Therapeutic Drug Monitoring of the Newer Anti-Epilepsy Medications

Abstract
1. Background on Therapeutic Drug Monitoring of Antiepileptic Medications
2. Reasons for Applying TDM to AEDS
3. The Newer Generation of AEDs
4. The Challenge of Establishing Reference Ranges for AEDs

{kind=link}
| Drug | Oral Bioavailability (%) | Serum protein binding (%) | Time to peak concentration (h) | Half-life in Absence of Concomitant Enzyme Inducersa | Half-life in Presence of Concomitant Enzyme Inducersa | Reference Range in Serum (mg/L)f |
|---|---|---|---|---|---|---|
| Eslicarbazepine acetate | ≥80 | 30 | 1–4 | 20–24 | 20–24 | Not established |
| Felbamate | >90 | 25 | 2–6 | 16–22 | 10–18 | 30–60 |
| Gabapentin | <60 | 0 | 2–3 | 5–9 | 5–9 | 2–20 |
| Lacosamide | ≥95 | 15 | 0.5–4 | 12–13 | 12–13 | 5–10 |
| Lamotrigine | ≥95 | 55 | 1–3 | 15–35b | 8–20 | 3–14 |
| Levetiracetam | ≥95 | 0 | 1 | 6–8 | 6–8 | 12–46 |
| Oxcarbazepinec | 90 | 40 | 3–6 | 8–15 | 7–12 | 3–35 |
| Pregabalin | ≥90 | 0 | 1–2 | 5–7 | 5–7 | 2.8–8.3 |
| Rufinamide | 85 | 30 | 5–6 | 8–12 | ≤8 | Not established |
| Stiripentold | ≥90 | 99 | 1–2 | Variablee | Variablee | 4–22 |
| Tiagabined | ≥90 | 96 | 1–2 | 5–9 | 2–4 | 0.02–0.2 |
| Topiramate | ≥80 | 15 | 2–4 | 20–30 | 10–15 | 5–20 |
| Vigabatrin | ≥60 | 0 | 1–2 | 5–8 | 5–8 | 0.8–36 |
| Zonisamide | ≥65 | 50 | 2–5 | 50–70 | 25–35 | 10–40 |
| Drug | Factors that Favor Use of TDM | Factors that May Limit Use of TDM and/or Complicate Interpretation |
|---|---|---|
| Eslicarbazepine acetate | Auto-induction with chronic dosing | Has active metabolite (oxcarbazepine) |
| Liver failure | ||
| Felbamate | Variable metabolism | Unclear toxic concentrations |
| Potential for severe toxicity | ||
| Gabapentin | Variable absorption | Wide range of clinically effective serum concentrations |
| Renal failure | ||
| Low incidence of toxicity | ||
| Lacosamide | Liver failure | Generally predictable pharmacokinetics Drug-drug interactions uncommon |
| Renal failure | ||
| Lamotrigine | Variable metabolism | |
| Common drug-drug interactions | ||
| Well-defined toxic concentrations | ||
| Common use in pregnancy | ||
| Levetiracetam | Renal failure | Wide range of clinically effective serum concentrations |
| Low incidence of toxicity | ||
| Oxcarbazepine | Variable metabolism | |
| Well-defined toxic concentrations | ||
| Pregabalin | Variable absorption | Wide range of clinically effective serum concentrations |
| Renal failure | ||
| Low incidence of toxicityShort half-life | ||
| Rufinamide | Variable absorption | |
| Common drug-drug interactions Renal failure | ||
| Stiripentol | Extensive first-pass metabolism High serum protein binding | High serum protein binding can complicate interpretation of total drug concentrations (free drug levels may be helpful) |
| Zero-order elimination kinetics | ||
| Tiagabine | High serum protein bindingLiver failure | High serum protein binding can complicate interpretation of total drug concentrations (free drug levels may be helpful) |
| Common drug-drug interactions | ||
| Topiramate | Common drug-drug interactions | |
| Vigabatrin | Renal failure | Poor correlation of serum concentrations and therapeutic effect (irreversible effect) |
| Zonisamide | Variable metabolism | |
| Common drug-drug interactions | ||
| Well-defined toxic concentrations |
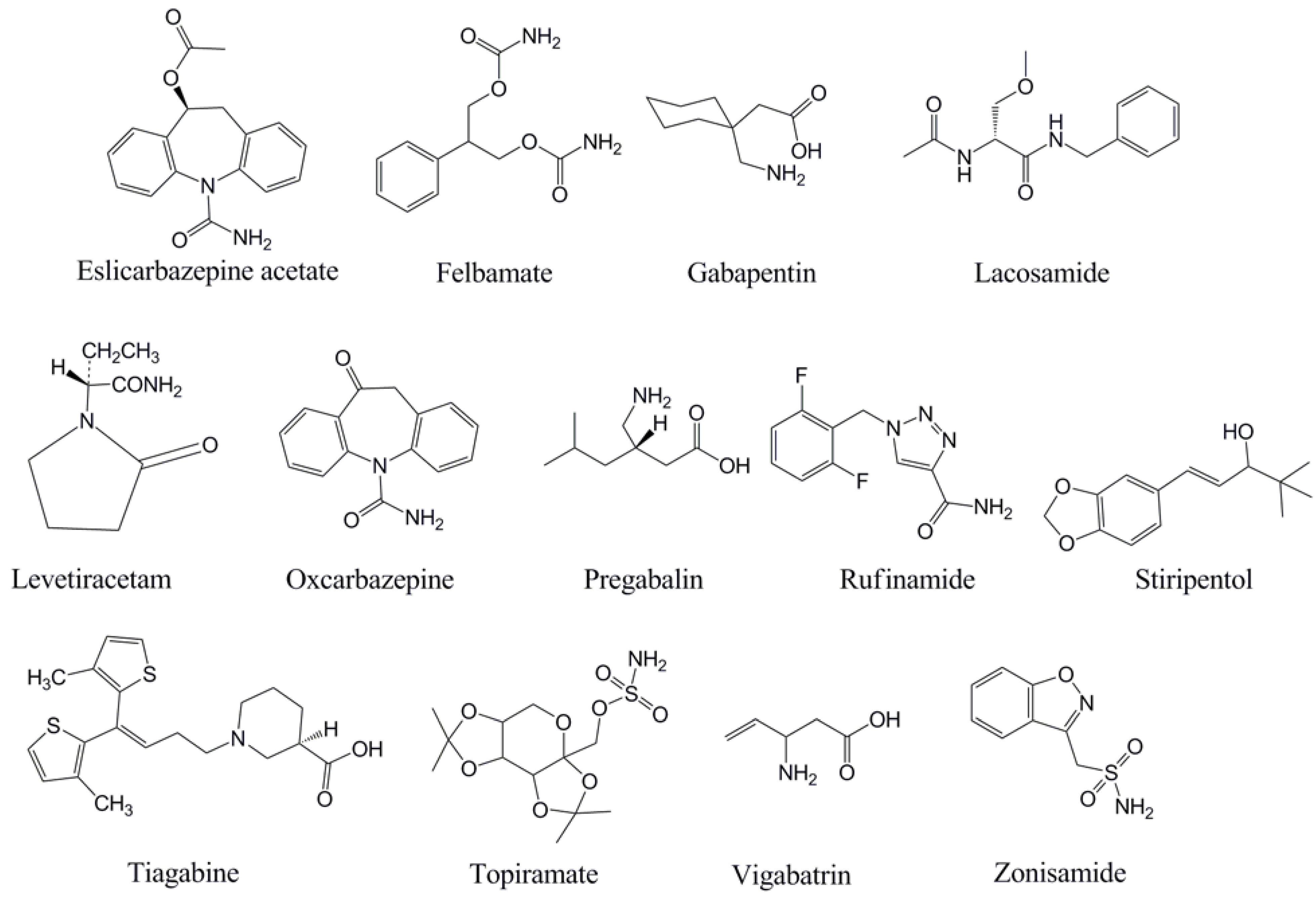
5. Eslicarbazepine Acetate
6. Felbamate
7. Gabapentin
8. Lacosamide
9. Lamotrigine
10. Levetiracetam
11. Oxcarbazepine
12. Pregabalin
13. Rufinamide
14. Stiripentol
15. Tiagabine
16. Topiramate
17. Vigabatrin
18. Zonisamide
19. Summary and Further Applications
Acknowledgements
References
- Neels, H.M.; Sierens, A.C.; Naelerts, K.; Scharpé, S.L.; Hatfield, G.M.; Lambert, W.E. Therapeutic drug monitoring of old and newer anti-epileptic drugs. Clin. Chem. Lab. Med. 2004, 42, 1228–1255. [Google Scholar]
- Patsalos, P.N.; Berry, D.J.; Bourgeois, B.F.D.; Cloyd, J.C.; Glauser, T.A.; Johannessen, S.I.; Tomson, T.; Perucca, E. Antiepileptic drugs—Best practice guidelines for therapeutic drug monitoring: A position paper by the subcommission on therapeutic drug monitoring, ILAE commission on therapeutic strategies. Epilepsia 2008, 49, 1239–1276. [Google Scholar]
- Fröscher, W.; Eichelbaum, M.; Gugler, R.; Hildebrand, G.; Penin, H. A prospective randomized trial on the effect of monitoring plasma anticonvulsant levels in epilepsy. J. Neurol. 1981, 224, 193–201. [Google Scholar]
- Januzzi, G.; Cian, P.; Fattore, C.; Gatti, G.; Bartoli, A.; Monaco, F.; Perucca, E. A multicenter randomized controlled trial on the clinical impact of therapeutic drug monitoring in patients with newly diagnosed epilepsy. Epilepsia 2000, 41, 222–230. [Google Scholar]
- Liu, H.; Delgado, M.R. Therapeutic drug concentration monitoring using saliva samples. Focus on anticonvulsants. Clin. Pharmacokinet. 1999, 36, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.J.; Beran, R.G.; Plunkeft, M.J.; Clarke, L.A.; Hung, W.T. The absorption of gabapentin following high dose escalation. Seizure 2003, 12, 28–36. [Google Scholar]
- Tsiropoulos, I.; Kristensen, O.; Klitgaard, N.A. Saliva and serum concentration of lamotrigine in patients with epilepsy. Ther. Drug. Monit. 2000, 22, 517–521. [Google Scholar]
- Mecarelli, O.; Li Voti, P.; Pro, S.; Romolo, F.S.; Rotolo, M.; Pulitano, P.; Accornero, N.; Vanacore, N. Saliva and serum levetiracetam concentrations in patients with epilepsy. Ther. Drug. Monit. 2007, 29, 313–318. [Google Scholar]
- Miles, M.V.; Tang, P.H.; Ryan, M.A.; Grim, S.A.; Fakhoury, T.A.; Strawsburg, R.H.; DeGrauw, T.J.; Baumann, R.J. Feasibility and limitations of oxcarbazepine monitoring using salivary monohydroxycarbamazepine (MHD). Ther. Drug. Monit. 2004, 26, 300–304. [Google Scholar]
- Miles, M.V.; Tang, P.H.; Glauser, T.A.; Ryan, M.A.; Grim, S.A.; Strawsburg, R.H.; deGrauw, T.J.; Baumann, R.J. Topiramate concentration in saliva: An alternative to serum monitoring. Pediatr. Neurol. 2003, 29, 143–147. [Google Scholar]
- Kumagai, N.; Seki, T.; Yamada, T.; Takuma, Y.; Hirai, K. Concentrations of zonisamide in serum, free fraction, mixed saliva and cerebrospinal fluid in epileptic children treated with monotherapy. Jpn. J. Psychiatry Neurol. 1993, 47, 291–292. [Google Scholar] [PubMed]
- Jones, M.D.; Ryan, M.; Miles, M.V.; Tang, P.H.; Fakhoury, T.A.; Degrauw, T.J.; Baumann, R.J. Stability of salivary concentrations of the newer antiepileptic drugs in the postal system. Ther. Drug Monit. 2005, 27, 576–579. [Google Scholar]
- Bialer, M. The pharmacokinetics and interactions of new antiepileptic drugs: An overview. Ther. Drug Monit. 2005, 27, 722–726. [Google Scholar]
- Perucca, E. Clinical pharmacokinetics of new-generation antiepileptic drugs at the extremes of age. Clin. Pharmacokinet. 2006, 45, 351–364. [Google Scholar]
- Perucca, E.; Bialer, M. The clinical pharmacokinetics of the newer antiepileptic drugs. Focus on topiramate, zonisamide and tiagabine. Clin. Pharmacokinet. 1996, 31, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, E.G. Induction of cytochromes P450. Curr. Drug Metab. 2001, 2, 139–147. [Google Scholar]
- Tien, E.S.; Negishi, M. Nuclear receptors CAR and PXR in the regulation of hepatic metabolism. Xenobiotica 2006, 36, 1152–1163. [Google Scholar]
- Zhang, B.; Xie, W.; Krasowski, M.D. PXR: A xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics 2008, 9, 1695–1709. [Google Scholar]
- Lacerda, G.; Krummel, T.; Sabourdy, C.; Ryvlin, P.; Hirsch, E. Optimizing therapy of seizures in patients with renal or hepatic dysfunction. Neurology 2006, 67, S28–S33. [Google Scholar]
- Pitlick, W.H.; Levy, R.H. Time-dependent kinetics I: Exponential autoinduction of carbamazepine in monkeys. J. Pharm. Sci. 1977, 66, 647–649. [Google Scholar]
- Dasgupta, A. Usefulness of monitoring free (unbound) concentrations of therapeutic drugs in patient management. Clin. Chim. Acta 2007, 377, 1–13. [Google Scholar]
- LaRoche, S.M.; Helmers, S.L. The new antiepileptic drugs: Clinical applications. JAMA 2004, 291, 615–620. [Google Scholar]
- Patsalos, P.N. New antiepileptic drugs. Ann. Clin. Biochem. 1999, 36, 10–19. [Google Scholar]
- Johannessen Landmark, C. Antiepileptic drugs in non-epilepsy disorders: Relations between mechanisms of action and clinical efficacy. CNS Drugs 2008, 22, 27–47. [Google Scholar]
- Perucca, E. Is there a role for therapeutic drug monitoring of new anticonvulsants? Clin. Pharmacokinet. 2000, 38, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, S.I.; Tomson, T. Pharmacokinetic variability of newer epileptic drugs? Clin. Pharmacokinet. 2006, 45, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, A.F.; Silva, A.P.; Malva, J.O.; Soares-da-Silva, P.; Carvalho, A.P.; Carvalho, C.M. Inhibition of glutamate release by BIA 2-093 and BIA 2-024, two novel derivatives of carbamazepine, due to blockade of sodium but not calcium channels. Biochem. Pharmacol. 2001, 61, 1271–1275. [Google Scholar] [PubMed]
- Maia, J.; Vaz-da-Silva, M.; Almeida, L.; Falcao, A.; Silveira, P.; Guimaraes, S.; Graziela, P.; Soares-da-Silva, P. Effect of food on the pharmacokinetic profile of eslicarbazepine acetate (BIA 2-093). Drugs R D 2005, 6, 201–206. [Google Scholar]
- Almeida, L.; Falcao, A.; Maia, J.; Mazur, D.; Gellert, M.; Soares-da-Silva, P. Single-dose and steady-state pharmacokinetics of eslicarbazepine acetate (BIA 2-093) in healthy elderly and young subjects. J. Clin. Pharmacol. 2005, 45, 1062–1066. [Google Scholar]
- Almeida, L.; Nunes, T.; Sicard, E.; Rocha, J.F.; Falcao, A.; Brunet, J.S.; Lefebvre, M.; Soares-da-Silva, P. Pharmacokinetic interaction study between eslicarbazepine acetate and lamotrigine in healthy subjects. Acta Neurol. Scand. 2010, 121, 257–264. [Google Scholar]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, H.S. Progress report on new antiepileptic drugs: A summary of the ninth eilat conference (EILAT IX). Epilepsy Res. 2009, 83, 1–43. [Google Scholar]
- Almeida, L.; Potgieter, J.H.; Maia, J.; Potgieter, M.A.; Mota, F.; Soares-da-Silva, P. Pharmacokinetics of eslicarbazepine acetate in patients with moderate hepatic impairment. Eur. J. Clin. Pharmacol. 2008, 64, 267–273. [Google Scholar]
- Maia, J.; Almeida, L.; Falcao, A.; Soares, E.; Mota, F.; Potgieter, M.A.; Potgieter, J.H.; Soares-da-Silva, P. Effect of renal impairment on the pharmacokinetics of eslicarbazepine acetate. Int. J. Clin. Pharmacol. Ther. 2008, 46, 119–130. [Google Scholar]
- Alves, G.; Figueiredo, I.; Castel-Branco, M.; Loureiro, A.; Fortuna, A.; Falcao, A.; Caramona, M. Enantioselective HPLC-UV method for determination of eslicarbazepine acetate (BIA 2-093) and its metabolites in human plasma. Biomed. Chromatogr. 2007, 21, 1127–1134. [Google Scholar]
- Popova, E.; Leighton, C.; Bernabarre, A.; Bernardo, M.; Vieta, E. Oxcarbazepine in the treatment of bipolar and schizoaffective disorders. Expert Rev. Neurother. 2007, 7, 617–626. [Google Scholar]
- Wagner, M.L.; Graves, N.M.; Marienau, K.; Holmes, G.B.; Remmel, R.P.; Leppik, I.E. Discontinuation of phenytoin and carbamazepine in patients receiving felbamate. Epilepsia 1991, 32, 398–406. [Google Scholar]
- Ramsay, R.E.; Pellock, J.M.; Garnett, W.R.; Sanchez, R.M.; Valakas, A.M.; Wargin, W.A.; Lai, A.A.; Hubbell, J.; Chern, W.H.; Allsup, T.; et al. Pharmacokinetics and safety of lamotrigine (Lamictal) in patients with epilepsy. Epilepsy Res. 1991, 10, 191–200. [Google Scholar] [CrossRef] [PubMed]
- May, T.W.; Korn-Merker, E.; Rambeck, B. Clinical pharmacokinetics of oxcarbazepine. Clin. Pharmacokinet. 2003, 42, 1023–1042. [Google Scholar]
- Perucca, E.; Cloyd, J.; Critchley, D.; Fuseau, E. Rufinamide: Clinical pharmacokinetics and concentration-response relationships in patients with epilepsy. Epilepsia 2008, 49, 1123–1141. [Google Scholar]
- So, E.L.; Wolff, D.; Graves, N.M.; Leppik, I.E.; Cascino, G.D.; Pixton, G.C.; Gustavson, L.E. Pharmacokinetics of tiagabine as add-on therapy in patients taking enzyme-inducing antiepilepsy drugs. Epilepsy Res. 1995, 22, 221–226. [Google Scholar]
- Britzi, M.; Perucca, E.; Soback, S.; Levy, R.H.; Fattore, C.; Crema, F.; Gatti, G.; Doose, D.R.; Maryanoff, B.E.; Bialer, M. Pharmacokinetic and metabolic investigation of topiramate disposition in healthy subjects in the absence and in the presence of enzyme induction by carbamazepine. Epilepsia 2005, 46, 378–384. [Google Scholar]
- Faught, E.; Sachdeo, R.C.; Remler, M.P.; Chayasirisobhon, S.; Iragui-Madoz, V.J.; Ramsay, R.E.; Sutula, T.P.; Kanner, A.; Harner, R.N.; Kuzniecky, R.; Kramer, L.D.; Karmin, M.; Rosenberg, A. Felbamate monotherapy for partial-onset seizures: An active-controlled trial. Neurology 1993, 43, 688–692. [Google Scholar]
- Sachdeo, R.C.; Kramer, L.D.; Rosenberg, A.; Sachdeo, S. Felbamate monotherapy: Controlled trial in patients with partial onset seizures. Ann. Neurol. 1992, 32, 386–392. [Google Scholar]
- Lindberger, M.; Luhr, O.; Johannessen, S.I.; Larsson, S.; Tomson, T. Serum concentrations and effects of gabapentin and vigabatrin: Observations from a dose titration study. Ther. Drug Monit. 2003, 25, 457–462. [Google Scholar]
- Kellinghaus, C. Lacosamide as treatment for partial epilepsy: Mechanisms of action, pharmacology, effects, and safety. Ther. Clin. Risk Manag. 2009, 5, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, A.; Guerrini, R.; Belmonte, A.; Alessandri, M.G.; Gatti, G.; Perucca, E. The influence of dosage, age, and comedication on steady state plasma lamotrigine concentrations in epileptic children: A prospective study with preliminary assessments of correlations with clinical response. Ther. Drug Monit. 1997, 19, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Leppik, I.E.; Rarick, J.O.; Walczak, T.S.; Tran, T.A.; White, J.R.; Gumnit, R.J. Effective levetiracetam doses and serum concentrations: Age effects. Epilepsia 2002, 43, 240. [Google Scholar]
- Friis, M.L.; Kristensen, O.; Boas, J.; Dalby, M.; Deth, S.H.; Gram, L.; Mikkelsen, M.; Pedersen, B.; Sabers, A.; Worm-Petersen, J. Therapeutic experiences with 947 epileptic out-patients in oxcarbazepine treatment. Acta Neurol. Scand. 1993, 87, 224–227. [Google Scholar]
- Farwell, J.R.; Anderson, G.D.; Kerr, B.M.; Tor, J.A.; Levy, R.H. Stiripentol in atypical absence seizures in children: An open trial. Epilepsia 1993, 34, 305–311. [Google Scholar]
- Uthman, B.M.; Rowan, A.J.; Ahmann, P.A.; Leppik, I.E.; Schachter, S.C.; Sommerville, K.W.; Shu, V. Tiagabine for complex partial seizures: A randomized, add-on, dose-response trial. Arch. Neurol. 1998, 55, 56–62. [Google Scholar] [PubMed]
- Johannessen, S.I.; Battino, D.; Berry, D.J.; Bialer, M.; Kramer, G.; Tomson, T.; Patsalos, P.N. Therapeutic drug monitoring of the newer antiepileptic drugs. Ther. Drug Monit. 2003, 25, 347–363. [Google Scholar]
- Mimaki, T. Clinical pharmacology and therapeutic drug monitoring of zonisamide. Ther. Drug Monit. 1998, 20, 593–597. [Google Scholar]
- Pellock, J.M.; Faught, E.; Leppik, I.E.; Shinnar, S.; Zupanc, M.L. Felbamate: Consensus of current clinical experience. Epilepsy Res. 2006, 71, 89–101. [Google Scholar]
- Thompson, C.D.; Barthen, M.T.; Hopper, D.W.; Miller, T.A.; Quigg, M.; Hudspeth, C.; Montouris, G.; Marsh, L.; Perhach, J.L.; Sofia, R.D.; Macdonald, T.L. Quantification in patient urine samples of felbamate and three metabolites: Acid carbamate and two mercapturic acids. Epilepsia 1999, 40, 769–776. [Google Scholar]
- Shumaker, R.C.; Fantel, C.; Kelton, E.; Wong, K.; Weliky, I. Evaluation of the elimination of (14C) felbamate in healthy men. Epilepsia 1990, 31, 642. [Google Scholar]
- Sachdeo, R.C.; Narang-Sachdeo, S.K.; Howard, J.R.; Dix, R.K.; Shumaker, R.C.; Perhach, J.L.; Rosenberg, A. Steady-state pharmacokinetics and dose-proportionality of felbamate after oral administration of 1200, 2400, and 3600 mg/day of felbamate. Epilepsia 1993, 34, 80. [Google Scholar]
- Ward, D.L.; Wagner, M.L.; Perhach, J.L.; Kramer, L.; Graves, N.; Leppik, I.; Shumaker, R.C. Felbamate steady-state pharmacokinetics during co-administration of valproate. Epilepsia 1991, 32, 8. [Google Scholar]
- Annesley, T.M.; Clayton, L.T. Determination of felbamate in human serum by high-performance liquid chromatography. Ther. Drug Monit. 1994, 16, 419–424. [Google Scholar]
- Behnke, C.E.; Reddy, M.N. Determination of felbamate concentration in pediatric samples by high-performance liquid chromatography. Ther. Drug Monit. 1997, 19, 301–306. [Google Scholar]
- Poquette, M.A. Isothermal gas chromatographic method for the rapid determination of felbamate concentration in human serum. Ther. Drug Monit. 1995, 17, 168–173. [Google Scholar]
- Shihabi, Z.K.; Oles, K.S. Felbamate measured in serum by two methods: HPLC and capillary electrophoresis. Clin. Chem. 1994, 40, 1904–1908. [Google Scholar]
- McLean, M.J. Gabapentin. Epilepsia 1995, 36, S57–S86. [Google Scholar]
- Vollmer, K.O.; von Hodenberg, A.; Kölle, E.U. Pharmacokinetics and metabolism of gabapentin in rat, dog and man. Arzneimittelforschung 1988, 36, 830–839. [Google Scholar]
- Gidal, B.E.; DeCerce, J.; Bockbrader, H.N.; Gonzalez, J.; Kruger, S.; Pitterle, M.E.; Rutecki, P.; Ramsay, R.E. Gabapentin bioavailability: Effect of dose and frequency of administration in adult patients with epilepsy. Epilepsy Res. 1998, 31, 91–99. [Google Scholar]
- Wong, M.O.; Eldon, M.A.; Keane, W.F.; Turck, D.; Bockbrader, H.N.; Underwood, B.A.; Sedman, A.J.; Halstenson, C.E. Disposition of gabapentin in anuric subjects on hemodialysis. J. Clin. Pharmacol. 1995, 35, 622–626. [Google Scholar]
- Armijo, J.A.; Perna, M.A.; Adin, J.; Vega-Gil, N. Association between patient age and gabapentin serum concentration-to-dose ratio: A preliminary multivariate analysis. Ther. Drug Monit. 2004, 26, 633–637. [Google Scholar]
- Sivenius, J.; Kälviäinen, R.; Ylinen, A.; Riekkinen, P. A double-blind study of gabapentin in the treatment of partial seizures. Epilepsia 1991, 32, 539–542. [Google Scholar]
- Bahrami, G.; Kiani, A. Sensitive high-performance liquid chromatographic quantitation of gabapentin in human serum using liquid-liquid extraction and pre-column derivatization with 9-fluorenylmethyl chloroformate. J. Chromatogr. B 2006, 835, 123–126. [Google Scholar]
- Juenke, J.M.; Brown, P.I.; McMillin, G.A.; Urry, F.M. Procedure for the monitoring of gabapentin with 2,4,6-trinitrobenzene sulfonic acid derivatization followed by HPLC with ultraviolet detection. Clin. Chem. 2003, 49, 1198–1201. [Google Scholar]
- Ifa, D.R.; Falci, M.; Moraes, M.E.; Bezerra, F.A.; Moraes, M.O.; de Nucci, G. Gabapentin quantification in human plasma by high-performance liquid chromatography coupled to electrospray tandem mass spectrometry. Application to bioequivalence study. J. Mass Spectrom. 2001, 36, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Borrey, D.C.; Godderis, K.O.; Engelrelst, V.I.; Bernard, D.R.; Langlois, M.R. Quantitative determination of vigabatrin and gabapentin in human serum by gas chromatography-mass spectrometry. Clin. Chim. Acta 2005, 354, 147–151. [Google Scholar]
- Gambelunghe, C.; Mariucci, G.; Tantucci, M.; Ambrosini, M.V. Gas chromatography-tandem mass spectrometry analysis of gabapentin in serum. Biomed. Chromatogr. 2005, 19, 63–67. [Google Scholar]
- Curia, G.; Biagini, G.; Perucca, E.; Avoli, M. Lacosamide: A new approach to target voltage-gated sodium currents in epileptic disorders. CNS Drugs 2009, 23, 555–568. [Google Scholar]
- Chung, S.; Sperling, M.R.; Biton, V.; Krauss, G.; Hebert, D.; Rudd, G.D.; Doty, P. Lacosamide as adjunctive therapy for partial-onset seizures: A randomized controlled trial. Epilepsia 2010, 51, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Ben-Menachem, E.; Biton, V.; Jatuzis, D.; Abou-Khalil, B.; Doty, P.; Rudd, G.D. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia 2007, 48, 1308–1317. [Google Scholar]
- Johannessen Landmark, C.; Patsalos, P.N. Drug interactions involving the new second- and third-generation antiepileptic drugs. Expert Rev. Neurother. 2010, 10, 119–140. [Google Scholar]
- Beydoun, A.; D'Souza, J.; Hebert, D.; Doty, P. Lacosamide: Pharmacology, mechanisms of action and pooled efficacy and safety data in partial-onset seizures. Expert Rev. Neurother. 2009, 9, 33–42. [Google Scholar]
- Gidal, B.E.; Privitera, M.D.; Sheth, R.D.; Gilman, J.T. Vigabatrin: A novel therapy for seizure disorders. Ann. Pharmacother. 1999, 33, 1277–1286. [Google Scholar]
- Cawello, W.; Nickel, B.; Eggert-Formella, A. No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. J. Clin. Pharmacol. 2010, 50, 459–471. [Google Scholar]
- Halford, J.J.; Lapointe, M. Clinical perspectives on lacosamide. Epilepsy Curr. 2009, 9, 1–9. [Google Scholar]
- Sabers, A.; Tomson, T. Managing antiepileptic drugs during pregnancy and lactation. Curr. Opin. Neurol. 2009, 22, 157–161. [Google Scholar]
- Tomson, T.; Battino, D. Pharmacokinetics and therapeutic drug monitoring of newer antiepileptic drugs during pregnancy and the puerperium. Clin. Pharmacokinet. 2007, 46, 209–219. [Google Scholar]
- Incecayir, T.; Agabeyoglu, I.; Gucuyener, K. Comparison of plasma and saliva concentrations of lamotrigine in healthy volunteers. Arzneimittelforschung 2007, 57, 517–521. [Google Scholar]
- Malone, S.A.; Eadie, M.J.; Addison, R.S.; Wright, A.W.; Dickinson, R.G. Monitoring salivary lamotrigine concentrations. J. Clin. Neurosci. 2006, 13, 902–907. [Google Scholar]
- Biton, V. Pharmacokinetics, toxicology and safety of lamotrigine in epilepsy. Expert Opin. Drug Metab. Toxicol. 2006, 2, 1009–1018. [Google Scholar]
- Hussein, Z.; Posner, J. Population pharmacokinetics of lamotrigine monotherapy in patients with epilepsy: Retrospective analysis of routine monitoring data. Br. J. Clin. Pharmacol. 1997, 43, 457–464. [Google Scholar]
- Reimers, A.; Helde, G.; Brodtkorb, E. Ethinyl estradiol, not progestogens, reduces lamotrigine serum concentrations. Epilepsia 2005, 46, 1414–1417. [Google Scholar] [CrossRef] [PubMed]
- Sabers, A.; Buchholt, J.M.; Uldall, P.; Hansen, E.L. Lamotrigine plasma levels reduced by oral contraceptives. Epilepsy Res. 2001, 47, 151–154. [Google Scholar]
- Fillastre, J.P.; Taburet, A.M.; Fialaire, A.; Etienne, I.; Bidault, R.; Singlas, E. Pharmacokinetics of lamotrigine in patients with renal impairment: Influence of haemodialysis. Drug. Exp. Clin. Res. 1993, 19, 25–32. [Google Scholar]
- Morris, R.G.; Black, A.B.; Harris, A.L.; Batty, A.B.; Sallustio, B.C. Lamotrigine and therapeutic drug monitoring: retrospective survey following the introduction of a routine service. Br. J. Clin. Pharmacol. 1998, 46, 547–551. [Google Scholar]
- Pennell, P.B.; Peng, L.; Newport, D.J.; Ritchie, J.C.; Koganti, A.; Holley, D.K.; Newman, M.; Stowe, Z.N. Lamotrigine in pregnancy: Clearance, therapeutic drug monitoring, and seizure frequency. Neurology 2008, 70, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Angelis-Stoforidis, P.; Morgan, D.J.; O'Brien, T.J.; Vajda, F.J. Determination of lamotrigine in human plasma by high-performance liquid chromatography. J. Chromatogr. B 1999, 727, 113–118. [Google Scholar]
- Forssblad, E.; Eriksson, A.S.; Beck, O. Liquid chromatographic determination of plasma lamotrigine in pediatric samples. J. Pharmaceut. Biomed. Anal. 1996, 14, 755–758. [Google Scholar]
- Greiner-Sosanko, E.; Lower, D.R.; Virji, M.A.; Krasowski, M.D. Simultaneous determination of lamotrigine, zonisamide, and carbamazepine in human plasma by high-performance liquid chromatography. Biomed. Chromatogr. 2007, 21, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Biddlecombe, R.A.; Dean, K.L.; Smith, C.D.; Jeal, S.C. Validation of a radioimmunoassay for the determination of human plasma concentrations of lamotrigine. J. Pharmaceut. Biomed. Anal. 1990, 8, 691–694. [Google Scholar]
- Westley, I.S.; Morris, R.G. Seradyn quantitative microsphere system lamotrigine immunoassay on a Hitachi 911 analyzer compared with HPLC-UV. Ther. Drug Monit. 2008, 30, 634–637. [Google Scholar]
- Sailstad, J.M.; Findlay, J.W. Immunofluorometric assay for lamotrigine (Lamictal) in human plasma. Ther. Drug Monit. 1991, 13, 433–442. [Google Scholar]
- Theurillat, R.; Kuhn, M.; Thormann, W. Therapeutic drug monitoring of lamotrigine using capillary electrophoresis. Evaluation of assay performance and quality assurance over a 4-year period in the routine arena. J. Chromatogr. A 2002, 979, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Jann, M.W.; Hon, Y.Y.; Shamsi, S.A. Development of capillary zone electrophoresis-electrospray ionization-mass spectrometry for the determination of lamotrigine in human plasma. Electrophoresis 2004, 25, 2033–2043. [Google Scholar]
- Watelle, M.; Demedts, P.; Franck, F.; De Deyn, P.P.; Wauters, A.; Neels, H. Analysis of the antiepileptic phenyltriazine compound lamotrigine using gas chromatography with nitrogen phosphorus detection. Ther. Drug Monit. 1997, 19, 460–464. [Google Scholar]
- Dasgupta, A.; Hart, A.P. Lamotrigine analysis in plasma by gas chromatography-mass spectrometry after conversion to a tert.-butyldimethylsilyl derivative. J. Chromatogr. B 1997, 693, 101–107. [Google Scholar] [CrossRef]
- Beck, O.; Ohman, I.; Nordgren, H.K. Determination of lamotrigine and its metabolites in human plasma by liquid chromatography-mass spectrometry. Ther. Drug Monit. 2006, 28, 603–607. [Google Scholar]
- Lee, W.; Kim, J.H.; Kim, H.S.; Kwon, O.H.; Lee, B.I.; Heo, K. Determination of lamotrigine in human serum by high-performance liquid chromatography-tandem mass spectrometry. Neurol. Sci. 2010, in press.. [Google Scholar]
- Pucci, V.; Bugamelli, F.; Baccini, C.; Raggi, M.A. Analysis of lamotrigine and its metabolites in human plasma and urine by micellar electrokinetic capillary chromatography. Electrophoresis 2005, 26, 935–942. [Google Scholar]
- Leppik, I.E. The place of levetiracetam in the treatment of epilepsy. Epilepsia 2001, 42, S44–S45. [Google Scholar]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA. 2004, 101, 9861–9866. [Google Scholar]
- Fay, M.A.; Sheth, R.D.; Gidal, B.E. Oral absorption kinetics of levetiracetam: The effect of mixing with food or enteral nutrition formulas. Clin. Ther. 2005, 27, 594–598. [Google Scholar]
- Lins, R.L.; Otoul, C.; De Smedt, F.; Coupez, R.; Stockis, A. Comparison of plasma and saliva concentrations of levetiracetam following administration orally as a tablet and as a solution in healthy adult volunteers. Int. J. Clin. Pharmacol. Ther. 2007, 45, 47–54. [Google Scholar]
- Grim, S.A.; Ryan, M.; Miles, M.V.; Tang, P.H.; Strawsburg, R.H.; de Grauw, T.J.; Fakhoury, T.A.; Baumann, R.J. Correlation of levetiracetam concentrations between serum and plasma. Ther. Drug Monit. 2003, 25, 61–66. [Google Scholar]
- Patsalos, P.N. Clinical pharmacokinetics of levetiracetam. Clin. Pharmacokinet. 2004, 43, 707–724. [Google Scholar]
- Patsalos, P.N.; Ghattaura, S.; Ratnaraj, N.; Sander, J.W. In situ metabolism of levetiracetam in blood of patients with epilepsy. Epilepsia 2006, 47, 1818–1821. [Google Scholar] [CrossRef] [PubMed]
- Pucci, V.; Bugamelli, F.; Mandrioli, R.; Ferranti, A.; Kenndler, E.; Raggi, M.A. High-performance liquid chromatographic determination of Levetiracetam in human plasma: Comparison of different sample clean-up procedures. Biomed. Chromatogr. 2004, 18, 37–44. [Google Scholar]
- Ratnaraj, N.; Doheny, H.C.; Patsalos, P.N. A micromethod for the determination of the new antiepileptic drug levetiracetam (ucb LO59) in serum or plasma by high performance liquid chromatography. Ther. Drug Monit. 1996, 18, 154–157. [Google Scholar]
- Greiner-Sosanko, E.; Giannoutsos, S.; Lower, D.R.; Virji, M.A.; Krasowski, M.D. Drug monitoring: Simultaneous analysis of lamotrigine, oxcarbazepine, 10-hydroxycarbazepine, and zonisamide by HPLC-UV and a rapid GC method using a nitrogen-phosphorus detector for levetiracetam. J. Chromatogr. Sci. 2007, 45, 616–622. [Google Scholar] [PubMed]
- Vermeij, T.A.; Edelbroek, P.M. High-performance liquid chromatographic and megabore gas-liquid chromatographic determination of levetiracetam (ucb L059) in human serum after solid-phase extraction. J. Chromatogr. B 1994, 662, 134–139. [Google Scholar]
- Isoherranen, N.; Roeder, M.; Soback, S.; Yagen, B.; Schurig, V.; Bialer, M. Enantioselective analysis of levetiracetam and its enantiomer R-alpha-ethyl-2-oxo-pyrrolidine acetamide using gas chromatography and ion trap mass spectrometric detection. J. Chromatogr. B 2000, 745, 325–332. [Google Scholar]
- Guo, T.; Oswald, L.M.; Mendu, D.R.; Soldin, S.J. Determination of levetiracetam in human plasma/serum/saliva by liquid chromatography-electrospray tandem mass spectrometry. Clin. Chim. Acta 2007, 375, 115–118. [Google Scholar]
- Ivanova, M.; Piunti, A.; Marziali, E.; Komarova, N.; Raggi, M.A.; Kenndler, E. Microemulsion electrokinetic chromatography applied for separation of levetiracetam from other antiepileptic drugs in polypharmacy. Electrophoresis 2003, 24, 992–998. [Google Scholar]
- Shihabi, Z.K.; Oles, K.; Hinsdale, M. Analysis of the antiepileptic drug keppra by capillary electrophoresis. J. Chromatogr. A 2003, 1004, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Radtke, R.A. Pharmacokinetics of levetiracetam. Epilepsia 2001, 42, 24–27. [Google Scholar]
- Larkin, J.G.; McKee, P.J.; Forrest, G.; Beastall, G.H.; Park, B.K.; Lowrie, J.I.; Lloyd, P.; Brodie, M.J. Lack of enzyme induction with oxcarbazepine (600 mg daily) in healthy subjects. Br. J. Clin. Pharmacol. 1991, 31, 65–71. [Google Scholar]
- Lloyd, P.; Flesch, G.; Dieterle, W. Clinical pharmacology and pharmacokinetics of oxcarbazepine. Epilepsia 2007, 35, S10–S13. [Google Scholar]
- Cardot, J.M.; Degen, P.; Flesch, G.; Menge, P.; Dieterle, W. Comparison of plasma and saliva concentrations of the active monohydroxy metabolite of oxcarbazepine in patients at steady state. Biopharm. Drug Dispos. 1995, 16, 603–614. [Google Scholar]
- Rouan, M.C.; Lecaillon, J.B.; Godbillon, J.; Menard, F.; Darragon, T.; Meyer, P.; Kourilsky, O.; Hillion, D.; Aldigier, J.C.; Jungers, P. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur. J. Clin. Pharmacol. 1994, 47, 161–167. [Google Scholar]
- Furlanut, M.; Franceschi, L.; Poz, D.; Silvestri, L.; Pecorari, M. Acute oxcarbazepine, benazepril, and hydrochlorothiazide overdose with alcohol. Ther. Drug Monit. 2006, 28, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, I.; Onat, F.Y.; Ozkara, C.; Atakli, D.; Specchio, L.M.; Neve, A.L.; Gatti, G.; Perucca, E. Changes in the disposition of oxcarbazepine and its metabolites during pregnancy and the puerperium. Epilepsia 2006, 47, 504–509. [Google Scholar]
- Battino, D.; Estienne, M.; Avanzini, G. Clinical pharmacokinetics of antiepileptic drugs in pediatric patients. Part II. Phenytoin, carbamazepine, sulthiame, lamotrigine, vigabatrin, oxcarbazepine and felbamate. Clin. Pharmacokinet. 1995, 29, 341–369. [Google Scholar] [CrossRef] [PubMed]
- Striano, S.; Striano, P.; Di Nocera, P.; Italiano, D.; Fasiello, C.; Ruosi, P.; Bilo, L.; Pisani, F. Relationship between serum mono-hydroxy-carbazepine concentrations and adverse effects in patients with epilepsy on high-dose oxcabazepine therapy. Epilepsy Res. 2006, 69, 170–176. [Google Scholar]
- von Unruh, G.E.; Paar, W.D. Gas chromatographic assay for oxcarbazepine and its main metabolites in plasma. J. Chromatogr. 1985, 345, 67–76. [Google Scholar]
- von Unruh, G.E.; Paar, W.D. Gas chromatographic/mass spectrometric assays for oxcarbazepine and its main metabolites, 10-hydroxy-carbazepine and carbazepine-10,11-trans-diol. Biol. Mass Spectrom. 1986, 13, 651–656. [Google Scholar]
- Juenke, J.M.; Brown, P.I.; Urry, F.M.; McMillin, G.A. Drug monitoring and toxicology: A procedure for the monitoring of oxcarbazepine metabolite by HPLC-UV. J. Chromatogr. Sci. 2006, 44, 45–48. [Google Scholar]
- Vermeij, T.A.; Edelbroek, P.M. Robust isocratic high performance liquid chromatographic method for simultaneous determination of seven antiepileptic drugs including lamotrigine, oxcarbazepine and zonisamide in serum after solid-phase extraction. J. Chromatogr. B 2007, 857, 40–46. [Google Scholar]
- Breton, H.; Cociglio, M.; Bressolle, F.; Peyriere, H.; Blayac, J.P.; Hillaire-Buys, D. Liquid chromatography-electrospray mass spectrometry determination of carbamazepine, oxcarbazepine and eight of their metabolites in human plasma. J. Chromatogr. B 2005, 828, 80–90. [Google Scholar]
- Paglia, G.; D'Apolito, O.; Garofalo, D.; Scarano, C.; Corso, G. Development and validation of a LC/MS/MS method for simultaneous quantification of oxcarbazepine and its main metabolites in human serum. J. Chromatogr. B 2007, 860, 153–159. [Google Scholar]
- Pucci, V.; Kenndler, E.; Raggi, M.A. Quantitation of oxcarbazepine and its metabolites in human plasma by micellar electrokinetic chromatography. Biomed. Chromatogr. 2003, 17, 231–238. [Google Scholar]
- Selak, I. Pregabalin (Pfizer). Curr. Opin. Invest. Drugs 2001, 2, 828–834. [Google Scholar]
- Busch, J.A.; Strand, J.C.; Posvar, E.L.; Bockbrader, H.N.; Radulovic, L.L. Pregabalin (CI-1008) single-dose pharmacokinetics and safety/tolerance in healthy subjects after oral administration of pregabalin solution or capsule doses. Epilepsia 1998, 39, 58. [Google Scholar]
- Corrigan, B.W.; Poole, W.F.; Posvar, E.L.; Strand, J.C.; Alvey, C.W.; Radulovic, L.L. Metabolic disposition of pregabalin in healthy volunteers. Clin. Pharmacol. Ther. 2001, 69, P18. [Google Scholar]
- Randinitis, E.J.; Posvar, E.L.; Alvey, C.W.; Sedman, A.J.; Cook, J.A.; Bockbrader, H.N. Pharmacokinetics of pregabalin in subjects with various degrees of renal functions. J. Clin. Pharmacol. 2003, 43, 277–283. [Google Scholar]
- Bockbrader, H.N.; Hunt, T.; Strand, J.; Posvar, E.L.; Sedman, A. Pregabalin pharmacokinetics and safety in health volunteers: Results from two phase I studies. Neurology 2000, 11, 412. [Google Scholar]
- Berry, D.; Millington, C. Analysis of pregabalin at therapeutic concentrations in human plasma/serum by reversed-phase HPLC. Ther. Drug Monit. 2005, 27, 451–456. [Google Scholar]
- Nirogi, R.; Kandikere, V.; Mudigonda, K.; Komarneni, P.; Aleti, R. Liquid chromatography atmospheric pressure chemical ionization tandem mass spectrometry method for the quantification of pregabalin in human plasma. J. Chromatogr. B 2009, 877, 3899–3906. [Google Scholar]
- Wheless, J.W.; Vazquez, B. Rufinamide: A novel broad-spectrum antiepileptic drug. Epilepsy Curr. 2010, 10, 1–6. [Google Scholar]
- Luszczki, J.J. Third-generation antiepileptic drugs: Mechanisms of action, pharmacokinetics and interactions. Pharmacol. Rep. 2009, 61, 197–216. [Google Scholar]
- Contin, M.; Mohamed, S.; Candela, C.; Albani, F.; Riva, R.; Baruzzi, A. Simultaneous HPLC-UV analysis of rufinamide, zonisamide, lamotrigine, oxcarbazepine monohydroxy derivative and felbamate in deproteinized plasma of patients with epilepsy. J. Chromatogr. B 2010, 878, 461–465. [Google Scholar] [CrossRef]
- Chiron, C. Stiripentol. Neurotherapeutics 2007, 4, 123–125. [Google Scholar]
- Fisher, J.L. The anti-convulsant stiripentol acts directly on the GABA(A) receptor as a positive allosteric modulator. Neuropharmacology 2009, 56, 190–197. [Google Scholar]
- Levy, R.H.; Lin, H.S.; Blehaut, H.M.; Tor, J.A. Pharmacokinetics of stiripentol in normal man: evidence of nonlinearity. J. Clin. Pharmacol. 1983, 23, 523–533. [Google Scholar]
- Levy, R.H.; Loiseau, P.; Guyot, M.; Blehaut, H.M.; Tor, J.; Moreland, T.A. Stiripentol kinetics in epilepsy: Nonlinearity and interactions. Clin. Pharmacol. Ther. 1984, 36, 661–669. [Google Scholar]
- Tran, A.; Rey, E.; Pons, G.; Rousseau, M.; d'Athis, P.; Olive, G.; Mather, G.G.; Bishop, F.E.; Wurden, C.J.; Labroo, R.; Trager, W.F.; Kunze, K.L.; Thummel, K.E.; Vincent, J.C.; Gillardin, J.M.; Lepage, F.; Levy, R.H. Influence of stiripentol on cytochrome P450-mediated metabolic pathways in humans: In vitro and in vivo comparison and calculation of in vivo inhibition constants. Clin. Pharmacol. Ther. 1997, 62, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Arends, R.H.; Zhang, K.; Levy, R.H.; Baillie, T.A.; Shen, D.D. Stereoselective pharmacokinetics of stiripentol: An explanation for the development of tolerance to anticonvulsant effect. Epilepsy Res. 1994, 18, 91–96. [Google Scholar]
- Schapel, G.; Chadwick, D. Tiagabine and non-convulsive status epilepticus. Seizure 1996, 5, 153–156. [Google Scholar]
- Balslev, T.; Uldall, P.; Buchholt, J. Provocation of non-convulsive status epilepticus by tiagabine in three adolescent patients. Eur. J. Paediatr. Neurol. 2000, 4, 169–170. [Google Scholar]
- Kellinghaus, C.; Dziewas, R.; Ludemann, P. Tiagabine-related non-convulsive status epilepticus in partial epilepsy: Three case reports and a review of the literature. Seizure 2002, 11, 243–249. [Google Scholar]
- Gustavson, L.E.; Mengel, H.B. Pharmacokinetics of tiagabine, a γ-aminobutyric acid-uptake inhibitor, in healthy subjects after single and multiple doses. Epilepsia 1995, 36, 605–611. [Google Scholar]
- Patsalos, P.N.; Elyas, A.A.; Ratnaraj, N.; Iley, J. Concentration-dependent displacement of tiagabine by valproic acid. Epilepsia 2002, 43, 143. [Google Scholar]
- Cato, A., III; Gustavson, L.E.; Qian, J.; El-Shourbagy, T.; Kelly, E.A. Effect of renal impairment on the pharmacokinetics and tolerability of tiagabine. Epilepsia 1998, 39, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.H.; Gustavson, L.E.; Sperelakis, R.; Lam, N.P.; El-Shourbagy, T.; Qian, J.X.; Layden, T. Pharmacokinetics and safety of tiagabine in subjects with various degrees of hepatic function. Epilepsia 1997, 38, 445–451. [Google Scholar]
- Gustavson, L.E.; Boellner, S.W.; Granneman, G.R.; Qian, J.X.; Guenther, H.J.; el-Shourbagy, T.; Sommerville, K.W. A single-dose study to define tiagabine pharmacokinetics in pediatric patients with complex partial seizures. Neurology 1997, 48, 1032–1037. [Google Scholar]
- Williams, J.; Bialer, M.; Johannessen, S.I.; Krämer, G.; Levy, R.; Mattson, R.H.; Perucca, E.; Patsalos, P.N.; Wilson, J.F. Interlaboratory variability in the quantification of new generation antiepileptic drugs based on external quality assessment data. Epilepsia 2003, 44, 40–45. [Google Scholar]
- Chollet, D.F.; Castella, E.; Goumaz, L.; Anderegg, G. Gas chromatography-mass spectrometry assay method for the therapeutic drug monitoring of the antiepileptic drug tiagabine. J. Pharm. Biomed. Anal. 1999, 21, 641–646. [Google Scholar]
- Wang, X.; Ratnaraj, N.; Patsalos, P.N. The pharmacokinetic inter-relationship of tiagabine in blood, cerebrospinal fluid and brain extracellular fluid (frontal cortex and hippocampus). Seizure 2004, 13, 574–581. [Google Scholar]
- Easterling, D.E.; Zakszewski, T.; Moyer, M.D.; Margul, B.L.; Marriott, T.B.; Nayak, R.K. Plasma pharmacokinetics of topiramate, a new anticonvulsants in humans. Epilepsia 1988, 29, 662. [Google Scholar]
- Mimrod, D.; Specchio, L.M.; Britzi, M.; Perucca, E.; Specchio, N.; La Neve, A.; Soback, S.; Levy, R.H.; Gatti, G.; Doose, D.R.; Maryanoff, B.E.; Bialer, M. A comparative study of the effect of carbamazepine and valproic acid on the pharmacokinetics and metabolic profile of topiramate at steady state in patients with epilepsy. Epilepsia 2005, 46, 1046–1054. [Google Scholar]
- Rosenfeld, W.E.; Doose, D.R.; Walker, S.A.; Baldassarre, J.S.; Reifer, R.A. A study of topiramate pharmacokinetics and tolerability in children with epilepsy. Pediatr. Neurol. 1999, 20, 339–344. [Google Scholar]
- Riffitts, J.M.; Gisclon, L.G.; Stubbs, R.J.; Palmer, M.E. A capillary gas chromatographic assay with nitrogen phosphorus detection for the quantification of topiramate in human plasma, urine and whole blood. J. Pharm. Biomed. Anal. 1999, 19, 363–371. [Google Scholar]
- Mozayani, A.; Carter, J.; Nix, R. Distribution of topiramate in a medical examiner's case. J. Anal. Toxicol. 1999, 23, 556–558. [Google Scholar]
- Bahrami, G.; Mirzaeei, S.; Kiani, A. Sensitive analytical method for Topiramate in human serum by HPLC with pre-column fluorescent derivatization and its application in human pharmacokinetic studies. J. Chromatogr. B 2004, 813, 175–180. [Google Scholar]
- Britzi, M.; Soback, S.; Isoherranen, N.; Levy, R.H.; Perucca, E.; Doose, D.R.; Maryanoff, B.E.; Bialer, M. Analysis of topiramate and its metabolites in plasma and urine of healthy subjects and patients with epilepsy by use of a novel liquid chromatography-mass spectrometry assay. Ther. Drug Monit. 2003, 25, 314–322. [Google Scholar]
- Contin, M.; Riva, R.; Albani, F.; Baruzzi, A. Simple and rapid liquid chromatographic-turbo ion spray mass spectrometric determination of topiramate in human plasma. J. Chromatogr. B 2001, 761, 133–137. [Google Scholar]
- Christensen, J.; Hojskov, C.S.; Poulsen, J.H. Liquid chromatography tandem mass spectrometry assay for topiramate analysis in plasma and cerebrospinal fluid: Validation and comparison with fluorescence-polarization immunoassay. Ther. Drug Monit. 2002, 24, 658–664. [Google Scholar]
- Berry, D.J.; Patsalos, P.N. Comparison of topiramate concentrations in plasma and serum by fluorescence polarization immunoassay. Ther. Drug Monit. 2000, 22, 460–464. [Google Scholar]
- Snozek, C.L.; Rollins, L.A.; Peterson, P.W.; Langman, L.J. Comparison of a new serum topiramate immunoassay to fluorescence polarization immunoassay. Ther. Drug Monit. 2010, 32, 107–111. [Google Scholar]
- Rey, E.; Pons, G.; Olive, G. Vigabatrin. Clinical pharmacokinetics. Clin. Pharmacokinet. 1992, 23, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Schechter, P.J. Clinical pharmacology of vigabatrin. Br. J. Clin. Pharmacol. 1989, 27, S19–S22. [Google Scholar]
- Durham, S.L.; Hoke, J.F.; Chen, T.M. Pharmacokinetics and metabolism of vigabatrin following a single oral dose of [14C]vigabatrin in healthy male volunteers. Drug Metab. Dispos. 1993, 21, 480–484. [Google Scholar]
- Jacqz-Aigrain, E.; Guillonneau, M.; Rey, E.; Macher, M.A.; Montes, C.; Chiron, C.; Loirat, C. Pharmacokinetics of the S(+) and R(-) enantiomers of vigabatrin during chronic dosing in a patient with renal failure. Br. J. Clin. Pharmacol. 1997, 44, 183–185. [Google Scholar]
- Chang, S.Y.; Lin, W.C. Determination of vigabatrin by capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. B 2003, 794, 17–22. [Google Scholar]
- Erturk, S.; Aktas, E.S.; Atmaca, S. Determination of vigabatrin in human plasma and urine by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B 2001, 760, 207–212. [Google Scholar]
- Buchanan, R.; Bockbrader, H.N.; Chang, T.; Sedman, A.J. Single- and multiple-dose pharmacokinetics of zonisamide. Epilepsia 1996, 37, 172. [Google Scholar]
- Ijiri, Y.; Inoue, T.; Fukuda, F.; Suzuki, K.; Kobayashi, T.; Shibahara, N.; Takenaka, H.; Tanaka, K. Dialyzability of the antiepileptic drug zonisamide in patients undergoing hemodialysis. Epilepsia 2004, 45, 924–927. [Google Scholar]
- Berent, S.; Sackellares, J.C.; Giordani, B.; Wagner, J.G.; Donofrio, P.D.; Abou-Khalil, B. Zonisamide (CI-912) and cognition:Results from preliminary study. Epilepsia 1987, 28, 61–67. [Google Scholar]
- Glauser, T.A.; Pippenger, C.E. Controversies in blood-level monitoring: re-examining its role in the treatment of epilepsy. Epilepsia 2000, 41, S6–S15. [Google Scholar]
- Juenke, J.; Brown, P.I.; Urry, F.M.; McMillin, G.A. Drug monitoring and toxicology: A procedure for the monitoring of levetiracetam and zonisamide by HPLC-UV. J. Anal. Toxicol. 2006, 30, 27–30. [Google Scholar]
- Nakamura, M.; Hirade, K.; Sugiyama, T.; Katagiri, Y. High-performance liquid chromatographic assay of zonisamide in human plasma using a non-porous silica column. J. Chromatogr. B 2001, 755, 337–341. [Google Scholar]
- Subramanian, M.; Birnbaum, A.K.; Remmel, R.P. High-speed simultaneous determination of nine antiepileptic drugs using liquid chromatography-mass spectrometry. Ther. Drug Monit. 2008, 30, 347–356. [Google Scholar]
- Makino, K.; Goto, Y.; Sueyasu, M.; Futagami, K.; Kataoka, Y.; Oishi, R. Micellar electrokinetic capillary chromatography for therapeutic drug monitoring of zonisamide. J. Chromatogr. B 1997, 695, 417–425. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krasowski, M.D. Therapeutic Drug Monitoring of the Newer Anti-Epilepsy Medications. Pharmaceuticals 2010, 3, 1909-1935. https://doi.org/10.3390/ph3061909
Krasowski MD. Therapeutic Drug Monitoring of the Newer Anti-Epilepsy Medications. Pharmaceuticals. 2010; 3(6):1909-1935. https://doi.org/10.3390/ph3061909
Chicago/Turabian StyleKrasowski, Matthew D. 2010. "Therapeutic Drug Monitoring of the Newer Anti-Epilepsy Medications" Pharmaceuticals 3, no. 6: 1909-1935. https://doi.org/10.3390/ph3061909
APA StyleKrasowski, M. D. (2010). Therapeutic Drug Monitoring of the Newer Anti-Epilepsy Medications. Pharmaceuticals, 3(6), 1909-1935. https://doi.org/10.3390/ph3061909