Unlocking the Door to Neuronal Woes in Alzheimer’s Disease: Aβ and Mitochondrial Permeability Transition Pore

{kind=link}
Abstract
:1. Introduction
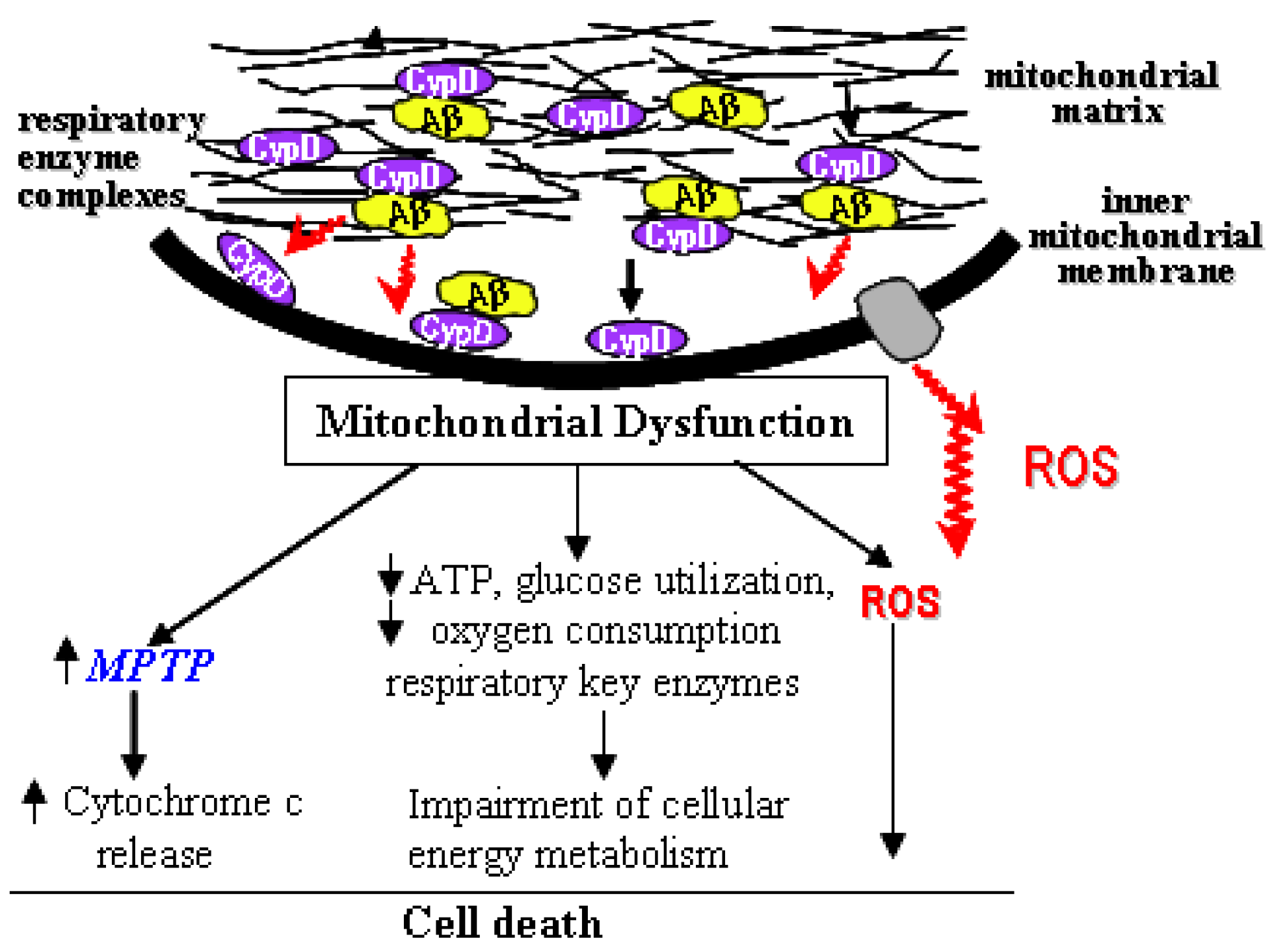
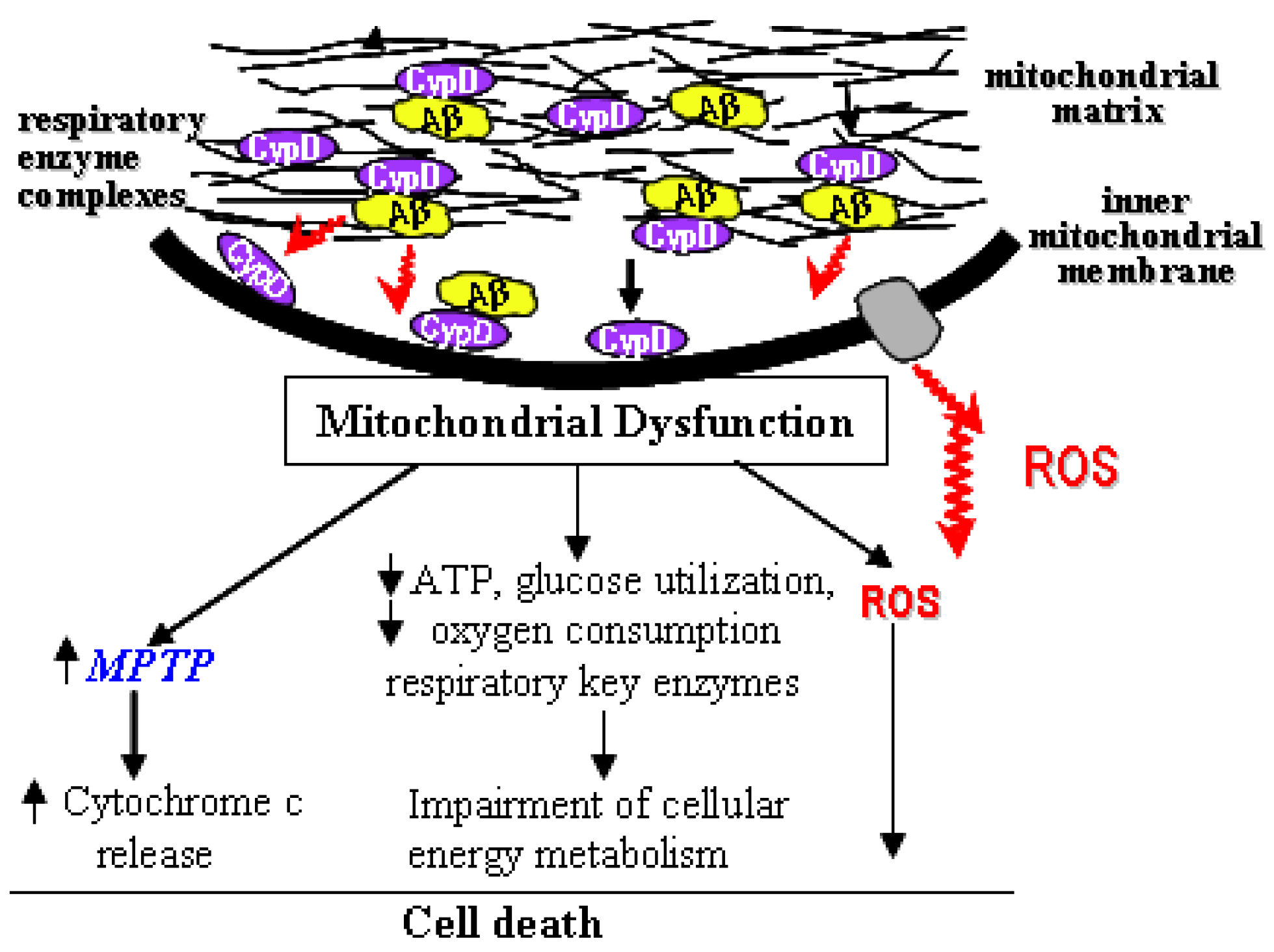
2. mPTP and Alzheimer’s Disease
3. The Interplay of Aβ and mPTP
4. Blockage of mPTP Attenuates Aβ-Mediated Neuronal and Mitochondrial Malfunction
5. Conclusions
Acknowledgements
References and Notes
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; Trinchese, F.; Liu, S.; Gunn-Moore, F.; Lue, L.F.; Walker, D.G.; Kuppusamy, P.; Zewier, Z.L.; Arancio, O.; Stern, D.; Yan, S.S.; Wu, H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 2004, 304, 448–452. [Google Scholar]
- Selkoe, D.J. Alzheimer's disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar]
- Cummings, B.J.; Pike, C.J.; Shankle, R.; Cotman, C.W. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol. Aging 1996, 17, 921–933. [Google Scholar]
- Gu, Z.; Liu, W.; Yan, Z. {beta}-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 2009, 284, 10639–10649. [Google Scholar] [PubMed]
- Takuma, K.; Fang, F.; Zhang, W.; Yan, S.; Fukuzaki, E.; Du, H.; Sosunov, A.; McKhann, G.; Funatsu, Y.; Nakamichi, N.; Nagai, T.; Mizoguchi, H.; Ibi, D.; Hori, O.; Ogawa, S.; Stern, D.M.; Yamada, K.; Yan, S.S. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 20021–20026. [Google Scholar]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; Gunn-Moore, F.J.; Vonsattel, J.P.; Arancio, O.; Chen, J.X.; Yan, S.D. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar]
- Wang, X.; Su, B.; Perry, G.; Smith, M.A.; Zhu, X. Insights into amyloid-beta-induced mitochondrial dysfunction in Alzheimer disease. Free Radical Biol. Med. 2007, 43, 1569–1573. [Google Scholar]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Rhein, V.; Muller-Spahn, F.; Gotz, J.; Muller, W.E. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neuro-Degenerative Dis. 2008, 5, 157–159. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; Ankarcrona, M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2009. [Google Scholar]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.C.; Stern, D.M.; Arancio, O.; Yan, S.S. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005, 19, 597–598. [Google Scholar]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar]
- Hauptmann, S.; Scherping, I.; Drose, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Muller, W.E. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar]
- Qiao, H.; Koya, R.C.; Nakagawa, K.; Tanaka, H.; Fujita, H.; Takimoto, M.; Kuzumaki, N. Inhibition of Alzheimer's amyloid-beta peptide-induced reduction of mitochondrial membrane potential and neurotoxicity by gelsolin. Neurobiol. Aging 2005, 26, 849–855. [Google Scholar]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Binder, D.R.; Bennett, J.P., Jr.; Di Iorio, G.; Golbe, L.I.; Parker, W.D., Jr. Biochemical analysis of cybrids expressing mitochondrial DNA from Contursi kindred Parkinson's subjects. Exp. Neurol. 2001, 169, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Wille, E.; Stack, C.; Calingasan, N.Y.; Beal, M.F.; Lin, M.T. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J. 2009, 23, 2459–2466. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Galvan, V.; Lange, M.B.; Tang, H.; Sowell, R.A.; Spilman, P.; Fombonne, J.; Gorostiza, O.; Zhang, J.; Sultana, R.; Bredesen, D.E. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic. Biol. Med. 48, 136–144. [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar]
- Suen, K.C.; Lin, K.F.; Elyaman, W.; So, K.F.; Chang, R.C.; Hugon, J. Reduction of calcium release from the endoplasmic reticulum could only provide partial neuroprotection against beta-amyloid peptide toxicity. J. Neurochem. 2003, 87, 1413–1426. [Google Scholar]
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 2008, 3, e2718. [Google Scholar]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar]
- Rui, Y.; Tiwari, P.; Xie, Z.; Zheng, J.Q. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J. Neurosci. 2006, 26, 10480–10487. [Google Scholar]
- Mancuso, M.; Orsucci, D.; Siciliano, G.; Murri, L. Mitochondria, mitochondrial DNA and Alzheimer's disease. What comes first? Curr. Alzheimer Res. 2008, 5, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondrial DNA--related mitochondrial dysfunction in neurodegenerative diseases. Arch. Pathol. Lab. Med. 2002, 126, 271–280. [Google Scholar]
- Bozner, P.; Grishko, V.; LeDoux, S.P.; Wilson, G.L.; Chyan, Y.C.; Pappolla, M.A. The amyloid beta protein induces oxidative damage of mitochondrial DNA. J. Neuropathol. Exp. Neurol. 1997, 56, 1356–1362. [Google Scholar]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Rego, A.C.; Oliveira, C. Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J. Neurosci. Res. 2002, 69, 257–267. [Google Scholar]
- Billups, B.; Forsythe, I.D. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 2002, 22, 5840–5847. [Google Scholar]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar]
- Li, G.; Zou, L.Y.; Cao, C.M.; Yang, E.S. Coenzyme Q10 protects SHSY5Y neuronal cells from beta amyloid toxicity and oxygen-glucose deprivation by inhibiting the opening of the mitochondrial permeability transition pore. Biofactors 2005, 25, 97–107. [Google Scholar]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Oliveira, C. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci. Rep. 2001, 21, 789–800. [Google Scholar]
- Shevtzova, E.F.; Kireeva, E.G.; Bachurin, S.O. Effect of beta-amyloid peptide fragment 25-35 on nonselective permeability of mitochondria. Bull. Exp. Biol. Med. 2001, 132, 1173–1176. [Google Scholar]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar]
- Halestrap, A. Biochemistry: a pore way to die. Nature 2005, 434, 578–579. [Google Scholar]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996, 378, 150–152. [Google Scholar]
- Clarke, S.J.; Khaliulin, I.; Das, M.; Parker, J.E.; Heesom, K.J.; Halestrap, A.P. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ. Res. 2008, 102, 1082–1090. [Google Scholar]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; Robbins, J.; Molkentin, J.D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar]
- Connern, C.P.; Halestrap, A.P. Chaotropic agents and increased matrix volume enhance binding of mitochondrial cyclophilin to the inner mitochondrial membrane and sensitize the mitochondrial permeability transition to [Ca2+]. Biochemistry 1996, 35, 8172–8180. [Google Scholar]
- Zheng, Y.; Shi, Y.; Tian, C.; Jiang, C.; Jin, H.; Chen, J.; Almasan, A.; Tang, H.; Chen, Q. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 2004, 23, 1239–1247. [Google Scholar]
- Pestana, C.R.; Silva, C.H.; Pardo-Andreu, G.L.; Rodrigues, F.P.; Santos, A.C.; Uyemura, S.A.; Curti, C. Ca(2+) binding to c-state of adenine nucleotide translocase (ANT)-surrounding cardiolipins enhances (ANT)-Cys(56) relative mobility: a computational-based mitochondrial permeability transition study. Biochim. Biophys. Acta 2009, 1787, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323. [Google Scholar]
- Li, V.; Brustovetsky, T.; Brustovetsky, N. Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp. Neurol. 2009, 218, 171–182. [Google Scholar]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar]
- Karlsson, J.; Fong, K.S.; Hansson, M.J.; Elmer, E.; Csiszar, K.; Keep, M.F. Life span extension and reduced neuronal death after weekly intraventricular cyclosporin injections in the G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosurg. 2004, 101, 128–137. [Google Scholar]
- Brustovetsky, N.; Brustovetsky, T.; Purl, K.J.; Capano, M.; Crompton, M.; Dubinsky, J.M. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J. Neurosci. 2003, 23, 4858–4867. [Google Scholar]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; Duchen, M.R.; Abramov, A.Y. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar]
- Wang, H.L.; Chou, A.H.; Yeh, T.H.; Li, A.H.; Chen, Y.L.; Kuo, Y.L.; Tsai, S.R.; Yu, S.T. PINK1 mutants associated with recessive Parkinson's disease are defective in inhibiting mitochondrial release of cytochrome c. Neurobiol. Dis. 2007, 28, 216–226. [Google Scholar]
- Brown, M.R.; Sullivan, P.G.; Geddes, J.W. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J. Biol. Chem. 2006, 281, 11658–11668. [Google Scholar]
- Rockenstein, E.M.; McConlogue, L.; Tan, H.; Power, M.; Masliah, E.; Mucke, L. Levels and alternative splicing of amyloid beta protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer's disease. J. Biol. Chem. 1995, 270, 28257–28267. [Google Scholar]
- Mucke, L.; Masliah, E.; Johnson, W.B.; Ruppe, M.D.; Alford, M.; Rockenstein, E.M.; Forss-Petter, S.; Pietropaolo, M.; Mallory, M.; Abraham, C.R. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994, 666, 151–167. [Google Scholar]
- Soskic, V.; Klemm, M.; Proikas-Cezanne, T.; Schwall, G.P.; Poznanovic, S.; Stegmann, W.; Groebe, K.; Zengerling, H.; Schoepf, R.; Burnet, M.; Schrattenholz, A. A connection between the mitochondrial permeability transition pore, autophagy, and cerebral amyloidogenesis. J. Proteome Res. 2008, 7, 2262–2269. [Google Scholar] [PubMed]
- Moreira, A.E.; Hueb, W.A.; Soares, P.R.; Meneghetti, J.C.; Jorge, M.C.; Chalela, W.A.; Martinez Filho, E.E.; Oliveira, S.A.; Jatene, F.B.; Ramires, J.A. Comparative study between the therapeutic effects of surgical myocardial revascularization and coronary angioplasty in equivalent ischemic situations: analysis through myocardial scintigraphy with 99mTc-Sestamibi. Arq. Bras. Cardiol. 2005, 85, 92–99. [Google Scholar]
- Chin, J.H.; Tse, F.W.; Harris, K.; Jhamandas, J.H. Beta-amyloid enhances intracellular calcium rises mediated by repeated activation of intracellular calcium stores and nicotinic receptors in acutely dissociated rat basal forebrain neurons. Brain Cell Biol. 2006, 35, 173–186. [Google Scholar]
- Brewer, G.J.; Lim, A.; Capps, N.G.; Torricelli, J.R. Age-related calcium changes, oxyradical damage, caspase activation and nuclear condensation in hippocampal neurons in response to glutamate and beta-amyloid. Exp. Gerontol. 2005, 40, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Schoneich, C.; Pogocki, D.; Hug, G.L.; Bobrowski, K. Free radical reactions of methionine in peptides: mechanisms relevant to beta-amyloid oxidation and Alzheimer's disease. J. Am. Chem. Soc. 2003, 125, 13700–13713. [Google Scholar]
- Morais Cardoso, S.; Swerdlow, R.H.; Oliveira, C.R. Induction of cytochrome c-mediated apoptosis by amyloid beta 25-35 requires functional mitochondria. Brain Res. 2002, 931, 117–125. [Google Scholar]
- Celsi, F.; Svedberg, M.; Unger, C.; Cotman, C.W.; Carri, M.T.; Ottersen, O.P.; Nordberg, A.; Torp, R. Beta-amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol. Dis. 2007, 26, 342–352. [Google Scholar]
- Montiel, T.; Quiroz-Baez, R.; Massieu, L.; Arias, C. Role of oxidative stress on beta-amyloid neurotoxicity elicited during impairment of energy metabolism in the hippocampus: protection by antioxidants. Exp. Neurol. 2006, 200, 496–508. [Google Scholar]
- Parks, J.K.; Smith, T.S.; Trimmer, P.A.; Bennett, J.P., Jr.; Parker, W.D., Jr. Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 2001, 76, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, J.H.; Lee, J.P.; Kim, E.M.; Chang, K.A.; Park, C.H.; Jeong, S.J.; Wittendorp, M.C.; Seo, J.H.; Choi, S.H.; Suh, Y.H. Amyloid beta peptide induces cytochrome C release from isolated mitochondria. Neuroreport 2002, 13, 1989–1993. [Google Scholar]
- Rodrigues, C.M.; Sola, S.; Brito, M.A.; Brondino, C.D.; Brites, D.; Moura, J.J. Amyloid beta-peptide disrupts mitochondrial membrane lipid and protein structure: protective role of tauroursodeoxycholate. Biochem. Biophys. Res. Commun. 2001, 281, 468–474. [Google Scholar]
- Zhang, S.; Zhang, Z.; Sandhu, G.; Ma, X.; Yang, X.; Geiger, J.D.; Kong, J. Evidence of oxidative stress-induced BNIP3 expression in amyloid beta neurotoxicity. Brain Res. 2007, 1138, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Suman, S.; Chandna, S.; Das, T.K. Possible role of amyloid-beta, adenine nucleotide translocase and cyclophilin-D interaction in mitochondrial dysfunction of Alzheimer's disease. Bioinformation 2009, 3, 440–445. [Google Scholar]
- Vlachos, P.; Nyman, U.; Hajji, N.; Joseph, B. The cell cycle inhibitor p57(Kip2) promotes cell death via the mitochondrial apoptotic pathway. Cell Death Differ. 2007, 14, 1497–1507. [Google Scholar]
- Vale, C.; Nicolaou, K.C.; Frederick, M.O.; Vieytes, M.R.; Botana, L.M. Cell volume decrease as a link between azaspiracid-induced cytotoxicity and c-Jun-N-terminal kinase activation in cultured neurons. Toxicol Sci. 113, 158–168. [PubMed]
- Xia, Z.; Tauskela, J.; Small, D.L. Disulfonic stilbenes prevent beta-amyloid (25-35) neuronal toxicity in rat cortical cultures. Neurosci. Lett. 2003, 340, 53–56. [Google Scholar]
- Clarke, S.J.; McStay, G.P.; Halestrap, A.P. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002, 277, 34793–34799. [Google Scholar]
- Halestrap, A.P.; Connern, C.P.; Griffiths, E.J.; Kerr, P.M. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell Biochem 1997, 174, 167–172. [Google Scholar]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Rahder, M.; Stem, K.; Bernardi, P.; Bourdette, D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar]
- Keep, M.; Elmer, E.; Fong, K.S.; Csiszar, K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001, 894, 327–331. [Google Scholar]
- Gold, B.G.; Voda, J.; Yu, X.; McKeon, G.; Bourdette, D.N. FK506 and a nonimmunosuppressant derivative reduce axonal and myelin damage in experimental autoimmune encephalomyelitis: neuroimmunophilin ligand-mediated neuroprotection in a model of multiple sclerosis. J. Neurosci. Res. 2004, 77, 367–377. [Google Scholar]
- Kumar, P.; Kumar, A. Neuroprotective effect of cyclosporine and FK506 against 3-nitropropionic acid induced cognitive dysfunction and glutathione redox in rat: possible role of nitric oxide. Neurosci. Res. 2009, 63, 302–314. [Google Scholar]
- Van Den Heuvel, C.; Donkin, J.J.; Finnie, J.W.; Blumbergs, P.C.; Kuchel, T.; Koszyca, B.; Manavis, J.; Jones, N.R.; Reilly, P.L.; Vink, R. Downregulation of amyloid precursor protein (APP) expression following post-traumatic cyclosporin-A administration. J. Neurotrauma 2004, 21, 1562–1572. [Google Scholar]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; Lacapere, J.J.; Massaad, C.; Schumacher, M.; Steidl, E.M.; Maux, D.; Delaage, M.; Henderson, C.E.; Pruss, R.M. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Du, H.; ShiDu Yan, S. Unlocking the Door to Neuronal Woes in Alzheimer’s Disease: Aβ and Mitochondrial Permeability Transition Pore. Pharmaceuticals 2010, 3, 1936-1948. https://doi.org/10.3390/ph3061936
Du H, ShiDu Yan S. Unlocking the Door to Neuronal Woes in Alzheimer’s Disease: Aβ and Mitochondrial Permeability Transition Pore. Pharmaceuticals. 2010; 3(6):1936-1948. https://doi.org/10.3390/ph3061936
Chicago/Turabian StyleDu, Heng, and Shirley ShiDu Yan. 2010. "Unlocking the Door to Neuronal Woes in Alzheimer’s Disease: Aβ and Mitochondrial Permeability Transition Pore" Pharmaceuticals 3, no. 6: 1936-1948. https://doi.org/10.3390/ph3061936
APA StyleDu, H., & ShiDu Yan, S. (2010). Unlocking the Door to Neuronal Woes in Alzheimer’s Disease: Aβ and Mitochondrial Permeability Transition Pore. Pharmaceuticals, 3(6), 1936-1948. https://doi.org/10.3390/ph3061936