Gene Therapy for the Treatment of Parkinson’s Disease: The Nature of the Biologics Expands the Future Indications

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. A Neurotherapeutic Framework for PD
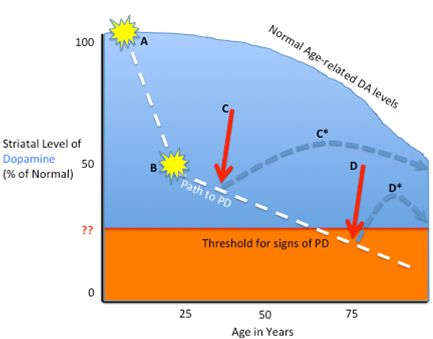
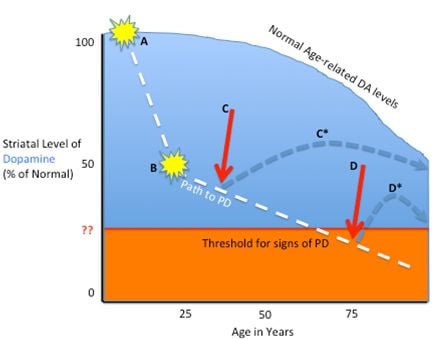
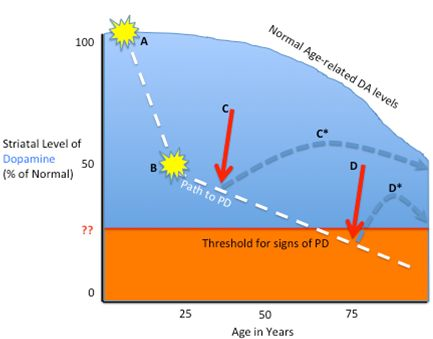
2.1. Pathophysiology

2.2. Medical Therapeutics for PD
2.3. Surgical Therapeutics for PD
2.4. Insights from Preclinical Gene Therapy Studies for PD
2.5. Insights from Clinical Gene Therapy Trials for PD
- The use of optimized CED techniques with real-time imaging and stereotactic guidance to confirm the targeting and distribution of the infusate, especially in large targets, such as PUT. There is no supportive evidence for using non-convective delivery methods (hand injection) for gene therapy within the CNS, even within a minute target such as STN. Postmortem assessments of targeted PUT show that non-convective delivery methods offer limited transduced volumes of distribution within large brain targets. Real-time image-guidance is necessary to not only improve safety, but also to confirm target acquisition and coverage.
- The importance in using a specifically designed, MRI-compatible, CED infusion cannula to minimize brain trauma, while optimizing reflux-free convection of therapeutics within the brain.
- Increasing the infusion volume for putamenal infusions in an attempt to cover and transduce >50% of the post-commissural putamenal volume.
- Since the vectors used (AAV2 and LV) and transgenes appear safe at increasing titers, we support using the higher vector titer found to be safe based on preclinical and Phase I studies.
2.6. Delivery Vectors
2.6.1. AAV
2.6.2. Lentivirus
2.6.3. Other Viral or Non-Viral Gene Therapy Delivery Platforms
2.7. Neurotrophic Factors (NTFs)
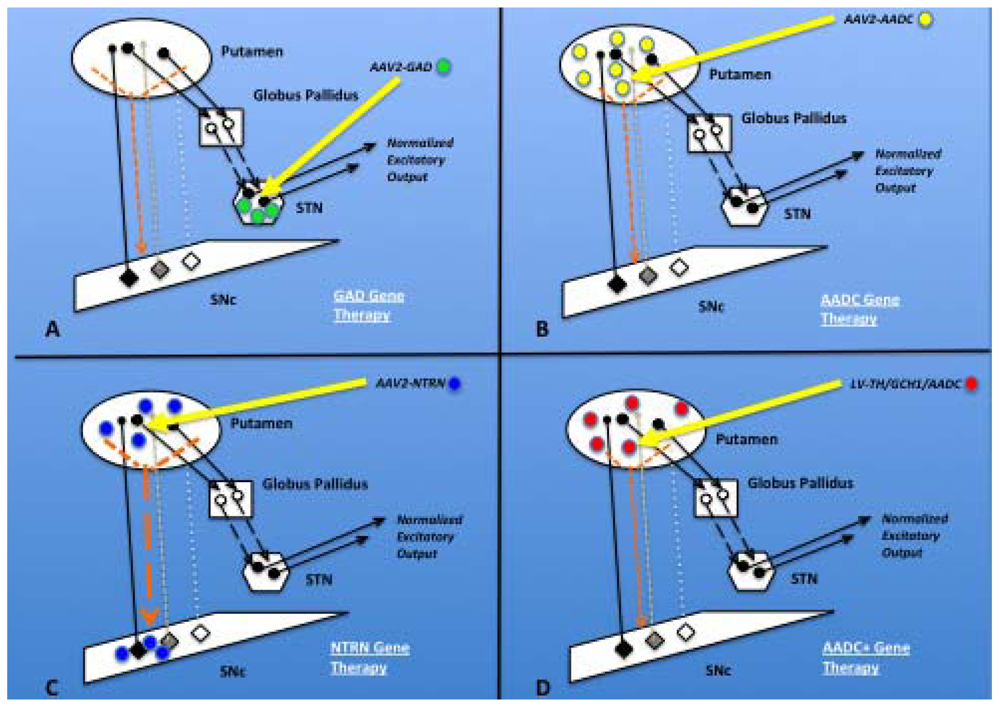
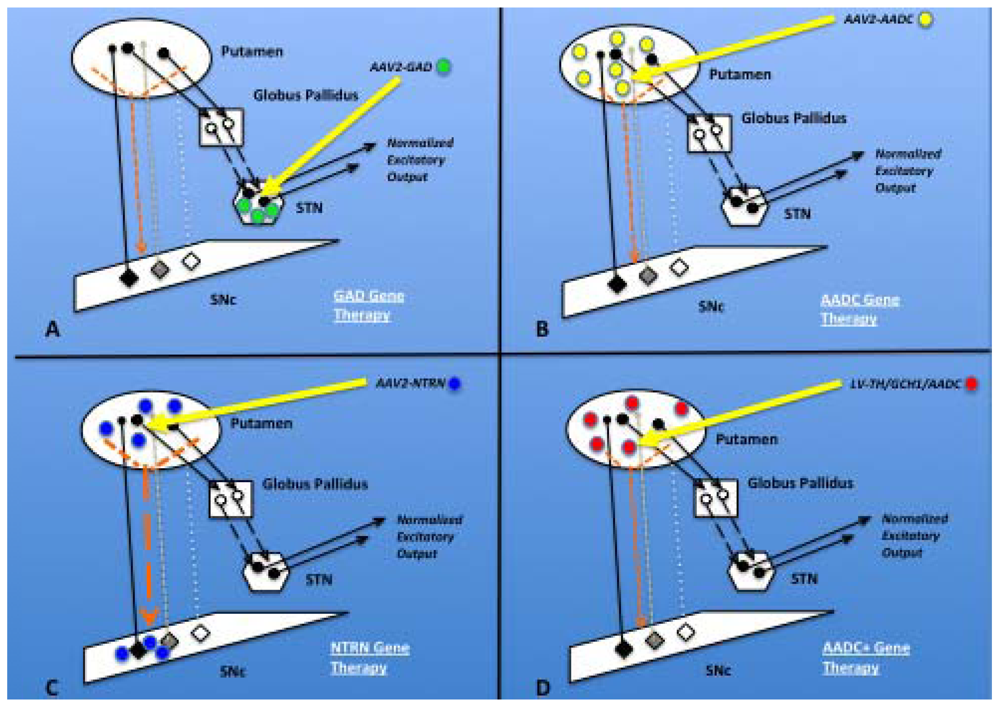
2.7.1. Neurturin (NTRN)
2.7.2. GDNF
2.8. Enzyme Delivery/Replacement
2.8.1. GAD
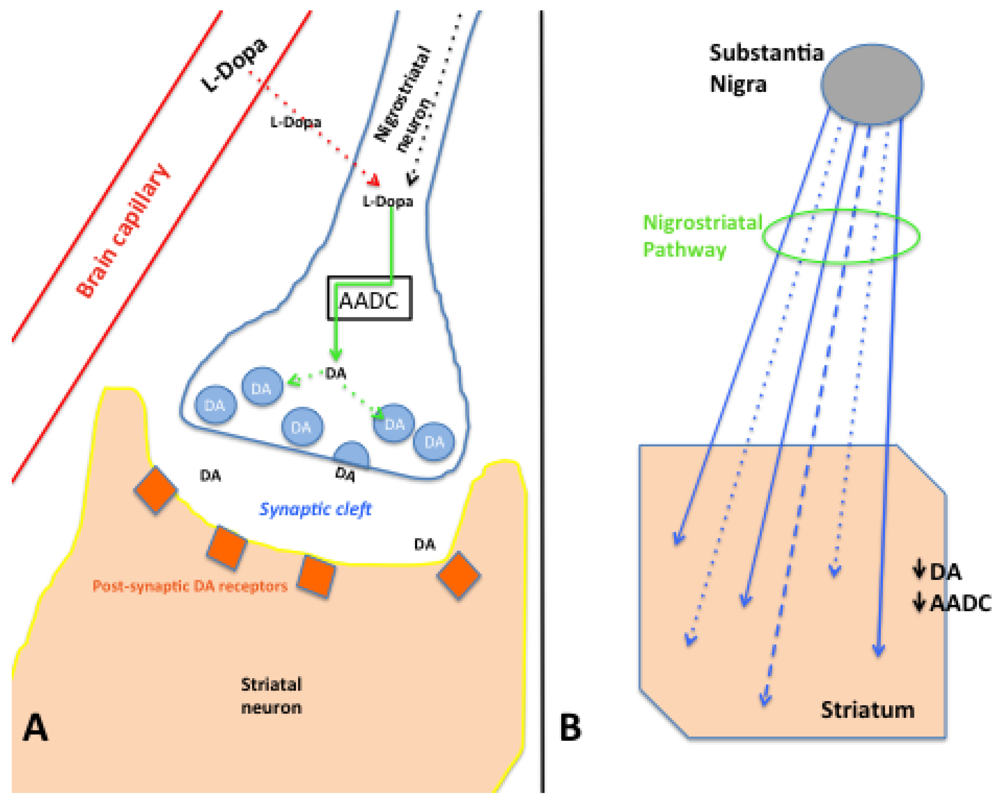
2.8.2. AADC
2.8.3. TH/GCH1/AADC
2.9. Non-Regulated versus Regulated NTF Gene Therapy
3. Conclusions
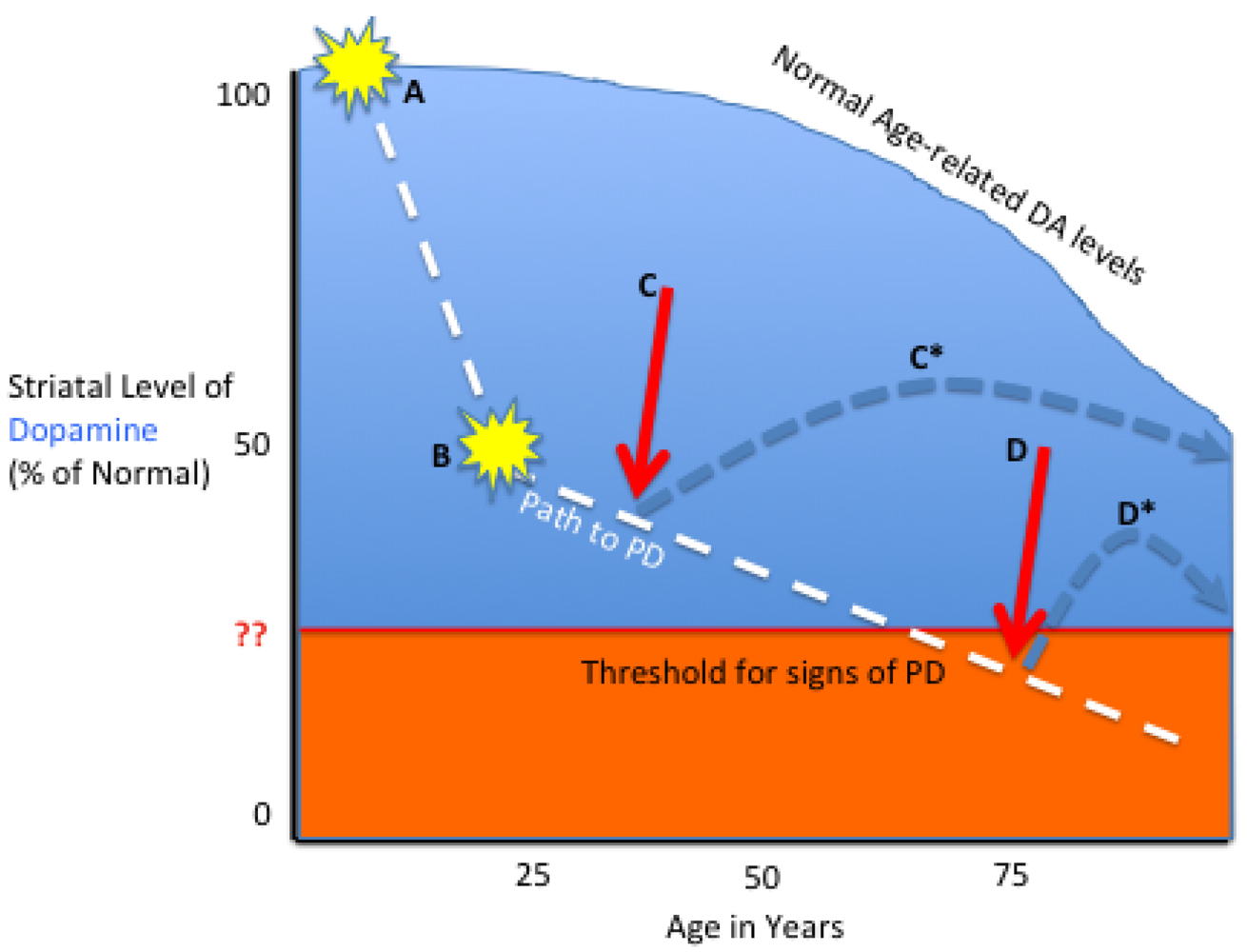
3.1. Tailoring the Biologic to the Particular PD Stage
3.2. Future Needs

Acknowledgments
Conflict of Interest
References
- Olanow, C.W.; Stern, M.B.; Sethi, K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology 2009, 72, S1–S136. [Google Scholar]
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, S1–S58. [Google Scholar]
- Lokk, J.; Borg, S.; Svensson, J.; Persson, U.; Ljunggren, G. Drug and treatment costs in Parkinson’s disease patients in Sweden. Acta Neurol. Scand. 2012, 125, 142–147. [Google Scholar] [CrossRef]
- Winter, Y.; von Campenhausen, S.; Reese, J.P.; Balzer-Geldsetzer, M.; Longo, K.; Spiga, G.; Boetzel, K.; Eggert, K.; Oertel, W.H.; Dodel, R.; et al. Costs of Parkinson’s disease and antiparkinsonian pharmacotherapy: An Italian cohort study. Neurodegener. Dis. 2010, 7, 365–372. [Google Scholar] [CrossRef]
- Winter, Y.; Balzer-Geldsetzer, M.; Spottke, A.; Reese, J.P.; Baum, E.; Klotsche, J.; Rieke, J.; Simonow, A.; Eggert, K.; Oertel, W.H.; et al. Longitudinal study of the socioeconomic burden of Parkinson’s disease in Germany. Eur. J. Neurol. 2010, 17, 1156–1163. [Google Scholar] [CrossRef]
- Winter, Y.; Balzer-Geldsetzer, M.; von Campenhausen, S.; Spottke, A.; Eggert, K.; Oertel, W.H.; Dodel, R. Trends in resource utilization for Parkinson’s disease in Germany. J. Neurol. Sci. 2010, 294, 18–22. [Google Scholar] [CrossRef]
- Vossius, C.; Nilsen, O.B.; Larsen, J.P. Parkinson’s disease and hospital admissions: Frequencies, diagnoses and costs. Acta Neurol. Scand. 2010, 121, 38–43. [Google Scholar] [CrossRef]
- Chen, J.J. Parkinson’s disease: Health-related quality of life, economic cost, and implications of early treatment. Am. J. Manag. Care 2010, 16, S87–S93. [Google Scholar]
- O’Brien, J.A.; Ward, A.; Michels, S.L.; Tzivelekis, S.; Brandt, N.J. Economic burden associated with Parkinson disease. Drug Benefit Trends 2009, 21, 179–190. [Google Scholar]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with Parkinson’s disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar]
- Maguire-Zeiss, K.A.; Mhyre, T.R.; Federoff, H.J. Gazing into the future: Parkinson’s disease gene therapeutics to modify natural history. Exp. Neurol. 2008, 209, 101–113. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy; Sherwood, Neely, and Jones, Paternoster Row: London, UK, 1817. [Google Scholar]
- Lewy, F.H. Die pathologische anatomie der paralysis agitans. Lewandowskys Handb. 1912, 3, 920. [Google Scholar]
- Hornykiewicz, O. Dopamine in the basal ganglia. Its role and therapeutic implications (including the clinical use of L-DOPA). Br. Med. Bull. 1973, 29, 172–178. [Google Scholar]
- Hornykiewicz, O. Parkinson’s disease: From brain homogenate to treatment. Fed. Proc. 1973, 32, 183–190. [Google Scholar]
- Bernheimer, H.; Birkmayer, W.; Hornykiewicz, O.; Jellinger, K.; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 1973, 20, 415–455. [Google Scholar] [CrossRef]
- Cotzias, G.C.; van Woert, M.H.; Schiffer, L.M. Aromatic amino acids and modification of parkinsonism. N. Engl. J. Med. 1967, 276, 374–379. [Google Scholar] [CrossRef]
- Lesser, R.P.; Fahn, S.; Snider, S.R.; Cote, L.J.; Isgreen, W.P.; Barrett, R.E. Analysis of the clinical problems in parkinsonism and the complications of long-term levodopa therapy. Neurology 1979, 29, 1253–1260. [Google Scholar] [CrossRef]
- Parkinson’s Disease: Diagnosis & Clinical Management, 2nd; Factor, S.A.; Weiner, W.J. (Eds.) Demos Medical Publishing LLC: New York, NY, USA, 2008; pp. 471–574, Section VII, Drugs, Chapters 38 to 45.
- Toulouse, A.; Sullivan, A.M. Progress in Parkinson’s disease-where do we stand? Prog. Neurobiol. 2008, 85, 376–392. [Google Scholar] [CrossRef]
- Lewy, F.H. Historical introduction: The basal ganglia and their diseases. In The Diseases of the Basal Ganglia; Putnam, T.J., Ed.; Hafner Publishing: New York, NY, USA, 1966; pp. 1–20. [Google Scholar]
- Gildenberg, P.L. Evolution of basal ganglia surgery for movement disorders. Stereotact. Funct. Neurosurg. 2006, 84, 131–135. [Google Scholar] [CrossRef]
- DeLong, M.R. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef]
- Bergman, H.; Wichmann, T.; DeLong, M.R. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 1990, 249, 1436–1438. [Google Scholar]
- Cooper, I.S. Parkinsonism: Its Medical and Surgical Therapy; Charles C. Thomas: Springfield, IL, USA, 1961. [Google Scholar]
- Iskander, B.J.; Nashold, J.B.S. History of functional neurosurgery. In Neurosurgery Clinics of North America; Gildenberg, P.L., Ed.; WB Saunders Company: Philadelphia, PA, USA, 1995. [Google Scholar]
- Madrazo, I.; Leon, V.; Torres, C.; Aguilera, M.C.; Varela, G.; Alvarez, F.; Fraga, A.; Drucker-Colin, R.; Ostrosky, F.; Skurovich, M.; et al. Transplantation of fetal substantia nigra and adrenal medulla to the caudate nucleus in two patients with Parkinson’s disease. N. Engl. J. Med. 1988, 318, 51. [Google Scholar]
- Spencer, D.D.; Robbins, R.J.; Naftolin, F.; Marek, K.L.; Vollmer, T.; Leranth, C.; Roth, R.H.; Price, L.H.; Gjedde, A.; Bunney, B.S.; et al. Unilateral transplantation of human fetal mesencephalic tissue into the caudate nucleus of patients with Parkinson’s disease. N. Engl. J. Med. 1992, 327, 1541–1548. [Google Scholar]
- Widner, H.; Tetrud, J.; Rehncrona, S.; Snow, B.; Brundin, P.; Gustavii, B.; Bjorklund, A.; Lindvall, O.; Langston, J.W. Bilateral fetal mesencephalic grafting in two patients with parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N. Engl. J. Med. 1992, 327, 1556–1563. [Google Scholar] [CrossRef]
- Redmond, D.E., Jr.; Robbins, R.J.; Naftolin, F.; Marek, K.L.; Vollmer, T.L.; Leranth, C.; Roth, R.H.; Price, L.H.; Gjedde, A.; Bunney, B.S.; et al. Cellular replacement of dopamine deficit in Parkinson’s disease using human fetal mesencephalic tissue: Preliminary results in four patients. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1993, 71, 325–359. [Google Scholar]
- Madrazo, I.M.; Drucker-Colin, R.; Diaz, V.; Martinez-Mata, J.; Torres, C.; Becerril, J.J. Open microsurgical autograft of adrenal medulla to the right caudate nucleus in two patients with intractable Parkinson’s disease. N. Engl. J. Med. 3161, 831–834. [Google Scholar]
- Allen, G.S.; Burns, R.S.; Tulipan, N.E.; Parker, R.A. Adrenal medullary transplantation into the caudate nucleus in Parkinson’s disease: Initial clinical results in 18 patients. Arch. Neurol. 1989, 46, 487–491. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kordower, J.H.; Hansen, J.T.; Jiao, S.S.; Gash, D.M. Adrenal medullary autografts into the basal ganglia of Cebus monkeys: Injury-induced regeneration. Exp. Neurol. 1988, 102, 76–91. [Google Scholar] [CrossRef]
- Plunkett, R.J.; Bankiewicz, K.S.; Cummins, A.C.; Miletich, R.S.; Schwartz, J.P.; Oldfield, E.H. Long-term evaluation of hemiparkinsonian monkeys after adrenal autografting or cavitation alone. J. Neurosurg. 1990, 73, 918–926. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Plunkett, R.J.; Jacobowitz, D.M.; Kopin, I.J.; Oldfield, E.H. Fetal nondopaminergic neural implants in parkinsonian primates. Histochemical and behavioral studies. J. Neurosurg. 1991, 74, 97–104. [Google Scholar] [CrossRef]
- Sheng, J.G.; McShane, L.M.; Plunkett, R.J.; Cummins, A.C.; Oldfield, E.H.; Kopin, I.J.; Palmatier, M.A. Dopaminergic neuronal sprouting and behavioral recovery in hemi-parkinsonian rats after implantation of amnion cells. Exp. Neurol. 1993, 123, 192–203. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Palmatier, M.; Plunkett, R.J.; Cummins, A.; Oldfield, E.H. Reversal of hemiparkinsonian syndrome in nonhuman primates by amnion implantation into caudate nucleus. J.Neurosurg. 1994, 81, 869–876. [Google Scholar] [CrossRef]
- Benabid, A.L.; Benazzouz, A.; Hoffmann, D.; Limousin, P.; Krack, P.; Pollak, P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov. Disord. 1998, 13, 119–125. [Google Scholar]
- Benabid, A.L. Deep brain stimulation for Parkinson’s disease. Curr. Opin. Neurobiol. 2003, 13, 696–706. [Google Scholar] [CrossRef]
- Gage, F.H.; Wolff, J.A.; Rosenberg, M.B.; Xu, L.; Yee, J.-K.; Shults, C.; Friedmann, T. Grafting genetically modified cells into the brain: Possibilities for the future. Neuroscience 1987, 23, 795–807. [Google Scholar] [CrossRef]
- Aebischer, P.; Winn, S.R.; Galletti, P.M. Transplantation of neural tissue in polymer capsules. Brain Res. 1988, 448, 36–48. [Google Scholar]
- Aebischer, P.; Goddard, M.; Signore, A.P.; Timpson, R.L. Functional recovery in hemiparkinsonian primates transplanted with polymer-encapsulated PC12 cells. Exp. Neurol. 1994, 126, 151–158. [Google Scholar] [CrossRef]
- Kordower, J.H.; Palfi, S.; Chen, E.Y.; Ma, S.Y.; Sendera, T.; Cochran, E.J.; Mufson, E.J.; Penn, R.; Goetz, C.G.; Comella, C.D. Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson’s disease. Ann. Neurol. 1999, 46, 419–424. [Google Scholar] [CrossRef]
- Nutt, J.G.; Burchiel, K.J.; Comella, C.L.; Jankovic, J.; Lang, A.E.; Laws, E.R., Jr.; Lozano, A.M.; Penn, R.D.; Simpson, R.K., Jr.; Stacy, M.; et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 2003, 60, 69–73. [Google Scholar]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar]
- Slevin, J.T.; Gerhardt, G.A.; Smith, C.D.; Gash, D.M.; Kryscio, R.; Young, B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J. Neurosurg. 2005, 102, 216–222. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson’s disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Vite, C.H.; Passini, M.A.; Haskins, M.E.; Wolfe, J.H. Adeno-associated virus vector-mediated transduction in the cat brain. Gene Ther. 2003, 10, 1874–1881. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Varenika, V.; Eberling, J.; McKnight, T.R.; Bringas, J.; Pivirotto, P.; Beyer, J.; Hadaczek, P.; Forsayeth, J.; Bowers, W.J.; et al. Real-time MR imaging of adeno-associated viral vector delivery to the primate brain. Neuroimage 2009, 47, T27–T35. [Google Scholar]
- Bankiewicz, K.S.; Eberling, J.L.; Kohutnicka, M.; Jagust, W.; Pivirotto, P.; Bringas, J.; Cunningham, J.; Budinger, T.F.; Harvey-White, J. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp. Neurol. 2000, 164, 2–14. [Google Scholar] [CrossRef]
- Forsayeth, J. Influence of the immune system on central nervous system gene transfer. In Gene Therapy of the Central Nervous System: From Bench to Bedside; Kaplitt, M.G., During, M.J., Eds.; Academic Press: Amsterdam; Boston, MA, USA, 2006; pp. 47–51. [Google Scholar]
- Zaiss, A.K.; Muruve, D.A. Immune responses to adeno-associated virus vectors. Curr. Gene Ther. 2005, 5, 323–331. [Google Scholar] [CrossRef]
- Wright, J.F.; Qu, G.; Tang, C.; Sommer, J.M. Recombinant adeno-associated virus: Formulation challenges and strategies for a gene therapy vector. Curr. Opin. Drug Discov. Devel. 2003, 6, 174–178. [Google Scholar]
- Bobo, R.H.; Laske, D.W.; Akbasak, A.; Morrison, P.F.; Dedrick, R.L.; Oldfield, E.H. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. USA 1994, 91, 2076–2080. [Google Scholar]
- Morrison, P.F.; Chen, M.Y.; Chadwick, R.S.; Lonser, R.R.; Oldfield, E.H. Focal delivery during direct infusion to brain: Role of flow rate, catheter diameter, and tissue mechanics. Am. J. Physiol. 1999, 277, R1218–R1229. [Google Scholar]
- Chen, M.Y.; Lonser, R.R.; Morrison, P.F.; Governale, L.S.; Oldfield, E.H. Variables affecting convection-enhanced delivery to the striatum: A systematic examination of rate of infusion, cannula size, infusate concentration, and tissue-cannula sealing time. J. Neurosurg. 1999, 90, 315–320. [Google Scholar] [CrossRef]
- Chen, M.Y.; Hoffer, A.; Morrison, P.F.; Hamilton, J.F.; Hughes, J.; Schlageter, K.S.; Lee, J.; Kelly, B.R.; Oldfield, E.H. Surface properties, more than size, limiting convective distribution of virus-sized particles and viruses in the central nervous system. J. Neurosurg. 2005, 103, 311–319. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Forsayeth, J.R.; Dickinson, P.J.; Bankiewicz, K.S. Image-guided convection-enhanced delivery platform in the treatment of neurological diseases. Neurotherapeutics 2008, 5, 123–127. [Google Scholar] [CrossRef]
- Richardson, R.M.; Kells, A.P.; Rosenbluth, K.H.; Salegio, E.A.; Fiandaca, M.S.; Larson, P.S.; Starr, P.A.; Martin, A.J.; Lonser, R.R.; Federoff, H.J.; et al. Interventional MRI-guided putaminal delivery of AAV2-GDNF for a planned clinical trial in Parkinson’s disease. Mol. Ther. 2011, 19, 1048–1057. [Google Scholar]
- Fung, L.K.; Ewend, M.G.; Sills, A.; Sipos, E.P.; Thompson, R.; Watts, M.; Colvin, O.M.; Brem, H.; Saltzman, W.M. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998, 58, 672–684. [Google Scholar]
- Salvatore, M.F.; Ai, Y.; Fischer, B.; Zhang, A.M.; Grondin, R.C.; Zhang, Z.; Gerhardt, G.A.; Gash, D.M. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp. Neurol. 2006, 202, 497–505. [Google Scholar] [CrossRef]
- Lonser, R.R.; Gogate, N.; Morrison, P.F.; Wood, J.D.; Oldfield, E.H. Direct convective delivery of macromolecules to the spinal cord. J. Neurosurg. 1998, 89, 616–622. [Google Scholar] [CrossRef]
- Lonser, R.R.; Walbridge, S.; Garmestani, K.; Butman, J.A.; Walters, H.A.; Vortmeyer, A.O.; Morrison, P.F.; Brechbiel, M.W.; Oldfield, E.H. Successful and safe perfusion of the primate brainstem: In vivo magnetic resonance imaging of macromolecular distribution during infusion. J. Neurosurg. 2002, 97, 905–913. [Google Scholar] [CrossRef]
- Murad, G.J.; Walbridge, S.; Morrison, P.F.; Garmestani, K.; Degen, J.W.; Brechbiel, M.W.; Oldfield, E.H.; Lonser, R.R. Real-time, image-guided, convection-enhanced delivery of interleukin 13 bound to pseudomonas exotoxin. Clin. Cancer Res. 2006, 12, 145–151. [Google Scholar]
- Murad, G.J.; Walbridge, S.; Morrison, P.F.; Szerlip, N.; Butman, J.A.; Oldfield, E.H.; Lonser, R.R. Image-guided convection-enhanced delivery of gemcitabine to the brainstem. J.Neurosurg. 2007, 106, 351–356. [Google Scholar] [CrossRef]
- Yin, D.; Richardson, R.M.; Fiandaca, M.S.; Bringas, J.; Forsayeth, J.; Berger, M.S.; Bankiewicz, K.S. Cannula placement for effective convection-enhanced delivery in the nonhuman primate thalamus and brainstem: Implications for clinical delivery of therapeutics. J. Neurosurg. 2010, 113, 240–248. [Google Scholar] [CrossRef]
- Yin, D.; Valles, F.E.; Fiandaca, M.S.; Bringas, J.; Gimenez, F.; Berger, M.S.; Forsayeth, J.; Bankiewicz, K.S. Optimal region of the putamen for image-guided convection-enhanced delivery of therapeutics in human and non-human primates. Neuroimage 2011, 54, S196–S203. [Google Scholar] [CrossRef]
- Krauze, M.T.; Saito, R.; Noble, C.; Tamas, M.; Bringas, J.; Park, J.W.; Berger, M.S.; Bankiewicz, K.S. Reflux-free cannula for convection-enhanced high-speed delivery of therapeutic agents. J. Neurosurg. 2005, 103, 923–929. [Google Scholar] [CrossRef]
- Krauze, M.T.; McKnight, T.R.; Yamashita, Y.; Bringas, J.; Noble, C.O.; Saito, R.; Geletneky, K.; Forsayeth, J.; Berger, M.S.; Jackson, P.; et al. Real-time visualization and characterization of liposomal delivery into the monkey brain by magnetic resonance imaging. Brain Res. Brain Res. Protoc. 2005, 16, 20–26. [Google Scholar] [CrossRef]
- Saito, R.; Krauze, M.T.; Bringas, J.R.; Noble, C.; McKnight, T.R.; Jackson, P.; Wendland, M.F.; Mamot, C.; Drummond, D.C.; Kirpotin, D.B.; et al. Gadolinium-loaded liposomes allow for real-time magnetic resonance imaging of convection-enhanced delivery in the primate brain. Exp. Neurol. 2005, 196, 381–389. [Google Scholar] [CrossRef]
- Krauze, M.T.; Forsayeth, J.; Park, J.W.; Bankiewicz, K.S. Real-time imaging and quantification of brain delivery of liposomes. Pharm. Res. 2006, 23, 2493–2504. [Google Scholar] [CrossRef]
- Lonser, R.R.; Schiffman, R.; Robison, R.A.; Butman, J.A.; Quezado, Z.; Walker, M.L.; Morrison, P.F.; Walbridge, S.; Murray, G.J.; Park, D.M.; et al. Image-guided, direct convective delivery of glucocerebrosidase for neuronopathic Gaucher disease. Neurology 2007, 68, 254–261. [Google Scholar]
- Lonser, R.R.; Warren, K.E.; Butman, J.A.; Quezado, Z.; Robison, R.A.; Walbridge, S.; Schiffman, R.; Merrill, M.; Walker, M.L.; Park, D.M.; et al. Real-time image-guided direct convective perfusion of intrinsic brainstem lesions. Technical note. J. Neurosurg. 2007, 107, 190–197. [Google Scholar] [CrossRef]
- Szerlip, N.J.; Walbridge, S.; Yang, L.; Morrison, P.F.; Degen, J.W.; Jarrell, S.T.; Kouri, J.; Kerr, P.B.; Kotin, R.; Oldfield, E.H.; et al. Real-time imaging of convection-enhanced delivery of viruses and virus-sized particles. J. Neurosurg. 2007, 107, 560–567. [Google Scholar] [CrossRef]
- Dickinson, P.J.; LeCouteur, R.A.; Higgins, R.J.; Bringas, J.R.; Roberts, B.; Larson, R.F.; Yamashita, Y.; Krauze, M.; Noble, C.O.; Drummond, D.; et al. Canine model of convection-enhanced delivery of liposomes containing CPT-11 monitored with real-time magnetic resonance imaging: Laboratory investigation. J. Neurosurg. 2008, 108, 989–998. [Google Scholar] [CrossRef]
- Krauze, M.T.; Vandenberg, S.R.; Yamashita, Y.; Saito, R.; Forsayeth, J.; Noble, C.; Park, J.; Bankiewicz, K.S. Safety of real-time convection-enhanced delivery of liposomes to primate brain: A long-term retrospective. Exp. Neurol. 2008, 210, 638–644. [Google Scholar] [CrossRef]
- Varenika, V.; Dickenson, P.; Bringas, J.; LeCouteur, R.; Higgins, R.; Park, J.; Fiandaca, M.; Berger, M.; Sampson, J.; Bankiewicz, K.S. Detection of infusate leakage in the brain using real-time imaging of convection-enhanced delivery. J. Neurosurg. 2008, 109, 874–880. [Google Scholar] [CrossRef]
- Heiss, J.D.; Walbridge, S.; Asthagiri, A.R.; Lonser, R.R. Image-guided convection-enhanced delivery of muscimol to the primate brain. J. Neurosurg. 2010, 112, 790–795. [Google Scholar] [CrossRef]
- Su, X.; Kells, A.P.; Aguilar-Salegio, E.; Richardson, R.M.; Hadaczek, P.; Beyer, J.; Bringas, J.; Pivirotto, P.; Forsayeth, J.; Bankiewicz, K.S. Real-time MR imaging with gadoteridol predicts distribution of transgenes after convection-enhanced delivery of AAV2 vectors. Mol. Ther. 2010, 18, 1490–1495. [Google Scholar] [CrossRef]
- Gimenez, F.; Krauze, M.T.; Valles, F.; Hadaczek, P.; Bringas, J.; Sharma, N.; Forsayeth, J.; Bankiewicz, K.S. Image-guided convection-enhanced delivery of GDNF protein into monkey putamen. Neuroimage 2011, 54, S189–S195. [Google Scholar] [CrossRef]
- Richardson, R.M.; Gimenez, F.; Salegio, E.A.; Su, X.; Bringas, J.; Berger, M.S.; Bankiewicz, K.S. T2 imaging in monitoring of intraparenchymal real-time convection-enhanced delivery. Neurosurgery 2011, 69, 154–163. [Google Scholar] [CrossRef]
- Richardson, R.M.; Kells, A.P.; Martin, A.J.; Larson, P.S.; Starr, P.A.; Piferi, P.G.; Bates, G.; Tansey, L.; Rosenbluth, K.H.; Bringas, J.R.; et al. Novel platform for MRI-guided convection-enhanced delivery of therapeutics: Preclinical validation in nonhuman primate brain. Stereotact. Funct. Neurosurg. 2011, 89, 141–151. [Google Scholar] [CrossRef]
- Saito, R.; Krauze, M.T.; Noble, C.O.; Tamas, M.; Drummond, D.C.; Kirpotin, D.B.; Berger, M.S.; Park, J.W.; Bankiewicz, K.S. Tissue affinity of the infusate affects the distribution volume during convection-enhanced delivery into rodent brains: Implications for local drug delivery. J. Neurosci. Methods 2006, 154, 225–232. [Google Scholar] [CrossRef]
- Kells, A.P.; Hadaczek, P.; Yin, D.; Bringas, J.; Varenika, V.; Forsayeth, J.; Bankiewicz, K.S. Efficient gene therapy-based method for the delivery of therapeutics to primate cortex. Proc. Natl. Acad. Sci. USA 2009, 106, 2407–2411. [Google Scholar]
- Fiandaca, M.S.; Bankiewicz, K.S. Gene therapy for Parkinson’s disease: From nonhuman primates to humans. Curr. Opin. Mol. Ther. 2010, 12, 519–529. [Google Scholar]
- Muramatsu, S.; Fujimoto, K.; Kato, S.; Mizukami, H.; Asari, S.; Ikeguchi, K.; Kawakami, T.; Urabe, M.; Kume, A.; Sato, T.; et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol. Ther. 2010, 18, 1731–1735. [Google Scholar] [CrossRef]
- Christine, C.W.; Starr, P.A.; Larson, P.S.; Eberling, J.L.; Jagust, W.J.; Hawkins, R.A.; VanBrocklin, H.F.; Wright, J.F.; Bankiewicz, K.S.; Aminoff, M.J. Safety and tolerability of putaminal gene therapy for Parkinson’s disease. Neurology 2009, 73, 1662–1669. [Google Scholar]
- Zrinzo, L.; Foltynie, T.; Limousin, P.; Hariz, M.I. Reducing hemorrhagic complications in functional neurosurgery: A large case series and systematic literature review. J. Neurosurg. 2012, 116, 84–94. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; Feigin, A.; Tang, C.; Fitzsimons, H.L.; Mattis, P.; Lawlor, P.A.; Bland, R.J.; Young, D.; Strybing, K.; Eidelberg, D.; et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 2007, 369, 2097–2105. [Google Scholar]
- LeWitt, P.A.; Rezai, A.R.; Leehey, M.A.; Ojemann, S.G.; Flaherty, A.W.; Eskandar, E.N.; Kostyk, S.K.; Thomas, K.; Sarkar, A.; Siddiqui, M.S.; et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011, 10, 309–319. [Google Scholar] [CrossRef]
- McCarthy, D. Neurologix Files to Liquidate Under Chapter 7 Bankruptcy. Available online: http://www.bloomberg.com/news/2012-03-16/neurologix-files-to-liquidate-under-chapter-7-bankruptcy-1-.html/ (accessed on 16 March 2012).
- Eberling, J.L.; Jagust, W.J.; Christine, C.W.; Starr, P.; Larson, P.; Bankiewicz, K.S.; Aminoff, M.J. Results from a phase I safety trial of hAADC gene therapy for Parkinson’s disease. Neurology 2008, 70, 1980–1983. [Google Scholar] [CrossRef]
- Valles, F.; Fiandaca, M.S.; Eberling, J.L.; Starr, P.A.; Larson, P.S.; Christine, C.W.; Forsayeth, J.; Richardson, R.M.; Su, X.; Aminoff, M.J.; et al. Qualitative imaging of adeno-associated virus serotype 2-human aromatic L-amino acid decarboxylase gene therapy in a phase I study for the treatment of Parkinson disease. Neurosurgery 2010, 67, 1377–1385. [Google Scholar]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Marks, W.J.; Ostrem, J.L.; Verhagen, L.; Starr, P.A.; Larson, P.S.; Bakay, R.A.E.; Taylor, R.; Cahn-Weiner, D.A.; Stoessl, A.J.; Olanow, C.W.; et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open label, phase I trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Ceregene. A phase I/II trial assessing the safety and efficacy of bilateral intraputaminal and intranigral administration of CERE-120 (Adeno-Associated Virus Serotype 2 [AAV2]-Neurturin [NTN]) in subjects with idiopathic Parkinson’s disease (06/17/2009). Available online: http://videocast.nih.gov/ram/rac061709.ram/ (accessed on 17 June 2009).
- Bartus, R.T.; Herzog, C.D.; Chu, Y.; Wilson, A.; Brown, L.; Siffert, J.; Johnson, E.M., Jr.; Olanow, C.W.; Mufson, E.J.; Kordower, J.H. Bioactivity of AAV2-neurturin gene therapy (CERE-120): Differences between Parkinson’s disease and nonhuman primate brains. Mov. Disord. 2011, 26, 27–36. [Google Scholar]
- Jarraya, B.; Boulet, S.; Ralph, G.S.; Jan, C.; Bonvento, G.; Azzouz, M.; Miskin, J.E.; Shin, M.; Delzescaux, T.; Drouot, X.; et al. Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci. Transl. Med. 2009, 1, 1–10. [Google Scholar]
- Biomedica, O. Oxford BioMedica Announces Interim Update On ProSavin® Phase I/II Study In Parkinson’s Disease—15/12/2011. Available online: http://www.oxfordbiomedica.co.uk/page.asp?pageid=59&newsid=302/ (accessed on 15 December 2011).
- Grieger, J.C.; Samulski, R.J. Adeno-associated virus as a gene therapy vector: Vector development, production and clinical applications. Adv. Biochem. Eng. Biotechnol. 2005, 99, 119–145. [Google Scholar]
- Berns, K.I.; Giraud, C. Adenovirus and adeno-associated virus as vectors for gene therapy. Ann. NY Acad. Sci. 1995, 772, 95–104. [Google Scholar] [CrossRef]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of infectious adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar]
- Edelstein, M. Vectors used in gene therapy clinical trials (updated June 2011). Available online: http://www.abedia.com/wiley/vectors.php/ (accessed on 30 June 2011).
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar]
- Surosky, R.T.; Urabe, M.; Godwin, S.G.; McQuiston, S.A.; Kurtzman, G.J.; Ozawa, K.; Natsoulis, G. Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J. Virol. 1997, 71, 7951–7959. [Google Scholar]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef]
- Ferrari, F.K.; Samulski, T.; Shenk, T.; Samulski, R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996, 70, 3227–3234. [Google Scholar]
- Hauck, B.; Zhao, W.; High, K.; Xiao, W. Intracellular viral processing, not single-stranded DNA accumulation, is crucial for recombinant adeno-associated virus transduction. J. Virol. 2004, 78, 13678–13686. [Google Scholar] [CrossRef]
- Qing, K.; Hansen, J.; Weigel-Kelley, K.A.; Tan, M.; Zhou, S.; Srivastava, A. Adeno-associated virus type 2-mediated gene transfer: Role of cellular FKBP52 protein in transgene expression. J. Virol. 2001, 75, 8968–8976. [Google Scholar]
- Qing, K.; Li, W.; Zhong, L.; Tan, M.; Hansen, J.; Weigel-Kelley, K.A.; Chen, L.; Yoder, M.C.; Srivastava, A. Adeno-associated virus type 2-mediated gene transfer: Role of cellular T-cell protein tyrosine phosphatase in transgene expression in established cell lines in vitro and transgenic mice in vivo. J. Virol. 2003, 77, 2741–2746. [Google Scholar]
- Zhong, L.; Zhao, W.; Wu, J.; Li, B.; Zolotukhin, S.; Govindasamy, L.; Agbandje-McKenna, M.; Srivastava, A. A dual role of EGFR protein tyrosine kinase signaling in ubiquitination of AAV2 capsids and viral second-strand DNA synthesis. Mol. Ther. 2007, 15, 1323–1330. [Google Scholar] [CrossRef]
- Walters, R.W.; Yi, S.M.; Keshavjee, S.; Brown, K.E.; Welsh, M.J.; Chiorini, J.A.; Zabner, J. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J. Biol. Chem. 2001, 276, 20610–20616. [Google Scholar]
- Summerford, C.; Samulski, R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar]
- Qing, K.; Mah, C.; Hansen, J.; Zhou, S.; Dwarki, V.; Srivastava, A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 1999, 5, 71–77. [Google Scholar] [CrossRef]
- Burger, C.; Gorbatyuk, O.S.; Velardo, M.J.; Peden, C.S.; Williams, P.; Zolotukhin, S.; Reier, P.J.; Mandel, R.J.; Muzyczka, N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 2004, 10, 302–317. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Alisky, J.M.; Hughes, S.M.; Sauter, S.L.; Jolly, D.; Dubensky, T.W., Jr.; Staber, P.D.; Chiorini, J.A.; Davidson, B.L. Transduction of murine cerebellar neurons with recombinant FIV and AAV5 vectors. Neuroreport 2000, 11, 2669–2673. [Google Scholar] [CrossRef]
- Manfredsson, F.P.; Rising, A.C.; Mandel, R.J. AAV9: A potential blood-brain barrier buster. Mol. Ther. 2009, 17, 403–405. [Google Scholar] [CrossRef]
- Davidson, B.L.; Stein, C.S.; Heth, J.A.; Kotin, R.M.; Darksen, T.A.; Ghodsi, Z.A.; Chiorini, J.A. Recombinant Adeno-associated virus type 2,4, and 5 vectors: Transduction of varient cell types and regions in the mammalian central nervous system. Proc. Natl. Acad. Sci. USA 2000, 97, 3428–3432. [Google Scholar]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef]
- Heilbronn, R.; Weger, S. Viral vectors for gene transfer: Current status of gene therapeutics. Handb. Exp. Pharmacol. 2010, 197, 143–170. [Google Scholar] [CrossRef]
- Amado, R.G.; Chen, I.S. Lentiviral vectors—The promise of gene therapy within reach? Science 1999, 674–676. [Google Scholar] [CrossRef]
- Olsen, J.C. Gene transfer vectors derived from equine infectious anemia virus. Gene Ther. 1998, 5, 1481–1487. [Google Scholar]
- Poeschla, E.M.; Wong-Staal, F.; Looney, D.J. Efficient transduction of nondividing human cells by feline immunodeficiency virus lentiviral vectors. Nat. Med. 1998, 4, 354–357. [Google Scholar] [CrossRef]
- Kumar, M.; Keller, B.; Makalou, N.; Sutton, R.E. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 2001, 12, 1893–1905. [Google Scholar] [CrossRef]
- Sinn, P.L.; Sauter, S.L.; McCray, P.B. Gene therapy progress and prospects: Development of improved lentiviral and retroviral vectors—Design, biosafety, and production. Gene Ther. 2005, 12, 1089–1098. [Google Scholar] [CrossRef]
- Wong, L.F.; Goodhead, L.; Prat, C.; Mitrophanous, K.A.; Kingsman, S.M.; Mazarakis, N.D. Lentivirus-mediated gene transfer to the central nervous system: Therapeutic and research applications. Hum. Gene Ther. 2006, 17, 1–9. [Google Scholar] [CrossRef]
- Schambachand, A.; Baum, C. Clinical applications of lentiviral vectors—Concepts and practice. Curr. Gene Ther. 2008, 8, 474–482. [Google Scholar] [CrossRef]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar]
- Azzouz, M.; Kingsman, S.M.; Mazarakis, N.D. Lentiviral vectors for treating and modeling human CNS disorders. J. Gene Med. 2004, 6, 951–962. [Google Scholar] [CrossRef]
- Valori, C.F.; Ning, K.; Wyles, M.; Azzouz, M. Development and applications of non-HIV-based lentiviral vectors in neurological disorders. Curr. Gene Ther. 2008, 8, 406–418. [Google Scholar] [CrossRef]
- Gartner, S.; Markovits, P.; Markovitz, D.M.; Betts, R.F.; Popovic, M. Virus isolation from and identification of HTLV-III/LAV-producing cells in brain tissue from a patient with AIDS. JAMA 1986, 256, 2365–2371. [Google Scholar]
- Watkins, B.A.; Dorn, H.H.; Kelly, W.B.; Armstrong, R.C.; Potts, B.J.; Michaels, F.; Kufta, C.V.; Dubois-Dalcq, M. Specific tropism of HIV-1 for microglial cells in primary human brain cultures. Science 1990, 249, 549–553. [Google Scholar]
- Azzouz, M.; Martin-Rendon, E.; Barber, R.D.; Mitrophanous, K.A.; Carter, E.E.; Rohll, J.B.; Kingsman, S.M.; Kingsman, A.J.; Mazarakis, N.D. Multicistronic lentiviral vector-mediated striatal gene transfer of aromatic L-amino acid decarboxylase, tyrosine hydroxylase, and GTP cyclohydrolase I induces sustained transgene expression, dopamine production, and functional improvement in a rat model of Parkinson’s disease. J. Neurosci. 2002, 22, 10302–10312. [Google Scholar]
- Biomedica, O. Press Release-Oxford BioMedica Initiates Phase I/II Trial of ProSavin® Gene-Based Treatment for Parkinson’s Disease—13 December 2007. Available online: http://www.oxfordbiomedica.co.uk/page.asp?pageid=59&newsid=114/ (accessed on 13 December 2007).
- Philpott, N.J.; Thrasher, A.J. Use of nonintegrating lentiviral vectors for gene therapy. Hum. Gene Ther. 2007, 18, 483–489. [Google Scholar] [CrossRef]
- Sarkis, C.; Philippe, S.; Mallet, J.; Serguera, C. Non-integrating lentiviral vectors. Curr. Gene Ther. 2008, 8, 430–437. [Google Scholar] [CrossRef]
- Rahim, A.A.; Wong, A.M.; Howe, S.J.; Buckley, S.M.; Acosta-Saltos, A.D.; Elston, K.E.; Ward, N.J.; Philpott, N.J.; Cooper, J.D.; Anderson, P.N.; et al. Efficient gene delivery to the adult and fetal CNS using pseudotyped non-integrating lentiviral vectors. Gene Ther. 2009, 16, 509–520. [Google Scholar] [CrossRef]
- Lim, S.T.; Airavaara, M.; Harvey, B.K. Viral vectors for neurotrophic factor delivery: A gene therapy approach for neurodegenerative diseases of the CNS. Pharmacol. Res. 2010, 61, 14–26. [Google Scholar] [CrossRef]
- Kennedy, P.G. Potential use of herpes simplex virus (HSV) vectors for gene therapy of neurological disorders. Brain 1997, 120, 1245–1259. [Google Scholar] [CrossRef]
- Berges, B.K.; Wolfe, J.H.; Fraser, N.W. Transduction of brain by herpes simplex virus vectors. Mol. Ther. 2007, 15, 20–29. [Google Scholar] [CrossRef]
- Goss, J.R.; Gold, M.S.; Glorioso, J.C. HSV vector-mediated modification of primary nociceptor afferents: An approach to inhibit chronic pain. Gene Ther. 2009, 16, 493–501. [Google Scholar] [CrossRef]
- Wolfe, D.; Mata, M.; Fink, D.J. A human trial of HSV-mediated gene transfer for the treatment of chronic pain. Gene Ther. 2009, 16, 455–460. [Google Scholar] [CrossRef]
- Hibbitt, O.C.; Wade-Martins, R. Delivery of large genomic DNA inserts >100 kb using HSV-1 amplicons. Curr. Gene Ther. 2006, 6, 325–336. [Google Scholar] [CrossRef]
- Oehmig, A.; Fraefel, C.; Breakefield, X.O. Update on herpesvirus amplicon vectors. Mol. Ther. 2004, 10, 630–643. [Google Scholar] [CrossRef]
- Todo, T. Oncolytic virus therapy using genetically engineered herpes simplex viruses. Front. Biosci. 2008, 13, 2060–2064. [Google Scholar] [CrossRef]
- Klein, R.L.; Lewis, M.H.; Muzyczka, N.; Meyer, E.M. Prevention of 6-hydroxydopamine-induced rotational behavior by BDNF somatic gene transfer. Brain Res. 1999, 847, 314–320. [Google Scholar] [CrossRef]
- Natsume, A.; Mata, M.; Goss, J.; Huang, S.; Wolfe, D.; Oligino, T.; Glorioso, J.; Fink, D.J. Bcl-2 and GDNF delivered by HSV-mediated gene transfer act additively to protect dopaminergic neurons from 6-OHDA-induced degeneration. Exp. Neurol. 2001, 169, 231–238. [Google Scholar]
- Harvey, B.K.; Chang, C.F.; Chiang, Y.H.; Bowers, W.J.; Morales, M.; Hoffer, B.J.; Wang, Y.; Federoff, H.J. HSV amplicon delivery of glial cell line-derived neurotrophic factor is neuroprotective against ischemic injury. Exp. Neurol. 2003, 183, 47–55. [Google Scholar] [CrossRef]
- Bowers, W.J.; Olschowka, J.A.; Federoff, H.J. Immune responses to replication-defective HSV-1 type vectors within the CNS: Implications for gene therapy. Gene Ther. 2003, 10, 941–945. [Google Scholar] [CrossRef]
- Sun, M.; Kong, L.; Wang, X.; Lu, X.G.; Gao, Q.; Geller, A.I. Comparison of the capability of GDNF, BDNF, or both, to protect nigrostriatal neurons in a rat model of Parkinson’s disease. Brain Res. 1052, 119–129. [Google Scholar]
- Lasic, D.D. Liposomes in Gene Delivery; CRC Press: Boca Raton, FL, USA, 1997; p. 295. [Google Scholar]
- Templeton, N.S. Cationic liposome-mediated gene delivery in vivo. Biosci. Rep. 2002, 22, 283–295. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Berger, M.S.; Bankiewicz, K.S. The use of convection-enhanced delivery with liposomal toxins in neurooncology. Toxins 2011, 3, 369–397. [Google Scholar] [CrossRef]
- Templeton, N.S.; Lasic, D.D.; Frederik, P.M.; Strey, H.H.; Roberts, D.D.; Pavlakis, G.N. Improved DNA: Liposome complexes for increased systemic delivery and gene expression. Nat. Biotechnol. 1997, 15, 647–652. [Google Scholar] [CrossRef]
- Ramesh, R.; Saeki, T.; Templeton, N.S.; Ji, L.; Stephens, L.C.; Ito, I.; Wilson, D.R.; Wu, Z.; Branch, C.D.; Minna, J.D.; et al. Successful treatment of primary and disseminated human lung cancers by systemic delivery of tumor suppressor genes using an improved liposome vector. Mol. Ther. 2001, 3, 337–350. [Google Scholar] [CrossRef]
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef]
- Szebeni, J.; Baranyi, L.; Savay, S.; Milosevits, J.; Bunger, R.; Laverman, P.; Metselaar, J.M.; Storm, G.; Chanan-Khan, A.; Liebes, L.; et al. Role of complement activation in hypersensitivity reactions to doxil and hynic PEG liposomes: Experimental and clinical studies. J. Liposome Res. 2002, 12, 165–172. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar] [CrossRef]
- Aron, L.; Klein, R. Repairing the parkinsonian brain with neurotrophic factors. Trends Neurosci. 2011, 34, 88–100. [Google Scholar] [CrossRef]
- Barbacid, M. The trk family of neurotrophin receptors. J. Neurobiol. 1994, 25, 1386–1403. [Google Scholar] [CrossRef]
- Barbacid, M. Neurotrophic factors and their receptors. Curr. Opin. Cell Biol. 1995, 7, 148–155. [Google Scholar] [CrossRef]
- Krieglstein, K. Factors promoting survival of mesencephalic dopaminergic neurons. Cell Tissue Res. 2004, 318, 73–80. [Google Scholar] [CrossRef]
- Andressoo, J.O.; Saarma, M. Signalling mechanisms underlying development and maintenance of dopamine neurons. Curr. Opin. Neurobiol. 2008, 18, 297–306. [Google Scholar] [CrossRef]
- Evans, J.R.; Barker, R.A. Neurotrophic factors as a therapeutic target for Parkinson’s disease. Expert Opin. Ther. Targets 2008, 12, 437–447. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Bespalov, M.M.; Saarma, M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol. Sci. 2007, 28, 68–74. [Google Scholar] [CrossRef]
- Lindholm, P.; Saarma, M. Novel CDNF/MANF family of neurotrophic factors. Dev. Neurobiol. 2010, 70, 360–371. [Google Scholar]
- Saarma, M.; Sariola, H. Other neurotrophic factors: Glial cell line-derived neurotrophic factor (GDNF). Microsc. Res. Tech. 1999, 45, 292–302. [Google Scholar] [CrossRef]
- Saarma, M. GDNF—A stranger in the TGF-beta superfamily? Eur. J. Biochem. 2000, 267, 6968–6971. [Google Scholar] [CrossRef]
- Roussa, E.; von Bohlen und Halbach, O.; Krieglstein, K. TGF-beta in dopamine neuron development, maintenance and neuroprotection. Adv. Exp. Med. Biol. 2009, 651, 81–90. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar]
- Kotzbauer, P.; Lampe, P.; Heuckeroth, R.; Golden, J.; Creedon, D.; Johnson, E., Jr. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 1996, 384, 467–470. [Google Scholar]
- Trupp, M.; Arenas, E.; Fainzilber, M.; Nilsson, A.-S.; Sieber, B.-A.; Grigoriou, M.; Kilkenny, C.; Salazar-Grueso, E.; Pachnis, V.; Arumae, U.; et al. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature 1996, 381, 785–789. [Google Scholar]
- Jing, S.; Wen, D.; Yu, Y.; Holst, P.L.; Luo, Y.; Fang, M.; Tamir, R.; Antonio, L.; Hu, Z.; Cupples, R.; et al. GDNF-Induced activation of the ret protein tyrosine kinase is mediated by GDNFR-a, a novel receptor for GDNF. Cell 1996, 85, 1113–1124. [Google Scholar] [CrossRef]
- Durbec, P.; Marcos-Gutierrez, C.V.; Kilkenny, C.; Grigoriou, M.; Wartiowaara, K.; Suvanto, P.; Smith, D.; Ponder, B.; Costantini, F.; Saarma, M.; et al. GDNF signalling through the ret receptor tyrosine kinase. Nature 1996, 381, 789–793. [Google Scholar]
- Treanor, J.J.S.; Goodman, L.; de Sauvage, F.; Stone, D.M.; Poulsen, K.T.; Beck, C.D.; Gray, C.; Armanini, M.P.; Pollock, R.A.; Hefti, F.; et al. Characterization of a multicomponent receptor for GDNF. Nature 1996, 382, 80–83. [Google Scholar]
- Vega, Q.; Worby, C.; Lechner, M.; Dixon, J.; Dressler, G. Glial cell line-derived neurotrophic factor activates the receptor tyrosine kinase RET and promotes kidney morphogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 10657–10661. [Google Scholar]
- Takahashi, M.; Ritz, J.; Cooper, G.M. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985, 42, 581–588. [Google Scholar] [CrossRef]
- Sanicola, M.; Hession, C.; Worley, D.; Carmillo, P.; Ehrenfels, C.; Walus, L.; Robinson, S.; Jaworski, G.; Wei, H.; TIzard, R.; et al. Glial cell line-derived neurotrophic factor-dependent RET activation can be mediated by two different cell-surface accessory proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 6238–6243. [Google Scholar]
- Golden, J.P.; Baloh, R.H.; Kotzbauer, P.T.; Lampe, P.A.; Osborne, P.A.; Milbrandt, J.; Johnson, E.M., Jr. Expression of neurturin, GDNF and their receptors in the adult mouse CNS. J. Comp. Neurol. 1998, 398, 139–150. [Google Scholar] [CrossRef]
- Cik, M.; Masure, S.; Lesage, A.S.; van der Linden, I.; van Gompel, P.; Pangalos, M.N.; Gordon, R.D.; Leysen, J.E. Binding of GDNF and neurturin to human GDNF family receptor alpha 1 and 2. Influence of cRET and cooperative interactions. J. Biol. Chem. 2000, 275, 27505–27512. [Google Scholar]
- Widenfalk, J.; Nosrat, C.; Tomac, A.; Westphal, H.; Hoffer, B.; Olson, L. Neurturin and glial cell line-derived neurotrophic factor receptor-beta (GDNFR-beta), novel proteins related to GDNF and GDNFR-alpha with specific cellular patterns of expression suggesting roles in the developing and adult nervous system and in peripheral organs. J. Neurosci. 1997, 17, 8506–8519. [Google Scholar]
- Leitner, M.L.; Molliver, D.C.; Osborne, P.A.; Vejsada, R.; Golden, J.P.; Lampe, P.A.; Kato, A.C.; Mildbrant, J.; Johnson, E.M., Jr. Analysis of the retrograde transport of glial cell line-derived neurotrophic factor (GDNF), neurturin, and persephin suggests that in vivo signaling for the GDNF family is GFRalpha coreceptor-specific. J. Neurosci. 1999, 19, 9322–9331. [Google Scholar]
- Schaar, D.G.; Sieber, B.A.; Dreyfus, C.F.; Black, I.B. Regional and cell-specific expression of GDNF in rat brain. Exp. Neurol. 1993, 124, 368–371. [Google Scholar] [CrossRef]
- Stromberg, I.; Bjorklund, L.; Johansson, M.; Tomac, A.; Collins, F.; Olson, L.; Hoffer, B.; Humpel, C. Glial cell line-derived neurotrophic factor is expressed in the developing but not adult striatum and stimulates developing dopamine neurons in vivo. Exp. Neurol. 1993, 124, 401–412. [Google Scholar] [CrossRef]
- Springer, J.; Bergmann, L.; Mu, X.; Trojanowski, J. Expression of GDNF mRNA in rat and human nervous tissue. Exp. Neurol. 1994, 127, 167–170. [Google Scholar] [CrossRef]
- Suvanto, P.; Hiltunen, J.; Moshnyakov, M.; Sariola, H.; Sainio, K.; Saarma, M. Localization of glial cell line-derived neurotrophic factor (GDNF) mRNA in embryonic rat by in situ hybridization. Eur. J. Neurosci. 1996, 8, 816–822. [Google Scholar] [CrossRef]
- Trupp, M.; Belluardo, N.; Funakoshi, H.; Ibanez, C. Complementary and overlapping expression of glial cell line-derived neurotrophic factor (GDNF), c-ret proto-oncogene, and GDNF receptor-alpha indicates multiple mechanisms of trophic actions in the adult rat CNS. J. Neurosci. 1997, 17, 3554–3567. [Google Scholar]
- Chauhan, N.B.; Siegel, G.J.; Lee, J.M. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson’s disease brain. J. Chem. Neuroanat. 2001, 21, 277–288. [Google Scholar] [CrossRef]
- Sarabi, A.; Hoffer, B.J.; Olson, L.; Morales, M. GFRalpha-1 mRNA in dopaminergic and nondopaminergic neurons in the substantia nigra and ventral tegmental area. J. Comp. Neurol. 2001, 441, 106–117. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Soderstrom, D.E.; Kordower, J.H. Trophic factors therapy in Parkinson’s disease. Prog. Brain Res. 2009, 175, 201–218. [Google Scholar] [CrossRef]
- Gasmi, M.; Herzog, C.D.; Brandon, E.P.; Cunningham, J.J.; Ramirez, G.A.; Ketchum, E.T.; Bartus, R.T. Striatal delivery of neurturin by CERE-120, an AAV2 vector for the treatment of dopaminergic neuron degeneration in Parkinson’s disease. Mol. Ther. 2007, 15, 62–68. [Google Scholar]
- Horger, B.A.; Nishimura, M.C.; Armanini, M.P.; Wang, L.C.; Poulsen, K.T.; Rosenblad, C.; Kirik, D.; Moffat, B.; Simmons, L.; Johnson, E., Jr.; et al. Neurturin exerts potent actions on survival and function of midbrain dopaminergic neurons. J. Neurosci. 1998, 18, 4929–4937. [Google Scholar]
- Akerud, P.; Alberch, J.; Eketjall, S.; Wagner, J.; Arenas, E. Differential effects of glial cell line-derived neurotrophic factor and neurturin on developing and adult substantia nigra dopaminergic neurons. J. Neurochem. 1999, 733, 70–78. [Google Scholar]
- Kordower, J.H.; Herzog, C.D.; Dass, B.; Bakay, R.A.; Stansell, J., 3rd; Gasmi, M.; Bartus, R.T. Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann. Neurol. 2006, 60, 706–715. [Google Scholar] [CrossRef]
- Eslamboli, A.; Georgievska, B.; Ridley, R.M.; Baker, H.F.; Muzyczka, N.; Burger, C.; Mandel, R.J.; Annett, L.; Kirik, D. Continuous low-level glial cell line-derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson’s disease. J. Neurosci. 2005, 25, 769–777. [Google Scholar]
- Gash, D.M.; Zhang, Z.; Ovadia, A.; Cass, W.A.; Yi, A.; Simmerman, L.; Russell, D.; Martin, D.; Lapchak, P.A.; Collins, F.; et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature 1996, 380, 252–255. [Google Scholar]
- Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.; Leventhal, L.; McBride, J.; Chen, E.Y.; Palfi, S.; Roitberg, B.Z.; et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 2000, 290, 767–773. [Google Scholar]
- Dass, B.; Kladis, T.; Chu, Y.; Kordower, J.H. RET expression does not change with age in the substantia nigra pars compacta of rhesus monkeys. Neurobiol. Aging 2006, 27, 857–861. [Google Scholar] [CrossRef]
- Herzog, C.D.; Dass, B.; Holden, J.E.; Stansell, J., 3rd; Gasmi, M.; Tuszynski, M.H.; Bartus, R.T.; Kordower, J.H. Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Mov. Disord. 2007, 22, 1124–1132. [Google Scholar] [CrossRef]
- Hidalgo-Figueroa, M.; Bonilla, S.; Gutierrez, F.; Pascual, A.; Lopez-Barneo, J. GDNF is predominantly expressed in the PV+ neostriatal interneuronal ensemble in normal mouse and after injury of the nigrostriatal pathway. J. Neurosci. 2012, 32, 864–872. [Google Scholar] [CrossRef]
- Backman, C.M.; Shan, L.; Zhang, Y.J.; Hoffer, B.J.; Leonard, S.; Troncoso, J.C.; Vonsatel, P.; Tomac, A.C. Gene expression patterns for GDNF and its receptors in the human putamen affected by Parkinson’s disease: A real-time PCR study. Mol. Cell Endocrinol. 2006, 252, 160–166. [Google Scholar] [CrossRef]
- Walker, D.G.; Beach, T.G.; Xu, R.; Lile, J.; Beck, K.D.; McGeer, E.G.; McGeer, P.L. Expression of the proto-oncogene Ret, a component of the GDNF receptor complex, persists in human substantia nigra neurons in Parkinson’s disease. Brain Res. 1998, 792, 207–217. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Zhang, J.L.; Large, D.M.; Wilson, P.E.; Gash, C.R.; Thomas, T.C.; Haycock, J.W.; Bing, G.; Stanford, J.A.; Gash, D.M.; et al. Striatal GDNF administration increases tyrosine hydroxylase phosphorylation in the rat striatum and substantia nigra. J. Neurochem. 2004, 90, 245–254. [Google Scholar] [CrossRef]
- Lapchak, P.A. A preclinical development strategy designed to optimize the use of glial cell line-derived neurotrophic factor in the treatment of Parkinson’s disease. Mov. Disord. 1998, 13, 49–54. [Google Scholar]
- Kordower, J.H. In vivo gene delivery of glial cell line--derived neurotrophic factor for Parkinson’s disease. Ann. Neurol. 2003, 53, S120–S132. [Google Scholar] [CrossRef]
- Yu, T.; Scully, S.; Yu, Y.; Fox, G.M.; Jing, S.; Zhou, R. Expression of GDNF family receptor components during development: Implications in the mechanisms of interaction. J. Neurosci. 1998, 18, 4684–4696. [Google Scholar]
- Love, S.; Plaha, P.; Patel, N.K.; Hotton, G.R.; Brooks, D.J.; Gill, S.S. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat. Med. 2005, 11, 703–705. [Google Scholar]
- Slevin, J.T.; Gash, D.M.; Smith, C.D.; Gerhardt, G.A.; Kryscio, R.; Chebrolu, H.; Walton, A.; Wagner, R.; Young, A.B. Unilateral intraputamenal glial cell line-derived neurotrophic factor in patients with Parkinson disease: Response to 1 year of treatment and 1 year of withdrawal. J. Neurosurg. 2007, 106, 614–620. [Google Scholar]
- Hovland, D.N.; Boyd, R.B.; Butt, M.T.; Engelhardt, J.A.; Moxness, M.S.; Ma, M.H.; Emery, M.G.; Ernst, N.B.; Reed, R.P.; Zeller, J.R.; et al. Six-month continuous intraputamenal infusion toxicity study of recombinant methionyl human glial cell line-derived neurotrophic factor (r-metHuGDNF) in rhesus monkeys. Toxicol. Pathol. 2007, 35, 676–692. [Google Scholar] [CrossRef]
- Barker, R.A. Continuing trials of GDNF in Parkinson’s disease. Lancet Neurol. 2006, 5, 285–286. [Google Scholar] [CrossRef]
- Penn, R.D.; Dalvi, A.; Slevin, J.; Young, B.; Gash, D.; Gerhardt, G.; Hutchinson, M. GDNF in treatment of Parkinson’s disease: Response to editorial. Lancet Neurol. 2006, 5, 202–203. [Google Scholar] [CrossRef]
- Lang, A.E.; Langston, J.W.; Stoessl, A.J.; Brodsky, M.; Brooks, D.J.; Dhawan, V.; Elias, W.J.; Lozano, A.M.; Moro, E.; Nutt, J.G.; et al. GDNF in treatment of Parkinson’s disease: Response to editorial. Lancet Neurol. 2006, 5, 200–202. [Google Scholar] [CrossRef]
- AMT. Press Release: AMT Obtains License to Amgen’s GDNF Gene to Develop Treatment for Parkinson’s Disease with AMT’s Proprietary Gene Therapy Platform (09/18/08). In Euronext News; NYSE Euronext: Amsterdam, The Netherland, 1998.
- Humphries, C. Keeping neurons alive in Parkinson’s patients. Available online: http://www.technologyreview.com/biomedicine/37708/?mod=chfeatured/ (accessed on 6 June 2011).
- Lonser, R.R. Personal communication, Surgical Neurology Branch: NINDS, Bethesda, MD, USA, 2 March 2012.
- Granholm, A.C.; Reyland, M.; Albeck, D.; Sanders, L.; Gerhardt, G.; Hoernig, G.; Shen, L.; Westphal, H.; Hoffer, B. Glial cell line-derived neurotrophic factor is essential for postnatal survival of midbrain dopamine neurons. J. Neurosci. 2000, 20, 3182–3190. [Google Scholar]
- Pascual, A.; Hidalgo-Figueroa, M.; Piruat, J.I.; Pintado, C.O.; Gomez-Diaz, R.; Lopez-Barneo, J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat. Neurosci. 2008, 11, 755–761. [Google Scholar]
- Choi-Lundberg, D.L.; Lin, Q.; Schallert, T.; Crippens, D.; Davidson, B.L.; Chang, Y.N.; Chiang, Y.L.; Qian, J.; Bardwaj, L.; Bohn, M.C. Behavioral and cellular protection of rat dopaminergic neurons by an adenoviral vector encoding glial cell line-derived neurotrophic factor. Exp. Neurol. 1998, 154, 261–275. [Google Scholar] [CrossRef]
- Connor, B. Adenoviral vector-mediated delivery of glial cell line-derived neurotrophic factor provides neuroprotection in the aged parkinsonian rat. Clin. Exp. Pharmacol. Physiol. 2001, 28, 896–900. [Google Scholar] [CrossRef]
- Eslamboli, A.; Cummings, R.M.; Ridley, R.M.; Baker, H.F.; Muzyczka, N.; Burger, C.; Mandel, R.J.; Kirik, D.; Annett, L.E. Recombinant adeno-associated viral vector (rAAV) delivery of GDNF provides protection against 6-OHDA lesion in the common marmoset monkey (Callithrix jacchus). Exp. Neurol. 2003, 184, 536–548. [Google Scholar] [CrossRef]
- Li, H.; He, Z.; Su, T.; Ma, Y.; Lu, S.; Dai, C.; Sun, M. Protective action of recombinant neurturin on dopaminergic neurons in substantia nigra in a rhesus monkey model of Parkinson’s disease. Neurol. Res. 2003, 25, 263–267. [Google Scholar] [CrossRef]
- Oiwa, Y.; Nakai, K.; Itakura, T. Histological effects of intraputaminal infusion of glial cell line-derived neurotrophic factor in Parkinson disease model macaque monkeys. Neurol. Med. Chir. (Tokyo) 2006, 46, 267–275. [Google Scholar] [CrossRef]
- Kells, A.P.; Eberling, J.; Su, X.; Pivirotto, P.; Bringas, J.; Hadaczek, P.; Narrow, W.C.; Bowers, W.J.; Federoff, H.J.; Forsayeth, J.; et al. Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J. Neurosci. 2010, 30, 9567–9577. [Google Scholar]
- Bankiewicz, K.S.; Oldfield, E.H.; Chiueh, C.C.; Doppman, J.L.; Jacobowitz, D.M.; Kopin, I.J. Hemiparkinsonism in monkeys after unilateral internal carotid artery infusion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Life Sci. 1986, 39, 7–16. [Google Scholar]
- Bankiewicz, K.S.; Sanchez-Pernaute, R.; Oiwa, Y.; Kohutnicka, M.; Cummins, A.; Eberling, J. Preclinical models of Parkinson’s disease. Curr. Protoc. Neurosci. 2001. Chapter 9, Unit 9.4.. [Google Scholar]
- Eberling, J.L.; Bankiewicz, K.S.; Jordan, S.; VanBrocklin, H.F.; Jagust, W.J. PET studies of functional compensation in a primate model of Parkinson’s disease. Neuroreport 1997, 8, 2727–2733. [Google Scholar] [CrossRef]
- Hadaczek, P.; Kohutnicka, M.; Krauze, M.T.; Bringas, J.; Pivirotto, P.; Cunningham, J.; Bankiewicz, K. Convection-enhanced delivery of adeno-associated virus type 2 (AAV2) into the striatum and transport of AAV2 within monkey brain. Hum. Gene Ther. 2006, 17, 291–302. [Google Scholar] [CrossRef]
- Su, X.; Kells, A.P.; Huang, E.J.; Lee, H.S.; Hadaczek, P.; Beyer, J.; Bringas, J.; Pivirotto, P.; Penticuff, J.; Eberling, J.; et al. Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Hum. Gene Ther. 2009, 20, 1627–1640. [Google Scholar] [CrossRef]
- Ciesielska, A.; Mittermeyer, G.; Hadaczek, P.; Kells, A.P.; Forsayeth, J.; Bankiewicz, K.S. Anterograde axonal transport of AAV2-GDNF in rat basal ganglia. Mol. Ther. 2011, 19, 922–927. [Google Scholar] [CrossRef]
- Kells, A.P.; Forsayeth, J.; Bankiewicz, K.S. Glial-derived neurotrophic factor gene transfer for Parkinson’s disease: Anterograde distribution of AAV2 vectors in the primate brain. Neurobiol. Dis. 2011, in press. [Google Scholar]
- Manfredsson, F.P.; Tumer, N.; Erdos, B.; Landa, T.; Broxson, C.S.; Sullivan, L.F.; Rising, A.C.; Foust, K.D.; Zhang, Y.; Muzyczka, N.; et al. Nigrostriatal rAAV-mediated GDNF overexpression induces robust weight loss in a rat model of age-related obesity. Mol. Ther. 2009, 17, 980–991. [Google Scholar] [CrossRef]
- Johnston, L.C.; Eberling, J.; Pivirotto, P.; Hadaczek, P.; Federoff, H.J.; Forsayeth, J.; Bankiewicz, K.S. Clinically relevant effects of convection-enhanced delivery of AAV2-GDNF on the dopaminergic nigrostriatal pathway in aged rhesus monkeys. Hum. Gene Ther. 2009, 20, 497–510. [Google Scholar] [CrossRef]
- Eberling, J.L.; Kells, A.P.; Pivirotto, P.; Beyer, J.; Bringas, J.; Federoff, H.J.; Forsayeth, J.; Bankiewicz, K.S. Functional effects of AAV2-GDNF on the dopaminergic nigrostriatal pathway in Parkinsonian rhesus monkeys. Hum. Gene Ther. 2009, 20, 511–518. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Salegio, E.A.; Yin, D.; Richardson, R.M.; Valles, F.E.; Larson, P.S.; Starr, P.A.; Lonser, R.R.; Bankiewicz, K.S. Human/nonhuman primate AC-PC ratio—Considerations for translational brain measurements. J. Neurosci. Methods 2011, 196, 124–130. [Google Scholar] [CrossRef]
- Watanabe, M.; Maemura, K.; Kanbara, K.; Tamayama, T.; Hayasaki, H. GABA and GABA receptors in the central nervous system and other organs. Int. Rev. Cytol. 2002, 213, 1–47. [Google Scholar] [CrossRef]
- Erlander, M.G.; Tobin, A.J. The structural and functional heterogeneity of glutamic acid decarboxylase: A review. Neurochem. Res. 1991, 16, 215–226. [Google Scholar] [CrossRef]
- Bu, D.F.; Erlander, M.G.; Hitz, B.C.; Tillakaratne, N.J.; Kaufman, D.L.; Wagner-McPherson, C.B.; Evans, G.A.; Tobin, A.J. Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc. Natl. Acad. Sci. USA 1992, 89, 2115–2119. [Google Scholar]
- Kaufman, D.L.; Houser, C.R.; Tobin, A.J. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J. Neurochem. 1991, 56, 720–723. [Google Scholar] [CrossRef]
- Martin, D.L.; Rimvall, K. Regulation of gamma-aminobutyric acid synthesis in the brain. J. Neurochem. 1993, 60, 395–407. [Google Scholar] [CrossRef]
- Pinal, C.S.; Tobin, A.J. Uniqueness and redundancy in GABA production. Perspect. Dev. Neurobiol. 1998, 5, 109–118. [Google Scholar]
- Waagepetersen, H.S.; Sonnewald, U.; Schousboe, A. The GABA paradox: Multiple roles as metabolite, neurotransmitter, and neurodifferentiative agent. J. Neurochem. 1999, 73, 1335–1342. [Google Scholar]
- Lamigeon, C.; Bellier, J.P.; Sacchettoni, S.; Rujano, M.; Jacquemont, B. Enhanced neuronal protection from oxidative stress by coculture with glutamic acid decarboxylase-expressing astrocytes. J. Neurochem. 2001, 77, 598–606. [Google Scholar]
- de Jong, P.J.; Lakke, J.P.; Teelken, A.W. CSF GABA levels in Parkinson’s disease. Adv. Neurol. 1984, 40, 427–430. [Google Scholar]
- Hutchison, W.D.; Allan, R.J.; Opitz, H.; Levy, R.; Dostrovsky, J.O.; Lang, A.E.; Lozano, A.M. Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson’s disease. Ann. Neurol. 1998, 44, 622–628. [Google Scholar] [CrossRef]
- Benazzouz, A.; Gao, D.M.; Ni, Z.G.; Piallat, B.; Bouali-Benazzouz, R.; Benabid, A.L. Effect of high-frequency stimulation of the subthalamic nucleus on the neuronal activities of the substantia nigra pars reticulata and ventrolateral nucleus of the thalamus in the rat. Neuroscience 2000, 99, 289–295. [Google Scholar] [CrossRef]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Rodriguez, M.; Macias, R.; Alvarez, L.; Guridi, J.; Vitek, J.; DeLong, M.R. Pathophysiologic basis of surgery for Parkinson’s disease. Neurology 2000, 55, S7–S12. [Google Scholar]
- Su, P.C.; Ma, Y.; Fukuda, M.; Mentis, M.J.; Tseng, H.M.; Yen, R.F.; Liu, H.M.; Moeller, J.R.; Eidelberg, D. Metabolic changes following subthalamotomy for advanced Parkinson’s disease. Ann. Neurol. 2001, 50, 514–520. [Google Scholar] [CrossRef]
- Gill, S.S.; Heywood, P. Bilateral dorsolateral subthalamotomy for advanced Parkinson’s disease. Lancet 1997, 350, 1224. [Google Scholar]
- Alvarez, L.; Macias, R.; Guridi, J.; Lopez, G.; Alvarez, E.; Maragoto, C.; Teijeiro, J.; Torres, A.; Pavon, N.; Rodriguez-Oroz, M.C.; et al. Dorsal subthalamotomy for Parkinson’s disease. Mov. Disord. 2001, 16, 72–78. [Google Scholar] [CrossRef]
- Limousin, P.; Pollak, P.; Benazzouz, A.; Hoffmann, D.; Le Bas, J.F.; Broussolle, E.; Perret, J.E.; Benabid, A.L. Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet 1995, 345, 91–95. [Google Scholar]
- Group, D.S. Deep-brain stimulation of the subthalamic nucleus or the pars interna of the globus pallidus in Parkinson’s disease. N. Engl. J. Med. 2001, 345, 956–963. [Google Scholar] [CrossRef]
- Levy, R.; Lang, A.E.; Dostrovsky, J.O.; Pahapill, P.; Romas, J.; Saint-Cyr, J.; Hutchison, W.D.; Lozano, A.M. Lidocaine and muscimol microinjections in subthalamic nucleus reverse Parkinsonian symptoms. Brain 2001, 124, 2105–2118. [Google Scholar] [CrossRef]
- Luo, J.; Kaplitt, M.G.; Fitzsimons, H.L.; Zuzga, D.S.; Liu, Y.; Oshinsky, M.L.; During, M.J. Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science 2002, 298, 425–429. [Google Scholar]
- Scherer, L.J.; McPherson, J.D.; Wasmuth, J.J.; Marsh, J.L. Human dopa decarboxylase: Localization to human chromosome 7p11 and characterization of hepatic cDNAs. Genomics 1992, 13, 469–471. [Google Scholar]
- Nagatsu, T. Molecular biology of dopamine systems. In Controversies in the Treatment of Parkinson’s Disease; Rinne, U.K., Yanagisawa, N., Eds.; PMSI: Tokyo, Japan, 1992; pp. 15–26. [Google Scholar]
- Lloyd, K.G.; Davidson, L.; Hornykiewicz, O. The neurochemistry of Parkinson’s disease: Effect of L-dopa therapy. J. Pharmacol. Exp. Ther. 1975, 195, 453–464. [Google Scholar]
- Marsden, C.D.; Parkes, J.D. Success and problems of long-term levodopa therapy in Parkinson’s disease. Lancet 1977, 1, 345–349. [Google Scholar]
- Bankiewicz, K.S.; Daadi, M.; Pivirotto, P.; Bringas, J.; Sanftner, L.; Cunningham, J.; Forsayeth, J.R.; Eberling, J.L. Focal striatal dopamine may potentiate dyskinesias in parkinsonian monkeys. Exp. Neurol. 2006, 197, 363–372. [Google Scholar] [CrossRef]
- di Stefano, A.; Sozio, P.; Cerasa, L.S. Antiparkinson prodrugs. Molecules 2008, 13, 46–68. [Google Scholar] [CrossRef]
- Sanchez-Pernaute, R.; Harvey-White, J.; Cunningham, J.; Bankiewicz, K.S. Functional effect of adeno-associated virus mediated gene transfer of aromatic L-amino acid decarboxylase into the striatum of 6-OHDA-lesioned rats. Mol. Ther. 2001, 4, 324–330. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Forsayeth, J.; Eberling, J.L.; Sanchez-Pernaute, R.; Pivirotto, P.; Bringas, J.; Herscovitch, P.; Carson, R.E.; Eckelman, W.; Reutter, B.; et al. Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol. Ther. 2006, 14, 564–570. [Google Scholar] [CrossRef]
- Eberling, J.L.; Jagust, W.J.; Taylor, S.; Bringas, J.; Pivirotto, P.; VanBrocklin, H.F.; Bankiewicz, K.S. A novel MPTP primate model of Parkinson’s disease: Neurochemical and clinical changes. Brain Res. 1998, 805, 259–262. [Google Scholar] [CrossRef]
- Doudet, D.J.; Chan, G.L.; Jivan, S.; DeJesus, O.T.; McGeer, E.G.; English, C.; Ruth, T.J.; Holden, J.E. Evaluation of dopaminergic presynaptic integrity: 6-[18F]Fluoro-L-dopa versus 6-[18F]fluoro-L-m-tyrosine. J. Cereb. Blood Flow Metab. 1999, 19, 278–287. [Google Scholar]
- Forsayeth, J.R.; Eberling, J.L.; Sanftner, L.M.; Zhen, Z.; Pivirotto, P.; Bringas, J.; Cunningham, J.; Bankiewicz, K.S. A dose-ranging study of AAV-hAADC therapy in Parkinsonian monkeys. Mol. Ther. 2006, 14, 571–577. [Google Scholar] [CrossRef]
- Bankiewicz, K.S. Personal communication, Department of Neurological Surgery: UCSF, San Francisco, CA, USA, 2012.
- Elsworth, J.D.; Roth, R.H. Dopamine synthesis, uptake, metabolism, and receptors: Relevance to gene therapy of Parkinson’s disease. Exp. Neurol. 1997, 144, 4–9. [Google Scholar] [CrossRef]
- Nagatsu, T.; Ichinose, H. GTP cyclohydrolase I gene, dystonia, juvenile parkinsonism, and Parkinson’s disease. J. Neural. Transm. Suppl. 1997, 49, 203–209. [Google Scholar]
- Nagatsu, T.; Ichinose, H. Regulation of pteridine-requiring enzymes by the cofactor tetrahydrobiopterin. Mol. Neurobiol. 1999, 19, 79–96. [Google Scholar] [CrossRef]
- Nagatsu, T.; Yamaguchi, T.; Rahman, M.K.; Trocewicz, J.; Oka, K.; Hirata, Y.; Nagatsu, I.; Narabayashi, H.; Kondo, T.; Iizuka, R. Catecholamine-related enzymes and the biopterin cofactor in Parkinson’s disease and related extrapyramidal diseases. Adv. Neurol. 1984, 40, 467–473. [Google Scholar]
- Wolff, J.A.; Fisher, L.J.; Xu, L. Grafting fibroblasts genetically modified to produce L-dopa in a rat model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 1989, 86, 9011–9014. [Google Scholar]
- Uchida, K.; Takamatsu, K.; Kaneda, N.; Toya, S.; Tsukada, Y.; Kurosawa, Y.; Fujita, K.; Nagatsu, T.; Kohsaka, S. Synthesis of L-3,4-dihydroxyphenylalanine by tyrosine hydroxylase cDNA-transfected C6 cells: Application for intracerebral grafting. J. Neurochem. 1989, 53, 728–732. [Google Scholar] [CrossRef]
- Horellou, P.; Brundin, P.; Kalén, P.; Mallet, J.; Björklund, A. In vivo release of DOPA and dopamine from genetically engineered cells grafted to the denervated rat striatum. Neuron 1990, 5, 393–402. [Google Scholar] [CrossRef]
- During, M.J.; Naegele, J.R.; O’Malley, K.L.; Geller, A.I. Long-term behavioral recovery in parkinsonian rats by an HSV vector expressing tyrosine hydroxylase. Science 1994, 266, 1399–1403. [Google Scholar]
- Kaplitt, M.G.; Leone, P.; Samulski, R.J.; Xiao, X.; Pfaff, D.W.; O’Malley, K.L.; During, M.J. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet. 1994, 8, 148–154. [Google Scholar] [CrossRef]
- Bencsics, C.; Wachtel, S.R.; Milstien, S.; Hatakeyama, K.; Becker, J.B.; Kang, U.J. Double transduction with GTP cyclohydrolase I and tyrosine hydroxylase is necessary for spontaneous synthesis of L-dopa by primary fibroblasts. J. Neurosci. 1996, 16, 4449–4456. [Google Scholar]
- Mandel, R.J.; Rendahl, K.G.; Spratt, S.K.; Snyder, R.O.; Cohen, L.K.; Leff, S.E. Characterization of intrastriatal recombinant adeno-associated virus-mediated gene transfer of human tyrosine hydroxylase and human GTP-cyclohydrolase I in a rat model of Parkinson’s disease. J. Neurosci. 1998, 18, 4271–4284. [Google Scholar]
- Shen, Y.; Muramatsu, S.I.; Ikeguchi, K.; Fujimoto, K.I.; Fan, D.S.; Ogawa, M.; Mizukami, H.; Urabe, M.; Kume, A.; Nagatsu, I.; et al. Triple transduction with adeno-associated virus vectors expressing tyrosine hydroxylase, aromatic-L-amino-acid decarboxylase, and GTP cyclohydrolase I for gene therapy of Parkinson’s disease. Hum. Gene Ther. 2000, 11, 1509–1519. [Google Scholar] [CrossRef]
- Sun, M.; Kong, L.; Wang, X.; Holmes, C.; Gao, Q.; Zhang, G.R.; Pfeilschifter, J.; Goldstein, D.S.; Geller, A.I. Coexpression of tyrosine hydroxylase, GTP cyclohydrolase I, aromatic amino acid decarboxylase, and vesicular monoamine transporter 2 from a helper virus-free herpes simplex virus type 1 vector supports high-level, long-term biochemical and behavioral correction of a rat model of Parkinson’s disease. Hum. Gene Ther. 2004, 15, 1177–1196. [Google Scholar] [CrossRef]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef]
- Rivera, V.M.; Clackson, T.; Natesan, S.; Pollock, R.; Amara, J.F.; Keenan, T.; Magari, S.R.; Phillips, T.; Courage, N.L.; Cerasoli, F., Jr.; et al. A humanized system for pharmacologic control of gene expression. Nat. Med. 1996, 2, 1028–1032. [Google Scholar] [CrossRef]
- Sanftner, L.M.; Rivera, V.M.; Suzuki, B.M.; Feng, L.; Berk, L.; Zhou, S.; Forsayeth, J.R.; Clackson, T.; Cunningham, J. Dimerizer regulation of AADC expression and behavioral response in AAV-transduced 6-OHDA lesioned rats. Mol. Ther. 2006, 13, 167–174. [Google Scholar]
- Manfredsson, F.P.; Burger, C.; Rising, A.C.; Zuobi-Hasona, K.; Sullivan, L.F.; Lewin, A.S.; Huang, J.; Piercefield, E.; Muzyczka, N.; Mandel, R.J. Tight long-term dynamic doxycycline responsive nigrostriatal GDNF using a single rAAV vector. Mol. Ther. 2009, 17, 1857–1867. [Google Scholar] [CrossRef]
- Hadaczek, P.; Beyer, J.; Kells, A.; Narrow, W.; Bowers, W.; Federoff, H.J.; Forsayeth, J.; Bankiewicz, K.S. Evaluation of an AAV2-Based Rapamycin-Regulated Glial Cell Line-Derived Neurotrophic Factor (GDNF) Expression Vector System. PloS One 2011, 6, e27728. [Google Scholar]
- Le Guiner, C.; Stieger, K.; Snyder, R.O.; Rolling, F.; Moullier, P. Immune responses to gene product of inducible promoters. Curr. Gene Ther. 2007, 7, 334–346. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar]
- Kordower, J.H.; Olanow, C.W. Regulateable promoters and gene therapy for Parkinson’s disease: Is the only thing to fear, fear itself? Exp. Neurol. 2008, 209, 34–40. [Google Scholar] [CrossRef]
- Cress, D.E. The need for regulatable vectors for gene therapy for Parkinson’s disease. Exp. Neurol. 2008, 209, 30–33. [Google Scholar] [CrossRef]
- Multiauthored Discussion: Regulated GDNF Vectors. 2009. Available online: http://www.pdonlineresearch.org/responses/10602/345/regulated-gdnf-vectors/ (accessed on 5 July 2009).
- Hadaczek, P.; Johnston, L.; Forsayeth, J.; Bankiewicz, K.S. Pharmacokinetics and bioactivity of glial cell line-derived factor (GDNF) and neurturin (NTN) infused in rat brain. Neuropharmacology 2010, 58, 1114–1121. [Google Scholar] [CrossRef]
- Patel, N.K.; Bunnage, M.; Plaha, P.; Svendsen, C.N.; Heywood, P.; Gill, S.S. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: A two-year outcome study. Ann. Neurol. 2005, 57, 298–302. [Google Scholar] [CrossRef]
- Auricchio, A.; Rivera, V.M.; Clackson, T.; O’Connor, E.E.; Maguire, A.M.; Tolentino, M.J.; Bennett, J.; Wilson, J.M. Pharmacological regulation of protein expression from adeno-associated viral vectors in the eye. Mol. Ther. 2002, 6, 238–242. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fiandaca, M.S.; Bankiewicz, K.S.; Federoff, H.J. Gene Therapy for the Treatment of Parkinson’s Disease: The Nature of the Biologics Expands the Future Indications. Pharmaceuticals 2012, 5, 553-590. https://doi.org/10.3390/ph5060553
Fiandaca MS, Bankiewicz KS, Federoff HJ. Gene Therapy for the Treatment of Parkinson’s Disease: The Nature of the Biologics Expands the Future Indications. Pharmaceuticals. 2012; 5(6):553-590. https://doi.org/10.3390/ph5060553
Chicago/Turabian StyleFiandaca, Massimo S., Krystof S. Bankiewicz, and Howard J. Federoff. 2012. "Gene Therapy for the Treatment of Parkinson’s Disease: The Nature of the Biologics Expands the Future Indications" Pharmaceuticals 5, no. 6: 553-590. https://doi.org/10.3390/ph5060553
APA StyleFiandaca, M. S., Bankiewicz, K. S., & Federoff, H. J. (2012). Gene Therapy for the Treatment of Parkinson’s Disease: The Nature of the Biologics Expands the Future Indications. Pharmaceuticals, 5(6), 553-590. https://doi.org/10.3390/ph5060553