FedExosomes: Engineering Therapeutic Biological Nanoparticles that Truly Deliver

Abstract
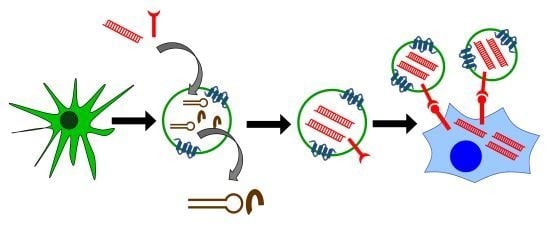
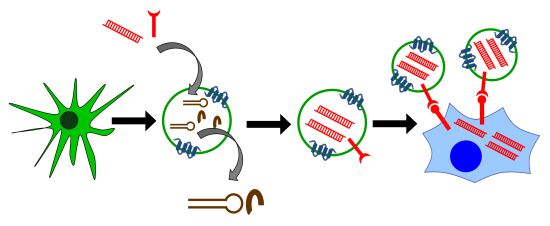
:
1. Introduction
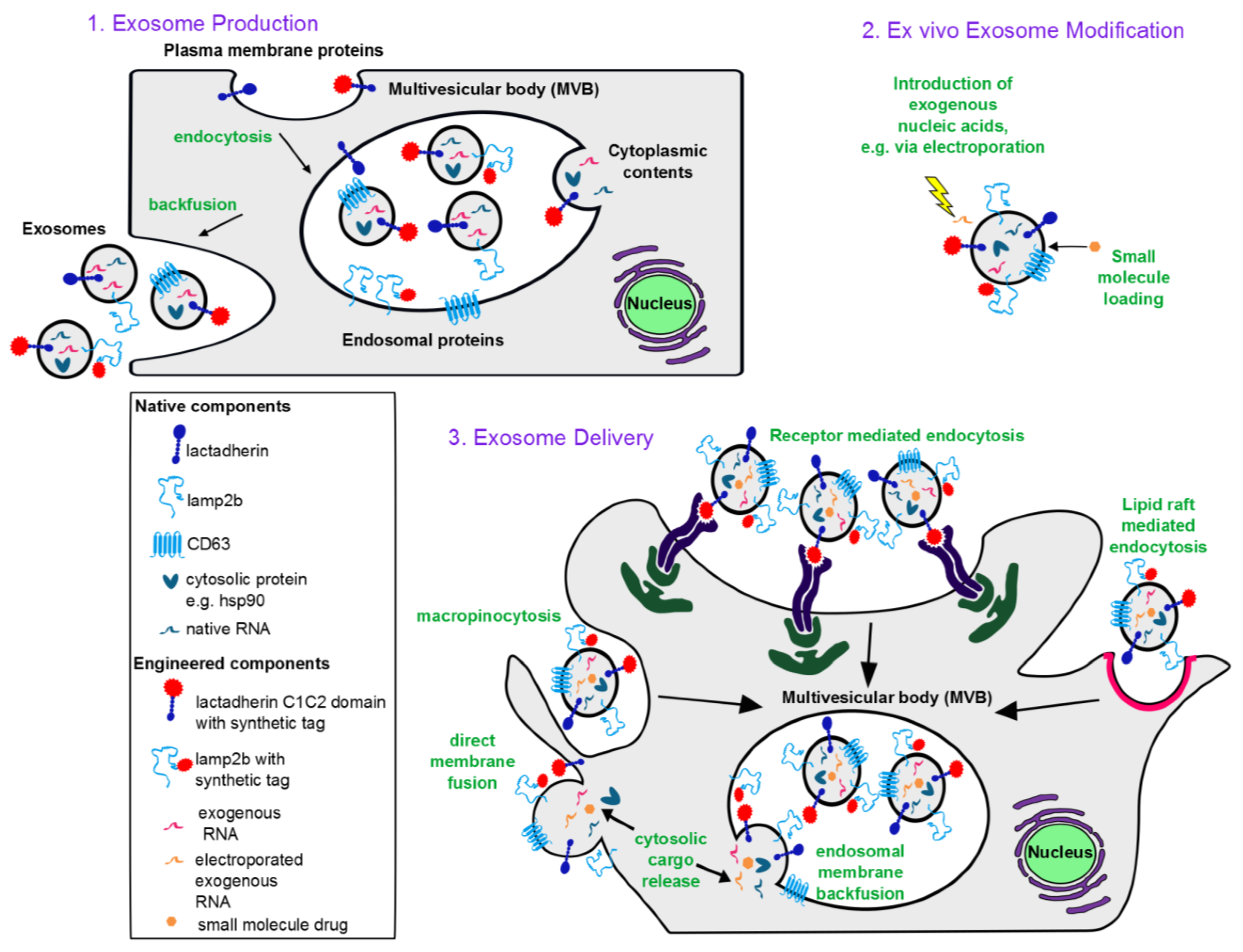
1.1. Exosome Biogenesis
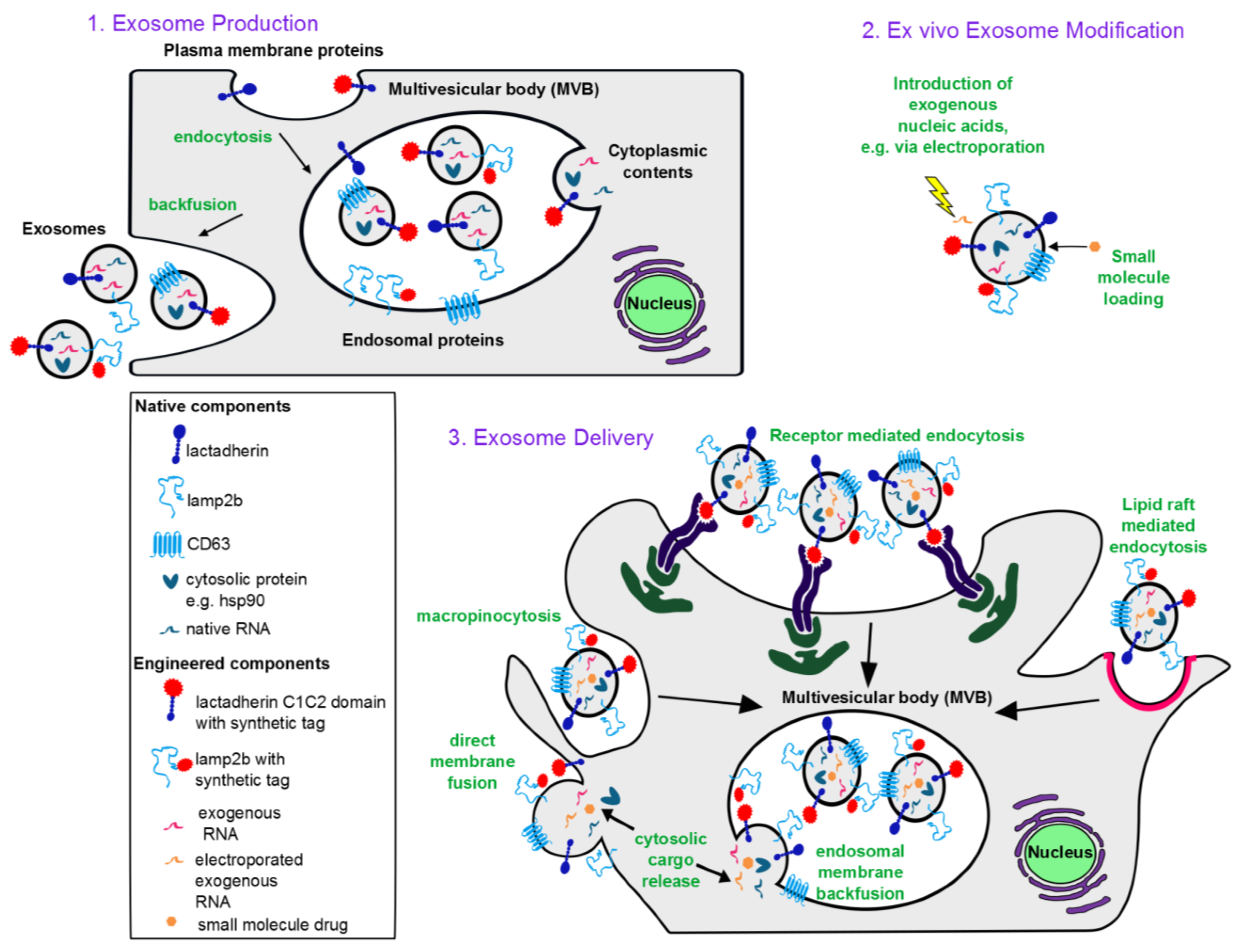
1.2. Characteristic Exosome Contents

{kind=link}
{kind=link}
| Exosome source | Recipient cell type | Cargo delivered | Functional consequences | Ref. |
|---|---|---|---|---|
| Immunosuppressive effects | ||||
| EBV transformed human B cells | Human Monocyte-derived DC | Viral miRNA | Down-regulate immune response to virus | [21] |
| Serum of pregnant human patients | Human Jurkat T cells | FasL | Suppress CD3ζ signaling and IL-2 production | [22] |
| Murine BMDC overexpressing IL-10 | Murine T cells | Antigen, presented on MHCII | Suppress T cell proliferation | [23] |
| Immunostimulatory effects | ||||
| Murine BMDC | Murine CD8+ and CD4+ T cells (in vitro and in vivo) | Antigen, presented on MHC | Induce T cell proliferation | [24,25] |
| CD28 stimulated human CD3+ T cells | Unstimulated human CD3+ T cells | Unidentified | T cell activation, induction of proliferation and cytokine production when co-delivered with IL-2 | [26] |
| Murine BMDC | Murine BMDC (allogeneic) | Antigen | Transfer of foreign antigen, followed by foreign antigen presentation to and activation of T cells | [14] |
| Therapeutic effects | ||||
| Human H9 CD4+ T cells | Human Jurkat T cells, Human PMBC | APOBEC3 protein (HIV replication inhibitor) | Reduce HIV replication | [27] |
| Human Endothelial cells | Human Aortic Smooth Muscle Cells | miR-143, miR-145 | Reduce atherosclerotic lesions | [28] |
| Murine MSC | Murine Primary Neurons | miR-133b | Neurite outgrowth after injury | [29] |
| Pathogenic effects | ||||
| Human B cell lymphoma cell lines | None | Bind and sequester rituximab (antibody used in B cell lymphoma immunotherapy) | [30] | |
| Human CSF | None | Phosphorylated tau | Transport of neurotoxic protein in Alzheimer’s disease | [31] |
| Human PMBC derived DC incubated with HIV | Jurkat T cell line expressing CCR5 | HIV viral particles | Delivery of functional HIV viral particles encapsulated in exosomes, leading to HIV infection of recipient cells | [32] |
2. Opportunities and Challenges in Harnessing Exosomes for Therapeutic Applications
2.1. Therapeutically Attractive Exosome Properties
2.1.1. Intrinsic Therapeutic Activity
2.1.2. Immunological Compatibility
2.1.3. Cargo Versatility
2.1.4. Experience in Clinical Trials
2.2. Challenges for Realizing Exosome-Mediated Therapeutics
2.2.1. Exosome Isolation and Purification
| Isolation method | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Differential centrifugation | Potentially high yields Potentially sterile | Time-consuming Subject to operator-based variability | [55,57] |
| HPLC + centrifugation | High throughput | Low yields | [56] |
| Affinity beads | High throughput Fewer steps than centrifugation methods | Selection of exosome population subset Difficulty in completely removing antibody from sample | [57] |
| Polymer-based precipitation | Potentially high yields Fewer steps than centrifugation methods | No method for removing polymer from exosome sample | [56] |
| Filtration + centrifugation | Potentially high yields Sterile | Time-consuming Subject to operator-based variability | [57] |
2.2.2. Selecting and Culturing Exosome-Producing Cells
3. Engineering Exosomes as Therapeutic Delivery Vehicles
3.1. Incorporating Therapeutic Molecular Cargo into Exosomes
3.1.1. Protein Cargo
3.1.2. RNA Cargo
3.2. Targeting Exosome Delivery
3.2.1. Targeting Exosomes to Specific Recipient Cells
3.2.2. Targeting Exosome-Mediated Delivery to Specific Subcellular Locations
4. Reverse Engineering Exosomes: Designing Exosomal Features into Synthetic Vectors
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Trams, E.G.; Lauter, C.J.; Salem, N., Jr.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta 1981, 645, 63–70. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Skokos, D.; le Panse, S.; Villa, I.; Rousselle, J.C.; Peronet, R.; David, B.; Namane, A.; Mecheri, S. Mast cell-dependent b and t lymphocyte activation is mediated by the secretion of immunologically active exosomes. J. Immunol. 2001, 166, 868–876. [Google Scholar]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Harding, C.V.; Heuser, J.E.; Stahl, P.D. Exosomes: Looking back three decades and into the future. J. Cell Biol. 2013, 200, 367–371. [Google Scholar] [CrossRef]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport rna and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional micrornas between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through met. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Cai, Z.; Yang, F.; Yu, L.; Yu, Z.; Jiang, L.; Wang, Q.; Yang, Y.; Wang, L.; Cao, X.; Wang, J. Activated t cell exosomes promote tumor invasion via fas signaling pathway. J. Immunol. 2012, 188, 5954–5961. [Google Scholar] [CrossRef]
- Sheldon, H.; Heikamp, E.; Turley, H.; Dragovic, R.; Thomas, P.; Oon, C.E.; Leek, R.; Edelmann, M.; Kessler, B.; Sainson, R.C.; et al. New mechanism for notch signaling to endothelium at a distance by delta-like 4 incorporation into exosomes. Blood 2010, 116, 2385–2394. [Google Scholar] [CrossRef]
- Mathivanan, S.; Fahner, C.J.; Reid, G.E.; Simpson, R.J. Exocarta 2012: Database of exosomal proteins, rna and lipids. Nucleic Acids Res. 2012, 40, D1241–D1244. [Google Scholar] [CrossRef]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Trau, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar]
- Kharaziha, P.; Ceder, S.; Li, Q.; Panaretakis, T. Tumor cell-derived exosomes: A message in a bottle. Biochim. Biophys. Acta 2012, 1826, 103–111. [Google Scholar]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral mirnas via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef]
- Taylor, D.D.; Akyol, S.; Gercel-Taylor, C. Pregnancy-associated exosomes and their modulation of t cell signaling. J. Immunol. 2006, 176, 1534–1542. [Google Scholar]
- Kim, S.H.; Lechman, E.R.; Bianco, N.; Menon, R.; Keravala, A.; Nash, J.; Mi, Z.; Watkins, S.C.; Gambotto, A.; Robbins, P.D. Exosomes derived from il-10-treated dendritic cells can suppress inflammation and collagen-induced arthritis. J. Immunol. 2005, 174, 6440–6448. [Google Scholar]
- Qazi, K.R.; Gehrmann, U.; Domange Jordo, E.; Karlsson, M.C.; Gabrielsson, S. Antigen-loaded exosomes alone induce th1-type memory through a b-cell-dependent mechanism. Blood 2009, 113, 2673–2683. [Google Scholar] [CrossRef]
- Luketic, L.; Delanghe, J.; Sobol, P.T.; Yang, P.; Frotten, E.; Mossman, K.L.; Gauldie, J.; Bramson, J.; Wan, Y. Antigen presentation by exosomes released from peptide-pulsed dendritic cells is not suppressed by the presence of active ctl. J. Immunol. 2007, 179, 5024–5032. [Google Scholar]
- Wahlgren, J.; Karlson Tde, L.; Glader, P.; Telemo, E.; Valadi, H. Activated human t cells secrete exosomes that participate in il-2 mediated immune response signaling. PloS One 2012, 7, e49723. [Google Scholar]
- Khatua, A.K.; Taylor, H.E.; Hildreth, J.E.; Popik, W. Exosomes packaging apobec3g confer human immunodeficiency virus resistance to recipient cells. J. Virol. 2009, 83, 512–521. [Google Scholar] [CrossRef]
- Hergenreider, E.; Heydt, S.; Treguer, K.; Boettger, T.; Horrevoets, A.J.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through mirnas. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-mediated transfer of mir-133b from multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012, 30, 1556–1564. [Google Scholar] [CrossRef]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal evasion of humoral immunotherapy in aggressive b-cell lymphoma modulated by atp-binding cassette transporter a3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef]
- Wiley, R.D.; Gummuluru, S. Immature dendritic cell-derived exosomes can mediate hiv-1 trans infection. Proc. Natl. Acad. Sci. USA 2006, 103, 738–743. [Google Scholar] [CrossRef]
- Colino, J.; Snapper, C.M. Exosomes from bone marrow dendritic cells pulsed with diphtheria toxoid preferentially induce type 1 antigen-specific igg responses in naive recipients in the absence of free antigen. J. Immunol. 2006, 177, 3757–3762. [Google Scholar]
- Schnitzer, J.K.; Berzel, S.; Fajardo-Moser, M.; Remer, K.A.; Moll, H. Fragments of antigen-loaded dendritic cells (dc) and dc-derived exosomes induce protective immunity against leishmania major. Vaccine 2010, 28, 5785–5793. [Google Scholar] [CrossRef]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase atp levels, decrease oxidative stress and activate pi3k/akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef]
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012, 126, 2601–2611. [Google Scholar] [CrossRef]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human nk cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef]
- Seow, Y.; Wood, M.J. Biological gene delivery vehicles: Beyond viral vectors. Mol. Ther. 2009, 17, 767–777. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of sirna to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to egfr deliver antitumor microrna to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef]
- Gould, S.J.; Booth, A.M.; Hildreth, J.E. The trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 10592–10597. [Google Scholar] [CrossRef]
- Monleon, I.; Martinez-Lorenzo, M.J.; Monteagudo, L.; Lasierra, P.; Taules, M.; Iturralde, M.; Pineiro, A.; Larrad, L.; Alava, M.A.; Naval, J.; et al. Differential secretion of fas ligand- or apo2 ligand/tnf-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human t cells. J. Immunol. 2001, 167, 6736–6744. [Google Scholar]
- Xie, Y.; Zhang, H.; Li, W.; Deng, Y.; Munegowda, M.A.; Chibbar, R.; Qureshi, M.; Xiang, J. Dendritic cells recruit t cell exosomes via exosomal lfa-1 leading to inhibition of cd8+ ctl responses through downregulation of peptide/mhc class i and fas ligand-mediated cytotoxicity. J. Immunol. 2010, 185, 5268–5278. [Google Scholar] [CrossRef]
- Li, X.; Li, J.J.; Yang, J.Y.; Wang, D.S.; Zhao, W.; Song, W.J.; Li, W.M.; Wang, J.F.; Han, W.; Zhang, Z.C.; et al. Tolerance induction by exosomes from immature dendritic cells and rapamycin in a mouse cardiac allograft model. PloS One 2012, 7, e44045. [Google Scholar] [CrossRef]
- Kim, S.H.; Bianco, N.; Menon, R.; Lechman, E.R.; Shufesky, W.J.; Morelli, A.E.; Robbins, P.D. Exosomes derived from genetically modified dc expressing fasl are anti-inflammatory and immunosuppressive. Mol. Ther. 2006, 13, 289–300. [Google Scholar] [CrossRef]
- Kim, S.H.; Bianco, N.R.; Shufesky, W.J.; Morelli, A.E.; Robbins, P.D. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express il-4. J. Immunol. 2007, 179, 2242–2249. [Google Scholar]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and glioblastoma cells release exosomes carrying mtdna. J. Neural Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef]
- Waldenstrom, A.; Genneback, N.; Hellman, U.; Ronquist, G. Cardiomyocyte microvesicles contain DNA/rna and convey biological messages to target cells. PloS One 2012, 7, e34653. [Google Scholar]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Takeshita, F.; Matsuki, Y.; Ochiya, T. Secretory mechanisms and intercellular transfer of micrornas in living cells. J. Biol. Chem. 2010, 285, 17442–17452. [Google Scholar] [CrossRef]
- Mignot, G.; Roux, S.; Thery, C.; Segura, E.; Zitvogel, L. Prospects for exosomes in immunotherapy of cancer. J. Cell. Mol. Med. 2006, 10, 376–388. [Google Scholar] [CrossRef]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (dc) derived-exosomes: Results of thefirst phase i clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase i study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase i clinical trial of autologous ascites-derived exosomes combined with gm-csf for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006. [Google Scholar]
- Taylor, D.D.; Zacharias, W.; Gercel-Taylor, C. Exosome isolation for proteomic analyses and rna profiling. Methods Mol. Biol. 2011, 728, 235–246. [Google Scholar] [CrossRef]
- Lamparski, H.G.; Metha-Damani, A.; Yao, J.Y.; Patel, S.; Hsu, D.H.; Ruegg, C.; Le Pecq, J.B. Production and characterization of clinical grade exosomes derived from dendritic cells. J. Immunol. Methods 2002, 270, 211–226. [Google Scholar] [CrossRef]
- Yeo, R.W.; Lai, R.C.; Zhang, B.; Tan, S.S.; Yin, Y.; Teh, B.J.; Lim, S.K. Mesenchymal stem cell: An efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 336–341. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, L.; Li, Y.; Zhang, X.; Gu, J.; Yan, Y.; Xu, X.; Wang, M.; Qian, H.; Xu, W. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012, 315, 28–37. [Google Scholar] [CrossRef]
- Shen, B.; Wu, N.; Yang, J.M.; Gould, S.J. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J. Biol. Chem. 2011, 286, 14383–14395. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Akao, Y.; Iio, A.; Itoh, T.; Noguchi, S.; Itoh, Y.; Ohtsuki, Y.; Naoe, T. Microvesicle-mediated rna molecule delivery system using monocytes/macrophages. Mol. Ther. 2011, 19, 395–399. [Google Scholar] [CrossRef]
- Zeelenberg, I.S.; Ostrowski, M.; Krumeich, S.; Bobrie, A.; Jancic, C.; Boissonnas, A.; Delcayre, A.; Le Pecq, J.B.; Combadiere, B.; Amigorena, S.; et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008, 68, 1228–1235. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Wei, J.; Glass, O.K.; Guo, H.; Lei, G.; Yang, X.Y.; Osada, T.; Hobeika, A.; Delcayre, A.; Le Pecq, J.B.; et al. Increasing vaccine potency through exosome antigen targeting. Vaccine 2011, 29, 9361–9367. [Google Scholar] [CrossRef]
- Wahlgren, J.; De, L.K.T.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering rna to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Lee, Y.; Lakhal-Littleton, S.; Li, J.; Seow, Y.; Gardiner, C.; Alvarez-Erviti, L.; Sargent, I.L.; Wood, M.J. Exosome-mediated delivery of sirna in vitro and in vivo. Nat. Protoc. 2012, 7, 2112–2126. [Google Scholar] [CrossRef]
- Rechavi, O.; Erlich, Y.; Amram, H.; Flomenblit, L.; Karginov, F.V.; Goldstein, I.; Hannon, G.J.; Kloog, Y. Cell contact-dependent acquisition of cellular and viral nonautonomously encoded small rnas. Genes Dev. 2009, 23, 1971–1979. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Hagiwara, K.; Takeshita, F.; Ochiya, T. Competitive interactions of cancer cells and normal cells via secretory micrornas. J. Biol. Chem. 2012, 287, 1397–1405. [Google Scholar]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Strobel, T.; Breakefield, X.O.; Saydam, O. Genetically engineered microvesicles carrying suicide mrna/protein inhibit schwannoma tumor growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef]
- Bolukbasi, M.F.; Mizrak, A.; Ozdener, G.B.; Madlener, S.; Strobel, T.; Erkan, E.P.; Fan, J.B.; Breakefield, X.O.; Saydam, O. Mir-1289 and “zipcode”-like sequence enrich mrnas in microvesicles. Molecular Ther. Nucleic Acids 2012, 1, e10. [Google Scholar] [CrossRef]
- Iguchi, H.; Kosaka, N.; Ochiya, T. Secretory micrornas as a versatile communication tool. Commun. Integr. Biol. 2010, 3, 478–481. [Google Scholar] [CrossRef]
- Ruiss, R.; Jochum, S.; Mocikat, R.; Hammerschmidt, W.; Zeidler, R. Ebv-gp350 confers b-cell tropism to tailored exosomes and is a neo-antigen in normal and malignant b cells--a new option for the treatment of b-cll. PloS One 2011, 6, e25294. [Google Scholar]
- Temchura, V.V.; Tenbusch, M.; Nchinda, G.; Nabi, G.; Tippler, B.; Zelenyuk, M.; Wildner, O.; Uberla, K.; Kuate, S. Enhancement of immunostimulatory properties of exosomal vaccines by incorporation of fusion-competent g protein of vesicular stomatitis virus. Vaccine 2008, 26, 3662–3672. [Google Scholar] [CrossRef]
- Pasquetto, M.V.; Vecchia, L.; Covini, D.; Digilio, R.; Scotti, C. Targeted drug delivery using immunoconjugates: Principles and applications. J. Immun. 2011, 34, 611–628. [Google Scholar] [CrossRef]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108. [Google Scholar] [CrossRef]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef]
- Lee, Y.S.; Pressman, S.; Andress, A.P.; Kim, K.; White, J.L.; Cassidy, J.J.; Li, X.; Lubell, K.; Lim do, H.; Cho, I.S.; et al. Silencing by small rnas is linked to endosomal trafficking. Nat. Cell Biol. 2009, 11, 1150–1156. [Google Scholar] [CrossRef]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of mirna effector complexes and modulate mirna activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef]
- Barres, C.; Blanc, L.; Bette-Bobillo, P.; Andre, S.; Mamoun, R.; Gabius, H.J.; Vidal, M. Galectin-5is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood 2010, 115, 696–705. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental ph is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Rajendran, L.; Knolker, H.J.; Simons, K. Subcellular targeting strategies for drug design and delivery. Nature Rev. Drug Discov. 2010, 9, 29–42. [Google Scholar] [CrossRef]
- Landesman-Milo, D.; Peer, D. Altering the immune response with lipid-based nanoparticles. J. Control. Release 2012, 161, 600–608. [Google Scholar] [CrossRef]
- Kooijmans, S.A.; Vader, P.; van Dommelen, S.M.; van Solinge, W.W.; Schiffelers, R.M. Exosome mimetics: A novel class of drug delivery systems. Int. J. Nanomed. 2012, 7, 1525–1541. [Google Scholar]
- Laulagnier, K.; Motta, C.; Hamdi, S.; Roy, S.; Fauvelle, F.; Pageaux, J.F.; Kobayashi, T.; Salles, J.P.; Perret, B.; Bonnerot, C.; et al. Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem. J. 2004, 380, 161–171. [Google Scholar] [CrossRef]
- Allen, T.M.; Austin, G.A.; Chonn, A.; Lin, L.; Lee, K.C. Uptake of liposomes by cultured mouse bone marrow macrophages: Influence of liposome composition and size. Biochim. Biophys. Acta 1991, 1061, 56–64. [Google Scholar]
- Clayton, A.; Harris, C.L.; Court, J.; Mason, M.D.; Morgan, B.P. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of cd55 and cd59. Eur. J. Immunol. 2003, 33, 522–531. [Google Scholar] [CrossRef]
- Szebeni, J.; Muggia, F.; Gabizon, A.; Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: Prediction and prevention. Adv. Drug Deliv. Rev. 2011, 63, 1020–1030. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Marcus, M.E.; Leonard, J.N. FedExosomes: Engineering Therapeutic Biological Nanoparticles that Truly Deliver. Pharmaceuticals 2013, 6, 659-680. https://doi.org/10.3390/ph6050659
Marcus ME, Leonard JN. FedExosomes: Engineering Therapeutic Biological Nanoparticles that Truly Deliver. Pharmaceuticals. 2013; 6(5):659-680. https://doi.org/10.3390/ph6050659
Chicago/Turabian StyleMarcus, Michelle E., and Joshua N. Leonard. 2013. "FedExosomes: Engineering Therapeutic Biological Nanoparticles that Truly Deliver" Pharmaceuticals 6, no. 5: 659-680. https://doi.org/10.3390/ph6050659
APA StyleMarcus, M. E., & Leonard, J. N. (2013). FedExosomes: Engineering Therapeutic Biological Nanoparticles that Truly Deliver. Pharmaceuticals, 6(5), 659-680. https://doi.org/10.3390/ph6050659