Non-Steroidal Anti-Inflammatory Drugs: An Overview of Cardiovascular Risks

Abstract
:1. Introduction
2. Methods
3. Results and Discussion
3.1. Prostaglandins and COX
3.1.1. Aspirin’s Affinity to COX
3.1.2. Aspirin: Benefits and Risks
3.2. The Family of Non-Steroidal Anti-Inflammatory Drugs
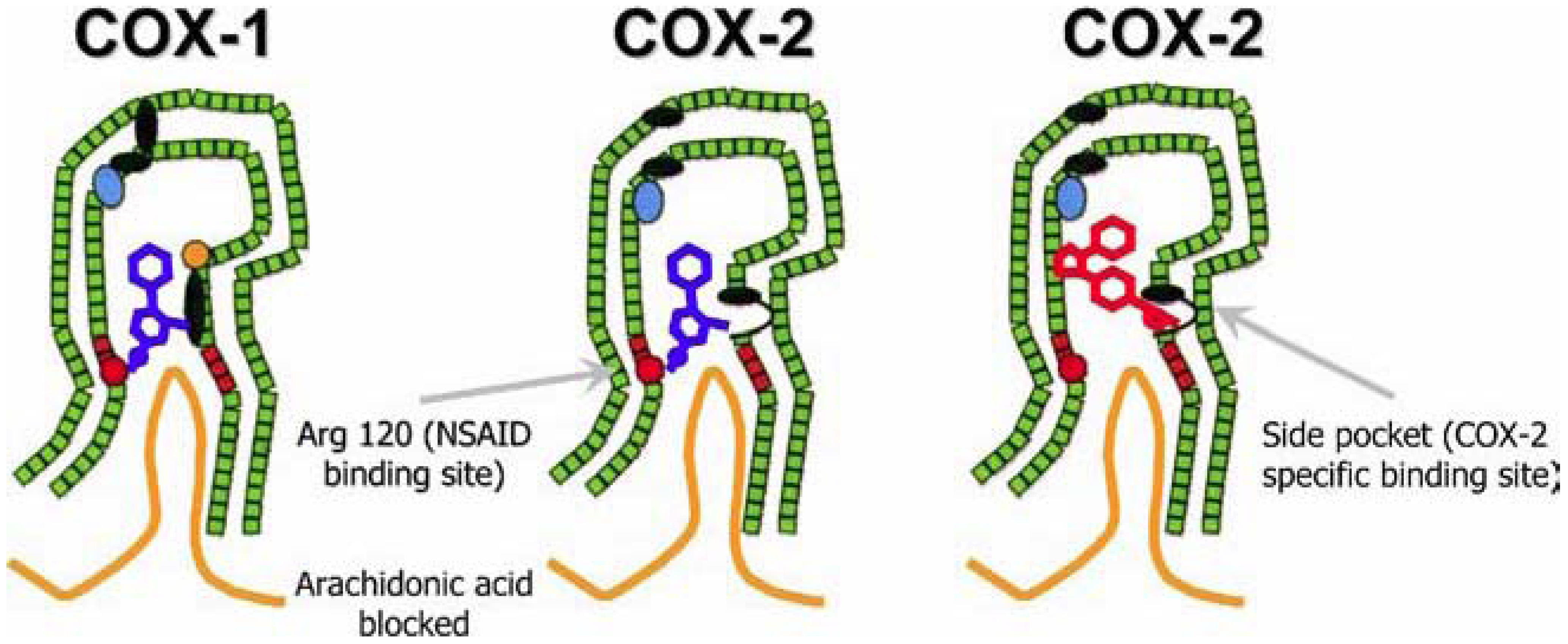
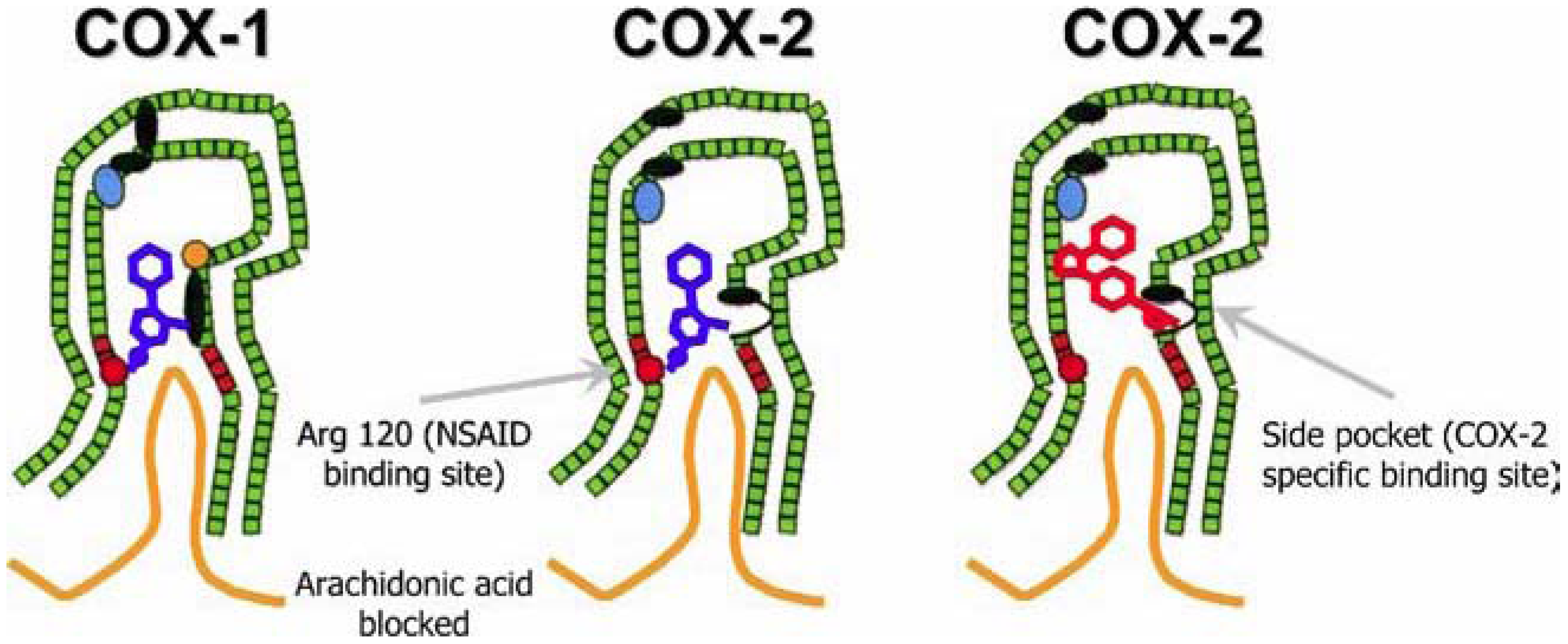
3.3. The Dangers Associated with Pain Relief

{kind=link}
| Low GI risk | Moderate GI risk (one or two risk factors) | High GI risk (more than two risk factors) | |
|---|---|---|---|
| Low CV risk | Non-selective NSAIDs | Non-selective NSAID + PPI or COX-2 + PPI | COX-2 + PPI |
| High CV risk | Naproxen + PPI | Naproxen + PPI | No NSAIDs |
3.4. Combining Aspirin and NSAIDs: the Devil in Disguise?
4. Conclusions
References
- Von Euler, U.S. Uber Die Specifische Blutdrucksenkende Substanz Des Menschlichen Prostate- Und Samenblasensekrets. Klin. Wochenschr. 1935, 14, 1182–1183. [Google Scholar]
- Goldblatt, M.W. Properties of Human Seminal Plasma. J. Physiol. 1935, 84, 208–218. [Google Scholar]
- Van, D.O.R.P.; Beerthuis, R.K.; Nugteren, D.H.; Vonkeman, H. Enzymatic conversion of all-cis-polyunsaturated fatty acids into prostaglandins. Nature 1964, 203, 839–841. [Google Scholar]
- Van, D.O.R.P.; Beerthuis, R.K.; Nugteren, D.H.; Vonkeman, H. The biosynthesis of prostaglandins. Biochim. Biophys. Acta 1964, 90, 204–207. [Google Scholar]
- Fu, J.Y.; Masferrer, J.L.; Seibert, K.; Raz, A.; Needleman, P. The Induction and Suppression of Prostaglandin H2 Synthase (Cyclooxygenase) in Human Monocytes. J. Biol. Chem. 1990, 265, 16737–16740. [Google Scholar]
- Picot, D.; Loll, P.J.; Garavito, R.M. The X-Ray Crystal Structure of the Membrane Protein Prostaglandin H2 Synthase-1. Nature 1994, 367, 243–249. [Google Scholar]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M.F. Flexibility of the NSAID Binding Site in the Structure of Human Cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. [Google Scholar]
- Smith, J.B.; Willis, A.L. Aspirin Selectively Inhibits Prostaglandin Production in Human Platelets. Nat. New Biol. 1971, 231, 235–237. [Google Scholar]
- Collier, J.G.; Flower, R.J. Effect of Aspirin on Human Seminal Prostaglandins. Lancet 1971, 2, 852–853. [Google Scholar]
- Vonkeman, H.E.; van de Laar, M.A. Nonsteroidal Anti-Inflammatory Drugs: Adverse Effects and Their Prevention. Semin. Arthritis Rheum. 2010, 39, 294–312. [Google Scholar]
- Catella-Lawson, F.; Crofford, L.J. Cyclooxygenase Inhibition and Thrombogenicity. Am. J. Med. 2001, 110 Suppl. 3A, 28–32. [Google Scholar] [PubMed]
- Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; Meade, T.; Patrono, C.; Roncaglioni, M.C.; Zanchetti, A. Aspirin in the Primary and Secondary Prevention of Vascular Disease: Collaborative Meta-Analysis of Individual Participant Data From Randomised Trials. Lancet 2009, 373, 1849–1860. [Google Scholar]
- McQuaid, K.R.; Laine, L. Systematic Review and Meta-Analysis of Adverse Events of Low-Dose Aspirin and Clopidogrel in Randomized Controlled Trials. Am. J. Med. 2006, 119, 624–638. [Google Scholar]
- Lanza, F.L. A Guideline for the Treatment and Prevention of NSAID-Induced Ulcers. Members of the Ad Hoc Committee on Practice Parameters of the American College of Gastroenterology. Am. J. Gastroenterol. 1998, 93, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Rosen, R.D. NSAID Induced Gastrointestinal Complications: the ARAMIS Perspective--1997. Arthritis, Rheumatism, and Aging Medical Information System. J. Rheumatol. Suppl 1998, 51, 8–16. [Google Scholar] [PubMed]
- Silverstein, F.E.; Graham, D.Y.; Senior, J.R.; Davies, H.W.; Struthers, B.J.; Bittman, R.M.; Geis, G.S. Misoprostol Reduces Serious Gastrointestinal Complications in Patients With Rheumatoid Arthritis Receiving Nonsteroidal Anti-Inflammatory Drugs. A Randomized, Double-Blind, Placebo-Controlled Trial. Ann. Intern. Med. 1995, 123, 241–249. [Google Scholar] [PubMed]
- Lai, K.C.; Lam, S.K.; Chu, K.M.; Wong, B.C.; Hui, W.M.; Hu, W.H.; Lau, G.K.; Wong, W.M.; Yuen, M.F.; Chan, A.O.; Lai, C.L.; Wong, J. Lansoprazole for the Prevention of Recurrences of Ulcer Complications From Long-Term Low-Dose Aspirin Use. N. Engl. J. Med. 2002, 346, 2033–2038. [Google Scholar]
- Chan, F.K.; Chung, S.C.; Suen, B.Y.; Lee, Y.T.; Leung, W.K.; Leung, V.K.; Wu, J.C.; Lau, J.Y.; Hui, Y.; Lai, M.S.; Chan, H.L.; Sung, J.J. Preventing Recurrent Upper Gastrointestinal Bleeding in Patients With Helicobacter Pylori Infection Who Are Taking Low-Dose Aspirin or Naproxen. N. Engl. J. Med. 2001, 344, 967–973. [Google Scholar]
- Adams, S.S. The Discovery of Brufen. Chem. Br. 1987, 23, 1193–1195. [Google Scholar]
- Jasani, M.K.; Downie, W.W.; Samuels, B.M.; Buchanan, W.W. Ibuprofen in Rheumatoid Arthritis. Clinical Study of Analgesic and Anti-Inflammatory Activity. Ann. Rheum. Dis. 1968, 27, 457–462. [Google Scholar] [PubMed]
- Flower, R.; Gryglewski, R.; Herbaczynska-Cedro, K.; Vane, J.R. Effects of Anti-Inflammatory Drugs on Prostaglandin Biosynthesis. Nat. New Biol. 1972, 238, 104–106. [Google Scholar]
- Van, H.A.; Schwartz, J.I.; Depre, M.; De, L., I.; Dallob, A.; Tanaka, W.; Wynants, K.; Buntinx, A.; Arnout, J.; Wong, P.H.; Ebel, D.L.; Gertz, B.J.; De Schepper, P.J. Comparative Inhibitory Activity of Rofecoxib, Meloxicam, Diclofenac, Ibuprofen, and Naproxen on COX-2 Versus COX-1 in Healthy Volunteers. J. Clin. Pharmacol. 2000, 40, 1109–1120. [Google Scholar] [PubMed]
- Hawkey, C.J. COX-2 Inhibitors. Lancet 1999, 353, 307–314. [Google Scholar]
- Lanzo, C.A.; Beechem, J.M.; Talley, J.; Marnett, L.J. Investigation of the Binding of Isoform-Selective Inhibitors to Prostaglandin Endoperoxide Synthases Using Fluorescence Spectroscopy. Biochemistry 1998, 37, 217–226. [Google Scholar]
- Brun, J.; Jones, R. Nonsteroidal Anti-Inflammatory Drug-Associated Dyspepsia: the Scale of the Problem. Am. J. Med. 2001, 110, 12S–13S. [Google Scholar]
- Langman, M.J.; Weil, J.; Wainwright, P.; Lawson, D.H.; Rawlins, M.D.; Logan, R.F.; Murphy, M.; Vessey, M.P.; Colin-Jones, D.G. Risks of Bleeding Peptic Ulcer Associated With Individual Non-Steroidal Anti-Inflammatory Drugs. Lancet 1994, 343, 1075–1078. [Google Scholar]
- Vonkeman, H.E.; Fernandes, R.W.; van de Laar, M.A. Under-Utilization of Gastroprotective Drugs in Patients With NSAID-Related Ulcers. Int. J. Clin. Pharmacol. Ther. 2007, 45, 281–288. [Google Scholar]
- Armstrong, C.P.; Blower, A.L. Non-Steroidal Anti-Inflammatory Drugs and Life Threatening Complications of Peptic Ulceration. Gut 1987, 28, 527–532. [Google Scholar]
- Rostom, A.; Dube, C.; Wells, G.; Tugwell, P.; Welch, V.; Jolicoeur, E.; McGowan, J. Prevention of NSAID-Induced Gastroduodenal Ulcers. Cochrane. Database. Syst. Rev. 2002, CD002296. [Google Scholar]
- Graham, D.Y.; Agrawal, N.M.; Campbell, D.R.; Haber, M.M.; Collis, C.; Lukasik, N.L.; Huang, B. Ulcer Prevention in Long-Term Users of Nonsteroidal Anti-Inflammatory Drugs: Results of a Double-Blind, Randomized, Multicenter, Active- and Placebo-Controlled Study of Misoprostol Vs Lansoprazole. Arch. Intern. Med. 2002, 162, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, P.G.; Lazzaroni, M.; Imbesi, V.; Montrone, F.; Santagada, T. Efficacy of Pantoprazole in the Prevention of Peptic Ulcers, Induced by Non-Steroidal Anti-Inflammatory Drugs: a Prospective, Placebo-Controlled, Double-Blind, Parallel-Group Study. Dig. Liver Dis. 2000, 32, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Dezi, A.; Ferrario, F.; Capurso, I. Prevention of Nonsteroidal Anti-Inflammatory Drug-Induced Gastrointestinal Mucosal Injury. A Meta-Analysis of Randomized Controlled Clinical Trials. Arch. Intern. Med. 1996, 156, 2321–2332. [Google Scholar] [CrossRef] [PubMed]
- Taha, A.S.; Hudson, N.; Hawkey, C.J.; Swannell, A.J.; Trye, P.N.; Cottrell, J.; Mann, S.G.; Simon, T.J.; Sturrock, R.D.; Russell, R.I. Famotidine for the Prevention of Gastric and Duodenal Ulcers Caused by Nonsteroidal Antiinflammatory Drugs. N. Engl. J. Med. 1996, 334, 1435–1439. [Google Scholar]
- Chan, F.K.; To, K.F.; Wu, J.C.; Yung, M.Y.; Leung, W.K.; Kwok, T.; Hui, Y.; Chan, H.L.; Chan, C.S.; Hui, E.; Woo, J.; Sung, J.J. Eradication of Helicobacter Pylori and Risk of Peptic Ulcers in Patients Starting Long-Term Treatment With Non-Steroidal Anti-Inflammatory Drugs: a Randomised Trial. Lancet 2002, 359, 9–13. [Google Scholar]
- Chan, F.K.; Sung, J.J.; Chung, S.C.; To, K.F.; Yung, M.Y.; Leung, V.K.; Lee, Y.T.; Chan, C.S.; Li, E.K.; Woo, J. Randomised Trial of Eradication of Helicobacter Pylori Before Non-Steroidal Anti-Inflammatory Drug Therapy to Prevent Peptic Ulcers. Lancet 1997, 350, 975–979. [Google Scholar]
- Fitzgerald, G.A. Cardiovascular Pharmacology of Nonselective Nonsteroidal Anti-Inflammatory Drugs and Coxibs: Clinical Considerations. Am. J. Cardiol. 2002, 89, 26D–32D. [Google Scholar]
- Vonkeman, H.E.; Brouwers, J.R.; van de Laar, M.A. Understanding the NSAID Related Risk of Vascular Events. BMJ 2006, 332, 895–898. [Google Scholar]
- Bresalier, R.S.; Sandler, R.S.; Quan, H.; Bolognese, J.A.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; Konstam, M.A.; Baron, J.A. Cardiovascular Events Associated With Rofecoxib in a Colorectal Adenoma Chemoprevention Trial. N. Engl. J. Med. 2005, 352, 1092–1102. [Google Scholar]
- Kearney, P.M.; Baigent, C.; Godwin, J.; Halls, H.; Emberson, J.R.; Patrono, C. Do Selective Cyclo-Oxygenase-2 Inhibitors and Traditional Non-Steroidal Anti-Inflammatory Drugs Increase the Risk of Atherothrombosis? Meta-Analysis of Randomised Trials. BMJ 2006, 332, 1302–1308. [Google Scholar]
- McGettigan, P.; Henry, D. Cardiovascular Risk and Inhibition of Cyclooxygenase: a Systematic Review of the Observational Studies of Selective and Nonselective Inhibitors of Cyclooxygenase 2. JAMA 2006, 296, 1633–1644. [Google Scholar]
- Scott, P.A.; Kingsley, G.H.; Smith, C.M.; Choy, E.H.; Scott, D.L. Non-Steroidal Anti-Inflammatory Drugs and Myocardial Infarctions: Comparative Systematic Review of Evidence From Observational Studies and Randomised Controlled Trials. Ann. Rheum. Dis. 2007, 66, 1296–1304. [Google Scholar]
- Solomon, S.D.; Wittes, J.; Finn, P.V.; Fowler, R.; Viner, J.; Bertagnolli, M.M.; Arber, N.; Levin, B.; Meinert, C.L.; Martin, B.; Pater, J.L.; Goss, P.E.; Lance, P.; Obara, S.; Chew, E.Y.; Kim, J.; Arndt, G.; Hawk, E. Cardiovascular Risk of Celecoxib in 6 Randomized Placebo-Controlled Trials: the Cross Trial Safety Analysis. Circulation 2008, 117, 2104–2113. [Google Scholar]
- Garcia Rodriguez, L.A.; Tacconelli, S.; Patrignani, P. Role of Dose Potency in the Prediction of Risk of Myocardial Infarction Associated With Nonsteroidal Anti-Inflammatory Drugs in the General Population. J. Am. Coll. Cardiol. 2008, 52, 1628–1636. [Google Scholar]
- Gislason, G.H.; Jacobsen, S.; Rasmussen, J.N.; Rasmussen, S.; Buch, P.; Friberg, J.; Schramm, T.K.; Abildstrom, S.Z.; Kober, L.; Madsen, M.; Torp-Pedersen, C. Risk of Death or Reinfarction Associated With the Use of Selective Cyclooxygenase-2 Inhibitors and Nonselective Nonsteroidal Antiinflammatory Drugs After Acute Myocardial Infarction. Circulation 2006, 113, 2906–2913. [Google Scholar]
- Fosbol, E.L.; Gislason, G.H.; Jacobsen, S.; Folke, F.; Hansen, M.L.; Schramm, T.K.; Sorensen, R.; Rasmussen, J.N.; Andersen, S.S.; Abildstrom, S.Z.; Traerup, J.; Poulsen, H.E.; Rasmussen, S.; Kober, L.; Torp-Pedersen, C. Risk of Myocardial Infarction and Death Associated With the Use of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) Among Healthy Individuals: a Nationwide Cohort Study. Clin. Pharmacol. Ther. 2009, 85, 190–197. [Google Scholar]
- FDA Public Health Advisory: Alert for Healthcare Professionals Non-Selective Non-Steroidal Anti-Inflammatory Drugs (NSAIDs). Available online: http://www.fda.gov/Drugs/DrugSafety/ (accessed on 7 July 2010).
- European Medicines Agency update on review of non-selective NSAIDs. Available online: http://www.ema.europa.eu/htms/human/drugalert/drugalert.htm (accessed on 7 July 2010).
- Chan, F.K. Primer: Managing NSAID-Induced Ulcer Complications--Balancing Gastrointestinal and Cardiovascular Risks. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 563–573. [Google Scholar]
- Bhatt, D.L.; Scheiman, J.; Abraham, N.S.; Antman, E.M.; Chan, F.K.; Furberg, C.D.; Johnson, D.A.; Mahaffey, K.W.; Quigley, E.M. ACCF/ACG/AHA 2008 Expert Consensus Document on Reducing the Gastrointestinal Risks of Antiplatelet Therapy and NSAID Use: a Report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. Circulation 2008, 118, 1894–1909. [Google Scholar]
- Antman, E.M.; Bennett, J.S.; Daugherty, A.; Furberg, C.; Roberts, H.; Taubert, K.A. Use of Nonsteroidal Antiinflammatory Drugs: an Update for Clinicians: a Scientific Statement From the American Heart Association. Circulation 2007, 115, 1634–1642. [Google Scholar]
- Solomon, D. Nonselective NSAIDs: cardiovascular effects. UpToDate. Available online: http://www.uptodate.com/ (accessed on 30 June 2010).
- Aw, T.J.; Haas, S.J.; Liew, D.; Krum, H. Meta-Analysis of Cyclooxygenase-2 Inhibitors and Their Effects on Blood Pressure. Arch. Intern. Med. 2005, 165, 490–496. [Google Scholar]
- Rose, B.D.; Post, T.W. NSAIDs: Acute renal failure and nephrotic syndrome. UpToDate. Available online: http://www.uptodate.com/ (accessed on 30 June 2010).
- Feenstra, J.; Heerdink, E.R.; Grobbee, D.E.; Stricker, B.H. Association of Nonsteroidal Anti-Inflammatory Drugs With First Occurrence of Heart Failure and With Relapsing Heart Failure: the Rotterdam Study. Arch. Intern. Med. 2002, 162, 265–270. [Google Scholar]
- Page, J.; Henry, D. Consumption of NSAIDs and the Development of Congestive Heart Failure in Elderly Patients: an Underrecognized Public Health Problem. Arch. Intern. Med. 2000, 160, 777–784. [Google Scholar]
- Gislason, G.H.; Rasmussen, J.N.; Abildstrom, S.Z.; Schramm, T.K.; Hansen, M.L.; Fosbol, E.L.; Sorensen, R.; Folke, F.; Buch, P.; Gadsboll, N.; Rasmussen, S.; Poulsen, H.E.; Kober, L.; Madsen, M.; Torp-Pedersen, C. Increased Mortality and Cardiovascular Morbidity Associated With Use of Nonsteroidal Anti-Inflammatory Drugs in Chronic Heart Failure. Arch. Intern. Med. 2009, 169, 141–149. [Google Scholar]
- Chan, C.C.; Reid, C.M.; Aw, T.J.; Liew, D.; Haas, S.J.; Krum, H. Do COX-2 Inhibitors Raise Blood Pressure More Than Nonselective NSAIDs and Placebo? An Updated Meta-Analysis. J. Hypertens. 2009, 27, 2332–2341. [Google Scholar]
- Swan, S.K.; Rudy, D.W.; Lasseter, K.C.; Ryan, C.F.; Buechel, K.L.; Lambrecht, L.J.; Pinto, M.B.; Dilzer, S.C.; Obrda, O.; Sundblad, K.J.; Gumbs, C.P.; Ebel, D.L.; Quan, H.; Larson, P.J.; Schwartz, J.I.; Musliner, T.A.; Gertz, B.J.; Brater, D.C.; Yao, S.L. Effect of Cyclooxygenase-2 Inhibition on Renal Function in Elderly Persons Receiving a Low-Salt Diet. A Randomized, Controlled Trial. Ann. Intern. Med. 2000, 133, 1–9. [Google Scholar] [PubMed]
- Schneider, V.; Levesque, L.E.; Zhang, B.; Hutchinson, T.; Brophy, J.M. Association of Selective and Conventional Nonsteroidal Antiinflammatory Drugs with Acute Renal Failure: A Population-Based, Nested Case-Control Analysis. Am. J. Epidemiol. 2006, 164, 881–889. [Google Scholar]
- Clive, D.M.; Stoff, J.S. Renal Syndromes Associated With Nonsteroidal Antiinflammatory Drugs. N. Engl. J. Med. 1984, 310, 563–572. [Google Scholar]
- Wallace, J.L.; Viappiani, S.; Bolla, M. Cyclooxygenase-Inhibiting Nitric Oxide Donators for Osteoarthritis. Trends Pharmacol. Sci. 2009, 30, 112–117. [Google Scholar]
- White, W.B.; Schnitzer, T.J.; Fleming, R.; Duquesroix, B.; Beekman, M. Effects of the Cyclooxygenase Inhibiting Nitric Oxide Donator Naproxcinod Versus Naproxen on Systemic Blood Pressure in Patients With Osteoarthritis. Am. J. Cardiol. 2009, 104, 840–845. [Google Scholar] [PubMed]
- Catella-Lawson, F.; Reilly, M.P.; Kapoor, S.C.; Cucchiara, A.J.; DeMarco, S.; Tournier, B.; Vyas, S.N.; Fitzgerald, G.A. Cyclooxygenase Inhibitors and the Antiplatelet Effects of Aspirin. N. Engl. J. Med. 2001, 345, 1809–1817. [Google Scholar]
- Capone, M.L.; Sciulli, M.G.; Tacconelli, S.; Grana, M.; Ricciotti, E.; Renda, G.; Di, G.P.; Merciaro, G.; Patrignani, P. Pharmacodynamic Interaction of Naproxen With Low-Dose Aspirin in Healthy Subjects. J. Am.. Coll. Cardiol. 2005, 45, 1295–1301. [Google Scholar]
- Patrono, C.; Garcia Rodriguez, L.A.; Landolfi, R.; Baigent, C. Low-Dose Aspirin for the Prevention of Atherothrombosis. N. Engl. J. Med. 2005, 353, 2373–2383. [Google Scholar]
- Fitzgerald, G.A. Parsing an Enigma: the Pharmacodynamics of Aspirin Resistance. Lancet 2003, 361, 542–544. [Google Scholar]
- Richardson, C.G.; Chalmers, A.; Llewellyn-Thomas, H.A.; Klinkhoff, A.; Carswell, A.; Kopec, J.A. Pain Relief in Osteoarthritis: Patients' Willingness to Risk Medication-Induced Gastrointestinal, Cardiovascular, and Cerebrovascular Complications. J. Rheumatol. 2007, 34, 1569–1575. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Meek, I.L.; Van de Laar, M.A.F.J.; E. Vonkeman, H. Non-Steroidal Anti-Inflammatory Drugs: An Overview of Cardiovascular Risks. Pharmaceuticals 2010, 3, 2146-2162. https://doi.org/10.3390/ph3072146
Meek IL, Van de Laar MAFJ, E. Vonkeman H. Non-Steroidal Anti-Inflammatory Drugs: An Overview of Cardiovascular Risks. Pharmaceuticals. 2010; 3(7):2146-2162. https://doi.org/10.3390/ph3072146
Chicago/Turabian StyleMeek, Inger L., Mart A.F.J. Van de Laar, and Harald E. Vonkeman. 2010. "Non-Steroidal Anti-Inflammatory Drugs: An Overview of Cardiovascular Risks" Pharmaceuticals 3, no. 7: 2146-2162. https://doi.org/10.3390/ph3072146
APA StyleMeek, I. L., Van de Laar, M. A. F. J., & E. Vonkeman, H. (2010). Non-Steroidal Anti-Inflammatory Drugs: An Overview of Cardiovascular Risks. Pharmaceuticals, 3(7), 2146-2162. https://doi.org/10.3390/ph3072146