Protein Kinases as Drug Development Targets for Heart Disease Therapy

Abstract
:1. Introduction
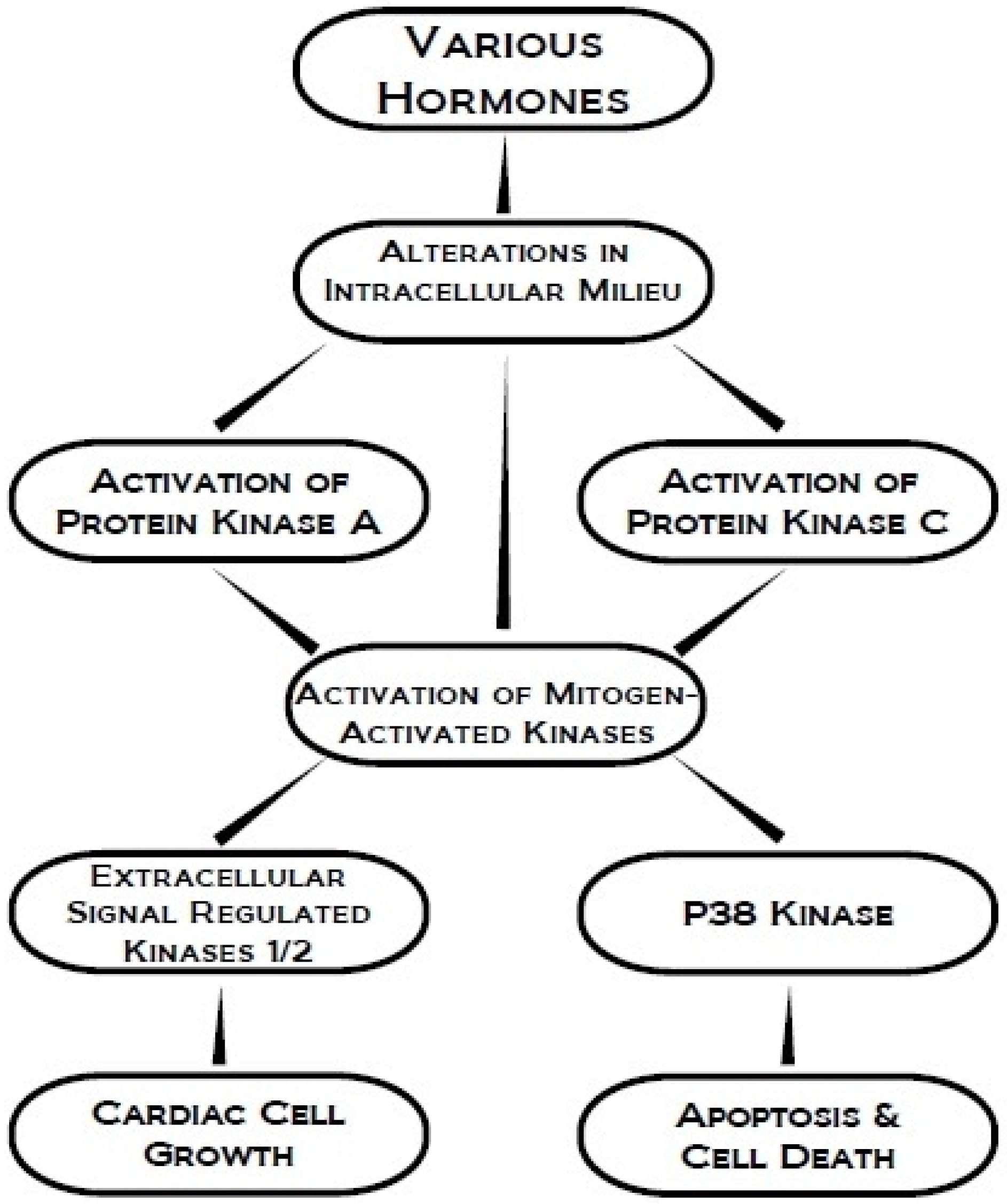
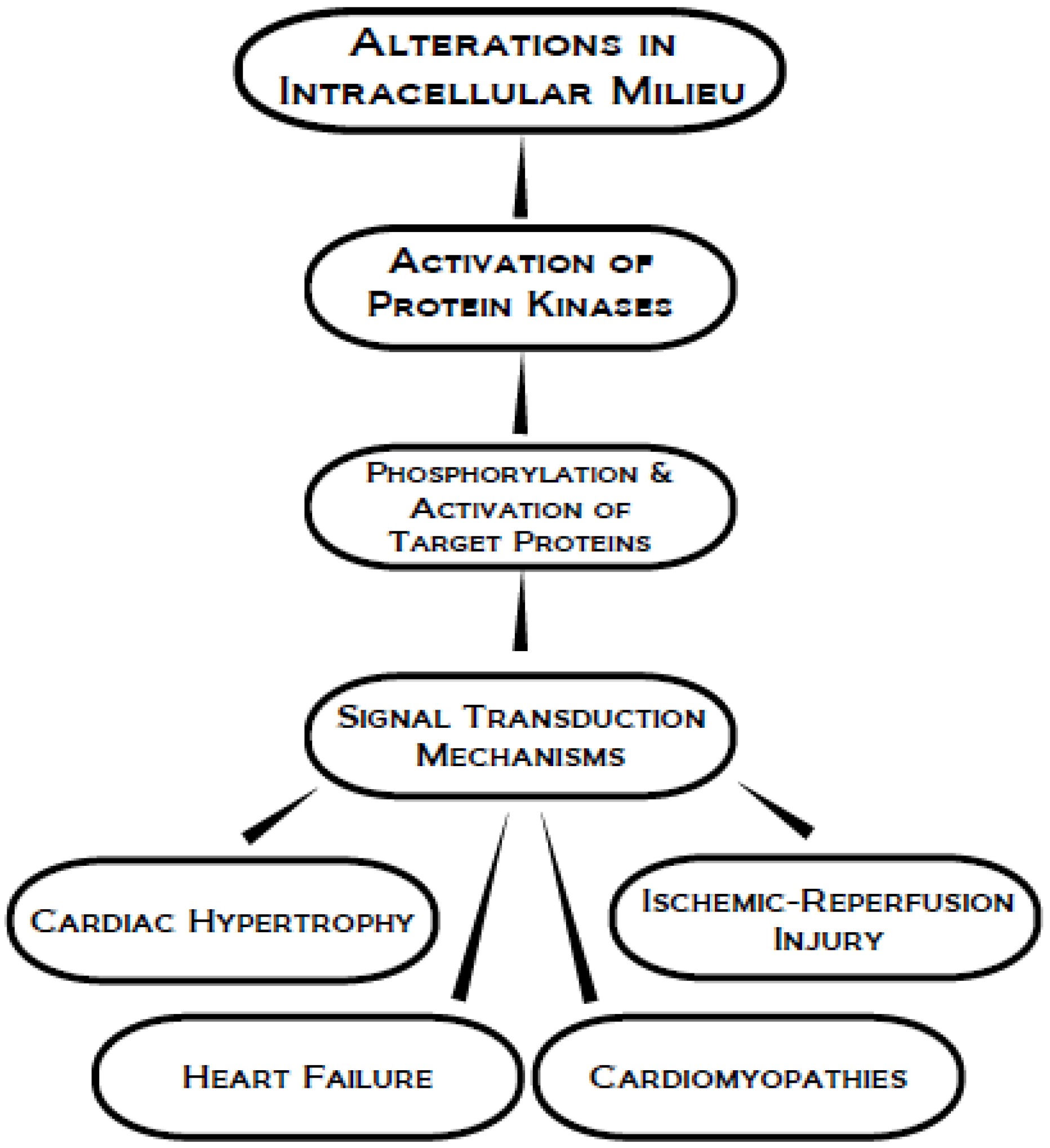
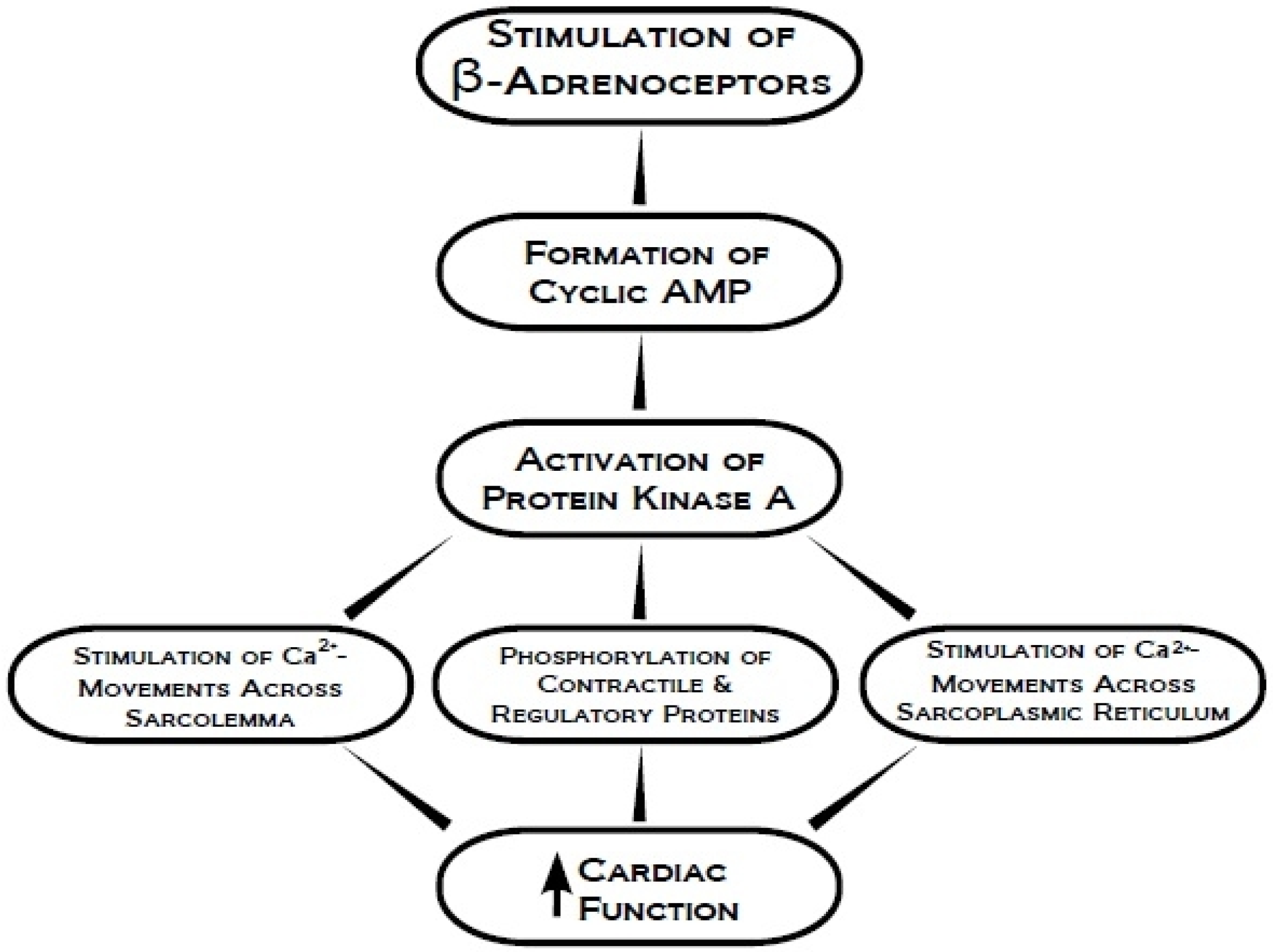
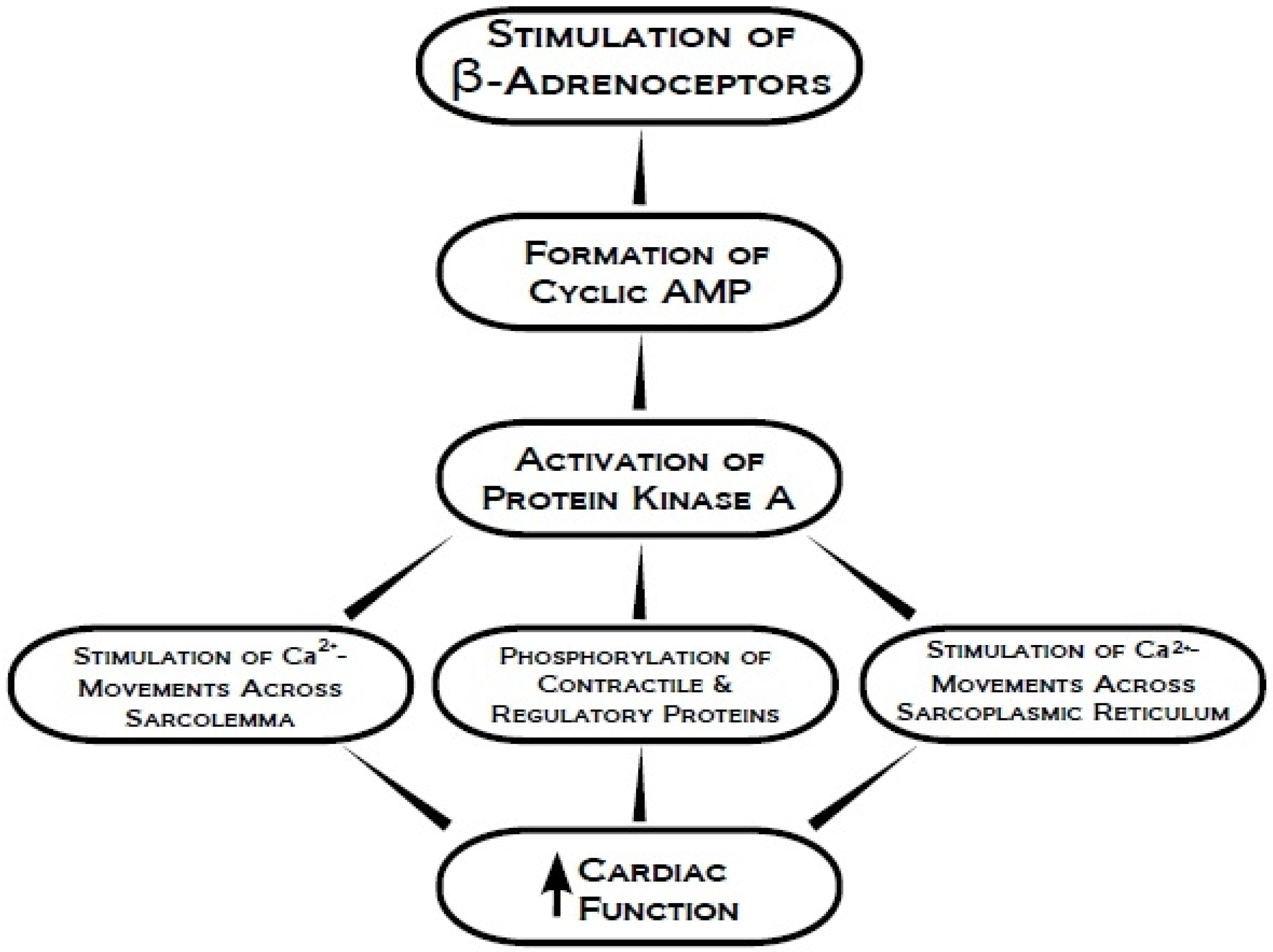
2. Protein Kinase A
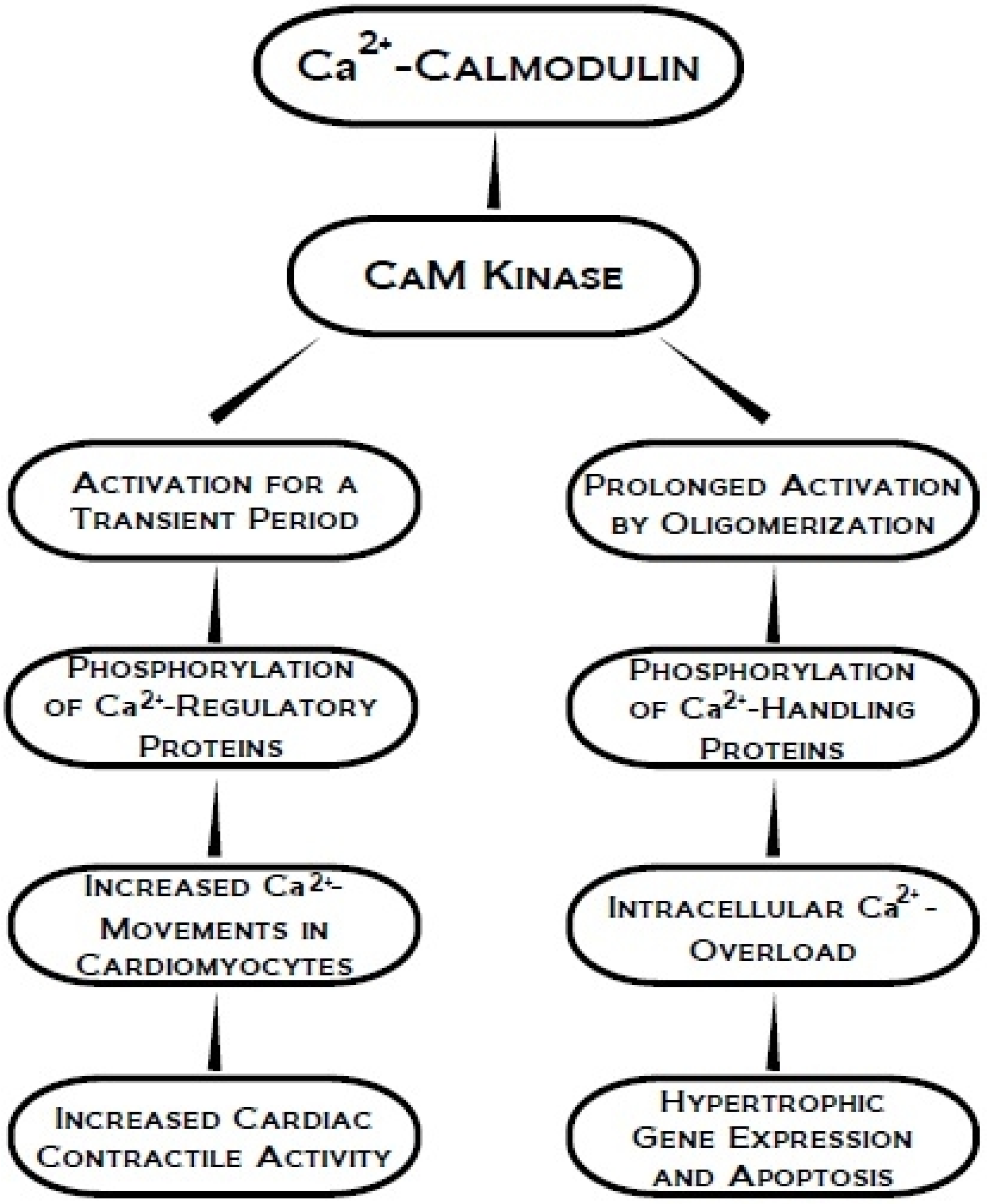
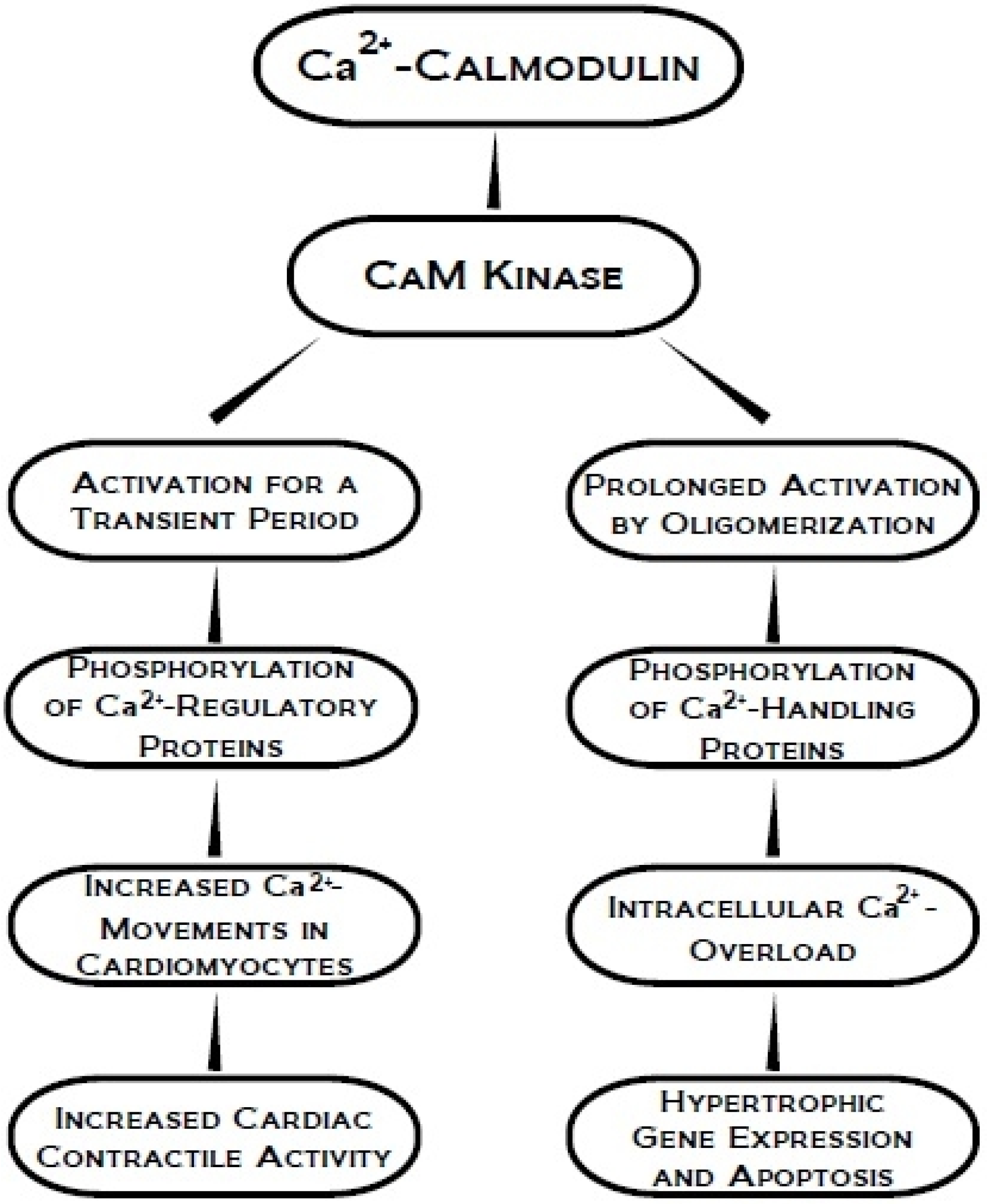
3. Ca2+-Calmodulin Dependent Kinase
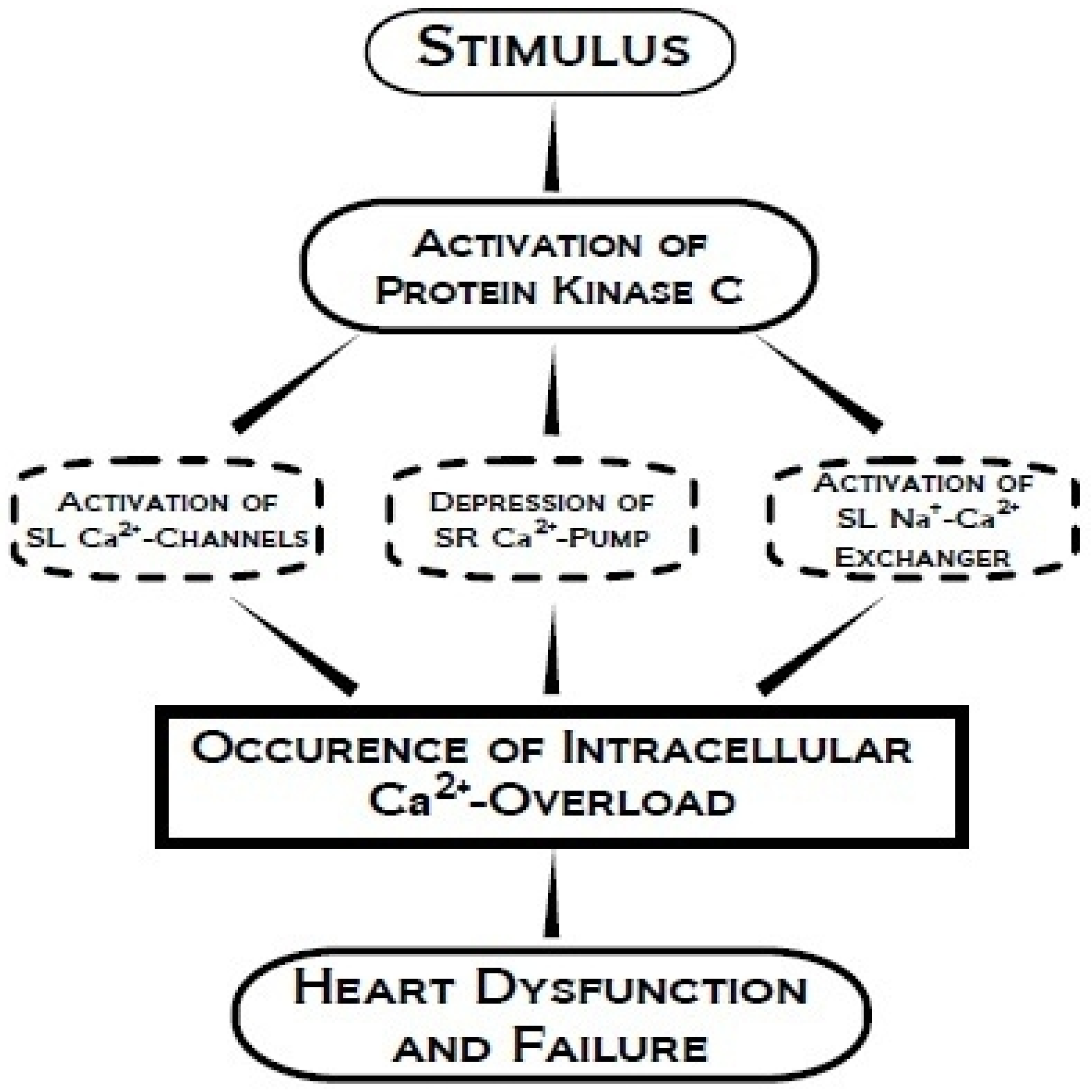
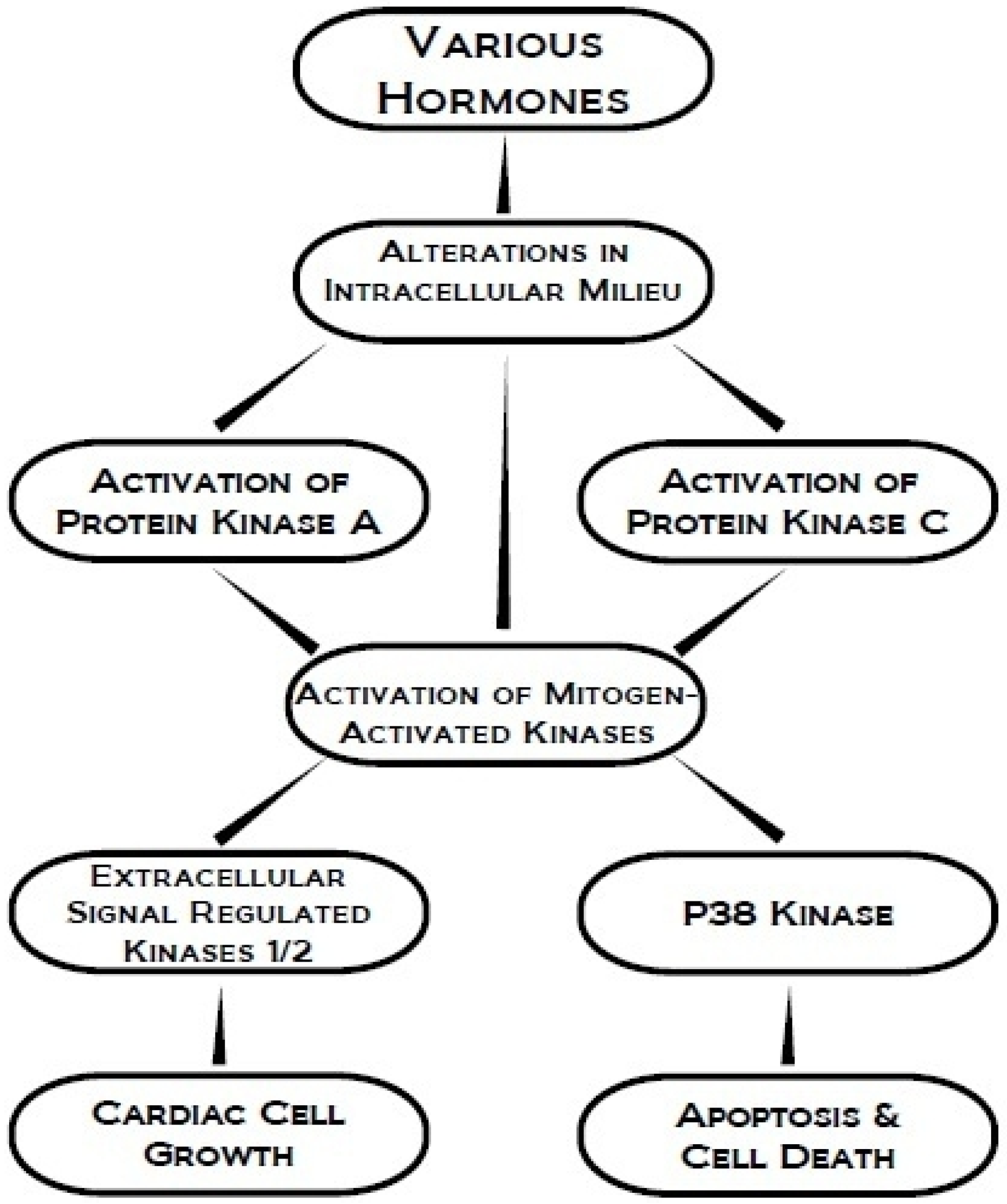
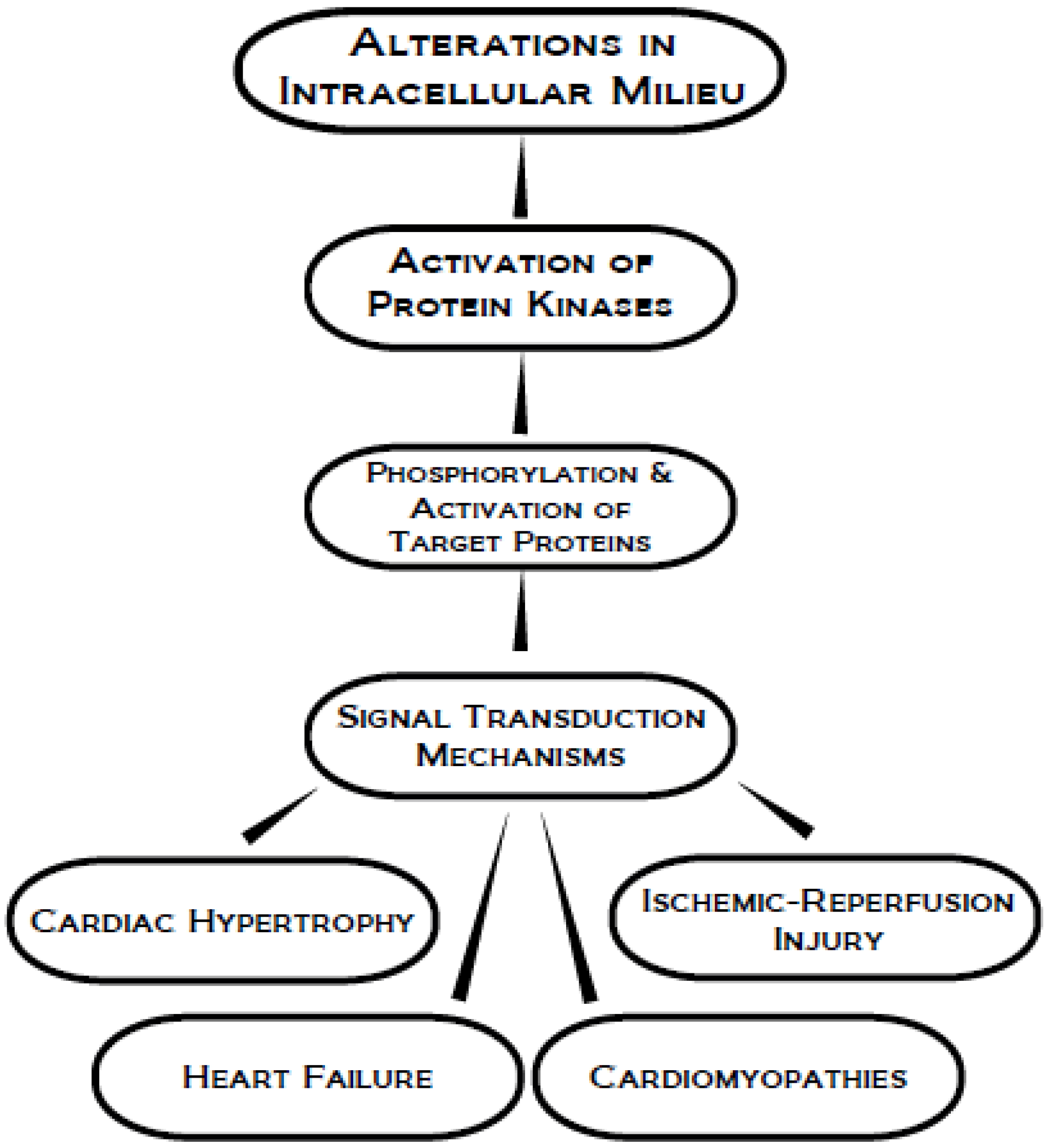
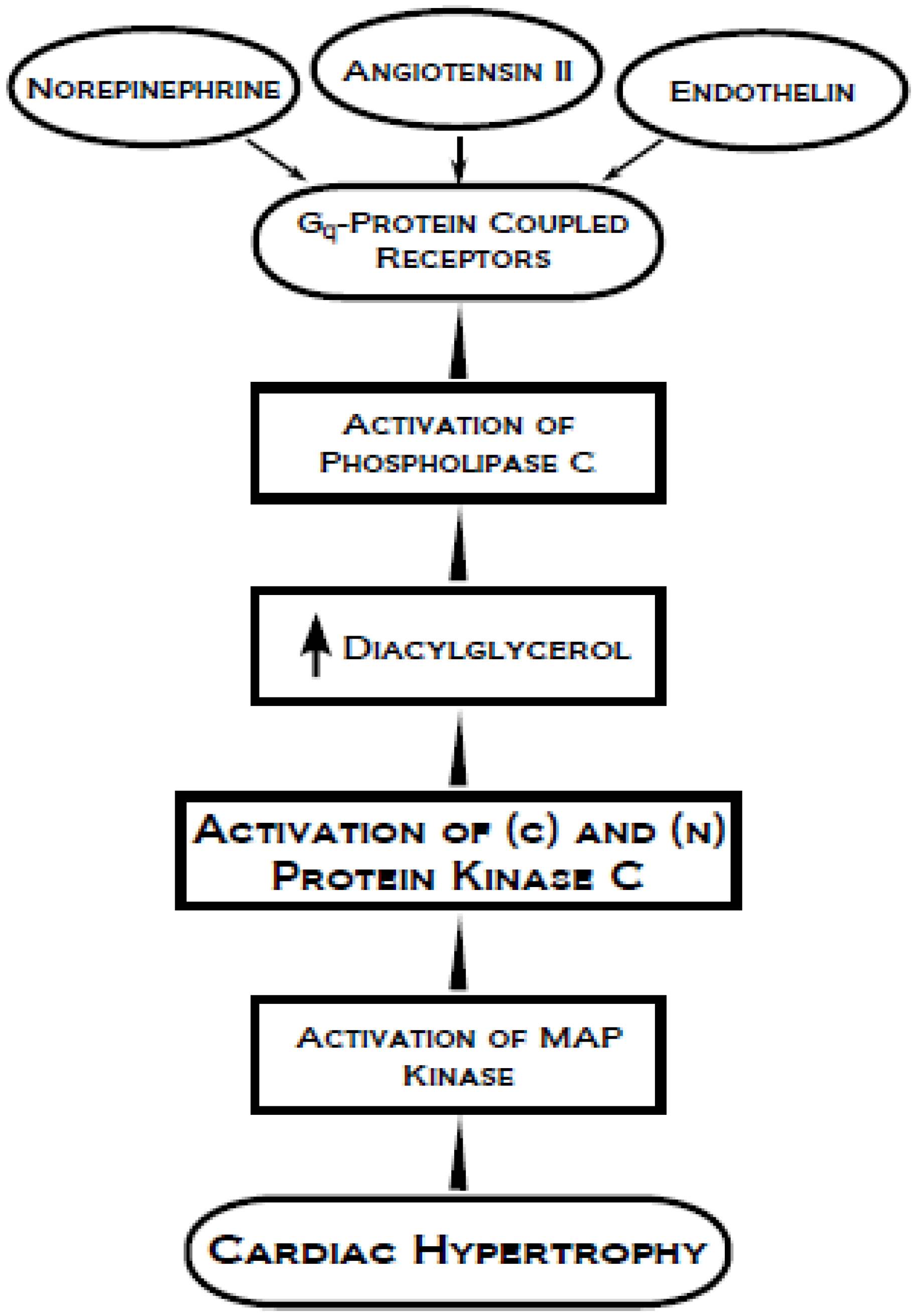
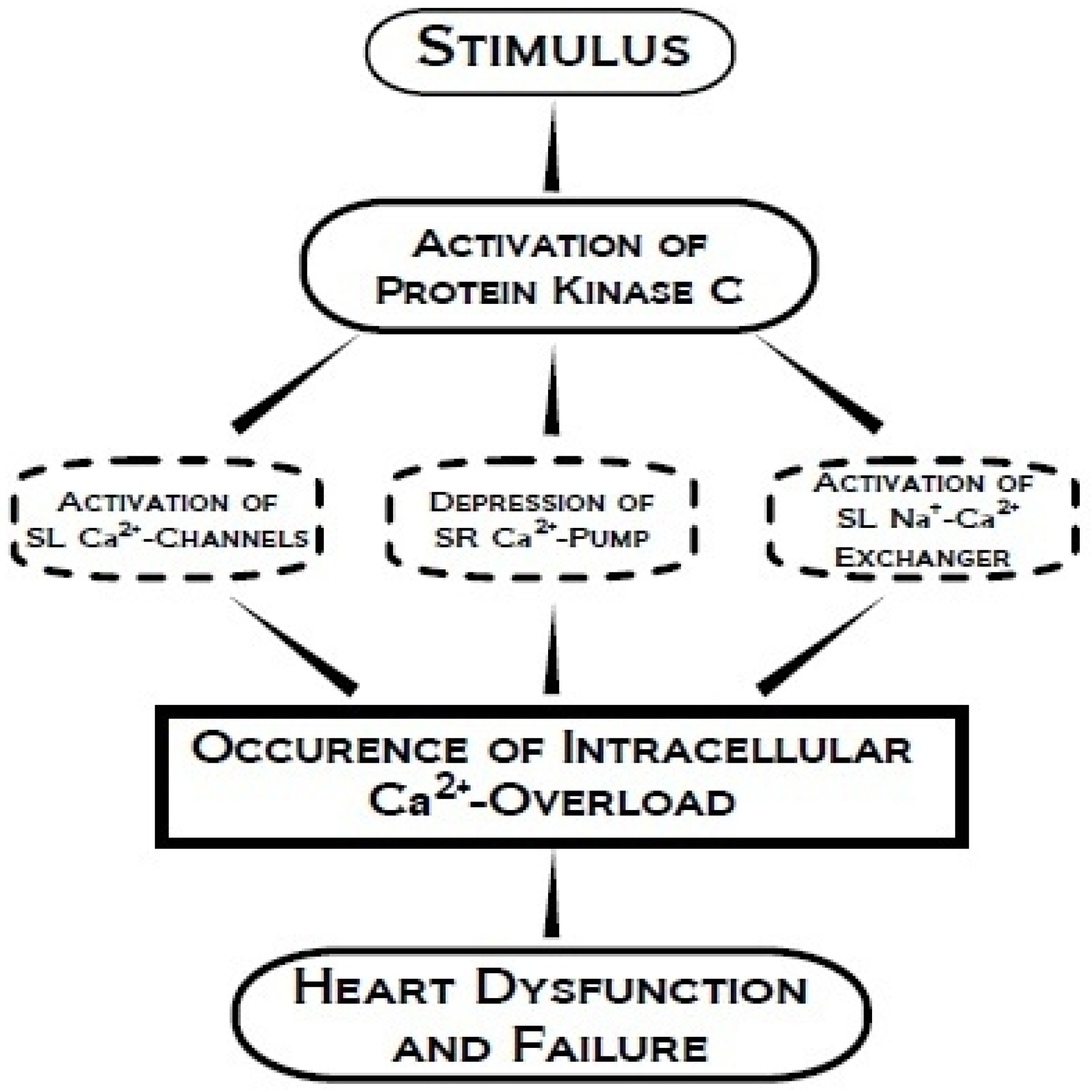
4. Protein Kinase C

5. Phosphoinositide 3-Kinase
6. P38 Mitogen-Activated Kinase
7. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Kinase | Substrates | References |
|---|---|---|
| Protein kinase A | PLB, RyR2, cTn1, cMyBP, L-type Ca2+ channels | [10,11,12,13,14] |
| Ca2+-calmodulin-dependent protein kinase | PLB, RyRs, L-type Ca2+ channels | [75,76,77] |
| Protein kinase C | AP-1, TGF-β, TnI, β-AR, adenylyl cyclase, GRK, L-type Ca2+ channel, RACKS, c-Raf | [236,237] |
| Phosphoinositide-3 kinase | Phosphatidylinositol, phosphatidylinositol (4,5)-bisphosphate | [238] |
| P38 mitogen-activatedprotein kinase | MKs, TFs, phospholipase A2, Tau, keratin 8, NHE-1 | [239] |
| Protein Kinase (Isoform) | Inhibitor | References |
|---|---|---|
| Protein kinase A | H89 | [58,41,68,69] |
| KT5720 | [42,43,44] | |
| Ca2+-calmodulin-dependent protein kinase | KN93 | [96,97,100,101] |
| Protein kinase C | Breviscapine | [120] |
| Chelerythine | [136] | |
| Gö6983 | [131,132] | |
| Protein kinase C(α) | Ro-320432 | [106] |
| Protein kinase C(α) | Ro-318110 | [106] |
| Protein kinase C(β) | Ruboxistaurin | [126,128,129] |
| PI3K | Wortmannin | [169,170,171,181] |
| P38 MAPK | SB203580 | [197,201,203,206,207,215,216,220,222,228,230] |
| SB202190 | [209,231,232] | |
| FR167653 | [214,216,233] | |
| SC409 | [235] |
Acknowledgements
References
- Nishizuka, Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984, 308, 693–698. [Google Scholar]
- Wood, J.S.; Yan, X.; Mendelow, M.; Corbin, J.D.; Francis, S.H.; Lawrence, D.S. Precision substrate targeting of protein kinases. The cGMP- and cAMP-dependent protein kinases. J. Biol. Chem. 1996, 271, 174–179. [Google Scholar] [PubMed]
- Dhalla, N.S.; Wang, J. Role of protein kinase C and protein kinase A in heart function in health and disease. Exp. Clin. Cardiol. 1999, 4, 7–14. [Google Scholar]
- Feuerstein, G.Z.; Young, P.R. Apoptosis in cardiac diseases: Stress- and mitogen-activated signaling pathways. Cardiovasc. Res. 2000, 45, 560–569. [Google Scholar]
- Sugden, P.H.; Bogoyevitch, M.A. Intracellular signaling through protein kinases in the heart. Cardiovasc. Res. 1995, 30, 478–492. [Google Scholar]
- Sadoshima, J.; Qiu, Z.; Morgan, J.P.; Izumo, S. Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes. The critical role of Ca2+-dependent signaling. Circ. Res. 1995, 76, 1–15. [Google Scholar] [PubMed]
- Kemp, B.E.; Bylund, D.B.; Huang, T.S.; Krebs, E.G. Substrate specificity of the cyclic AMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1975, 72, 3448–3452. [Google Scholar]
- Taylor, S.S.; Buechler, J.A.; Yonemoto, W. cAMP-dependent protein kinase: Framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem. 1990, 59, 971–1005. [Google Scholar]
- Opie, L.H. Receptors and signal transduction. In The Heart, Physiology and Metabolism; Opie, L.H., Ed.; Raven Press: New York, NY, USA, 1991; pp. 147–175. [Google Scholar]
- Barefield, D.; Sadayappan, S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 2010, 48, 866–875. [Google Scholar]
- Baryshnikova, O.K.; Li, M.X.; Sykes, B.D. Modulation of cardiac troponin C function by the cardiac-specific N-terminus of troponin I: Influence of PKA phosphorylation and involvement in cardiomyopathies. J. Mol. Biol. 2008, 375, 735–751. [Google Scholar]
- Jones, P.P.; Meng, X.; Xiao, B.; Cai, S.; Bolstad, J.; Wagenknecht, T.; Liu, Z.; Chen, S.R. Localization of PKA phosphorylation site, Ser(2030), in the three-dimensional structure of cardiac ryanodine receptor. Biochem. J. 2008, 410, 261–270. [Google Scholar] [PubMed]
- Li, J.; Bigelow, D.J.; Squier, T.C. Conformational changes within the cytosolic portion of phospholamban upon release of Ca-ATPase inhibition. Biochemistry 2004, 43, 3870–3879. [Google Scholar]
- Wolff, M.R.; Buck, S.H.; Stoker, S.W.; Greaser, M.L.; Mentzer, R.M. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: Role of altered beta-adrenergically mediated protein phosphorylation. J. Clin. Invest. 1996, 98, 167–176. [Google Scholar]
- Bogoyevitch, M.A.; Fuller, S.J.; Sugden, P.H. cAMP and protein synthesis in isolated adult rat heart preparations. Am. J. Physiol. Cell. Physiol. 1993, 265, C1247–C1257. [Google Scholar]
- Bohm, M.; Reiger, B.; Schwinger, R.H.; Erdmann, E. cAMP concentrations, cAMP dependent protein kinase activity, and phospholamban in non-failing and failing myocardium. Cardiovasc. Res. 1994, 28, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Karczewski, P.; Beckert, R.; Krause, E.G. Phosphorylation of the L-type calcium channel beta subunit is involved in beta-adrenergic signal transduction in canine myocardium. FEBS Lett. 1993, 335, 217–222. [Google Scholar]
- Perets, T.; Blumenstein, Y.; Shistik, E.; Lotan, I.; Dascal, N. A potential site of functional modulation by protein kinase A in the cardiac Ca2+ channel alpha 1C subunit. FEBS Lett. 1996, 384, 189–192. [Google Scholar]
- Milano, C.A.; Allen, L.F.; Rockman, H.A.; Dolber, P.C.; McMinn, T.R.; Chien, K.R.; Johnson, T.D.; Bond, R.A.; Lefkowitz, R.J. Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science 1994, 264, 582–586. [Google Scholar]
- Goldspink, P.H.; Russell, B. The cAMP response element binding protein is expressed and phosphorylated in cardiac myocytes. Circ. Res. 1994, 74, 1042–1049. [Google Scholar]
- Phrommintikul, A.; Chattipakorn, N. Roles of cardiac ryanodine receptor in heart failure and sudden cardiac death. Int. J. Cardiol. 2006, 112, 142–152. [Google Scholar]
- Mattiazzi, A.; Hove-Madsen, L.; Bers, D.M. Protein kinase inhibitors reduce SR Ca transport in permeabilized cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H812–H820. [Google Scholar]
- Sichelschmidt, O.J.; Hahnefeld, C.; Hohlfeld, T.; Herberg, F.W.; Schror, K. Trapidil protects ischemic hearts from reperfusion injury by stimulating PKAII activity. Cardiovasc. Res. 2003, 58, 602–610. [Google Scholar]
- Schmitt, J.P.; Ahmad, F.; Lorenz, K.; Hein, L.; Schulz, S.; Asahi, M.; Maclennan, D.H.; Seidman, C.E.; Seidman, J.G.; Lohse, M.J. Alterations of phospholamban function can exhibit cardiotoxic effects independent of excessive sarcoplasmic reticulum Ca2+-ATPase inhibition. Circulation 2009, 119, 436–444. [Google Scholar]
- Xiao, B.; Zhong, G.; Obayashi, M.; Yang, D.; Chen, K.; Walsh, M.P.; Shimoni, Y.; Cheng, H.; Ter Keurs, H.; Chen, S.R. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem. J. 2006, 396, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; J, O.U.; Kawai, M.; Hoshina, T.; Kusakari, Y.; Komukai, K.; Sasaki, H.; Hongo, K.; Kurihara, S. Protein kinase A-dependent phosphorylation of ryanodine receptors increases Ca2+ leak in mouse heart. Biochem. Biophys. Res. Commun. 2009, 390, 87–92. [Google Scholar]
- Pogwizd, S.M.; Bers, D.M. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 2004, 14, 61–66. [Google Scholar]
- Zakhary, D.R.; Moravec, C.S.; Bond, M. Regulation of PKA binding to AKAPs in the heart: alterations in human heart failure. Circulation 2000, 101, 1459–1464. [Google Scholar]
- Zakhary, D.R.; Moravec, C.S.; Stewart, R.W.; Bond, M. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation 1999, 99, 505–510. [Google Scholar]
- Piddo, A.M.; Sanchez, M.I.; Sapag-Hagar, M.; Corbalan, R.; Foncea, R.; Ebensperger, R.; Godoy, I.; Melendez, J.; Jalil, J.E.; Lavandero, S. Cyclic AMP-dependent protein kinase and mechanical heart function in ventricular hypertrophy induced by pressure overload or secondary to myocardial infarction. J. Mol. Cell. Cardiol. 1996, 28, 1073–1083. [Google Scholar]
- Dong, W.J.; Jayasundar, J.J.; An, J.; Xing, J.; Cheung, H.C. Effects of PKA phosphorylation of cardiac troponin I and strong crossbridge on conformational transitions of the N-domain of cardiac troponin C in regulated thin filaments. Biochemistry 2007, 46, 9752–9761. [Google Scholar]
- Tong, C.W.; Stelzer, J.E.; Greaser, M.L.; Powers, P.A.; Moss, R.L. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ. Res. 2008, 103, 974–982. [Google Scholar]
- El-Armouche, A.; Boknik, P.; Eschenhagen, T.; Carrier, L.; Knaut, M.; Ravens, U.; Dobrev, D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 2006, 114, 670–680. [Google Scholar]
- Jacques, A.M.; Copeland, O.; Messer, A.E.; Gallon, C.E.; King, K.; McKenna, W.J.; Tsang, V.T.; Marston, S.B. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J. Mol. Cell. Cardiol. 2008, 45, 209–216. [Google Scholar]
- van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.; van der Velden, J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [PubMed]
- Ray, K.P.; England, P.J. Phosphorylation of the inhibitory subunit of troponin and its effect on the calcium dependence of cardiac myofibril adenosine triphosphatase. FEBS Lett. 1976, 70, 11–16. [Google Scholar]
- Holroyde, M.J.; Potter, J.D.; Solaro, R.J. The calcium binding properties of phosphorylated and unphosphorylated cardiac and skeletal myosins. J. Biol. Chem. 1979, 254, 6478–6482. [Google Scholar]
- Kentish, J.C.; McCloskey, D.T.; Layland, J.; Palmer, S.; Leiden, J.M.; Martin, A.F.; Solaro, R.J. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ. Res. 2001, 88, 1059–1065. [Google Scholar]
- Herron, T.J.; Korte, F.S.; McDonald, K.S. Power output is increased after phosphorylation of myofibrillar proteins in rat skinned cardiac myocytes. Circ. Res. 2001, 89, 1184–1190. [Google Scholar]
- VanBuren, P.; Okada, Y. Thin filament remodeling in failing myocardium. Heart Fail. Rev. 2005, 10, 199–209. [Google Scholar]
- Makaula, S.; Lochner, A.; Genade, S.; Sack, M.N.; Awan, M.M.; Opie, L.H. H-89, a non-specific inhibitor of protein kinase A, promotes post-ischemic cardiac contractile recovery and reduces infarct size. J. Cardiovasc. Pharmacol. 2005, 45, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Oddis, C.V.; Simmons, R.L.; Hattler, B.G.; Finkel, M.S. Protein kinase A activation is required for IL-1-induced nitric oxide production by cardiac myocytes. Am. J. Physiol. Cell. Physiol. 1996, 271, C429–434. [Google Scholar]
- Zhang, X.H.; Li, G.R.; Bourreau, J.P. The effect of adrenomedullin on the L-type calcium current in myocytes from septic shock rats: signaling pathway. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2888–2893. [Google Scholar]
- Fontes-Sousa, A.P.; Pires, A.L.; Carneiro, C.S.; Bras-Silva, C.; Leite-Moreira, A.F. Effects of adrenomedullin on systolic and diastolic myocardial function. Peptides 2009, 30, 796–802. [Google Scholar]
- Ishimitsu, T.; Nishikimi, T.; Saito, Y.; Kitamura, K.; Eto, T.; Kangawa, K.; Matsuo, H.; Omae, T.; Matsuoka, H. Plasma levels of adrenomedullin, a newly identified hypotensive peptide, in patients with hypertension and renal failure. J. Clin. Invest. 1994, 94, 2158–2161. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, T.; Saito, Y.; Kitamura, K.; Ishimitsu, T.; Eto, T.; Kangawa, K.; Matsuo, H.; Omae, T.; Matsuoka, H. Increased plasma levels of adrenomedullin in patients with heart failure. J. Am. Coll. Cardiol. 1995, 26, 1424–1431. [Google Scholar]
- Jougasaki, M.; Wei, C.M.; McKinley, L.J.; Burnett, J.C., Jr. Elevation of circulating and ventricular adrenomedullin in human congestive heart failure. Circulation 1995, 92, 286–289. [Google Scholar]
- Yoshitomi, Y.; Nishikimi, T.; Kojima, S.; Kuramochi, M.; Takishita, S.; Matsuoka, H.; Miyata, A.; Matsuo, H.; Kangawa, K. Plasma levels of adrenomedullin in patients with acute myocardial infarction. Clin. Sci. 1998, 94, 135–139. [Google Scholar]
- Nishida, H.; Sato, T.; Miyazaki, M.; Nakaya, H. Infarct size limitation by adrenomedullin: Protein kinase A but not PI3-kinase is linked to mitochondrial KCa channels. Cardiovasc. Res. 2008, 77, 398–405. [Google Scholar]
- Mery, P.F.; Pavoine, C.; Belhassen, L.; Pecker, F.; Fischmeister, R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J. Biol. Chem. 1993, 268, 26286–26295. [Google Scholar] [PubMed]
- Shah, A.M.; Spurgeon, H.A.; Sollott, S.J.; Talo, A.; Lakatta, E.G. 8-bromo-cGMP reduces the myofilament response to Ca2+ in intact cardiac myocytes. Circ. Res. 1994, 74, 970–978. [Google Scholar]
- Wahler, G.M.; Dollinger, S.J. Nitric oxide donor SIN-1 inhibits mammalian cardiac calcium current through cGMP-dependent protein kinase. Am. J. Physiol. Cell. Physiol. 1995, 268, C45–C54. [Google Scholar]
- Olson, R.E. Myocardial metabolism in congestive heart failure. J. Chronic Dis. 1959, 9, 442–464. [Google Scholar]
- Weber, K.T.; Brilla, C.G. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991, 83, 1849–1865. [Google Scholar] [PubMed]
- Saini, H.K.; Xu, Y.J.; Zhang, M.; Liu, P.P.; Kirshenbaum, L.A.; Dhalla, N.S. Role of tumour necrosis factor-alpha and other cytokines in ischemia-reperfusion-induced injury in the heart. Exp. Clin. Cardiol. 2005, 10, 213–222. [Google Scholar]
- Murray, P.A.; Baig, H.; Fishbein, M.C.; Vatner, S.F. Effects of experimental right ventricular hypertrophy on myocardial blood flow in conscious dogs. J. Clin. Invest. 1979, 64, 421–427. [Google Scholar]
- Yu, Q.J.; Si, R.; Zhou, N.; Zhang, H.F.; Guo, W.Y.; Wang, H.C.; Gao, F. Insulin inhibits beta-adrenergic action in ischemic/reperfused heart: A novel mechanism of insulin in cardioprotection. Apoptosis 2008, 13, 305–317. [Google Scholar]
- Asai, M.; Tsukamoto, O.; Minamino, T.; Asanuma, H.; Fujita, M.; Asano, Y.; Takahama, H.; Sasaki, H.; Higo, S.; Asakura, M.; Takashima, S.; Hori, M.; Kitakaze, M. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J. Mol. Cell. Cardiol. 2009, 46, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Freude, B.; Masters, T.N.; Robicsek, F.; Fokin, A.; Kostin, S.; Zimmermann, R.; Ullmann, C.; Lorenz-Meyer, S.; Schaper, J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J. Mol. Cell. Cardiol. 2000, 32, 197–208. [Google Scholar]
- Scarabelli, T.; Stephanou, A.; Rayment, N.; Pasini, E.; Comini, L.; Curello, S.; Ferrari, R.; Knight, R.; Latchman, D. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation 2001, 104, 253–256. [Google Scholar]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Invest. 1994, 94, 1621–1628. [Google Scholar]
- Kajstura, J.; Cheng, W.; Reiss, K.; Clark, W.A.; Sonnenblick, E.H.; Krajewski, S.; Reed, J.C.; Olivetti, G.; Anversa, P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab. Invest. 1996, 74, 86–107. [Google Scholar]
- Guerra, S.; Leri, A.; Wang, X.; Finato, N.; Di Loreto, C.; Beltrami, C.A.; Kajstura, J.; Anversa, P. Myocyte death in the failing human heart is gender dependent. Circ. Res. 1999, 85, 856–866. [Google Scholar]
- Olivetti, G.; Abbi, R.; Quaini, F.; Kajstura, J.; Cheng, W.; Nitahara, J.A.; Quaini, E.; Di Loreto, C.; Beltrami, C.A.; Krajewski, S.; Reed, J.C.; Anversa, P. Apoptosis in the failing human heart. N. Engl. J. Med. 1997, 336, 1131–1141. [Google Scholar]
- Granata, R.; Trovato, L.; Gallo, M.P.; Destefanis, S.; Settanni, F.; Scarlatti, F.; Brero, A.; Ramella, R.; Volante, M.; Isgaard, J.; Levi, R.; Papotti, M.; Alloatti, G.; Ghigo, E. Growth hormone-releasing hormone promotes survival of cardiac myocytes in vitro and protects against ischaemia-reperfusion injury in rat heart. Cardiovasc. Res. 2009, 83, 303–312. [Google Scholar] [PubMed]
- Kwak, H.J.; Park, K.M.; Choi, H.E.; Chung, K.S.; Lim, H.J.; Park, H.Y. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell. Signal. 2008, 20, 803–814. [Google Scholar]
- Robinet, A.; Hoizey, G.; Millart, H. PI 3-kinase, protein kinase C, and protein kinase A are involved in the trigger phase of beta1-adrenergic preconditioning. Cardiovasc. Res. 2005, 66, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.J.; Chae, S.W.; Kim, H.R. Cyclic adenosine monophosphate inhibits nitric oxide-induced apoptosis of cardiac muscle cells in a c-Jun N-terminal kinase-dependent manner. Immunopharmacol. Immunotoxicol. 2004, 26, 249–263. [Google Scholar]
- Prabu, S.K.; Anandatheerthavarada, H.K.; Raza, H.; Srinivasan, S.; Spear, J.F.; Avadhani, N.G. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J. Biol. Chem. 2006, 281, 2061–2070. [Google Scholar]
- Tombes, R.M.; Faison, M.O.; Turbeville, J.M. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene 2003, 322, 17–31. [Google Scholar]
- Hudmon, A.; Schulman, H. Neuronal Ca2+/calmodulin-dependent protein kinase II: The role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 2002, 71, 473–510. [Google Scholar]
- Hudmon, A.; Schulman, H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 2002, 364, 593–611. [Google Scholar]
- De Koninck, P.; Schulman, H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 1998, 279, 227–230. [Google Scholar]
- Lantsman, K.; Tombes, R.M. CaMK-II oligomerization potential determined using CFP/YFP FRET. Biochim. Biophys. Acta 2005, 1746, 45–54. [Google Scholar]
- Hook, S.S.; Means, A.R. Ca(2+)/CaM-dependent kinases: From activation to function. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 471–505. [Google Scholar]
- Dzhura, I.; Wu, Y.; Colbran, R.J.; Balser, J.R.; Anderson, M.E. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat. Cell. Biol. 2000, 2, 173–177. [Google Scholar]
- Maier, L.S.; Bers, D.M. Calcium, calmodulin, and calcium-calmodulin kinase II: Heartbeat to heartbeat and beyond. J. Mol. Cell. Cardiol. 2002, 34, 919–939. [Google Scholar] [CrossRef] [PubMed]
- Hain, J.; Onoue, H.; Mayrleitner, M.; Fleischer, S.; Schindler, H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J. Biol. Chem. 1995, 270, 2074–2081. [Google Scholar]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar]
- Kolodziej, S.J.; Hudmon, A.; Waxham, M.N.; Stoops, J.K. Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase II-alpha and truncated CaM kinase II-alpha reveal a unique organization for its structural core and functional domains. J. Biol. Chem. 2000, 275, 14354–14359. [Google Scholar]
- Gaertner, T.R.; Kolodziej, S.J.; Wang, D.; Kobayashi, R.; Koomen, J.M.; Stoops, J.K.; Waxham, M.N. Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J. Biol. Chem. 2004, 279, 12484–12494. [Google Scholar] [PubMed]
- Kanaseki, T.; Ikeuchi, Y.; Sugiura, H.; Yamauchi, T. Structural features of Ca2+/calmodulin-dependent protein kinase II revealed by electron microscopy. J. Cell. Biol. 1991, 115, 1049–1060. [Google Scholar]
- Palomeque, J.; Rueda, O.V.; Sapia, L.; Valverde, C.A.; Salas, M.; Petroff, M.V.; Mattiazzi, A. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ. Res. 2009, 105, 1204–1212. [Google Scholar]
- Xie, L.H.; Chen, F.; Karagueuzian, H.S.; Weiss, J.N. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 2009, 104, 79–86. [Google Scholar]
- Maier, L.S.; Bers, D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar]
- Wu, Y.; Temple, J.; Zhang, R.; Dzhura, I.; Zhang, W.; Trimble, R.; Roden, D.M.; Passier, R.; Olson, E.N.; Colbran, R.J.; Anderson, M.E. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 2002, 106, 1288–1293. [Google Scholar]
- Sag, C.M.; Wadsack, D.P.; Khabbazzadeh, S.; Abesser, M.; Grefe, C.; Neumann, K.; Opiela, M.K.; Backs, J.; Olson, E.N.; Brown, J.H.; Neef, S.; Maier, S.K.; Maier, L.S. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ. Heart Fail. 2009, 2, 664–675. [Google Scholar]
- Yurukova, S.; Kilic, A.; Volker, K.; Leineweber, K.; Dybkova, N.; Maier, L.S.; Brodde, O.E.; Kuhn, M. CaMKII-mediated increased lusitropic responses to beta-adrenoreceptor stimulation in ANP-receptor deficient mice. Cardiovasc. Res. 2007, 73, 678–688. [Google Scholar]
- Chelu, M.G.; Sarma, S.; Sood, S.; Wang, S.; van Oort, R.J.; Skapura, D.G.; Li, N.; Santonastasi, M.; Müller, F.U.; Schmitz, W.; Schotten, U.; Anderson, M.E.; Valderrábano, M.; Dobrev, D.; Wehrens, X.H. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 2009, 119, 1940–1951. [Google Scholar]
- Saito, T.; Fukuzawa, J.; Osaki, J.; Sakuragi, H.; Yao, N.; Haneda, T.; Fujino, T.; Wakamiya, N.; Kikuchi, K.; Hasebe, N. Roles of calcineurin and calcium/calmodulin-dependent protein kinase II in pressure overload-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2003, 35, 1153–1160. [Google Scholar]
- Backs, J.; Backs, T.; Neef, S.; Kreusser, M.M.; Lehmann, L.H.; Patrick, D.M.; Grueter, C.E.; Qi, X.; Richardson, J.A.; Hill, J.A.; Katus, H.A.; Bassel-Duby, R.; Maier, L.S.; Olson, E.N. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA. 2009, 106, 2342–2347. [Google Scholar]
- Colomer, J.M.; Mao, L.; Rockman, H.A.; Means, A.R. Pressure overload selectively up-regulates Ca2+/calmodulin-dependent protein kinase II in vivo. Mol. Endocrinol. 2003, 17, 183–192. [Google Scholar] [PubMed]
- Peng, W.; Zhang, Y.; Zheng, M.; Cheng, H.; Zhu, W.; Cao, C.M.; Xiao, R.P. Cardioprotection by CaMKII-deltaB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ. Res. 2010, 106, 102–110. [Google Scholar]
- Srinivasan, M.; Edman, C.F.; Schulman, H. Alternative splicing introduces a nuclear localization signal that targets multifunctional CaM kinase to the nucleus. J. Cell. Biol. 1994, 126, 839–852. [Google Scholar]
- Kohlhaas, M.; Zhang, T.; Seidler, T.; Zibrova, D.; Dybkova, N.; Steen, A.; Wagner, S.; Chen, L.; Brown, J.H.; Bers, D.M.; Maier, L.S. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ. Res. 2006, 98, 235–244. [Google Scholar]
- Kashiwase, K.; Higuchi, Y.; Hirotani, S.; Yamaguchi, O.; Hikoso, S.; Takeda, T.; Watanabe, T.; Taniike, M.; Nakai, A.; Tsujimoto, I.; Matsumura, Y.; Ueno, H.; Nishida, K.; Hori, M.; Otsu, K. CaMKII activates ASK1 and NF-kappaB to induce cardiomyocyte hypertrophy. Biochem. Biophys. Res. Commun. 2005, 327, 136–142. [Google Scholar]
- Passier, R.; Zeng, H.; Frey, N.; Naya, F.J.; Nicol, R.L.; McKinsey, T.A.; Overbeek, P.; Richardson, J.A.; Grant, S.R.; Olson, E.N. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Invest. 2000, 105, 1395–1406. [Google Scholar] [PubMed]
- Woischwill, C.; Karczewski, P.; Bartsch, H.; Luther, H.P.; Kott, M.; Haase, H.; Morano, I. Regulation of the human atrial myosin light chain 1 promoter by Ca2+-calmodulin-dependent signaling pathways. FASEB J. 2005, 19, 503–511. [Google Scholar]
- Benter, I.F.; Juggi, J.S.; Khan, I.; Yousif, M.H.; Canatan, H.; Akhtar, S. Signal transduction mechanisms involved in cardiac preconditioning: Role of Ras-GTPase, Ca2+/calmodulin-dependent protein kinase II and epidermal growth factor receptor. Mol. Cell. Biochem. 2005, 268, 175–183. [Google Scholar]
- Osada, M.; Netticadan, T.; Kawabata, K.; Tamura, K.; Dhalla, N.S. Ischemic preconditioning prevents I/R-induced alterations in SR calcium-calmodulin protein kinase II. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1791–1798. [Google Scholar]
- Netticadan, T.; Temsah, R.M.; Kawabata, K.; Dhalla, N.S. Sarcoplasmic reticulum Ca(2+)/Calmodulin-dependent protein kinase is altered in heart failure. Circ. Res. 2000, 86, 596–605. [Google Scholar]
- MacDonnell, S.M.; Weisser-Thomas, J.; Kubo, H.; Hanscome, M.; Liu, Q.; Jaleel, N.; Berretta, R.; Chen, X.; Brown, J.H.; Sabri, A.K.; Molkentin, J.D.; Houser, S.R. CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ. Res. 2009, 105, 316–325. [Google Scholar]
- Nishizuka, Y. Studies and perspectives of protein kinase C. Science 1986, 233, 305–312. [Google Scholar]
- Bogoyevitch, M.A.; Parker, P.J.; Sugden, P.H. Characterization of protein kinase C isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ. Res. 1993, 72, 757–767. [Google Scholar] [PubMed]
- Hambleton, M.; Hahn, H.; Pleger, S.T.; Kuhn, M.C.; Klevitsky, R.; Carr, A.N.; Kimball, T.F.; Hewett, T.E.; Dorn, G.W., 2nd; Koch, W.J.; Molkentin, J.D. Pharmacological- and gene therapy-based inhibition of protein kinase C-alpha/beta enhances cardiac contractility and attenuates heart failure. Circulation 2006, 114, 574–582. [Google Scholar] [PubMed]
- Kraft, A.S.; Anderson, W.B. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature 1983, 301, 621–623. [Google Scholar]
- Mellor, H.; Parker, P.J. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar]
- Nishizuka, Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 1992, 258, 607–614. [Google Scholar]
- Toker, A.; Newton, A.C. Cellular Signaling: Pivoting around PDK-1. Cell 2000, 103, 185–188. [Google Scholar]
- Liu, X.; Wang, J.; Takeda, N.; Binaglia, L.; Panagia, V.; Dhalla, N.S. Changes in cardiac protein kinase C activities and isozymes in streptozotocin-induced diabetes. Am. J. Physiol. Cell. Physiol. 1999, 277, E798–E804. [Google Scholar]
- Gu, X.; Bishop, S.P. Increased protein kinase C and isozyme redistribution in pressure-overload cardiac hypertrophy in the rat. Circ. Res. 1994, 75, 926–931. [Google Scholar]
- Braun, M.U.; LaRosee, P.; Simonis, G.; Borst, M.M.; Strasser, R.H. Regulation of protein kinase C isozymes in volume overload cardiac hypertrophy. Mol. Cell. Biochem. 2004, 262, 135–143. [Google Scholar]
- Paul, K.; Ball, N.A.; Dorn, G.W., II.; Walsh, R.A. Left ventricular stretch stimulates angiotensin II--mediated phosphatidylinositol hydrolysis and protein kinase C epsilon isoform translocation in adult guinea pig hearts. Circ. Res. 1997, 81, 643–650. [Google Scholar] [PubMed]
- Morgan, H.E.; Baker, K.M. Cardiac hypertrophy. Mechanical, neural, and endocrine dependence. Circulation 1991, 83, 13–25. [Google Scholar] [PubMed]
- Komuro, I.; Katoh, Y.; Kaida, T.; Shibazaki, Y.; Kurabayashi, M.; Hoh, E.; Takaku, F.; Yazaki, Y. Mechanical loading stimulates cell hypertrophy and specific gene expression in cultured rat cardiac myocytes. Possible role of protein kinase C activation. J. Biol. Chem. 1991, 266, 1265–1268. [Google Scholar] [PubMed]
- Sadoshima, J.; Jahn, L.; Takahashi, T.; Kulik, T.J.; Izumo, S. Molecular characterization of the stretch-induced adaptation of cultured cardiac cells. An in vitro model of load-induced cardiac hypertrophy. J. Biol. Chem. 1992, 267, 10551–10560. [Google Scholar] [PubMed]
- Bayer, A.L.; Heidkamp, M.C.; Patel, N.; Porter, M.; Engman, S.; Samarel, A.M. Alterations in protein kinase C isoenzyme expression and autophosphorylation during the progression of pressure overload-induced left ventricular hypertrophy. Mol. Cell. Biochem. 2003, 242, 145–152. [Google Scholar]
- Davidoff, A.J.; Davidson, M.B.; Carmody, M.W.; Davis, M.E.; Ren, J. Diabetic cardiomyocyte dysfunction and myocyte insulin resistance: Role of glucose-induced PKC activity. Mol. Cell. Biochem. 2004, 262, 155–163. [Google Scholar]
- Wang, M.; Zhang, W.B.; Zhu, J.H.; Fu, G.S.; Zhou, B.Q. Breviscapine ameliorates cardiac dysfunction and regulates the myocardial Ca(2+)-cycling proteins in streptozotocin-induced diabetic rats. Acta Diabetol. 2009. [Google Scholar]
- Beckman, J.A.; Goldfine, A.B.; Gordon, M.B.; Garrett, L.A.; Creager, M.A. Inhibition of protein kinase C-beta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ. Res. 2002, 90, 107–111. [Google Scholar]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; Bodi, I.; Wang, S.; Schwartz, A.; Lakatta, E.G.; DePaoli-Roach, A.A.; Robbins, J.; Hewett, T.E.; Bibb, J.A.; Westfall, M.V.; Kranias, E.G.; Molkentin, J.D. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nature Med. 2004, 10, 248–254. [Google Scholar]
- Hahn, H.S.; Marreez, Y.; Odley, A.; Sterbling, A.; Yussman, M.G.; Hilty, K.C.; Bodi, I.; Liggett, S.B.; Schwartz, A.; Dorn, G.W., II. Protein kinase Calpha negatively regulates systolic and diastolic function in pathological hypertrophy. Circ. Res. 2003, 93, 1111–1119. [Google Scholar]
- Cotter, M.A.; Jack, A.M.; Cameron, N.E. Effects of the protein kinase C beta inhibitor LY333531 on neural and vascular function in rats with streptozotocin-induced diabetes. Clin. Sci. 2002, 103, 311–321. [Google Scholar]
- Wu, Y.; Wu, G.; Qi, X.; Lin, H.; Qian, H.; Shen, J.; Lin, S. Protein kinase C beta inhibitor LY333531 attenuates intercellular adhesion molecule-1 and monocyte chemotactic protein-1 expression in the kidney in diabetic rats. J. Pharmacol. Sci. 2006, 101, 335–343. [Google Scholar]
- Connelly, K.A.; Kelly, D.J.; Zhang, Y.; Prior, D.L.; Advani, A.; Cox, A.J.; Thai, K.; Krum, H.; Gilbert, R.E. Inhibition of protein kinase C-beta by ruboxistaurin preserves cardiac function and reduces extracellular matrix production in diabetic cardiomyopathy. Circ. Heart Fail. 2009, 2, 129–137. [Google Scholar]
- Fujita, M.; Minamino, T.; Asanuma, H.; Sanada, S.; Hirata, A.; Wakeno, M.; Myoishi, M.; Okuda, H.; Ogai, A.; Okada, K.; Tsukamoto, O.; Koyama, H.; Hori, M.; Kitakaze, M. Aldosterone nongenomically worsens ischemia via protein kinase C-dependent pathways in hypoperfused canine hearts. Hypertension 2005, 46, 113–117. [Google Scholar]
- Boyle, A.J.; Kelly, D.J.; Zhang, Y.; Cox, A.J.; Gow, R.M.; Way, K.; Itescu, S.; Krum, H.; Gilbert, R.E. Inhibition of protein kinase C reduces left ventricular fibrosis and dysfunction following myocardial infarction. J. Mol. Cell. Cardiol. 2005, 39, 213–221. [Google Scholar]
- Wang, J.; Liu, X.; Sentex, E.; Takeda, N.; Dhalla, N.S. Increased expression of protein kinase C isoforms in heart failure due to myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2277–H2287. [Google Scholar]
- Liu, Q.; Chen, X.; Macdonnell, S.M.; Kranias, E.G.; Lorenz, J.N.; Leitges, M.; Houser, S.R.; Molkentin, J.D. Protein kinase Cα, but not PKCβ or PKCγ, regulates contractility and heart failure susceptibility: Implications for ruboxistaurin as a novel therapeutic approach. Circ. Res. 2009, 105, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Young, L.H.; Balin, B.J.; Weis, M.T. Go 6983: a fast acting protein kinase C inhibitor that attenuates myocardial ischemia/reperfusion injury. Cardiovasc. Drug Rev. 2005, 23, 255–272. [Google Scholar]
- Peterman, E.E.; Taormina, P., II.; Harvey, M.; Young, L.H. Go 6983 exerts cardioprotective effects in myocardial ischemia/reperfusion. J. Cardiovasc. Pharmacol. 2004, 43, 645–656. [Google Scholar] [PubMed]
- Omiyi, D.; Brue, R.J.; Taormina, P., II.; Harvey, M.; Atkinson, N.; Young, L.H. Protein kinase C βII peptide inhibitor exerts cardioprotective effects in rat cardiac ischemia/reperfusion injury. J. Pharmacol. Exp. Ther. 2005, 314, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, A.; Peterman, E.E.; Taormina, P., Jr.; Harvey, M.; Brue, R.J.; Atkinson, N.; Omiyi, D.; Chukwu, U.; Young, L.H. Protein kinase C-ζ inhibition exerts cardioprotective effects in ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H898–H907. [Google Scholar]
- Hahn, H.S.; Yussman, M.G.; Toyokawa, T.; Marreez, Y.; Barrett, T.J.; Hilty, K.C.; Osinska, H.; Robbins, J.; Dorn, G.W., II. Ischemic protection and myofibrillar cardiomyopathy: Dose-dependent effects of in vivo delta-PKC inhibition. Circ. Res. 2002, 91, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Fantinelli, J.C.; Mosca, S.M. Comparative effects of ischemic pre and postconditioning on ischemia-reperfusion injury in spontaneously hypertensive rats (SHR). Mol. Cell. Biochem. 2007, 296, 45–51. [Google Scholar]
- Cave, A.C.; Apstein, C.S. Polymyxin B, a protein kinase C inhibitor, abolishes preconditioning-induced protection against contractile dysfunction in the isolated blood perfused rat hear. J. Mol. Cell. Cardiol. 1996, 28, 977–987. [Google Scholar]
- Liu, Y.; Tsuchida, A.; Cohen, M.V.; Downey, J.M. Pretreatment with angiotensin II activates protein kinase C and limits myocardial infarction in isolated rabbit hearts. J. Mol. Cell. Cardiol. 1995, 27, 883–892. [Google Scholar]
- Mochly-Rosen, D.; Wu, G.; Hahn, H.; Osinska, H.; Liron, T.; Lorenz, J.N.; Yatani, A.; Robbins, J.; Dorn, G.W., II. Cardiotrophic effects of protein kinase C epsilon: Analysis by in vivo modulation of PKCepsilon translocation. Circ. Res. 2000, 86, 1173–1179. [Google Scholar]
- Ping, P.; Takano, H.; Zhang, J.; Tang, X.L.; Qiu, Y.; Li, R.C.; Banerjee, S.; Dawn, B.; Balafonova, Z.; Bolli, R. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: A signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ. Res. 1999, 84, 587–604. [Google Scholar]
- Inagaki, K.; Hahn, H.S.; Dorn, G.W., 2nd; Mochly-Rosen, D. Additive protection of the ischemic heart ex vivo by combined treatment with δ-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation 2003, 108, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.C.; Kay, H.; Chen, Q.; Adams, J.S.; Grilli, C.; Guglielmello, G.; Zambrano, C.; Krass, S.; Bell, A.; Young, L.H. Mechanisms related to the cardioprotective effects of protein kinase C epsilon (PKC epsilon) peptide activator or inhibitor in rat ischemia/reperfusion injury. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 1–15. [Google Scholar]
- Yu, Q.; Nguyen, T.; Ogbi, M.; Caldwell, R.W.; Johnson, J.A. Differential loss of cytochrome-c oxidase subunits in ischemia-reperfusion injury: Exacerbation of COI subunit loss by PKC-epsilon inhibition. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2637–2645. [Google Scholar]
- Wang, G.Y.; Zhou, J.J.; Shan, J.; Wong, T.M. Protein kinase C-epsilon is a trigger of delayed cardioprotection against myocardial ischemia of κ-opioid receptor stimulation in rat ventricular myocytes. J. Pharmacol. Exp. Ther. 2001, 299, 603–610. [Google Scholar]
- Malhotra, A.; Kang, B.P.; Hashmi, S.; Meggs, L.G. PKC-epsilon inhibits the hyperglycemia-induced apoptosis signal in adult rat ventricular myocytes. Mol. Cell. Biochem. 2005, 268, 169–173. [Google Scholar]
- Malhotra, A.; Begley, R.; Kang, B.P.; Rana, I.; Liu, J.; Yang, G.; Mochly-Rosen, D.; Meggs, L.G. PKC-ε-dependent survival signals in diabetic hearts. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1343–H1350. [Google Scholar]
- House, S.L.; Melhorn, S.J.; Newman, G.; Doetschman, T.; Schultz Jel, J. The protein kinase C pathway mediates cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H354–H365. [Google Scholar]
- Das, A.; Ockaili, R.; Salloum, F.; Kukreja, R.C. Protein kinase C plays an essential role in sildenafil-induced cardioprotection in rabbits. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1455–H1460. [Google Scholar]
- Chen, L.; Hahn, H.; Wu, G.; Chen, C.H.; Liron, T.; Schechtman, D.; Cavallaro, G.; Banci, L.; Guo, Y.; Bolli, R.; Dorn, G.W., II.; Mochly-Rosen, D. Opposing cardioprotective actions and parallel hypertrophic effects of δPKC and εPKC. Proc. Natl. Acad. Sci. USA. 2001, 98, 11114–11119. [Google Scholar]
- Carpenter, C.L.; Duckworth, B.C.; Auger, K.R.; Cohen, B.; Schaffhausen, B.S.; Cantley, L.C. Purification and characterization of phosphoinositide 3-kinase from rat liver. J. Biol. Chem. 1990, 265, 19704–19711. [Google Scholar]
- Escobedo, J.A.; Navankasattusas, S.; Kavanaugh, W.M.; Milfay, D.; Fried, V.A.; Williams, L.T. cDNA cloning of a novel 85 kd protein that has SH2 domains and regulates binding of PI3-kinase to the PDGF β-receptor. Cell 1991, 65, 75–82. [Google Scholar]
- Otsu, M.; Hiles, I.; Gout, I.; Fry, M.J.; Ruiz-Larrea, F.; Panayotou, G.; Thompson, A.; Dhand, R.; Hsuan, J.; Totty, N.; et al. Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell 1991, 65, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Skolnik, E.Y.; Margolis, B.; Mohammadi, M.; Lowenstein, E.; Fischer, R.; Drepps, A.; Ullrich, A.; Schlessinger, J. Cloning of PI3 kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell 1991, 65, 83–90. [Google Scholar]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar]
- Walker, E.H.; Perisic, O.; Ried, C.; Stephens, L.; Williams, R.L. Structural insights into phosphoinositide 3-kinase catalysis and signaling. Nature 1999, 402, 313–320. [Google Scholar]
- Williams, R.; Berndt, A.; Miller, S.; Hon, W.C.; Zhang, X. Form and flexibility in phosphoinositide 3-kinases. Biochem. Soc. Trans. 2009, 37, 615–626. [Google Scholar]
- Knight, Z.A.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; Balla, T.; Weiss, W.A.; Williams, R.L.; Shokat, K.M. A pharmacological map of the PI3-K family defines a role for p110-α in insulin signaling. Cell 2006, 125, 733–747. [Google Scholar]
- Crackower, M.A.; Oudit, G.Y.; Kozieradzki, I.; Sarao, R.; Sun, H.; Sasaki, T.; Hirsch, E.; Suzuki, A.; Shioi, T.; Irie-Sasaki, J.; Sah, R.; Cheng, H.Y.; Rybin, V.O.; Lembo, G.; Fratta, L.; Oliveira-dos-Santos, A.J.; Benovic, J.L.; Kahn, C.R.; Izumo, S.; Steinberg, S.F.; Wymann, M.P.; Backx, P.H.; Penninger, J.M. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 2002, 110, 737–749. [Google Scholar]
- Rose, R.A.; Kabir, M.G.; Backx, P.H. Altered heart rate and sinoatrial node function in mice lacking the cAMP regulator phosphoinositide 3-kinase-γ. Circ. Res. 2007, 101, 1274–1282. [Google Scholar]
- Ravingerova, T.; Matejikova, J.; Neckar, J.; Andelova, E.; Kolar, F. Differential role of PI3K/Akt pathway in the infarct size limitation and antiarrhythmic protection in the rat heart. Mol. Cell. Biochem. 2007, 297, 111–120. [Google Scholar]
- Siragusa, M.; Katare, R.; Meloni, M.; Damilano, F.; Hirsch, E.; Emanueli, C.; Madeddu, P. Involvement of phosphoinositide 3-kinase γ in angiogenesis and healing of experimental myocardial infarction in mice. Circ. Res. 2010, 106, 757–768. [Google Scholar]
- Perrino, C.; Rockman, H.A.; Chiariello, M. Targeted inhibition of phosphoinositide 3-kinase activity as a novel strategy to normalize β-adrenergic receptor function in heart failure. Vascul. Pharmacol. 2006, 45, 77–85. [Google Scholar]
- Perrino, C.; Naga Prasad, S.V.; Patel, M.; Wolf, M.J.; Rockman, H.A. Targeted inhibition of β-adrenergic receptor kinase-1-associated phosphoinositide-3 kinase activity preserves β-adrenergic receptor signaling and prolongs survival in heart failure induced by calsequestrin overexpression. J. Am. Coll. Cardiol. 2005, 45, 1862–1870. [Google Scholar]
- Perrino, C.; Naga Prasad, S.V.; Schroder, J.N.; Hata, J.A.; Milano, C.; Rockman, H.A. Restoration of β-adrenergic receptor signaling and contractile function in heart failure by disruption of the β-ARK1/phosphoinositide 3-kinase complex. Circulation 2005, 111, 2579–2587. [Google Scholar]
- Nienaber, J.J.; Tachibana, H.; Naga Prasad, S.V.; Esposito, G.; Wu, D.; Mao, L.; Rockman, H.A. Inhibition of receptor-localized PI3K preserves cardiac β-adrenergic receptor function and ameliorates pressure overload heart failure. J. Clin. Invest. 2003, 112, 1067–1079. [Google Scholar]
- Awad, A.E.; Kandalam, V.; Chakrabarti, S.; Wang, X.; Penninger, J.M.; Davidge, S.T.; Oudit, G.Y.; Kassiri, Z. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3K-gamma-dependent manner. Am. J. Physiol. Cell. Physiol. 2010, 298, C679–C692. [Google Scholar]
- Perrino, C.; Naga Prasad, S.V.; Mao, L.; Noma, T.; Yan, Z.; Kim, H.S.; Smithies, O.; Rockman, H.A. Intermittent pressure overload triggers hypertrophy-independent cardiac dysfunction and vascular rarefaction. J. Clin. Invest. 2006, 116, 1547–1560. [Google Scholar]
- Luo, J.; McMullen, J.R.; Sobkiw, C.L.; Zhang, L.; Dorfman, A.L.; Sherwood, M.C.; Logsdon, M.N.; Horner, J.W.; DePinho, R.A.; Izumo, S.; Cantley, L.C. Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol. Cell. Biol. 2005, 25, 9491–9502. [Google Scholar]
- Deschamps, A.M.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [PubMed]
- Chaudhary, K.R.; Batchu, S.N.; Das, D.; Suresh, M.R.; Falck, J.R.; Graves, J.P.; Zeldin, D.C.; Seubert, J.M. Role of B-type natriuretic peptide in epoxyeicosatrienoic acid-mediated improved post-ischaemic recovery of heart contractile function. Cardiovasc. Res. 2009, 83, 362–370. [Google Scholar]
- Okumura, H.; Nagaya, N.; Itoh, T.; Okano, I.; Hino, J.; Mori, K.; Tsukamoto, Y.; Ishibashi-Ueda, H.; Miwa, S.; Tambara, K.; Toyokuni, S.; Yutani, C.; Kangawa, K. Adrenomedullin infusion attenuates myocardial ischemia/reperfusion injury through the phosphatidylinositol 3-kinase/Akt-dependent pathway. Circulation 2004, 109, 242–248. [Google Scholar]
- Pretorius, L.; Du, X.J.; Woodcock, E.A.; Kiriazis, H.; Lin, R.C.; Marasco, S.; Medcalf, R.L.; Ming, Z.; Head, G.A.; Tan, J.W.; Cemerlang, N.; Sadoshima, J.; Shioi, T.; Izumo, S.; Lukoshkova, E.V.; Dart, A.M.; Jennings, G.L.; McMullen, J.R. Reduced phosphoinositide 3-kinase (p110-α) activation increases the susceptibility to atrial fibrillation. Am. J. Pathol. 2009, 175, 998–1009. [Google Scholar]
- Lu, Z.; Jiang, Y.P.; Wang, W.; Xu, X.H.; Mathias, R.T.; Entcheva, E.; Ballou, L.M.; Cohen, I.S.; Lin, R.Z. Loss of cardiac phosphoinositide 3-kinase p110 α results in contractile dysfunction. Circulation 2009, 120, 318–325. [Google Scholar]
- McMullen, J.R.; Amirahmadi, F.; Woodcock, E.A.; Schinke-Braun, M.; Bouwman, R.D.; Hewitt, K.A.; Mollica, J.P.; Zhang, L.; Zhang, Y.; Shioi, T.; Buerger, A.; Izumo, S.; Jay, P.Y.; Jennings, G.L. Protective effects of exercise and phosphoinositide 3-kinase(p110α) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 612–617. [Google Scholar]
- McMullen, J.R.; Shioi, T.; Zhang, L.; Tarnavski, O.; Sherwood, M.C.; Kang, P.M.; Izumo, S. Phosphoinositide 3-kinase(p110-α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 12355–12360. [Google Scholar]
- Takenaka, H.; Horiba, M.; Ishiguro, H.; Sumida, A.; Hojo, M.; Usui, A.; Akita, T.; Sakuma, S.; Ueda, Y.; Kodama, I.; Kadomatsu, K. Midkine prevents ventricular remodeling and improves long-term survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H462–469. [Google Scholar]
- Mao, W.; Iwai, C.; Liu, J.; Sheu, S.S.; Fu, M.; Liang, C.S. Darbepoetin alfa exerts a cardioprotective effect in autoimmune cardiomyopathy via reduction of ER stress and activation of the PI3K/Akt and STAT3 pathways. J. Mol. Cell. Cardiol. 2008, 45, 250–260. [Google Scholar]
- Curcio, A.; Noma, T.; Naga Prasad, S.V.; Wolf, M.J.; Lemaire, A.; Perrino, C.; Mao, L.; Rockman, H.A. Competitive displacement of phosphoinositide 3-kinase from β-adrenergic receptor kinase-1 improves postinfarction adverse myocardial remodeling. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1754–H1760. [Google Scholar]
- Wang, W.; Peng, Y.; Wang, Y.; Zhao, X.; Yuan, Z. The anti-apoptotic effect of heat shock protein 90 on hypoxia-mediated cardiomyocyte damage through the Pi3k/Akt pathway. Clin. Exp. Pharmacol. Physiol. 2009. [Google Scholar]
- Wang, M.J.; Wang, Y.; Weil, B.; Abarbanell, A.; Herrmann, J.; Tan, J.N.; Kelly, M.; Meldrum, D.R. Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am. J. Physiol. Regul. Integr. Compar. Physiol. 2009, 296, R972–R978. [Google Scholar]
- Bulhak, A.A.; Jung, C.; Ostenson, C.G.; Lundberg, J.O.; Sjoquist, P.O.; Pernow, J. PPAR-α activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: Involvement of the PI3-Kinase/Akt and NO pathway. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H719–727. [Google Scholar]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Carr, R.D.; Yellon, D.M. Preconditioning the diabetic heart: The importance of Akt phosphorylation. Diabetes 2005, 54, 2360–2364. [Google Scholar]
- Baines, C.P.; Wang, L.; Cohen, M.V.; Downey, J.M. Myocardial protection by insulin is dependent on phospatidylinositol 3-kinase but not protein kinase C or KATP channels in the isolated rabbit heart. Basic Res. Cardiol. 1999, 94, 188–198. [Google Scholar]
- Kim, K.H.; Oudit, G.Y.; Backx, P.H. Erythropoietin protects against doxorubicin-induced cardiomyopathy via a phosphatidylinositol 3-kinase-dependent pathway. J. Pharmacol. Exp. Ther. 2008, 324, 160–169. [Google Scholar]
- Hu, Y.; Chen, X.; Pan, T.T.; Neo, K.L.; Lee, S.W.; Khin, E.S.; Moore, P.K.; Bian, J.S. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch. 2008, 455, 607–616. [Google Scholar]
- Nagoshi, T.; Matsui, T.; Aoyama, T.; Leri, A.; Anversa, P.; Li, L.; Ogawa, W.; del Monte, F.; Gwathmey, J.K.; Grazette, L.; Hemmings, B.A.; Kass, D.A.; Champion, H.C.; Rosenzweig, A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J. Clin. Invest. 2005, 115, 2128–2138. [Google Scholar]
- Chiari, P.C.; Bienengraeber, M.W.; Pagel, P.S.; Krolikowski, J.G.; Kersten, J.R.; Warltier, D.C. Isoflurane protects against myocardial infarction during early reperfusion by activation of phosphatidylinositol-3-kinase signal transduction: Evidence for anesthetic-induced postconditioning in rabbits. Anesthesiology 2005, 102, 102–109. [Google Scholar]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Yellon, D.M. Postconditioning: A form of "modified reperfusion" protects the myocardium by activating the phosphatidylinositol 3-kinase-Akt pathway. Circ. Res. 2004, 95, 230–232. [Google Scholar]
- Tang, J.; Wang, J.; Kong, X.; Yang, J.; Guo, L.; Zheng, F.; Zhang, L.; Huang, Y.; Wan, Y. Vascular endothelial growth factor promotes cardiac stem cell migration via the PI3K/Akt pathway. Exp. Cell Res. 2009, 315, 3521–3531. [Google Scholar]
- Chan, T.O.; Rittenhouse, S.E.; Tsichlis, P.N. AKT/PKB and other D3 phosphoinositide-regulated kinases: Kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 1999, 68, 965–1014. [Google Scholar]
- DeBosch, B.; Treskov, I.; Lupu, T.S.; Weinheimer, C.; Kovacs, A.; Courtois, M.; Muslin, A.J. Akt1 is required for physiological cardiac growth. Circulation 2006, 113, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.; Sambandam, N.; Weinheimer, C.; Courtois, M.; Muslin, A.J. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J. Biol. Chem. 2006, 281, 32841–32851. [Google Scholar]
- Schindler, J.F.; Monahan, J.B.; Smith, W.G. p38 pathway kinases as anti-inflammatory drug targets. J. Dent. Res. 2007, 86, 800–811. [Google Scholar]
- Xu, Y.J.; Saini, H.K.; Zhang, M.; Elimban, V.; Dhalla, N.S. MAPK activation and apoptotic alterations in hearts subjected to calcium paradox are attenuated by taurine. Cardiovasc. Res. 2006, 72, 163–174. [Google Scholar]
- Wang, Y.; Huang, S.; Sah, V.P.; Ross, J., Jr.; Brown, J.H.; Han, J.; Chien, K.R. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem. 1998, 273, 2161–2168. [Google Scholar]
- See, F.; Thomas, W.; Way, K.; Tzanidis, A.; Kompa, A.; Lewis, D.; Itescu, S.; Krum, H. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J. Am. Coll. Cardiol. 2004, 44, 1679–1689. [Google Scholar]
- Barancik, M.; Htun, P.; Strohm, C.; Kilian, S.; Schaper, W. Inhibition of the cardiac p38-MAPK pathway by SB203580 delays ischemic cell death. J. Cardiovasc. Pharmacol. 2000, 35, 474–483. [Google Scholar]
- Pombo, C.M.; Bonventre, J.V.; Avruch, J.; Woodgett, J.R.; Kyriakis, J.M.; Force, T. The stress-activated protein kinases are major c-Jun amino-terminal kinases activated by ischemia and reperfusion. J. Biol. Chem. 1994, 269, 26546–26551. [Google Scholar]
- Nagarkatti, D.S.; Sha'afi, R.I. Role of p38 MAP kinase in myocardial stress. J. Mol. Cell. Cardiol. 1998, 30, 1651–1664. [Google Scholar]
- Saurin, A.T.; Martin, J.L.; Heads, R.J.; Foley, C.; Mockridge, J.W.; Wright, M.J.; Wang, Y.; Marber, M.S. The role of differential activation of p38-mitogen-activated protein kinase in preconditioned ventricular myocytes. FASEB J. 2000, 14, 2237–2246. [Google Scholar]
- Ma, X.L.; Kumar, S.; Gao, F.; Louden, C.S.; Lopez, B.L.; Christopher, T.A.; Wang, C.; Lee, J.C.; Feuerstein, G.Z.; Yue, T.L. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation 1999, 99, 1685–1691. [Google Scholar]
- Mackay, K.; Mochly-Rosen, D. An inhibitor of p38 mitogen-activated protein kinase protects neonatal cardiac myocytes from ischemia. J. Biol. Chem. 1999, 274, 6272–6279. [Google Scholar]
- Martin, J.L.; Avkiran, M.; Quinlan, R.A.; Cohen, P.; Marber, M.S. Antiischemic effects of SB203580 are mediated through the inhibition of p38-α mitogen-activated protein kinase: Evidence from ectopic expression of an inhibition-resistant kinase. Circ. Res. 2001, 89, 750–752. [Google Scholar]
- Otsu, K.; Yamashita, N.; Nishida, K.; Hirotani, S.; Yamaguchi, O.; Watanabe, T.; Hikoso, S.; Higuchi, Y.; Matsumura, Y.; Maruyama, M.; Sudo, T.; Osada, H.; Hori, M. Disruption of a single copy of the p38-α MAP kinase gene leads to cardioprotection against ischemia-reperfusion. Biochem. Biophys. Res. Commun. 2003, 302, 56–60. [Google Scholar]
- Willette, R.N.; Eybye, M.E.; Olzinski, A.R.; Behm, D.J.; Aiyar, N.; Maniscalco, K.; Bentley, R.G.; Coatney, R.W.; Zhao, S.; Westfall, T.D.; Doe, C.P. Differential effects of p38 mitogen-activated protein kinase and cyclooxygenase 2 inhibitors in a model of cardiovascular disease. J. Pharmacol. Exp. Ther. 2009, 330, 964–970. [Google Scholar]
- Shi, J.; Guan, J.; Jiang, B.; Brenner, D.A.; Del Monte, F.; Ward, J.E.; Connors, L.H.; Sawyer, D.B.; Semigran, M.J.; Macgillivray, T.E.; Seldin, D.C.; Falk, R.; Liao, R. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38-α MAPK pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4188–4193. [Google Scholar]
- Yin, H.; Zhang, J.; Lin, H.; Wang, R.; Qiao, Y.; Wang, B.; Liu, F. p38 mitogen-activated protein kinase inhibition decreases TNF-α secretion and protects against left ventricular remodeling in rats with myocardial ischemia. Inflammation 2008, 31, 65–73. [Google Scholar]
- Liu, Z.; Cao, W. p38 mitogen-activated protein kinase: A critical node linking insulin resistance and cardiovascular diseases in type 2 diabetes mellitus. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 38–46. [Google Scholar]
- Wenzel, S.; Soltanpour, G.; Schluter, K.D. No correlation between the p38 MAPK pathway and the contractile dysfunction in diabetic cardiomyocytes: Hyperglycaemia-induced signaling and contractile function. Pflugers Arch. 2005, 451, 328–337. [Google Scholar]
- Ogut, O.; Brozovich, F.V. The potential role of MLC phosphatase and MAPK signaling in the pathogenesis of vascular dysfunction in heart failure. J. Cell. Mol. Med. 2008, 12, 2158–2164. [Google Scholar]
- Fan, L.; Sawbridge, D.; George, V.; Teng, L.; Bailey, A.; Kitchen, I.; Li, J.M. Chronic cocaine-induced cardiac oxidative stress and mitogen-activated protein kinase activation: The role of Nox2 oxidase. J. Pharmacol. Exp. Ther. 2009, 328, 99–106. [Google Scholar]
- Muslin, A.J. MAPK signaling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [Google Scholar]
- Bao, W.; Behm, D.J.; Nerurkar, S.S.; Ao, Z.; Bentley, R.; Mirabile, R.C.; Johns, D.G.; Woods, T.N.; Doe, C.P.; Coatney, R.W.; Ohlstein, J.F.; Douglas, S.A.; Willette, R.N.; Yue, T.L. Effects of p38 MAPK Inhibitor on angiotensin II-dependent hypertension, organ damage, and superoxide anion production. J. Cardiovasc. Pharmacol. 2007, 49, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Tanaka, T.; Kasai, K.; Kita, T.; Tanaka, N. Role of p38 mitogen-activated protein kinase on cardiac dysfunction after hemorrhagic shock in rats. Shock 2007, 28, 291–299. [Google Scholar]
- Westermann, D.; Rutschow, S.; Van Linthout, S.; Linderer, A.; Bucker-Gartner, C.; Sobirey, M.; Riad, A.; Pauschinger, M.; Schultheiss, H.P.; Tschope, C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia 2006, 49, 2507–2513. [Google Scholar]
- Kyoi, S.; Otani, H.; Matsuhisa, S.; Akita, Y.; Tatsumi, K.; Enoki, C.; Fujiwara, H.; Imamura, H.; Kamihata, H.; Iwasaka, T. Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc. Res. 2006, 69, 888–898. [Google Scholar]
- Li, Z.; Ma, J.Y.; Kerr, I.; Chakravarty, S.; Dugar, S.; Schreiner, G.; Protter, A.A. Selective inhibition of p38-alpha MAPK improves cardiac function and reduces myocardial apoptosis in rat model of myocardial injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1972–H1977. [Google Scholar]
- Wang, M.; Sankula, R.; Tsai, B.M.; Meldrum, K.K.; Turrentine, M.; March, K.L.; Brown, J.W.; Dinarello, C.A.; Meldrum, D.R. P38 MAPK mediates myocardial proinflammatory cytokine production and endotoxin-induced contractile suppression. Shock 2004, 21, 170–174. [Google Scholar]
- Peng, T.; Lu, X.; Lei, M.; Moe, G.W.; Feng, Q. Inhibition of p38 MAPK decreases myocardial TNF-alpha expression and improves myocardial function and survival in endotoxemia. Cardiovasc. Res. 2003, 59, 893–900. [Google Scholar]
- Cain, B.S.; Meldrum, D.R.; Meng, X.; Dinarello, C.A.; Shames, B.D.; Banerjee, A.; Harken, A.H. p38 MAPK inhibition decreases TNF-α production and enhances postischemic human myocardial function. J. Surg. Res. 1999, 83, 7–12. [Google Scholar]
- Li, G.; Ali, I.S.; Currie, R.W. Insulin-induced myocardial protection in isolated ischemic rat hearts requires p38 MAPK phosphorylation of Hsp27. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H74–H87. [Google Scholar]
- Yoshimura, Y.; Kristo, G.; Keith, B.J.; Jahania, S.A.; Mentzer, R.M., Jr.; Lasley, R.D. The p38 MAPK inhibitor SB203580 blocks adenosine A(1) receptor-induced attenuation of in vivo myocardial stunning. Cardiovasc. Drugs Ther. 2004, 18, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Kristo, G.; Yoshimura, Y.; Keith, B.J.; Stevens, R.M.; Jahania, S.A.; Mentzer, R.M., Jr.; Lasley, R.D. Adenosine A1/A2a receptor agonist AMP-579 induces acute and delayed preconditioning against in vivo myocardial stunning. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2746–H2753. [Google Scholar] [PubMed]
- Cameron, S.J.; Itoh, S.; Baines, C.P.; Zhang, C.; Ohta, S.; Che, W.; Glassman, M.; Lee, J.D.; Yan, C.; Yang, J.; Abe, J. Activation of big MAP kinase 1 (BMK1/ERK5) inhibits cardiac injury after myocardial ischemia and reperfusion. FEBS Lett. 2004, 566, 255–260. [Google Scholar]
- Yasaka, A.; Hayashida, W. Alterations of load-induced p38 MAP kinase activation in failing rat hearts. Biochem. Biophys. Res. Commun. 2001, 285, 503–507. [Google Scholar]
- Braz, J.C.; Bueno, O.F.; Liang, Q.; Wilkins, B.J.; Dai, Y.S.; Parsons, S.; Braunwart, J.; Glascock, B.J.; Klevitsky, R.; Kimball, T.F.; Hewett, T.E.; Molkentin, J.D. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J. Clin. Invest. 2003, 111, 1475–1486. [Google Scholar]
- Liu, J.; Sadoshima, J.; Zhai, P.; Hong, C.; Yang, G.; Chen, W.; Yan, L.; Wang, Y.; Vatner, S.F.; Vatner, D.E. Pressure overload induces greater hypertrophy and mortality in female mice with p38-α MAPK inhibition. J. Mol. Cell. Cardiol. 2006, 41, 680–688. [Google Scholar]
- Mocanu, M.M.; Baxter, G.F.; Yue, Y.; Critz, S.D.; Yellon, D.M. The p38 MAPK inhibitor, SB203580, abrogates ischaemic preconditioning in rat heart but timing of administration is critical. Basic. Res. Cardiol. 2000, 95, 472–478. [Google Scholar] [PubMed]
- Iliodromitis, E.K.; Aggeli, I.K.; Gaitanaki, C.; Tsiafoutis, I.; Zoga, A.; Beis, I.; Kremastinos, D.T. p38-MAPK is involved in restoration of the lost protection of preconditioning by nicorandil in vivo. Eur. J. Pharmacol. 2008, 579, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.R.; Eaton, P.; Shattock, M.J. Role of p38-mitogen-activated protein kinase in ischaemic preconditioning in rat heart. Clin. Exp. Pharmacol. Physiol. 2008, 35, 126–134. [Google Scholar]
- Jaswal, J.S.; Gandhi, M.; Finegan, B.A.; Dyck, J.R.; Clanachan, A.S. Inhibition of p38 MAPK and AMPK restores adenosine-induced cardioprotection in hearts stressed by antecedent ischemia by altering glucose utilization. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1107–H1114. [Google Scholar]
- Wu, X.; Liu, X.; Zhu, X.; Tang, C. Hypoxic preconditioning induces delayed cardioprotection through p38 MAPK-mediated calreticulin upregulation. Shock 2007, 27, 572–577. [Google Scholar]
- Ruusalepp, A.; Czibik, G.; Flatebo, T.; Vaage, J.; Valen, G. Myocardial protection evoked by hyperoxic exposure involves signaling through nitric oxide and mitogen activated protein kinases. Basic Res. Cardiol. 2007, 102, 318–326. [Google Scholar]
- House, S.L.; Branch, K.; Newman, G.; Doetschman, T.; Schultz Jel, J. Cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2 is mediated by the MAPK cascade. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2167–H2175. [Google Scholar]
- Liu, Y.H.; Wang, D.; Rhaleb, N.E.; Yang, X.P.; Xu, J.; Sankey, S.S.; Rudolph, A.E.; Carretero, O.A. Inhibition of p38 mitogen-activated protein kinase protects the heart against cardiac remodeling in mice with heart failure resulting from myocardial infarction. J. Card. Fail. 2005, 11, 74–81. [Google Scholar]
- Weinbrenner, C.; Liu, G.S.; Cohen, M.V.; Downey, J.M. Phosphorylation of tyrosine 182 of p38 mitogen-activated protein kinase correlates with the protection of preconditioning in the rabbit heart. J. Mol. Cell. Cardiol. 1997, 29, 2383–2391. [Google Scholar]
- Bogoyevitch, M.A.; Sugden, P.H. The role of protein kinases in adaptational growth of the heart. Int. J. Biochem. Cell Biol. 1996, 28, 1–12. [Google Scholar]
- Malhotra, A.; Kang, B.P.; Opawumi, D.; Belizaire, W.; Meggs, L.G. Molecular biology of protein kinase C signaling in cardiac myocytes. Mol. Cell. Biochem. 2001, 225, 97–107. [Google Scholar]
- Djordjevic, S.; Driscoll, P.C. Structural insight into substrate specificity and regulatory mechanisms of phosphoinositide 3-kinases. Trends Biochem. Sci. 2002, 27, 426–432. [Google Scholar]
- Krishna, M.; Narang, H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell. Mol. Life Sci. 2008, 65, 3525–3544. [Google Scholar]
- Martindale, J.J.; Wall, J.A.; Martinez-Longoria, D.M.; Aryal, P.; Rockman, H.A.; Guo, Y.; Bolli, R.; Glembotski, C.C. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J. Biol. Chem. 2005, 280, 669–676. [Google Scholar] [PubMed]
- Bassi, R.; Heads, R.; Marber, M.S.; Clark, J.E. Targeting p38-MAPK in the ischaemic heart: Kill or cure? Curr. Opin. Pharmacol. 2008, 8, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Elimban, V.; Xu, Y.J.; Zhang, M.; Nijjar, M.S.; Dhalla, N.S. Alterations of cardiac ERK1/2 expression and activity due to volume overload were attenuated by the blockade of RAS. J. Cardiovasc. Pharmacol. Ther 2010, 15, 84–92. [Google Scholar]
- Sharma, V.; Abraham, T.; So, A.; Allard, M.F.; McNeill, J.H. Functional effects of protein kinases and peroxynitrite on cardiac carnitine palmitoyltransferase-1 in isolated mitochondria. Mol. Cell. Biochem. 2010, 337, 223–237. [Google Scholar]
- Fryer, R.M.; Wang, Y.; Hsu, A.K.; Nagase, H.; Gross, G.J. Dependence of β1-opioid receptor-induced cardioprotection on a tyrosine kinase-dependent but not a Src-dependent pathway. J. Pharmacol. Exp. Ther. 2001, 299, 477–482. [Google Scholar]
- Fryer, R.M.; Schultz, J.E.; Hsu, A.K.; Gross, G.J. Importance of PKC and tyrosine kinase in single or multiple cycles of preconditioning in rat hearts. Am. J. Physiol. Cell. Physiol. 1999, 276, H1229–H1235. [Google Scholar]
- Okubo, S.; Tanabe, Y.; Takeda, K.; Kitayama, M.; Kanemitsu, S.; Kukreja, R.C.; Takekoshi, N. Pretreatment with tyrosine kinase inhibitor attenuates the reduction of apoptosis 24 h after ischemic preconditioning. Jpn. J. Physiol. 2004, 54, 143–151. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dhalla, N.S.; Müller, A.L. Protein Kinases as Drug Development Targets for Heart Disease Therapy. Pharmaceuticals 2010, 3, 2111-2145. https://doi.org/10.3390/ph3072111
Dhalla NS, Müller AL. Protein Kinases as Drug Development Targets for Heart Disease Therapy. Pharmaceuticals. 2010; 3(7):2111-2145. https://doi.org/10.3390/ph3072111
Chicago/Turabian StyleDhalla, Naranjan S., and Alison L. Müller. 2010. "Protein Kinases as Drug Development Targets for Heart Disease Therapy" Pharmaceuticals 3, no. 7: 2111-2145. https://doi.org/10.3390/ph3072111
APA StyleDhalla, N. S., & Müller, A. L. (2010). Protein Kinases as Drug Development Targets for Heart Disease Therapy. Pharmaceuticals, 3(7), 2111-2145. https://doi.org/10.3390/ph3072111