrossRef]
- Bazan, E.; Campbell, A.K.; Rapoport, R.M. Protein kinase C activity in blood vessels from normotensive and spontaneously hypertensive rats. Eur. J. Pharmacol. 1992, 227, 343–348. [Google Scholar] [CrossRef]
- Sauro, M.D.; Hadden, J.W. Gamma-interferon corrects aberrant protein kinase C levels and immunosuppression in the spontaneously hypertensive rat. Int. J. Immunopharmacol. 1992, 14, 1421–1427. [Google Scholar] [CrossRef]
- Sasajima, H.; Shima, H.; Toyoda, Y.; Kimura, K.; Yoshikawa, A.; Hano, T.; Nishio, I. Increased Ca2+ sensitivity of contractile elements via protein kinase C in alpha-toxin permeabilized SMA from young spontaneously hypertensive rats. Cardiovasc. Res. 1997, 36, 86–91. [Google Scholar] [CrossRef]
- Rosen, B.; Barg, J.; Zimlichman, R. The effects of angiotensin II, endothelin-1, and protein kinase C inhibitor on DNA synthesis and intracellular calcium mobilization in vascular smooth muscle cells from young normotensive and spontaneously hypertensive rats. Am. J. Hypertens. 1999, 12, 1243–1251. [Google Scholar] [CrossRef]
- Bilder, G.E.; Kasiewski, C.J.; Perrone, M.H. Phorbol-12,13-dibutyrate-induced vasoconstriction in vivo: characterization of response in genetic hypertension. J. Pharmacol. Exp. Ther. 1990, 252, 526–530. [Google Scholar]
- Kanashiro, C.A.; Khalil, R.A. Gender-related distinctions in protein kinase C activity in rat vascular smooth muscle. Am. J. Physiol. Cell. Physiol. 2001, 280, C34–C45. [Google Scholar]
- Touyz, R.M.; Schiffrin, E.L. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J. Hypertens. 2001, 19, 1245–1254. [Google Scholar] [CrossRef]
- Escriba, P.V.; Sanchez-Dominguez, J.M.; Alemany, R.; Perona, J.S.; Ruiz-Gutierrez, V. Alteration of lipids, G proteins, and PKC in cell membranes of elderly hypertensives. Hypertension 2003, 41, 176–182. [Google Scholar] [CrossRef]
- Gu, X.; Bishop, S.P. Increased protein kinase C and isozyme redistribution in pressure-overload cardiac hypertrophy in the rat. Circ. Res. 1994, 75, 926–931. [Google Scholar] [CrossRef]
- Fatehi-Hassanabad, Z.; Fatehi, M.; Shahidi, M.I. Endothelial dysfunction in aortic rings and mesenteric beds isolated from deoxycorticosterone acetate hypertensive rats: Possible involvement of protein kinase C. Eur. J. Pharmacol. 2004, 494, 199–204. [Google Scholar] [CrossRef]
- Soloviev, A.I.; Parshikov, A.V.; Stefanov, A.V. Evidence for the involvement of protein kinase C in depression of endothelium-dependent vascular responses in spontaneously hypertensive rats. J. Vasc. Res. 1998, 35, 325–331. [Google Scholar] [CrossRef]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar]
- Michell, B.J.; Chen, Z.; Tiganis, T.; Stapleton, D.; Katsis, F.; Power, D.A.; Sim, A.T.; Kemp, B.E. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 2001, 276, 17625–17628. [Google Scholar]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, E68–E75. [Google Scholar] [CrossRef]
- Motley, E.D.; Eguchi, K.; Patterson, M.M.; Palmer, P.D.; Suzuki, H.; Eguchi, S. Mechanism of endothelial nitric oxide synthase phosphorylation and activation by thrombin. Hypertension 2007, 49, 577–583. [Google Scholar] [CrossRef]
- Partovian, C.; Zhuang, Z.; Moodie, K.; Lin, M.; Ouchi, N.; Sessa, W.C.; Walsh, K.; Simons, M. PKCalpha activates eNOS and increases arterial blood flow in vivo. Circ. Res. 2005, 97, 482–487. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef]
- Xia, Y.; Tsai, A.L.; Berka, V.; Zweier, J.L. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem. 1998, 273, 25804–25808. [Google Scholar] [CrossRef]
- Li, H.; Witte, K.; August, M.; Brausch, I.; Godtel-Armbrust, U.; Habermeier, A.; Closs, E.I.; Oelze, M.; Munzel, T.; Forstermann, U. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 2006, 47, 2536–2544. [Google Scholar] [CrossRef]
- Dai, X.; Cao, X.; Kreulen, D.L. Superoxide anion is elevated in sympathetic neurons in DOCA-salt hypertension via activation of NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1019–H1026. [Google Scholar]
- Fedorova, O.V.; Talan, M.I.; Agalakova, N.I.; Droy-Lefaix, M.T.; Lakatta, E.G.; Bagrov, A.Y. Myocardial PKC beta2 and the sensitivity of Na/K-ATPase to marinobufagenin are reduced by cicletanine in Dahl hypertension. Hypertension 2003, 41, 505–511. [Google Scholar] [CrossRef]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef]
- Benjamin, M.M.; Khalil, R.A. Matrix metalloproteinase inhibitors as investigative tools in the pathogenesis and management of vascular disease. Experientia. Supplementum. 2012, 103, 209–279. [Google Scholar] [CrossRef]
- Chew, D.K.; Conte, M.S.; Khalil, R.A. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J. Vasc. Surg. 2004, 40, 1001–1010. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Ross, R.L.; Khalil, R.A. Matrix metalloproteinase 2-induced venous dilation via hyperpolarization and activation of K+ channels: Relevance to varicose vein formation. J. Vasc. Surg. 2007, 45, 373–380. [Google Scholar] [CrossRef]
- Derosa, G.; D’Angelo, A.; Ciccarelli, L.; Piccinni, M.N.; Pricolo, F.; Salvadeo, S.; Montagna, L.; Gravina, A.; Ferrari, I.; Galli, S.; et al. Matrix metalloproteinase-2, -9, and tissue inhibitor of metalloproteinase-1 in patients with hypertension. Endothelium 2006, 13, 227–231. [Google Scholar] [CrossRef]
- Zervoudaki, A.; Economou, E.; Stefanadis, C.; Pitsavos, C.; Tsioufis, K.; Aggeli, C.; Vasiliadou, K.; Toutouza, M.; Toutouzas, P. Plasma levels of active extracellular matrix metalloproteinases 2 and 9 in patients with essential hypertension before and after antihypertensive treatment. J. Hum. Hypertens. 2003, 17, 119–124. [Google Scholar] [CrossRef]
- Flamant, M.; Placier, S.; Dubroca, C.; Esposito, B.; Lopes, I.; Chatziantoniou, C.; Tedgui, A.; Dussaule, J.C.; Lehoux, S. Role of matrix metalloproteinases in early hypertensive vascular remodeling. Hypertension 2007, 50, 212–218. [Google Scholar] [CrossRef]
- Papadimitriou, E.; Waters, C.R.; Manolopoulos, V.G.; Unsworth, B.R.; Maragoudakis, M.E.; Lelkes, P.I. Regulation of extracellular matrix remodeling and MMP-2 activation in cultured rat adrenal medullary endothelial cells. Endothelium 2001, 8, 243–253. [Google Scholar]
- Li, D.; Liu, L.; Chen, H.; Sawamura, T.; Ranganathan, S.; Mehta, J.L. LOX-1 mediates oxidized low-density lipoprotein-induced expression of matrix metalloproteinases in human coronary artery endothelial cells. Circulation 2003, 107, 612–617. [Google Scholar] [CrossRef]
- Smith, L.; Payne, J.A.; Sedeek, M.H.; Granger, J.P.; Khalil, R.A. Endothelin-induced increases in Ca2+ entry mechanisms of vascular contraction are enhanced during high-salt diet. Hypertension 2003, 41, 787–793. [Google Scholar] [CrossRef]
- Khalil, R.A. Dietary salt and hypertension: new molecular targets add more spice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R509–R513. [Google Scholar] [CrossRef]
- Fareh, J.; Touyz, R.M.; Schiffrin, E.L.; Thibault, G. Altered cardiac endothelin receptors and protein kinase C in deoxycorticosterone-salt hypertensive rats. J. Mol. Cell. Cardiol. 2000, 32, 665–676. [Google Scholar] [CrossRef]
- Kim, J.; Lee, Y.R.; Lee, C.H.; Choi, W.H.; Lee, C.K.; Bae, Y.M.; Cho, S.; Kim, B. Mitogen-activated protein kinase contributes to elevated basal tone in aortic smooth muscle from hypertensive rats. Eur. J. Pharmacol. 2005, 514, 209–215. [Google Scholar] [CrossRef]
- Sirous, Z.N.; Fleming, J.B.; Khalil, R.A. Endothelin-1 enhances eicosanoids-induced coronary smooth muscle contraction by activating specific protein kinase C isoforms. Hypertension 2001, 37, 497–504. [Google Scholar] [CrossRef]
- Cain, A.E.; Tanner, D.M.; Khalil, R.A. Endothelin-1--induced enhancement of coronary smooth muscle contraction via MAPK-dependent and MAPK-independent [Ca(2+)](i) sensitization pathways. Hypertension 2002, 39, 543–549. [Google Scholar] [CrossRef]
- Khalil, R.A. Modulators of the vascular endothelin receptor in blood pressure regulation and hypertension. Curr. Mol. Pharmacol. 2011, 4, 176–186. [Google Scholar]
- Schiffrin, E.L. Endothelin: potential role in hypertension and vascular hypertrophy. Hypertension 1995, 25, 1135–1143. [Google Scholar] [CrossRef]
- Kanayama, Y.; Negoro, N.; Okamura, M.; Konishi, Y.; Nishimura, M.; Umetani, N.; Inoue, T.; Takeda, T. Modulation of protein kinase C in aorta of spontaneously hypertensive rats with enalapril treatment. Osaka City Med. J. 1994, 40, 83–97. [Google Scholar]
- Bell, P.D.; Mashburn, N.; Unlap, M.T. Renal sodium/calcium exchange; a vasodilator that is defective in salt-sensitive hypertension. Acta. Physiol. Scand. 2000, 168, 209–214. [Google Scholar] [CrossRef]
- Nowicki, S.; Kruse, M.S.; Brismar, H.; Aperia, A. Dopamine-induced translocation of protein kinase C isoforms visualized in renal epithelial cells. Am. J. Physiol. Cell. Physiol. 2000, 279, C1812–C1818. [Google Scholar]
- Ridge, K.M.; Dada, L.; Lecuona, E.; Bertorello, A.M.; Katz, A.I.; Mochly-Rosen, D.; Sznajder, J.I. Dopamine-induced exocytosis of Na,K-ATPase is dependent on activation of protein kinase C-epsilon and -delta. Mol. Biol. Cell. 2002, 13, 1381–1389. [Google Scholar] [CrossRef]
- Banday, A.A.; Fazili, F.R.; Lokhandwala, M.F. Oxidative stress causes renal dopamine D1 receptor dysfunction and hypertension via mechanisms that involve nuclear factor-kappaB and protein kinase C. J. Am. Soc. Nephrol. 2007, 18, 1446–1457. [Google Scholar] [CrossRef]
- Hughes-Darden, C.A.; Wachira, S.J.; Denaro, F.J.; Taylor, C.V.; Brunson, K.J.; Ochillo, R.; Robinson, T.J. Expression and distribution of protein kinase C isozymes in brain tissue of spontaneous hypertensive rats. Cell. Mol. Biol. (Noisy-le-grand) 2001, 47, 1077–1088. [Google Scholar]
- Rao, F.; Zhang, L.; Wessel, J.; Zhang, K.; Wen, G.; Kennedy, B.P.; Rana, B.K.; Das, M.; Rodriguez-Flores, J.L.; Smith, D.W.; et al. Tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis: discovery of common human genetic variants governing transcription, autonomic activity, and blood pressure in vivo. Circulation 2007, 116, 993–1006. [Google Scholar] [CrossRef]
- Hempel, A.; Maasch, C.; Heintze, U.; Lindschau, C.; Dietz, R.; Luft, F.C.; Haller, H. High glucose concentrations increase endothelial cell permeability via activation of protein kinase C alpha. Circ. Res. 1997, 81, 363–371. [Google Scholar] [CrossRef]
- Williams, B.; Howard, R.L. Glucose-induced changes in Na+/H+ antiport activity and gene expression in cultured vascular smooth muscle cells. Role of protein kinase C. J. Clin. Invest. 1994, 93, 2623–2631. [Google Scholar] [CrossRef]
- Farese, R.V.; Standaert, M.L.; Ishizuka, T.; Yu, B.; Hernandez, H.; Waldron, C.; Watson, J.; Farese, J.P.; Cooper, D.R.; Wickstrom, E. Antisense DNA downregulates protein kinase C isozymes (beta and alpha) and insulin-stimulated 2-deoxyglucose uptake in rat adipocytes. Antisense Res. Dev. 1991, 1, 35–42. [Google Scholar]
- Ishii, H.; Jirousek, M.R.; Koya, D.; Takagi, C.; Xia, P.; Clermont, A.; Bursell, S.E.; Kern, T.S.; Ballas, L.M.; Heath, W.F.; et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 1996, 272, 728–731. [Google Scholar]
- Young, J.L.; Libby, P.; Schonbeck, U. Cytokines in the pathogenesis of atherosclerosis. Thromb. Haemost. 2002, 88, 554–567. [Google Scholar]
- Waehre, T.; Yndestad, A.; Smith, C.; Haug, T.; Tunheim, S.H.; Gullestad, L.; Froland, S.S.; Semb, A.G.; Aukrust, P.; Damas, J.K. Increased expression of interleukin-1 in coronary artery disease with downregulatory effects of HMG-CoA reductase inhibitors. Circulation 2004, 109, 1966–1972. [Google Scholar] [CrossRef]
- Sardella, G.; Mariani, P.; D’Alessandro, M.; De Luca, L.; Pierro, M.; Mancone, M.; Porretta, A.; Accapezzato, D.; Fedele, F.; Paroli, M. Early elevation of interleukin-1beta and interleukin-6 levels after bare or drug-eluting stent implantation in patients with stable angina. Thromb. Res. 2006, 117, 659–664. [Google Scholar] [CrossRef]
- Lubrano, V.; Cocci, F.; Battaglia, D.; Papa, A.; Marraccini, P.; Zucchelli, G.C. Usefulness of high-sensitivity IL-6 measurement for clinical characterization of patients with coronary artery disease. J. Clin. Lab. Anal. 2005, 19, 110–114. [Google Scholar] [CrossRef]
- Funayama, H.; Ishikawa, S.E.; Kubo, N.; Katayama, T.; Yasu, T.; Saito, M.; Kawakami, M. Increases in interleukin-6 and matrix metalloproteinase-9 in the infarct-related coronary artery of acute myocardial infarction. Circ. J. 2004, 68, 451–454. [Google Scholar] [CrossRef]
- Lee, D.L.; Sturgis, L.C.; Labazi, H.; Osborne, J.B., Jr.; Fleming, C.; Pollock, J.S.; Manhiani, M.; Imig, J.D.; Brands, M.W. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H935–H940. [Google Scholar]
- Das, M.; Dempsey, E.C.; Bouchey, D.; Reyland, M.E.; Stenmark, K.R. Chronic hypoxia induces exaggerated growth responses in pulmonary artery adventitial fibroblasts: potential contribution of specific protein kinase c isozymes. Am. J. Respir. Cell. Mol. Biol. 2000, 22, 15–25. [Google Scholar]
- Littler, C.M.; Morris, K.G., Jr.; Fagan, K.A.; McMurtry, I.F.; Messing, R.O.; Dempsey, E.C. Protein kinase C-epsilon-null mice have decreased hypoxic pulmonary vasoconstriction. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1321–H1331. [Google Scholar]
- Ito, T.; Ozawa, K.; Shimada, K. Current drug targets and future therapy of pulmonary arterial hypertension. Curr. Med. Chem. 2007, 14, 719–733. [Google Scholar] [CrossRef]
- Puri, A.; McGoon, M.D.; Kushwaha, S.S. Pulmonary arterial hypertension: current therapeutic strategies. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 319–329. [Google Scholar] [CrossRef]
- Barman, S.A. Vasoconstrictor effect of endothelin-1 on hypertensive pulmonary arterial smooth muscle involves Rho-kinase and protein kinase C. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L472–L479. [Google Scholar] [CrossRef]
- Khalil, R.A.; Granger, J.P. Vascular mechanisms of increased arterial pressure in preeclampsia: Lessons from animal models. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R29–R45. [Google Scholar]
- Sheppard, S.J.; Khalil, R.A. Risk factors and mediators of the vascular dysfunction associated with hypertension in pregnancy. Cardiovasc. Hematol. Disord. Drug Targets 2010, 10, 33–52. [Google Scholar] [CrossRef]
- Magness, R.R.; Rosenfeld, C.R.; Carr, B.R. Protein kinase C in uterine and systemic arteries during ovarian cycle and pregnancy. Am. J. Physiol. 1991, 260, E464–E470. [Google Scholar]
- Kanashiro, C.A.; Cockrell, K.L.; Alexander, B.T.; Granger, J.P.; Khalil, R.A. Pregnancy-associated reduction in vascular protein kinase C activity rebounds during inhibition of NO synthesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 278, R295–R303. [Google Scholar]
- Kanashiro, C.A.; Alexander, B.T.; Granger, J.P.; Khalil, R.A. Ca(2+)-insensitive vascular protein kinase C during pregnancy and NOS inhibition. Hypertension 1999, 34, 924–930. [Google Scholar] [CrossRef]
- Khalil, R.A.; Crews, J.K.; Novak, J.; Kassab, S.; Granger, J.P. Enhanced vascular reactivity during inhibition of nitric oxide synthesis in pregnant rats. Hypertension 1998, 31, 1065–1069. [Google Scholar] [CrossRef]
- Crews, J.K.; Novak, J.; Granger, J.P.; Khalil, R.A. Stimulated mechanisms of Ca2+ entry into vascular smooth muscle during NO synthesis inhibition in pregnant rats. Am. J. Physiol. 1999, 276, R530–R538. [Google Scholar]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jupner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Invest. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Kupferminc, M.J.; Peaceman, A.M.; Wigton, T.R.; Rehnberg, K.A.; Socol, M.L. Tumor necrosis factor-alpha is elevated in plasma and amniotic fluid of patients with severe preeclampsia. Am. J. Obstet. Gynecol. 1994, 170, 1752–1757, discussion 1757–1759. [Google Scholar]
- Vince, G.S.; Starkey, P.M.; Austgulen, R.; Kwiatkowski, D.; Redman, C.W. Interleukin-6, tumour necrosis factor and soluble tumour necrosis factor receptors in women with pre-eclampsia. Br. J. Obstet. Gynaecol. 1995, 102, 20–25. [Google Scholar] [CrossRef]
- Conrad, K.P.; Benyo, D.F. Placental cytokines and the pathogenesis of preeclampsia. Am. J. Reprod. Immunol. 1997, 37, 240–249. [Google Scholar] [CrossRef]
- Williams, M.A.; Mahomed, K.; Farrand, A.; Woelk, G.B.; Mudzamiri, S.; Madzime, S.; King, I.B.; McDonald, G.B. Plasma tumor necrosis factor-alpha soluble receptor p55 (sTNFp55) concentrations in eclamptic, preeclamptic and normotensive pregnant Zimbabwean women. J. Reprod. Immunol. 1998, 40, 159–173. [Google Scholar] [CrossRef]
- LaMarca, B.D.; Ryan, M.J.; Gilbert, J.S.; Murphy, S.R.; Granger, J.P. Inflammatory cytokines in the pathophysiology of hypertension during preeclampsia. Curr. Hypertens. Rep. 2007, 9, 480–485. [Google Scholar] [CrossRef]
- Benyo, D.F.; Smarason, A.; Redman, C.W.; Sims, C.; Conrad, K.P. Expression of inflammatory cytokines in placentas from women with preeclampsia. J. Clin. Endocrinol. Metab. 2001, 86, 2505–2512. [Google Scholar] [CrossRef]
- Davis, J.R.; Giardina, J.B.; Green, G.M.; Alexander, B.T.; Granger, J.P.; Khalil, R.A. Reduced endothelial NO-cGMP vascular relaxation pathway during TNF-alpha-induced hypertension in pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R390–R399. [Google Scholar]
- Orshal, J.M.; Khalil, R.A. Reduced endothelial NO-cGMP-mediated vascular relaxation and hypertension in IL-6-infused pregnant rats. Hypertension 2004, 43, 434–444. [Google Scholar] [CrossRef]
- Giardina, J.B.; Green, G.M.; Cockrell, K.L.; Granger, J.P.; Khalil, R.A. TNF-alpha enhances contraction and inhibits endothelial NO-cGMP relaxation in systemic vessels of pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R130–R143. [Google Scholar]
- Orshal, J.M.; Khalil, R.A. Interleukin-6 impairs endothelium-dependent NO-cGMP-mediated relaxation and enhances contraction in systemic vessels of pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R1013–R1023. [Google Scholar] [CrossRef]
- Gilbert, J.S.; Babcock, S.A.; Granger, J.P. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 2007, 50, 1142–1147. [Google Scholar] [CrossRef]
- Gilbert, J.S.; Gilbert, S.A.; Arany, M.; Granger, J.P. Hypertension produced by placental ischemia in pregnant rats is associated with increased soluble endoglin expression. Hypertension 2009, 53, 399–403. [Google Scholar]
- Bagrov, A.Y.; Dmitrieva, R.I.; Dorofeeva, N.A.; Fedorova, O.V.; Lopatin, D.A.; Lakatta, E.G.; Droy-Lefaix, M.T. Cicletanine reverses vasoconstriction induced by the endogenous sodium pump ligand, marinobufagenin, via a protein kinase C dependent mechanis. J. Hypertens. 2000, 18, 209–215. [Google Scholar] [CrossRef]
- Seko, T.; Ito, M.; Kureishi, Y.; Okamoto, R.; Moriki, N.; Onishi, K.; Isaka, N.; Hartshorne, D.J.; Nakano, T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ. Res. 2003, 92, 411–418. [Google Scholar] [CrossRef]
- McCarty, M.F. Up-regulation of intracellular signalling pathways may play a central pathogenic role in hypertension, atherogenesis, insulin resistance, and cancer promotion—The “PKC syndrome”. Med. Hypotheses 1996, 46, 191–221. [Google Scholar] [CrossRef]
- Davis, M.D.; Sheetz, M.J.; Aiello, L.P.; Milton, R.C.; Danis, R.P.; Zhi, X.; Girach, A.; Jimenez, M.C.; Vignati, L. Effect of ruboxistaurin on the visual acuity decline associated with long-standing diabetic macular edema. Invest. Ophthalmol. Vis. Sci. 2009, 50, 1–4. [Google Scholar]
- Aiello, L.P.; Vignati, L.; Sheetz, M.J.; Zhi, X.; Girach, A.; Davis, M.D.; Wolka, A.M.; Shahri, N.; Milton, R.C. Oral protein kinase C beta inhibition using ruboxistaurin: efficacy, safety, and causes of vision loss among 813 patients (1,392 eyes) with diabetic retinopathy in the protein kinase C beta inhibitor-diabetic retinopathy study and the protein kinase C beta inhibitor-diabetic retinopathy study 2. Retina 2011, 31, 2084–2094. [Google Scholar] [CrossRef]
- Joy, S.V.; Scates, A.C.; Bearelly, S.; Dar, M.; Taulien, C.A.; Goebel, J.A.; Cooney, M.J. Ruboxistaurin, a protein kinase C beta inhibitor, as an emerging treatment for diabetes microvascular complications. Ann. Pharmacother. 2005, 39, 1693–1699. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khalil, R.A. Protein Kinase C Inhibitors as Modulators of Vascular Function and Their Application in Vascular Disease. Pharmaceuticals 2013, 6, 407-439. https://doi.org/10.3390/ph6030407
Khalil RA. Protein Kinase C Inhibitors as Modulators of Vascular Function and Their Application in Vascular Disease. Pharmaceuticals. 2013; 6(3):407-439. https://doi.org/10.3390/ph6030407
Chicago/Turabian StyleKhalil, Raouf A. 2013. "Protein Kinase C Inhibitors as Modulators of Vascular Function and Their Application in Vascular Disease" Pharmaceuticals 6, no. 3: 407-439. https://doi.org/10.3390/ph6030407
APA StyleKhalil, R. A. (2013). Protein Kinase C Inhibitors as Modulators of Vascular Function and Their Application in Vascular Disease. Pharmaceuticals, 6(3), 407-439. https://doi.org/10.3390/ph6030407