A Review of Therapeutic Aptamer Conjugates with Emphasis on New Approaches

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Strategies for Conferring Greater Stability and Pharmacokinetic Lifetimes to Aptamers
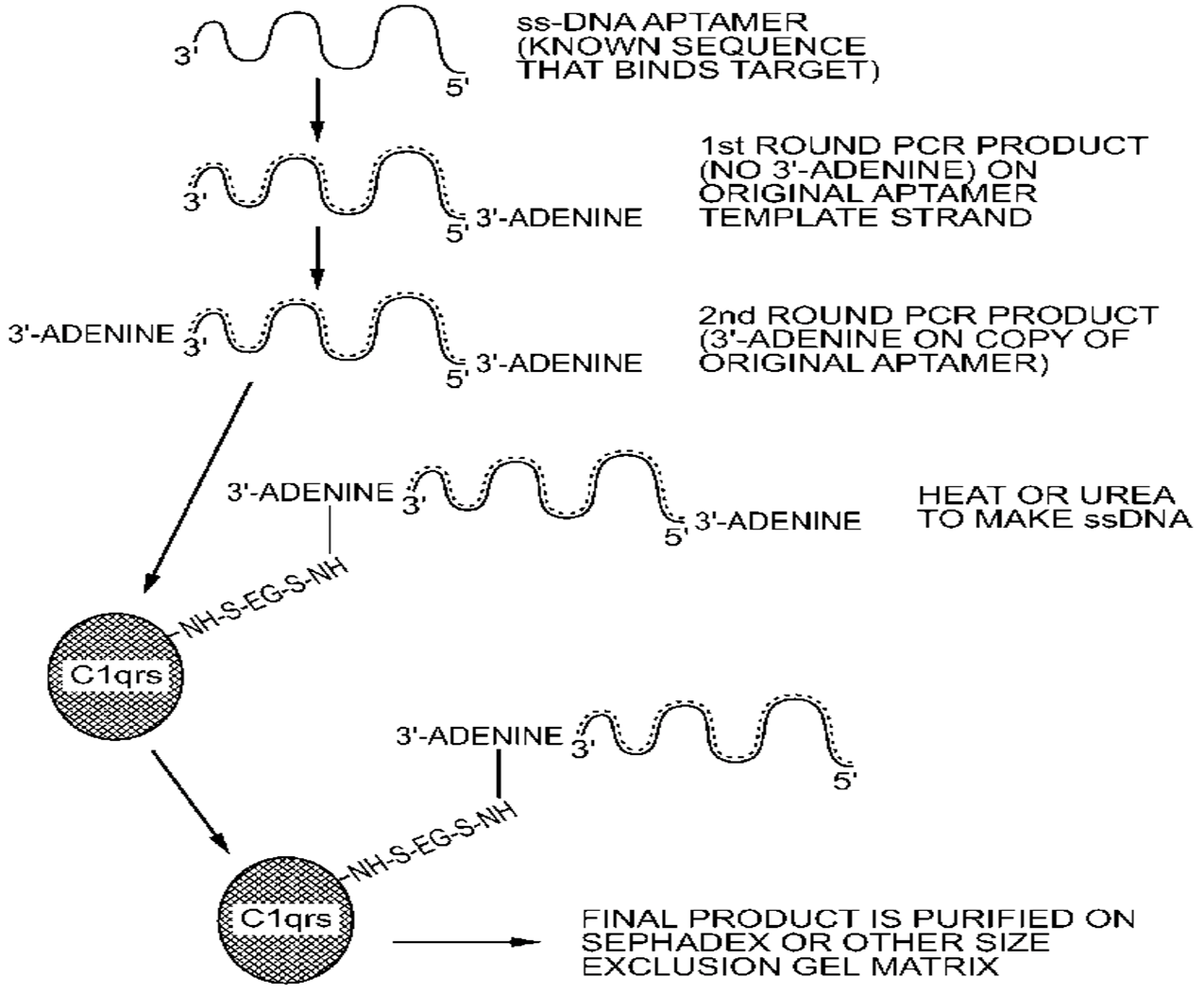
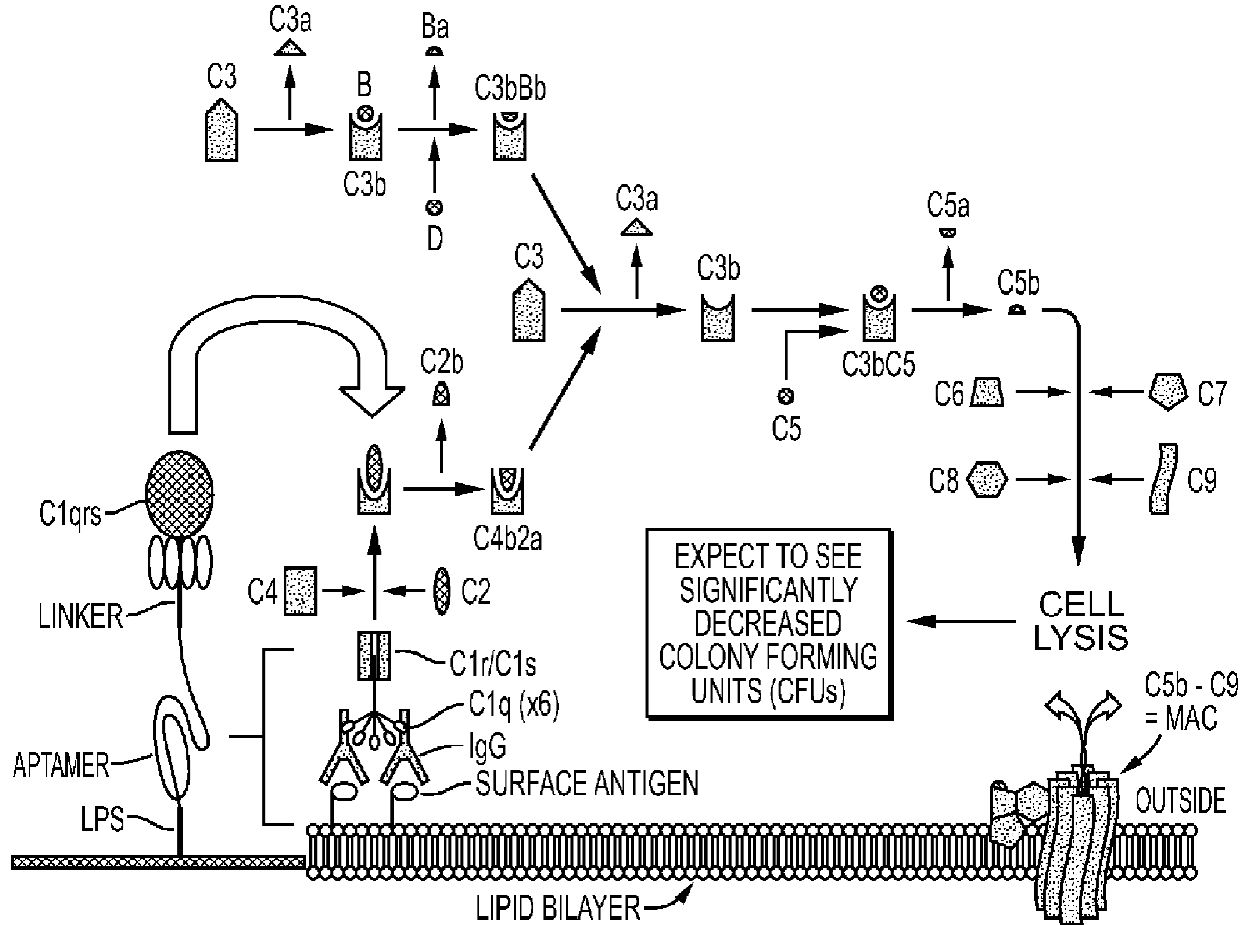

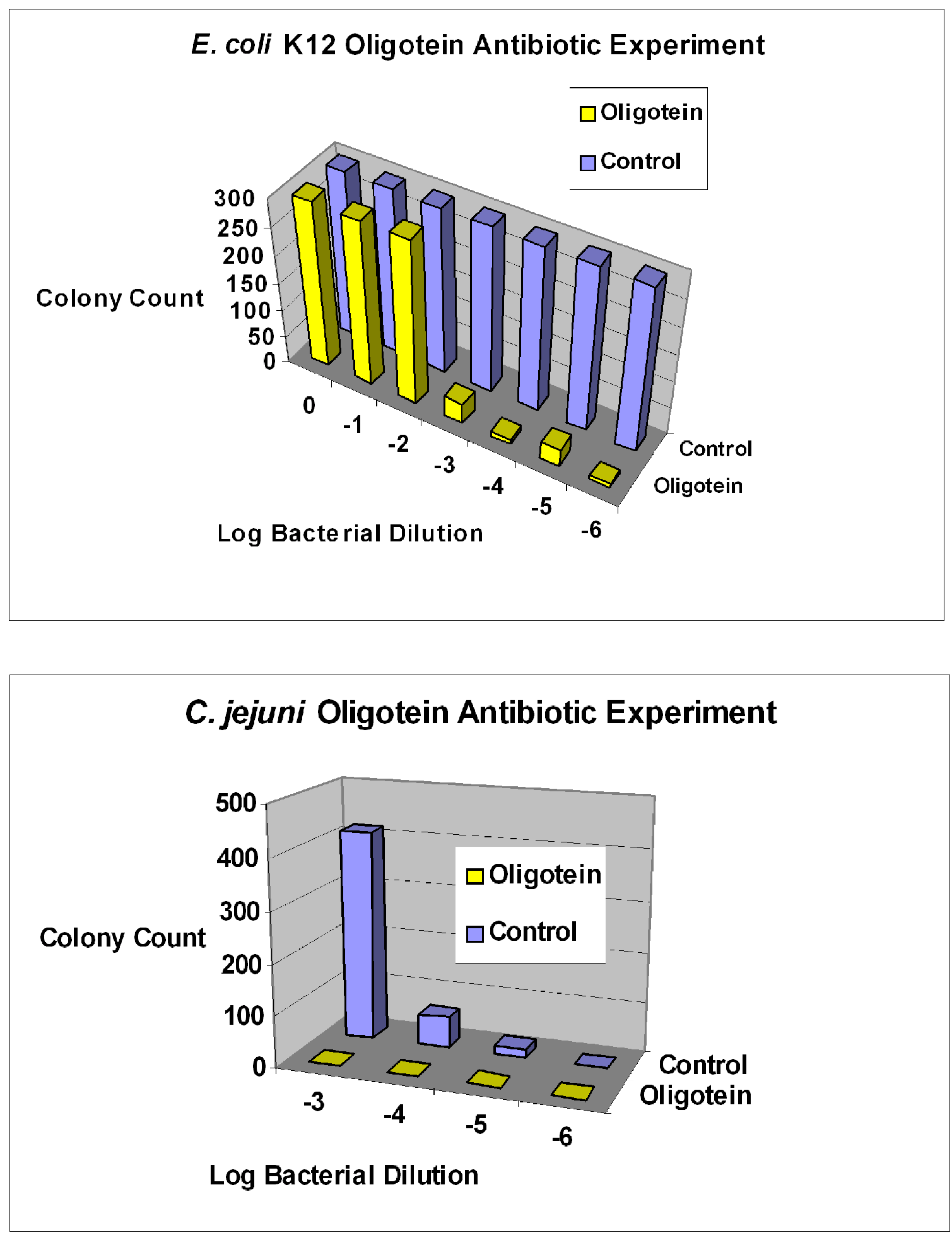
3. General and Novel Attachment Chemistries
4. Aptamer-Nanoparticle Conjugates
4.1. Aptamer-Magnetic Nanoparticle (MNP) Conjugates
4.2. Aptamer-Quantum Dot (QD) and Photosensitizer Conjugates
4.3. Aptamer-Carbon Nanotube (CNT) Conjugates
4.4. Aptamer-Gold Nanoparticle Conjugates
4.5 Aptamer-Chitosan Nanoparticle Conjugates
5. Aptamer-Drug and Toxin Conjugates
6. Other Aptamer Conjugates
7. Conclusions and Future Directions
Acknowledgments
Conflict of Interest
References
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Hedden, L.; O’Reilly, S.; Lohrisch, C.; Chia, S.; Speers, C.; Kovacic, L.; Taylor, S.; Peacock, S. Assessing the real-world cost-effectiveness of adjuvant trastuzumab in HER-2/neu positive breast cancer. Oncologist 2012, 17, 164–171. [Google Scholar] [CrossRef]
- Jeyakumar, A.; Younis, T. Trastuzumab for HER2-positive metastatic breast cancer: clinical and economic considerations. Clin. Med. Insights Oncol. 2012, 6, 179–187. [Google Scholar]
- Perez-Ellis, C.; Goncalves, A.; Jacquemier, J.; Marty, M.; Girre, V.; Roché, H.; Brain, E.; Moatti, J.P.; Viens, P.; le Corroller-Soriano, A.G. Cost-effectiveness analysis of trastuzumab (Herceptin) in HER2-overexpressed metastatic breast cancer. Am. J. Clin. Oncol. 2009, 32, 492–498. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef]
- Meng, L.; Yang, L.; Zhao, X.; Zhang, L.; Zhu, L.; Zhu, H.; Liu, C.; Tan, W. Targeted delivery of chemotherapy agents using a liver cancer-specific aptamer. PLoS One 2012, 7, e33434. [Google Scholar]
- Bolhassani, A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim. Biophys. Acta 2011, 1816, 232–246. [Google Scholar]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Lehto, T.; Kurrikoff, K.; Langel, U. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836. [Google Scholar] [CrossRef]
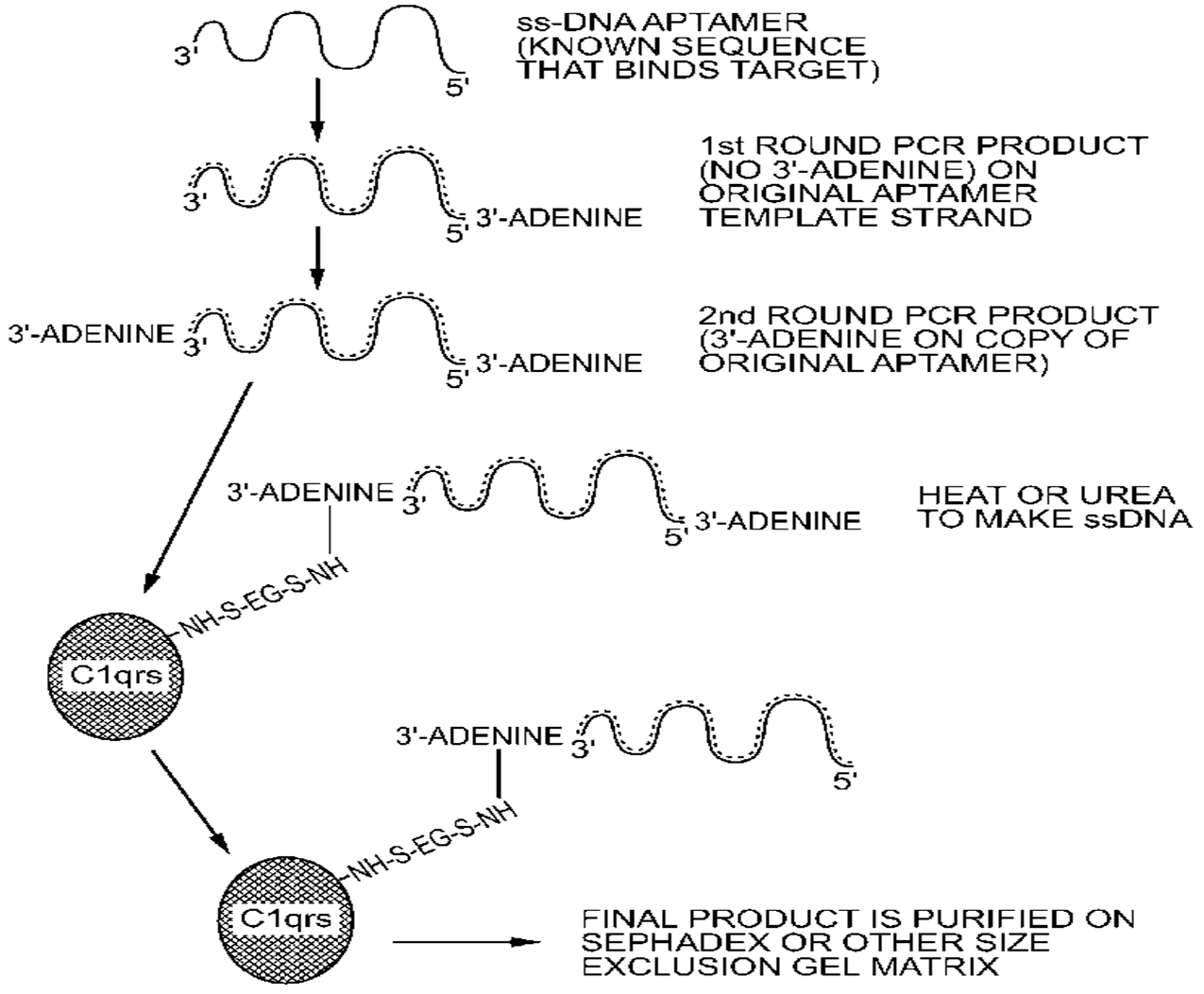
- Bruno, J.G.; Carrillo, M.P.; Crowell, R. Preliminary development of DNA aptamer-Fc conjugate opsonins. J. Biomed. Mat. Res. A 2009, 90, 1152–1161. [Google Scholar]
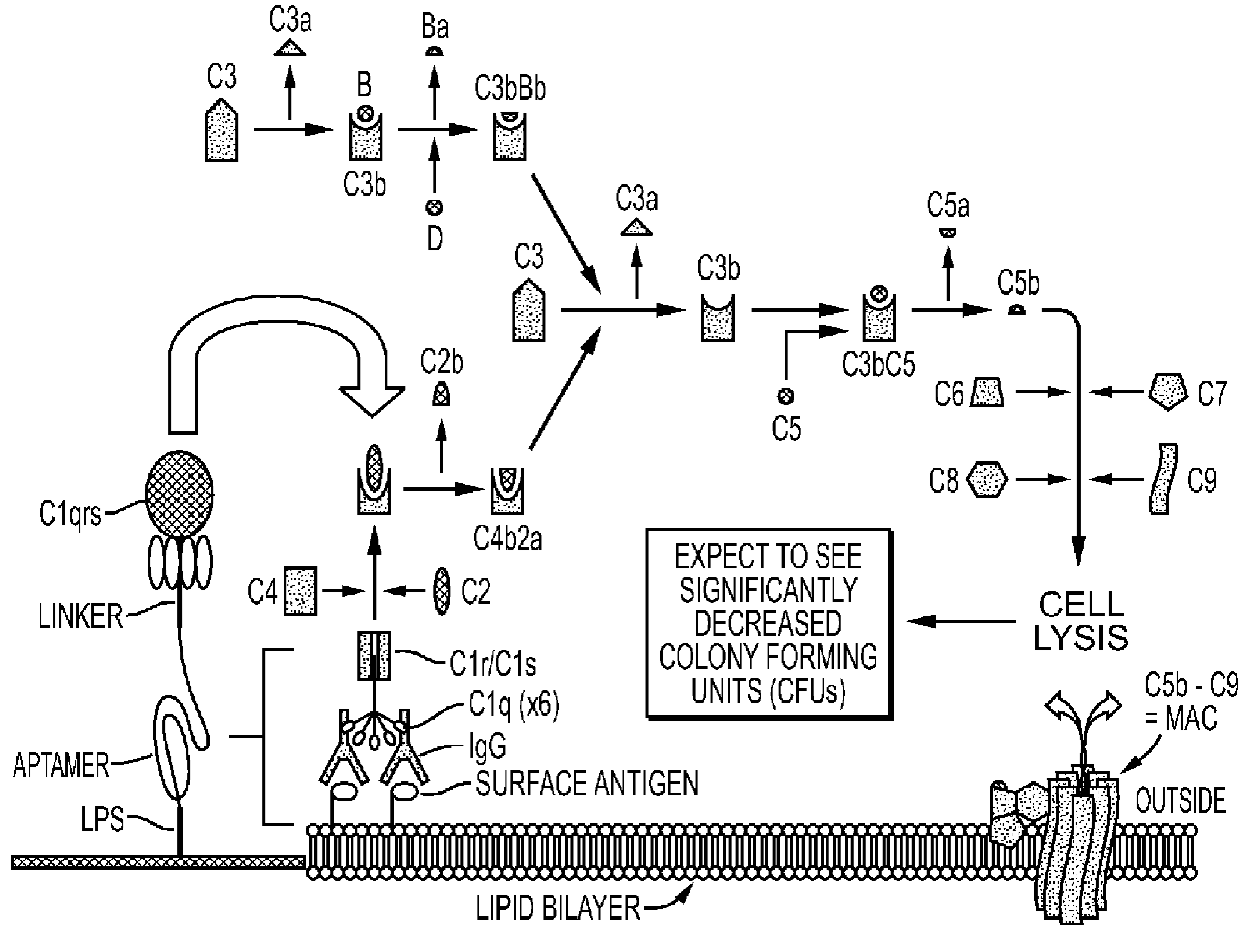
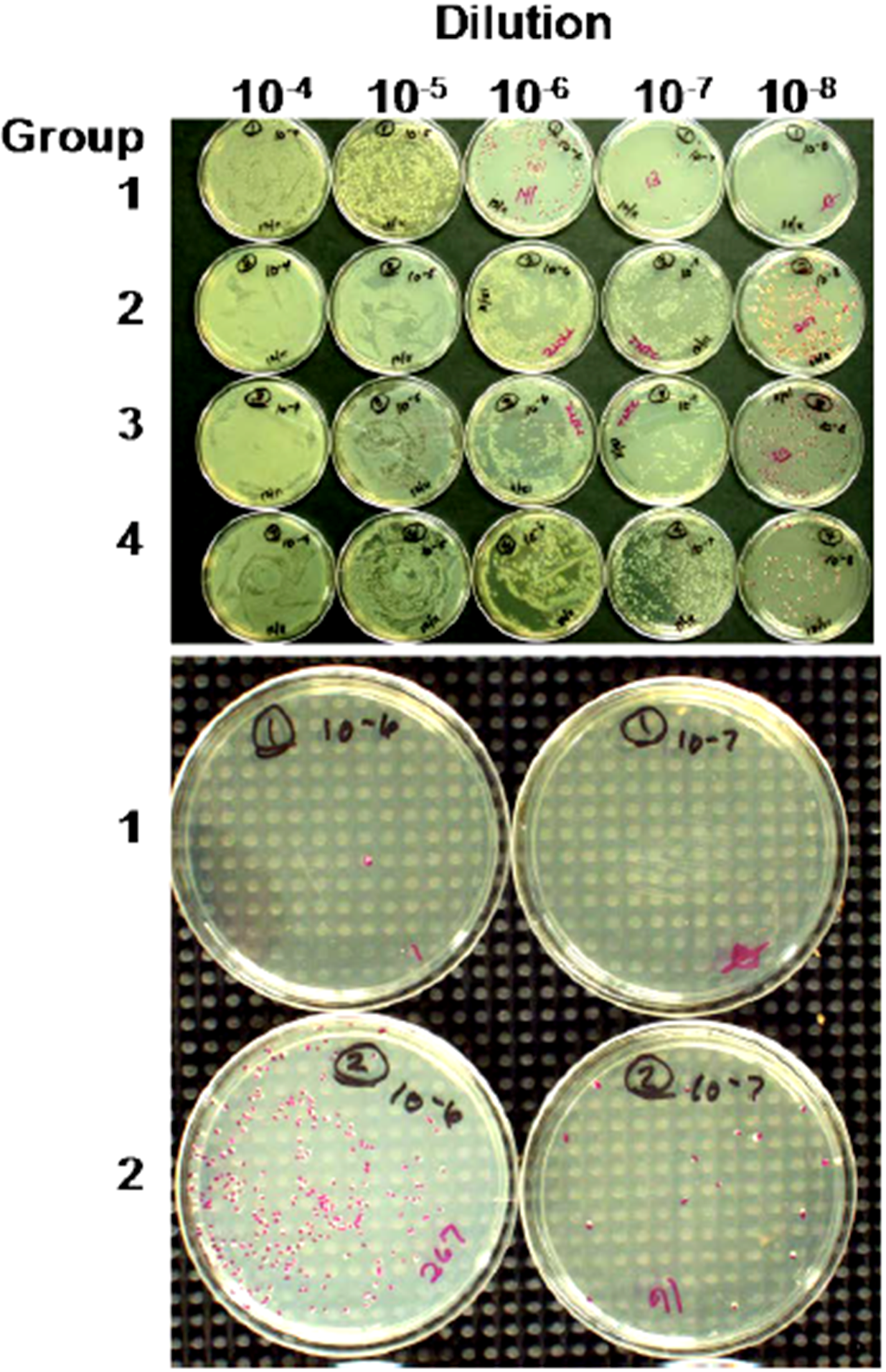
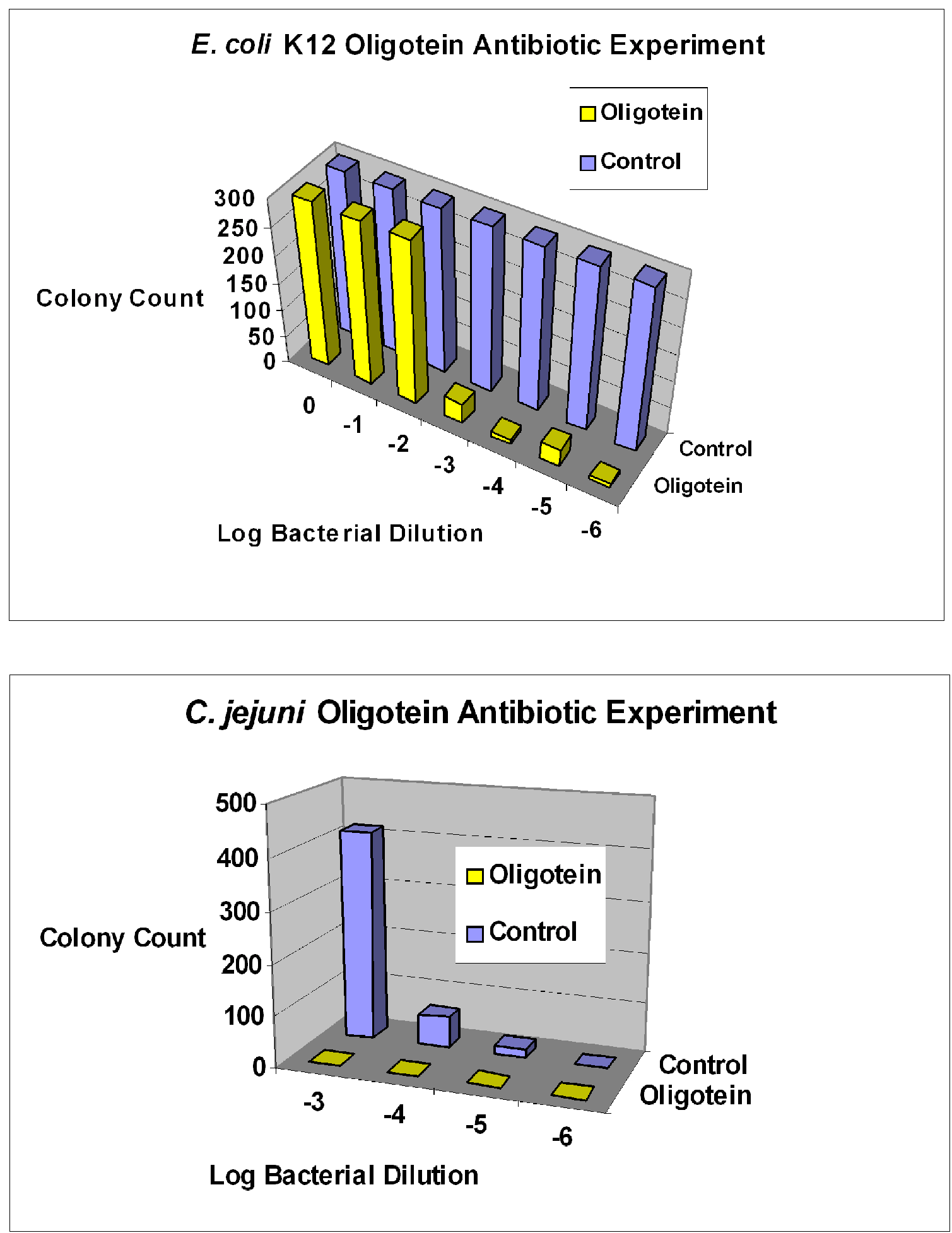
- Bruno, J.G.; Carrillo, M.P.; Phillips, T. In vitro antibacterial effects of anti-lipopolysaccharide DNA aptamer-C1qrs complexes. Folia Microbiol. 2008, 53, 295–302. [Google Scholar] [CrossRef]
- Bruno, J.G. Aptamer-biotin-streptavidin-C1q complexes can trigger the classical complement pathway to kill cancer cells. In Vitro Cell. Dev. Biol. 2010, 46, 107–113. [Google Scholar] [CrossRef]
- Bruno, J.G.; Miner, J.C. Therapeutic nucleic acid-3'—Conjugates. U.S. Patent Nos. 7,910,297, 8,318,920, and 8,389,710, 5 March 2013. [Google Scholar]
- Hakulinen, J.; Meri, S. Complement-mediated killing of microtumors in vitro. Am. J. Path. 1998, 153, 845–855. [Google Scholar]
- Stecker, J.R.; Savage, A.; Bruno, J.G.; Garcia, D.M.; Koke, J.R. Dynamics and visualization of MCF7 adenocarcinoma cell death by aptamer-C1q-mediated membrane attack. Nucleic Acid Ther. 2012, 22, 275–282. [Google Scholar]
- Chu, T.C.; Marks, J.W.; Lavery, L.A.; Faulkner, S.; Rosenblum, M.G.; Ellington, A.D.; Levy, M. Aptamer:toxin conjugates that specifically target prostate tumor cells. Cancer Res. 2006, 66, 5989–5992. [Google Scholar]
- Bagalkot, V.; Farokhzad, O.C.; Langer, R.; Jon, S. An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew. Chem. Int. Ed. Eng. 2006, 45, 8149–8152. [Google Scholar] [CrossRef]
- Huang, Y.F.; Shangguan, D.; Liu, H.; Phillips, J.A.; Zhang, X.; Chen, Y.; Tan, W. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem. 2009, 10, 862–868. [Google Scholar]
- Tan, W.; Wang, H.; Chen, Y.; Zhang, X.; Zhu, H.; Yang, C.; Yang, R.; Liu, C. Molecular aptamers for drug delivery. Trends Biotechnol. 2011, 29, 634–640. [Google Scholar] [CrossRef]
- Borbas, K.E.; Ferreira, C.S.M.; Perkins, A.; Bruce, J.I.; Missailidis, S. Design and synthesis of mono- and multimeric targeted radiopharmaceuticals based on novel cyclen ligands coupled to anti-MUC1 aptamers for the diagnostic imaging and targeted radiotherapy of cancer. Bioconj. Chem. 2007, 18, 1205–1212. [Google Scholar] [CrossRef]
- Da Pieve, C.D.; Perkins, A.C.; Missailidis, S. Anti-MUC1 aptamers: radiolabelling with (99m) Tc and biodistribution in MCF-7 tumour-bearing mice. Nucl. Med. Biol. 2009, 36, 703–710. [Google Scholar] [CrossRef]
- Bakalova, R.; Ohba, H.; Zhelev, Z.; Ishikawa, M.; Baba, Y. Quantum dots as photosensitizers? Nat. Biotechnol. 2004, 22, 1360–1361. [Google Scholar]
- Dwarakanath, S.; Bruno, J.G.; Athmaram, T.N.; Bali, G.; Vattem, D.; Rao, P. Antibody-quantum dot conjugates exhibit enhanced antibacterial effect vs. unconjugated quantum dots. Folia Microbiol. 2007, 52, 31–34. [Google Scholar] [CrossRef]
- Ferreira, C.S.; Cheung, M.C.; Missailidis, S.; Bisland, S.; Gariépy, J. Phototoxic aptamers selectively enter and kill epithelial cancer cells. Nucleic Acids Res. 2009, 37, 866–876. [Google Scholar] [CrossRef]
- Jin, T.; Sun, D.; Su, J.Y.; Zhang, H.; Sue, H.J. Antimicrobial efficacy of zinc oxide quantum dots against Listeria monocytogenes, Salmonella enteritidis, and Escherichia coli O157:H7. J. Food Sci. 2009, 74, 46–52. [Google Scholar]
- Samia, A.C.; Dayal, S.; Burda, C. Quantum dot-based energy transfer: perspectives and potential for applications in photodynamic therapy. Photochem. Photobiol. 2006, 82, 617–625. [Google Scholar] [CrossRef]
- Shi, L.; Hernandez, B.; Selke, M. Singlet oxygen generation from water-soluble quantum dot-organic dye nanocomposites. J. Am. Chem. Soc. 2006, 128, 6278–6279. [Google Scholar] [CrossRef]
- Yang, X.; Huang, J.; Wang, K.; Li, W.; Cui, L.; Li, X. Angiogenin-mediated photosensitizer-aptamer conjugate for photodynamic therapy. Chem. Med. Chem. 2011, 6, 1778–1780. [Google Scholar]
- Yang, X.; Liu, X.; Liu, Z.; Pu, F.; Ren, J.; Qu, X. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer-gated nanovehicles. Adv. Mater. 2012, 24, 2890–2895. [Google Scholar]
- Beqa, L.; Fan, Z.; Singh, A.K.; Senapati, D.; Ray, P.C. Gold nano-popcorn attached SWCNT hybrid nanomaterial for targeted diagnosis and photothermal therapy of human breast cancer cells. ACS Appl. Mater. Interfaces 2011, 3, 3316–3324. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar]
- Huang, Y.F.; Chang, H.T.; Tan, W. Cancer cell targeting using multiple aptamers conjugated on nanorods. Anal. Chem. 2008, 80, 567–572. [Google Scholar] [CrossRef]
- Huang, Y.F.; Sefah, K.; Bamrungsap, S.; Chang, H.T.; Tan, W. Selective photothermal therapy for mixed cancer cells using aptamer-conjugated nanorods. Langmuir 2008, 24, 11860–11865. [Google Scholar] [CrossRef]
- Li, L.L.; Yin, Q.; Cheng, J.; Lu, Y. Polyvalent mesoporous silica nanoparticle-aptamer bioconjugates target breast cancer cells. Adv. Health Mater. 2012, 1, 567–572. [Google Scholar]
- Liu, S.; Wei, L.; Hao, L.; Fang, N.; Wook Chang, M.; Xu, R.; Yang, Y.; Chen, Y. Sharper and faster “nano darts” kill more bacteria: A study of antibacterial activity of individually dispersed pristine single-walled carbon nanotube. ACS Nano 2009, 3, 3891–3902. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, G.; You, M.; Song, E.; Shukoor, M.I.; Zhang, K.; Altman, M.B.; Chen, Y.; Zhu, Z.; Huang, C.Z.; et al. Assembly of aptamer switch probes and photosensitizer on gold nanorods for targeted photothermal and photodynamic cancer therapy. ACS Nano 2012, 6, 5070–5077. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, X.; Ye, M.; Jiang, J.; Yang, R.; Fu, T.; Chen, Y.; Wang, K.; Liu, C.; Tan, W. Aptamer-conjugated nanomaterials and their applications. Adv. Drug Deliv. Rev. 2011, 63, 1361–1370. [Google Scholar] [CrossRef]
- Berezhnoy, A.; Brenneman, R.; Bajgelman, M.; Seales, D.; Gilboa, E. Thermal stability of siRNA modulates aptamer-conjugated siRNA inhibition. Mol. Ther. Nucl. Acids 2012, 1, e51. [Google Scholar] [CrossRef]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O.; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar]
- Neff, C.P.; Zhou, J.; Remling, L.; Kuruvilla, J.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Rossi, J.J.; Akkina, R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4 (+) T cell decline in humanized mice. Sci. Transl. Med. 2011, 3, 66ra6. [Google Scholar] [CrossRef]
- Zhou, J.; Bobbin, M.L.; Burnett, J.C.; Rossi, J.J. Current progress of RNA aptamer-based therapeutics. Front. Genet. 2012, 3, 1–14. [Google Scholar]
- Zhou, J.; Rossi, J.J. Aptamer-targeted cell-specific RNA interference. Silence 2010, 1, 4. [Google Scholar] [CrossRef]
- Bruno, J.G.; Crowell, R. Selective glutaraldehyde-mediated coupling of proteins to the 3' adenine terminus of Polymerase Chain Reaction products. J. Biomolec. Techn. 2008, 19, 177–183. [Google Scholar]
- Bruno, J.G.; Stecker, J.R.; Carrillo, M.P.; Phillips, T.; Savage, A.; Garcia, D.M.; Koke, J.R. Chapter 4: Novel aptamer-based therapeutic strategies. In Biomedical Applications of Aptamers; Bruno, J.G., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2013; pp. 55–72. [Google Scholar]
- Aarons, L.; Grennan, D.M.; Siddiqui, M. The binding of ibuprofen to plasma proteins. Eur. J. Clin. Pharmacol. 1983, 25, 815–818. [Google Scholar] [CrossRef]
- Cheruvallath, V.K.; Riley, C.M.; Narayanan, S.R.; Lindenbaum, S.; Perrin, J.H. A quantitative circular dichroic investigation of the binding of the enantiomers of ibuprofen and naproxen to human serum albumin. J. Pharm. Biomed. Anal. 1997, 15, 1719–1724. [Google Scholar] [CrossRef]
- Manoharan, M.; Rajeev, K.G.; Kesavan, V. Single-stranded and double-stranded oligonucleotides comprising a 2-arylpropyl moiety. U.S. Patent Application No. 12/604,897, filed, 23 October 2009. [Google Scholar]
- Stasiak, P.; Sznitowska, M.; Ehrhardt, C.; Luczyk-Juzwa, M.; Grieb, P. In vivo assessment of parenteral formulations of oligo (3-hydroxybutyric acid) conjugates with the model compound ibuprofen. AAPS Pharm. Sci. Tech. 2010, 11, 1636–1641. [Google Scholar] [CrossRef]
- Zion, T.C.; Lancaster, T.M. Polynucleotide aptamers-based cross-linked materials and uses thereof. U.S. Patent Application No. 13/145,531, 20 July 2011. [Google Scholar]
- Zsila, F.; Bikadi, Z.; Malik, D.; Hari, P.; Pechan, I.; Berces, A.; Hazai, E. Evaluation of drug-human serum albumin binding interactions with support vector machine aided online automated docking. Bioinformatics 2011, 27, 1806–1813. [Google Scholar]
- Wright, S.E. Immunotherapy of breast cancer. Expert Opin. Biol. Ther. 2012, 12, 479–490. [Google Scholar] [CrossRef]
- Wu, A.M.; Senter, P.D. Arming antibodies: prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar]
- Bruno, J.G.; Carrillo, M.P.; Richarte, A.M.; Phillips, T.; Andrews, C.; Lee, J.S. Development, screening, and analysis of a small DNA aptamer library potentially useful for diagnosis and passive immunity of arboviruses. BMC Res. Notes 2012, 5, 633. [Google Scholar] [CrossRef]
- Chen, F.; Zhou, J.; Huang, Y.H.; Huang, F.Y.; Liu, Q.; Fang, Z.; Yang, S.; Xiong, M.; Lin, Y.Y.; Tan, G.H. Function of ssDNA aptamer and aptamer pool against Mycobacterium tuberculosis in a mouse model. Mol. Med. Rep. 2013, 7, 669–773. [Google Scholar]
- Cheng, C.; Dong, J.; Yao, L.; Chen, A.; Jia, R.; Huan, L.; Guo, J.; Shu, Y.; Zhang, Z. Potent inhibition of human influenza H5N1 virus by oligonucleotides derived by SELEX. Biochem. Biophys. Res. Commun. 2008, 366, 670–674. [Google Scholar] [CrossRef]
- Mullis, K.B. Chemically programmable immunity. U.S. Patent Nos. 7,422,746, 7,645,743, 7,850,795, 8,236,321, and 8,263,082.
- Kiel, J.L.; Kiel, A.L. Chapter 1: Fundamental performance differences between antibodies and aptamers. In Biomedical Applications of Aptamers; Bruno, J.G., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2013; pp. 1–14. [Google Scholar]
- Fan, S.; Wu, F.; Martiniuk, F.; Hale, M.L.; Ellington, A.D.; Tchou-Wong, K.M. Protective effects of anti-ricin A-chain RNA aptamer against ricin toxicity. World J. Gastroenterol. 2008, 14, 6360–6365. [Google Scholar] [CrossRef]
- Lauridsen, L.H.; Veedu, R.N. Nucleic acid aptamers against biotoxins: A new paradigm toward the treatment and diagnostic approach. Nucleic Acid Ther. 2012, 22, 371–399. [Google Scholar]
- Dobler, R.K.; Maki, W.C. Mars health care delivery systems: Aptamers provide critical technology. In 12th NASA Symposium of VLSA Design, Coeur d'Alene, ID, USA, 4–5 October, 2005.
- Healy, J.M.; Lewis, S.D.; Kurz, M.; Boomer, R.M.; Thompson, K.M.; Wilson, C.; McCauley, T.G. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm. Res. 2004, 21, 2234–2246. [Google Scholar] [CrossRef]
- King, D.J.; Ventura, D.A.; Brasier, A.R.; Gorenstein, D.G. Novel combinatorial selection of phosphorothioate oligonucleotide aptamers. Biochemistry 1998, 37, 16489–16493. [Google Scholar] [CrossRef]
- Cload, S.T.; McCauley, T.G.; Keefe, A.D.; Healy, J.M.; Wilson, C. Chapter 17: Properties of therapeutic aptamers. In The Aptamer. Handbook; Klussmann, S., Ed.; Wiley-VCH, Verlag GmBH & Co.: Weinheim, Germany, 2006; pp. 363–416. [Google Scholar]
- Eulberg, D.; Klussmann, S. Spiegelmers: Biostable aptamers. Chembiochem 2003, 4, 979–983. [Google Scholar] [CrossRef]
- Eulberg, D.; Jarosch, F.; Vanhoff, S.; Klussmann, S. Chap 18: Spiegelmers for therapeutic applications—Use of chiral principles in evolutionary selection techniques. In The Aptamer. Handbook; Klussmann, S., Ed.; Wiley-VCH, Verlag GmBH & Co.: Weinheim, Germany, 2006; pp. 417–442. [Google Scholar]
- Boomer, R.M.; Lewis, S.D.; Healy, J.M.; Kurz, M.; Wilson, C.; McCauley, T.G. Conjugation to polyethylene glycol polymer promotes aptamer biodistribution to healthy and inflamed tissues. Oligonucleotides 2005, 15, 183–195. [Google Scholar] [CrossRef]
- Da Pieve, C.; Blackshaw, E.; Missailidis, S.; Perkins, A.C. PEGylation and biodistribution of an anti-MUC1 aptamer in MCF-7 tumor-bearing mice. Bioconjug. Chem. 2012, 237, 1377–1381. [Google Scholar]
- Jäschke, A.; Fürste, J.P.; Nordhoff, E.; Hillenkamp, F.; Cech, D.; Erdmann, V.A. Synthesis and properties of oligodeoxyribonucleotide-polyethylene glycol conjugates. Nucleic Acids Res. 1994, 22, 4810–4817. [Google Scholar] [CrossRef]
- Dougan, H.; Lyster, D.M.; Vo, C.V.; Stafford, A.; Weitz, J.I.; Hobbs, J.B. Extending the lifetime of anticoagulant oligodeoxynucleotide aptamers in blood. Nucl. Med. Biol. 2000, 27, 289–297. [Google Scholar] [CrossRef]
- Farkas, I.; Baranyi, L.; Ishikawa, Y.; Okada, N.; Bohata, C.; Budai, D.; Fukuda, A.; Imai, M.; Okada, H. CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. J. Physiol. 2002, 539, 537–545. [Google Scholar] [CrossRef]
- Dominguez, M.; Moreno, I.; Aispurua, C.; Torano, A. Early mechanisms of Leishmania. infection in human blood. Microbes Infect. 2003, 5, 507–513. [Google Scholar] [CrossRef]
- Carter, J.D.; LaBean, T.H. Coupling strategies for the synthesis of peptide-oligonucleotide conjugates for patterned synthetic biomineralization. J. Nucleic Acids. 2011, 2011, 926595. [Google Scholar]
- Hasegawa, H.; Taira, K.; Sode, K.; Ikebukuro, K. Improvement of aptamer affinity by dimerization. Sensors 2008, 8, 1090–1098. [Google Scholar] [CrossRef]
- Mallikaratchy, P.R.; Ruggiero, A.; Gardner, J.R.; Kuryavyi, V.; Maguire, W.F.; Heaney, M.L.; McDevitt, M.R.; Patel, D.J.; Scheinberg, D.A. A multivalent DNA aptamer specific for the B-cell receptor on human lymphoma and leukemia. Nucleic Acids Res. 2011, 39, 2458–2469. [Google Scholar] [CrossRef]
- Tian, L.; Heyduk, T. Bivalent ligands with long nanometer-scale flexible linkers. Biochemistry 2009, 48, 264–275. [Google Scholar] [CrossRef]
- Yang, L.; Meng, L.; Zhang, X.; Chen, Y.; Zhu, G.; Liu, H.; Xiong, X.; Sefah, K.; Tan, W. Engineering polymeric aptamers for selective cytotoxicity. J. Am. Chem. Soc. 2011, 133, 13380–13386. [Google Scholar]
- Murray, E.; Gregg, D.A.; Norton, M.L.; Swick, J.T.; Towler, W.I. Method for a continuous rapid thermal cycle system. U.S. Patent No. 8,163,489, 24 April 2012. [Google Scholar]
- Wahajuddin, S.A.; Arora, S. Superparamagnetic iron oxide nanoparticles: Magnetic nanoplatforms as drug carriers. Int. J. Nanomed. 2012, 7, 3445–3471. [Google Scholar] [CrossRef]
- Nair, B.G.; Nagaoka, Y.; Morimoto, H.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Aptamer conjugated magnetic nanoparticles as nanosurgeons. Nanotechnology 2010, 21, 455102. [Google Scholar] [CrossRef]
- Silva, A.C.; Oliveira, T.R.; Mamani, J.B.; Malheiros, S.M.; Malavolta, L.; Pavon, L.F.; Sibov, T.T.; Amaro, E.; Tannús, A.; Vidoto, E.L.; et al. Application of hyperthermia induced by superparamagnetic iron oxide nanoparticles in glioma treatment. Int. J. Nanomed. 2011, 6, 591–603. [Google Scholar]
- Hilger, I.; Kaiser, W.A. Iron oxide-based nanostructures for MRI and magnetic hyperthermia. Nanomedicine 2012, 7, 1443–1459. [Google Scholar] [CrossRef]
- Kobayashi, T. Cancer hyperthermia using magnetic nanoparticles. Biotechnol. J. 2011, 6, 1342–1347. [Google Scholar] [CrossRef]
- Cavaliere, R.; Ciocatto, E.C.; Giovanella, B.C.; Heidelberger, C.; Johnson, R.O.; Margottini, M.; Mondovi, B.; Moricca, G.; Rossi-Fanelli, A. Selective heat sensitivity of cancer cells. Biochemical and clinical studies. Cancer 1967, 20, 1351–1381. [Google Scholar] [CrossRef]
- Christophi, C.; Winkworth, A.; Muralihdaran, V.; Evans, P. The treatment of malignancy by hyperthermia. Surg. Oncol. 1998, 7, 83–90. [Google Scholar] [CrossRef]
- Sellins, K.S.; Cohen, J.J. Hyperthermia induces apoptosis in thymocytes. Radiat. Res. 1991, 126, 88–95. [Google Scholar] [CrossRef]
- Glazer, E.S.; Curley, S.A. Non-invasive radiofrequency ablation of malignancies mediated by quantum dots, gold nanoparticles and carbon nanotubes. Ther. Deliv. 2011, 2, 1325–1330. [Google Scholar] [CrossRef]
- Glazer, E.S.; Curley, S.A. Radiofrequency field-induced thermal cytotoxicity in cancer cells treated with fluorescent nanoparticles. Cancer 2010, 116, 3285–3293. [Google Scholar] [CrossRef]
- Glazer, E.S.; Zhu, C.; Massey, K.L.; Thompson, C.S.; Kaluarachchi, W.D.; Hamir, A.N.; Curley, S.A. Noninvasive radiofrequency field destruction of pancreatic adenocarcinoma xenografts treated with targeted gold nanoparticles. Clin. Cancer Res. 2010, 16, 5712–5721. [Google Scholar] [CrossRef]
- Wang, Q.; Fang, T.; Liu, P.; Min, X.; Li, X. Study of the bioeffects of CdTe quantum dots on Escherichia coli cells. J. Coll. Interface Sci. 2011, 363, 476–480. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, S.; Wang, L.; Qu, C.; Zhang, C.; Hong, L.; Yuan, L.; Huang, Z.; Wang, Z.; Liu, S.; Jiang, G. CdSe quantum dot (QD)-induced morphological and functional impairments to liver in mice. PLoS One 2011, 6, e24406. [Google Scholar]
- Li, K.G.; Chen, J.T.; Bai, S.S.; Wen, X.; Song, S.Y.; Yu, Q.; Li, J.; Wang, Y.Q. Intracellular oxidative stress and cadmium ions release induce cytotoxicity of unmodified cadmium sulfide quantum dots. Toxicol. In Vitro. 2009, 23, 1007–1013. [Google Scholar] [CrossRef]
- Tsay, J.M.; Trzoss, M.; Shi, L.; Kong, X.; Selke, M.; Jung, M.E.; Weiss, S. Singlet oxygen production by peptide-coated quantum dot-photosensitizer conjugates. J. Am. Chem. Soc. 2007, 129, 6865–6871. [Google Scholar]
- Yaghini, E.; Seifalian, A.M.; MacRobert, A.J. Quantum dots and their potential biomedical applications in photosensitization for photodynamic therapy. Nanomedicine 2009, 4, 353–363. [Google Scholar] [CrossRef]
- Savla, R.; Taratula, O.; Garbuzenko, O.; Minko, T. Tumor targeted quantum dot-mucin 1 aptamer-doxorubicin conjugate for imaging and treatment of cancer. J. Control. Release 2011, 153, 16–22. [Google Scholar] [CrossRef]
- Taghdisi, S.M.; Lavaee, P.; Ramezani, M.; Abnous, K. Reversible targeting and controlled release delivery of daunorubicin to cancer cells by aptamer-wrapped carbon nanotubes. Eur. J. Pharm. Biopharm. 2011, 77, 200–206. [Google Scholar] [CrossRef]
- Tan, W.; Wang, H.; Chen, Y.; Zhang, X.; Zhu, H.; Yang, C.; Yang, R.; Liu, C. Molecular aptamers for drug delivery. Trends Biotechnol. 2011, 9, 634–640. [Google Scholar]
- Lu, W.; Singh, A.K.; Khan, S.A.; Senapati, D.; Yu, H.; Ray, P.C. Gold nano-popcorn-based targeted diagnosis, nanotherapy treatment, and in situ monitoring of photothermal therapy response of prostate cancer cells using surface-enhanced Raman spectroscopy. J. Am. Chem. Soc. 2010, 132, 18103–18114. [Google Scholar] [CrossRef]
- Jarmila, V.; Vavríková, E. Chitosan derivatives with antimicrobial, antitumour and antioxidant activities-a review. Curr. Pharm. Des. 2011, 17, 3596–3607. [Google Scholar] [CrossRef]
- Patel, M.P.; Patel, R.R.; Patel, J.K. Chitosan mediated targeted drug delivery system: A review. J. Pharm. Pharmaceut. Sci. 2010, 13, 536–557. [Google Scholar]
- Bowman, K.; Leong, K.W. Chitosan nanoparticles for oral drug and gene delivery. Int. J. Nanomedicine 2006, 1, 117–128. [Google Scholar] [CrossRef]
- Hassan, E.E.; Gallo, J.M. Targeting anticancer drugs to the brain. I: Enhanced brain delivery of oxantrazole following administration in magnetic cationic microspheres. J. Drug Target. 1993, 1, 7–14. [Google Scholar] [CrossRef]
- Tallury, P.; Kar, S.; Bamrungsap, S.; Huang, Y.F.; Tan, W.; Santra, S. Ultra-small water dispersible fluorescent chitosan nanoparticles: Synthesis, characterization and specific targeting. Chem. Commun. 2009, 7, 2347–2349. [Google Scholar]
- Mann, A.P.; Bhavane, R.C.; Somasunderam, A.; Montalvo-Ortiz, B.; Ghaghada, K.B.; Volk, D.; Nieves-Alicea, R.; Suh, K.S.; Ferrari, M.; Annapragada, A.; et al. Thioaptamer conjugated liposomes for tumor vasculature targeting. Oncotarget 2011, 2, 298–304. [Google Scholar]
- Kolhe, P.; Khandare, J.; Pillai, O.; Kannan, S.; Lieh-Lai, M.; Kannan, R. Hyperbranched polymer-drug conjugates with high drug payload for enhanced cellular delivery. Pharm. Res. 2004, 21, 2185–2195. [Google Scholar] [CrossRef]
- Kolhe, P.; Khandare, J.; Pillai, O.; Kannan, S.; Lieh-Lai, M.; Kannan, R.M. Preparation, cellular transport, and activity of polyamidoamine-based dendritic nanodevices with a high drug payload. Biomaterials 2006, 27, 660–669. [Google Scholar] [CrossRef]
- Kim, Y.; Cao, Z.; Tan, W. Molecular assembly for high-performance bivalent nucleic acid inhibitor. Proc. Natl. Acad. Sci. USA 2008, 105, 5664–5669. [Google Scholar]
- McNamara, J.O.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008, 118, 376–386. [Google Scholar] [CrossRef]
- Wachowius, F.; Höbartner, C. Probing essential nucleobase functional groups in aptamers and deoxyribozymes by nucleotide analogue interference mapping of DNA. J. Am. Chem. Soc. 2011, 133, 14888–14891. [Google Scholar] [CrossRef]
- Hollenstein, M. Expanding the catalytic repertoire of DNAzymes by modified nucleosides. ChimiaInt. J. Chem. 2011, 65, 770–775. [Google Scholar] [CrossRef]
- Sanghvi, Y.S. A status update of modified oligonucleotides for chemotherapeutics applications. Curr. Protoc. Nucleic Acid Chem. 2011, 46, 4.1.1–4.1.22. [Google Scholar]
- Sundaram, P.; Kurniawan, H.; Byrne, M.E.; Wower, J. Therapeutic RNA aptamers in clinical trials. Eur. J. Pharm. Sci. 2013, 48, 259–271. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bruno, J.G. A Review of Therapeutic Aptamer Conjugates with Emphasis on New Approaches. Pharmaceuticals 2013, 6, 340-357. https://doi.org/10.3390/ph6030340
Bruno JG. A Review of Therapeutic Aptamer Conjugates with Emphasis on New Approaches. Pharmaceuticals. 2013; 6(3):340-357. https://doi.org/10.3390/ph6030340
Chicago/Turabian StyleBruno, John G. 2013. "A Review of Therapeutic Aptamer Conjugates with Emphasis on New Approaches" Pharmaceuticals 6, no. 3: 340-357. https://doi.org/10.3390/ph6030340
APA StyleBruno, J. G. (2013). A Review of Therapeutic Aptamer Conjugates with Emphasis on New Approaches. Pharmaceuticals, 6(3), 340-357. https://doi.org/10.3390/ph6030340