Evolution of Biologics Screening Technologies

Abstract
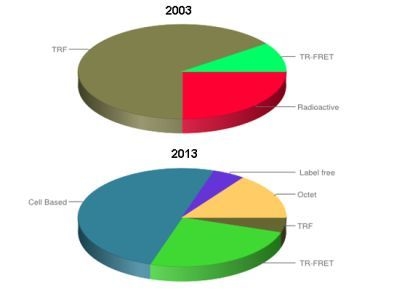
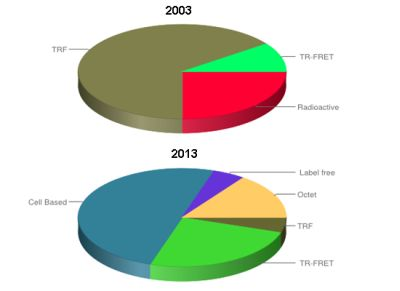
:
{kind=link}
{kind=link}
1. Introduction
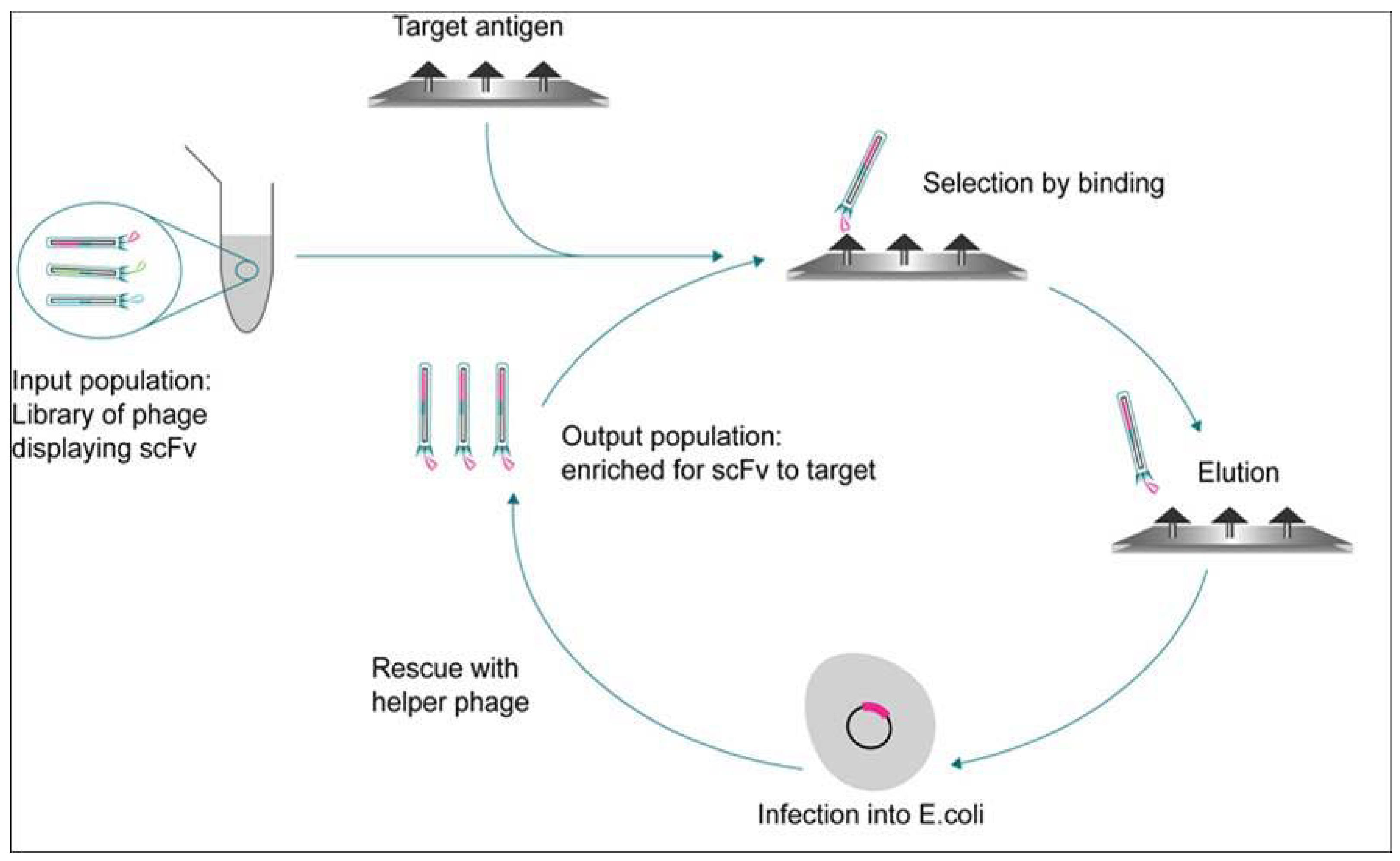
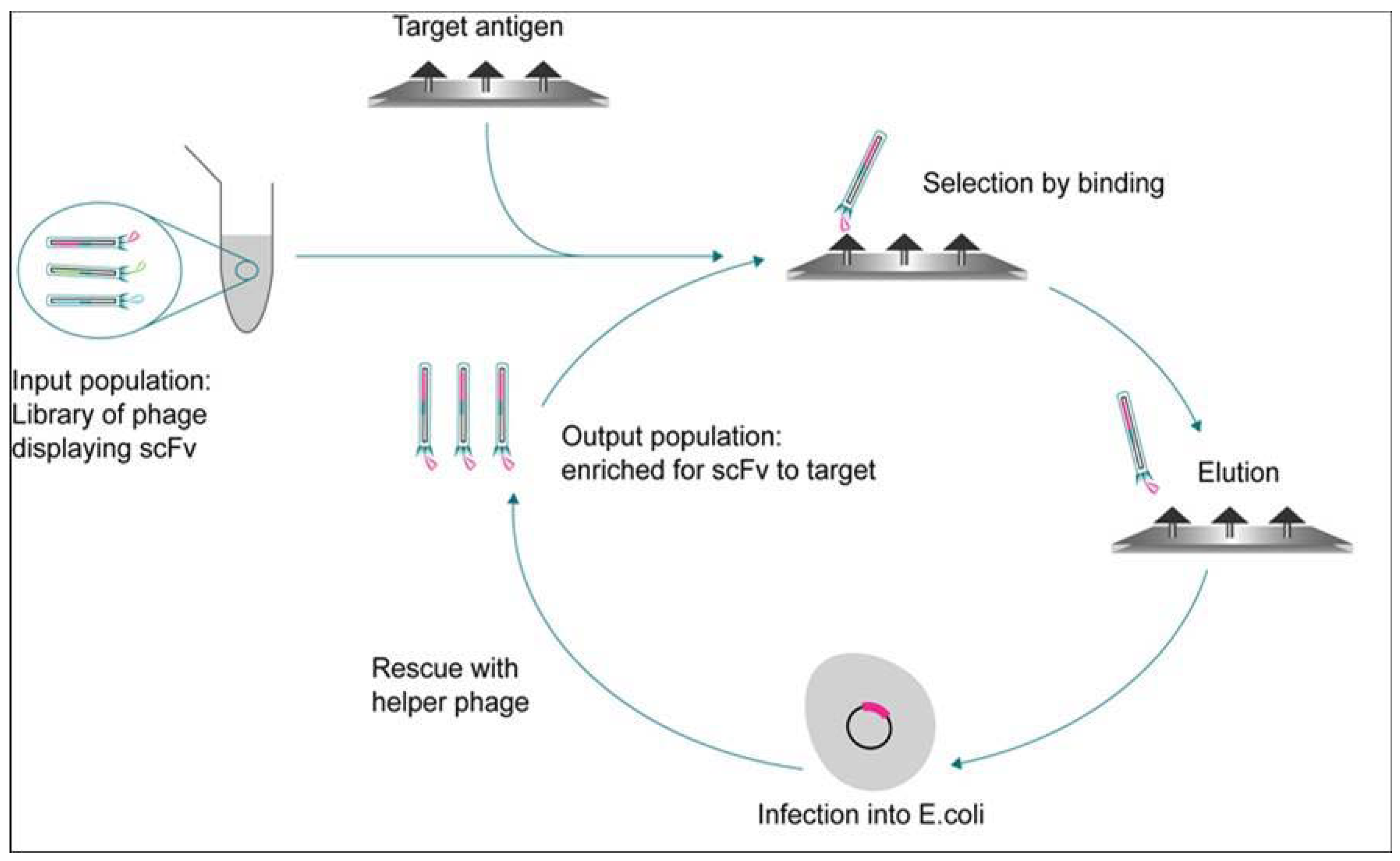
2. Heterogeneous Biochemical Assay Formats
3. Homogeneous Biochemical Assay Formats
4. Cell Based Binding Assay Platforms
5. Functional Screening Assays
6. Label Free Assays
7. High Throughput Affinity Screens
8. Conclusions
Conflict of Interest
References
- Lloyd, C.; Lowe, D.; Edwards, B.; Welsh, F.; Dilks, T.; Hardman, C.; Vaughan, T. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng. Des. Sel. 2009, 22, 159–168. [Google Scholar]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Lequin, R.M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef]
- Hemmila, I.; Dakubu, S.; Mukkala, V.M.; Siitari, H.; Lovgren, T. Europium as a label in time-resolved immunofluorometric assays. Anal. Biochem. 1984, 137, 335–343. [Google Scholar] [CrossRef]
- Pakard Instrument Company. Two methods of harvesting receptor binding assays. TopCount Topics 1992, TCA-010.
- Bosse, R.; Garlick, R.; Brown, B.; Menard, L. Development of nonseparation binding and functionalassays for G protein-coupled receptors for high throughput screening: Pharmacological characterization of the immobilized CCR5 receptor on flashplate. J. Biomol. Screening 1998, 3, 285–292. [Google Scholar] [CrossRef]
- Cook, N.D. Scintillation proximity assay: a versatile high-throughput screening technology. Drug Discov. Today 1996, 1, 287–294. [Google Scholar] [CrossRef]
- Kolb, J.M.; Yamanaka, G.; Manly, S.P. Use of a Novel Homogeneous Fluorescent Technology in High Throughput Screening. J. Biomol. Screening 1996, 1, 203–210. [Google Scholar] [CrossRef]
- Dickson, E.F.; Pollak, A.; Diamandis, E.P. Ultrasensitive bioanalytical assays using time-resolved fluorescence detection. Pharmacol. Ther. 1995, 66, 207–235. [Google Scholar] [CrossRef]
- Ullman, E.F.; Kirakossian, H.; Singh, S.; Wu, Z.P.; Irvin, B.R.; Pease, J.S.; Switchenko, A.C.; Irvine, J.D.; Dafforn, A.; Skold, C.N. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proc. Natl. Acad. Sci. USA. 1994, 91, 5426–5430. [Google Scholar] [CrossRef]
- Zwier, J.M.; Roux, T.; Cottet, M.; Durroux, T.; Douzon, S.; Bdioui, S.; Gregor, N.; Bourrier, M.; Oueslati, N.; Nicolas, L.; et al. A fluorescent ligand-binding alternative using tag-lite® technology. J. Biomol. Screening 2010, 15, 1248–1259. [Google Scholar] [CrossRef]
- Yan, H.; Gu, W.; Yang, J.; Bi, V.; Shen, Y.; Lee, E.; Winters, K.A.; Komorowski, R.; Zhang, C.; Patel, J.J.; et al. Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. Pharmacol. Ther. 2009, 329, 102–111. [Google Scholar]
- Miraglia, S.; Swartzman, E.E.; Mellentin-Michelotti, J.; Evangelista, L.; Smith, C.; Gunawan, I.I.; Lohman, K.; Goldberg, E.M.; Manian, B.; Yuan, P.M. Homogeneous cell- and bead-based assays for high throughput screening using fluorometric microvolume assay technology. J. Biomol. Screening 1999, 4, 193–204. [Google Scholar] [CrossRef]
- Black, C.B.; Duesing, T.B.; Trinkle, L.S.; Dunlay, T.R. Cell-Based screening using high-throughput flow cytometry. Assay Drug Dev. Technol. 2011, 9, 13–20. [Google Scholar] [CrossRef]
- Zhao, X.; Jones, A.; Olson, K.R.; Peng, K.; Wehrman, T.; Park, A.; Mallari, R.; Nebalasca, D.; Young, S.W.; Xiao, S.H. A homogeneous enzyme fragment complementation-based beta-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J. Biomol. Screening 2008, 13, 737–747. [Google Scholar] [CrossRef]
- Hanson, B.J.; Wetter, J.; Bercher, M.R.; Kopp, L.; Fuerstenau-Sharp, M.; Vedvik, K.L.; Zielinski, T.; Doucette, C.; Whitney, P.J.; Revankar, C. A homogeneous fluorescent live-cell assay for measuring 7-transmembrane receptor activity and agonist functional selectivity through beta-arrestin recruitment. J. Biomol. Screening 2009, 14, 798–810. [Google Scholar] [CrossRef]
- Asphahani, F.; Zhang, M. Cellular impedance biosensors for drug screening and toxin detection. Analyst 2007, 132, 835–841. [Google Scholar]
- Lee, P.H.; Gao, A.; van Staden, C.; Ly, J.; Salon, J.; Xu, A.; Fang, Y.; Verkleeren, R. Evaluation of dynamic mass redistribution technology for pharmacological studies of recombinant and endogenously expressed g protein-coupled receptors. Assay Drug Dev. Technol. 2008, 6, 83–94. [Google Scholar]
- Karlsson, R.; Michaelsson, A.; Mattsson, L. Kinetic analysis of monoclonal antibody-antigen interactions with a new biosensor based analytical system. J. Immunol. Methods 1991, 145, 229–240. [Google Scholar] [CrossRef]
- Abdiche, Y.; Malashock, D.; Pinkerton, A.; Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 2008, 377, 209–217. [Google Scholar] [CrossRef]
- Glaser, V. High throughput screening retools for the future. Available online: http://www.bio-itworld.com/BioIT_Article.aspx?id=86836/ (accessed on 23 April 2013).
- McCoy, P.J., Jr. High-content screening: getting more from less. Nat. Methods. 2011, 8, 390–391. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cariuk, P.; Gardener, M.J.; Vaughan, T.J. Evolution of Biologics Screening Technologies. Pharmaceuticals 2013, 6, 681-688. https://doi.org/10.3390/ph6050681
Cariuk P, Gardener MJ, Vaughan TJ. Evolution of Biologics Screening Technologies. Pharmaceuticals. 2013; 6(5):681-688. https://doi.org/10.3390/ph6050681
Chicago/Turabian StyleCariuk, Peter, Matthew J. Gardener, and Tristan J. Vaughan. 2013. "Evolution of Biologics Screening Technologies" Pharmaceuticals 6, no. 5: 681-688. https://doi.org/10.3390/ph6050681
APA StyleCariuk, P., Gardener, M. J., & Vaughan, T. J. (2013). Evolution of Biologics Screening Technologies. Pharmaceuticals, 6(5), 681-688. https://doi.org/10.3390/ph6050681