Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases

Abstract
Introduction
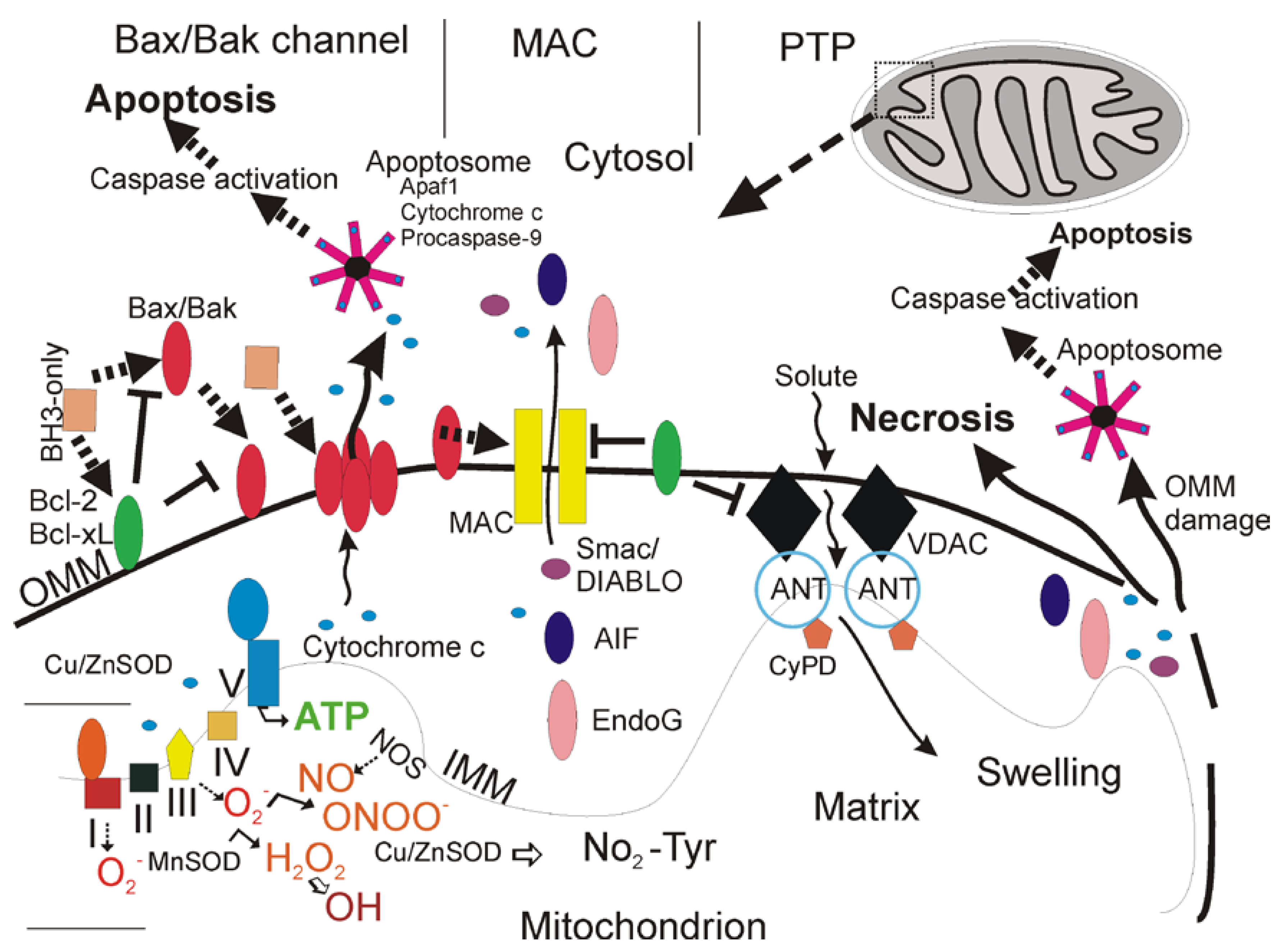
Types of Cell Death
Cell Necrosis and the Mitochondrial Permeability Transition Pore

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function |
|---|---|
| Bcl-2* | Anti-apoptotic, blocks Bax/Bak channel formation |
| Bcl-XL | Anti-apoptotic, blocks Bax/Bak channel formation |
| Bax* | Pro-apoptotic, forms pores for cytochrome c release |
| Bak* | Pro-apoptotic, forms pores for cytochrome c release |
| Bad | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| Bid | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| Noxa | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| Puma | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| p53* | Antagonizes activity of Bcl-2/Bcl-XL, promotes Bax/Bak oligomerization |
| Cytochrome c | Activator of apoptosome |
| Smac/DIABLO | IAP inhibitor |
| AIF | Antioxidant flavoprotein/released from mitochondria to promote nuclear DNA fragmentation |
| Endonuclease G | Released from mitochondria to promote nuclear DNA fragmentation |
| HtrA2/Omi | IAP inhibitor |
| VDAC | mPTP component in outer mitochondrial membrane |
| ANT+ | mPTP component in inner mitochondrial membrane |
| Cyclophilin D+ | mPTP component in mitochondrial matrix |
| TSPO (peripheral benzodiazepine receptor) | Modulator of mPTP |
| Hexokinase | Modulator of VDAC |
Apoptosis
Autophagy
Cellular and Molecular Regulation of Apoptosis
| Bcl-2 Family | Caspase Family | IAP Family | Tumor Suppressor | |
| Anti-apoptotic proteins | Pro-apoptotic proteins | |||
| Bcl-2 | Bax | Apoptosis “initiators”: caspase-2, 8, 9, 10 | NAIP | p53 |
| Bcl-xL | Bak1 | Apollon | p63 | |
| Mcl-1 | Bcl-xS | Survivin | p73 | |
| Boo/Diva | Bad | Apoptosis “executioners”: caspase-2, 3, 6, 7 | IAP1 | |
| Bid | IAP2 | |||
| Bik | XIAP | |||
| Bim | Cytokine processors: caspase-1, 4, 5, 11, 12, 14 | |||
| Noxa | ||||
| Puma | ||||
Bcl-2 family of Cell Survival and Cell Death Proteins
Caspase Family of Cell Demolition Proteases
Inhibitor of Apoptosis Protein (IAP) Family
Apoptosis Inducing Factor (AIF)
p53/p63/p73 Family of Tumor Suppressors
Cell Surface Death Receptors
Excitotoxic Neuronal Cell Death
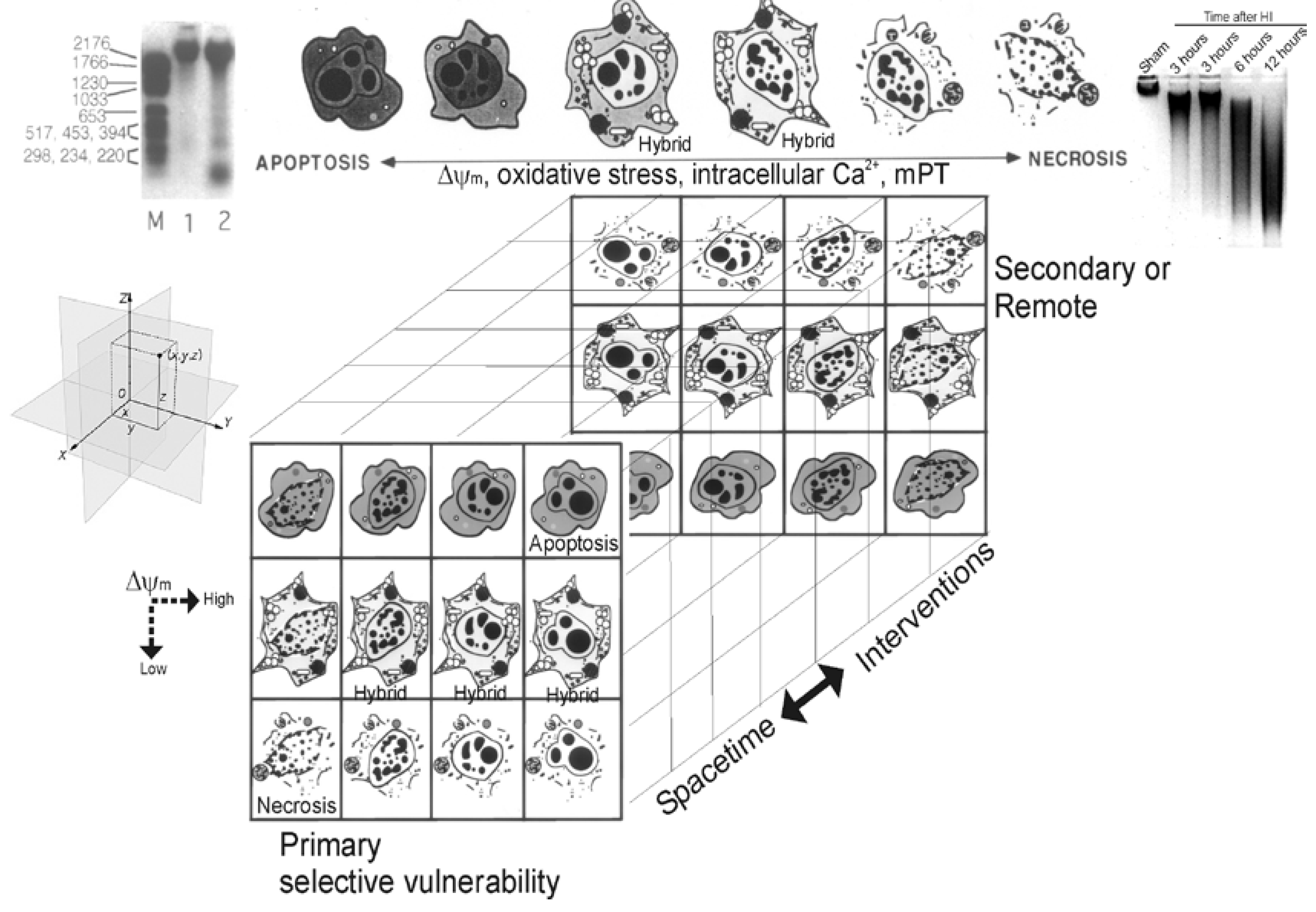
The Cell Death Continuum
The Cell Death Matrix
Cell Death in Human Neurodegenerative Diseases
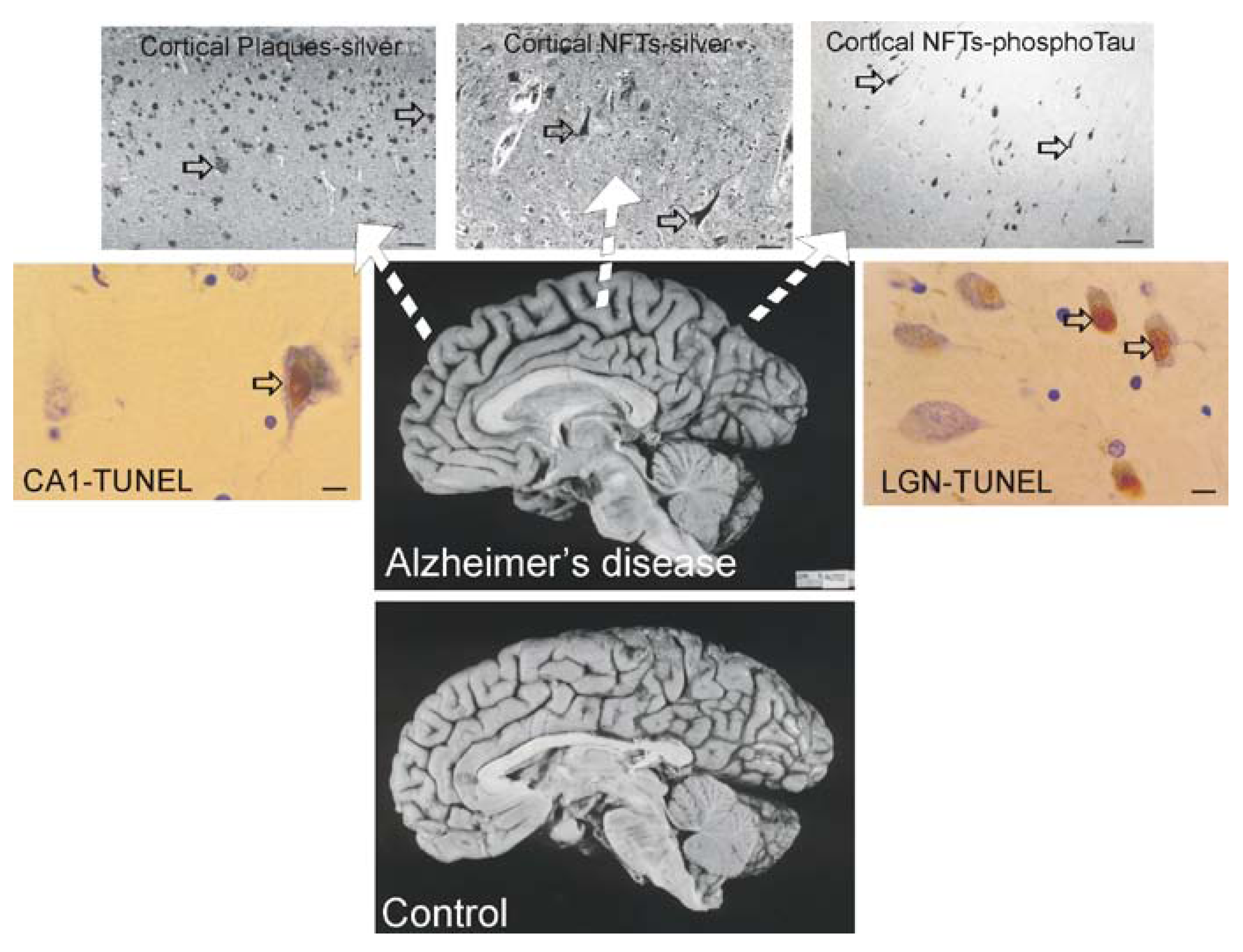
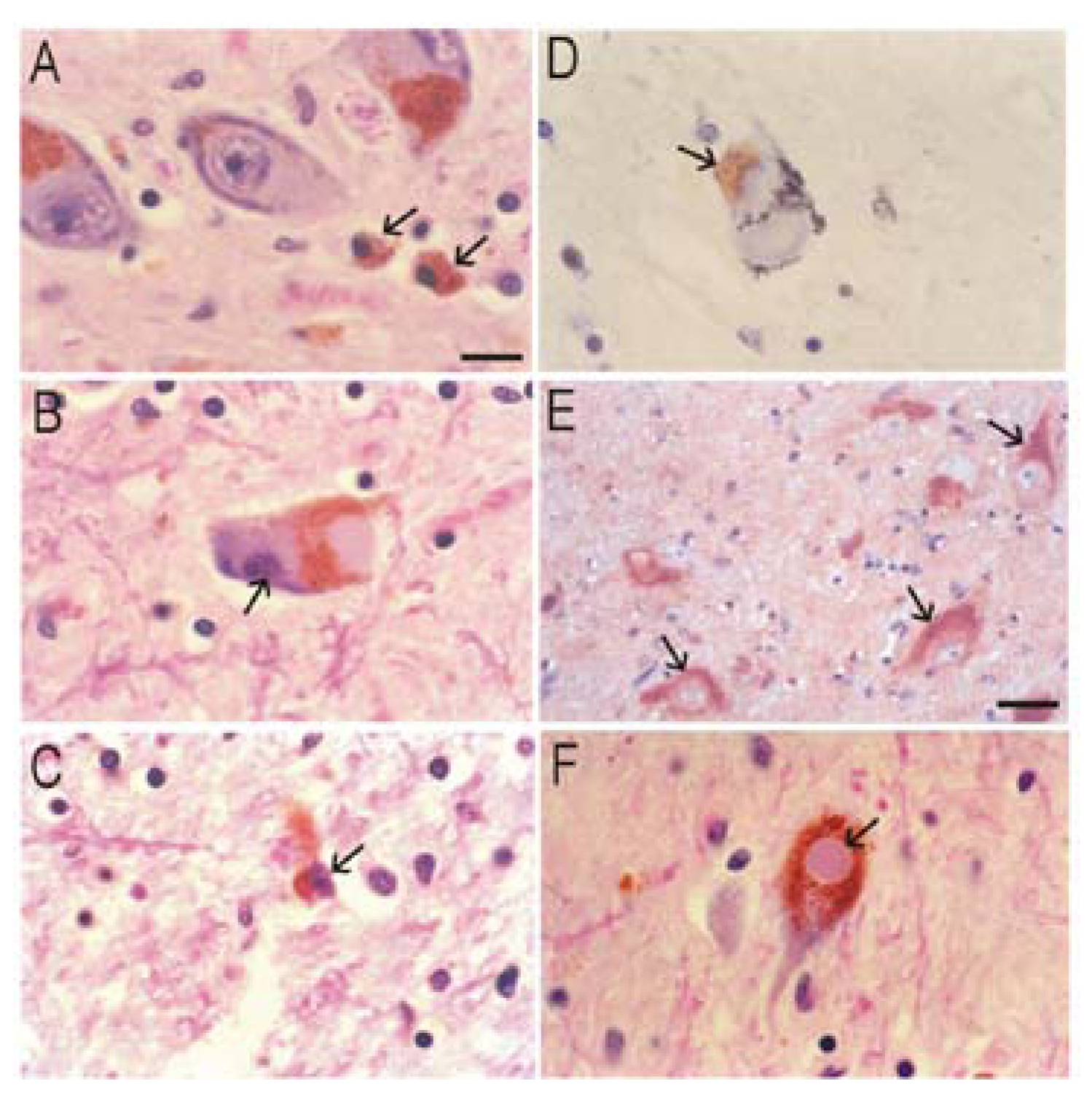
Alzheimer’s disease (AD)
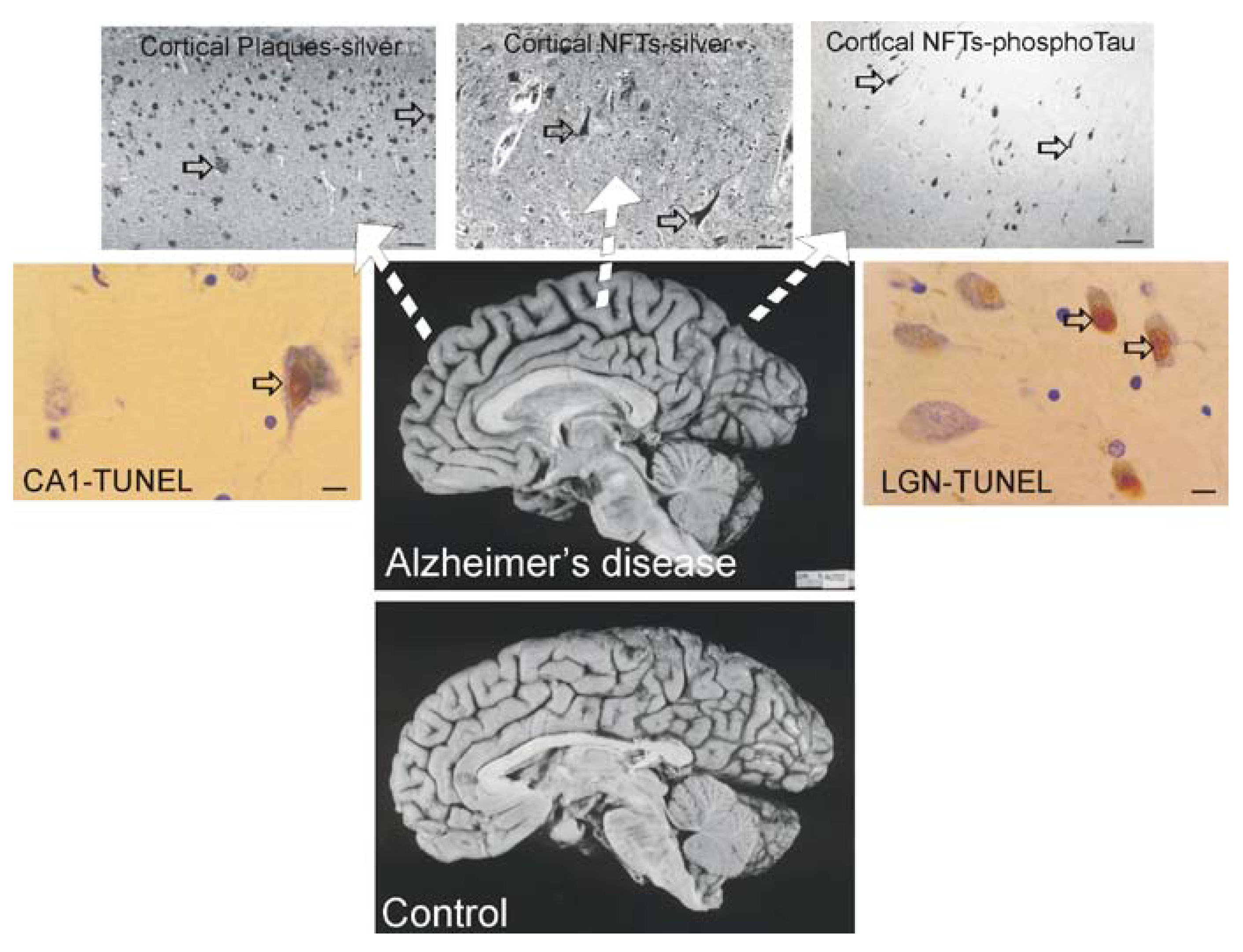
Most Tg Mouse Models of AD are not Useful to Study Neuronal Cell Death
Cell Culture Models of Cortical and Hippocampal Neuron Cell Death and Interactions between APP, Aβ, Tau, and Caspases
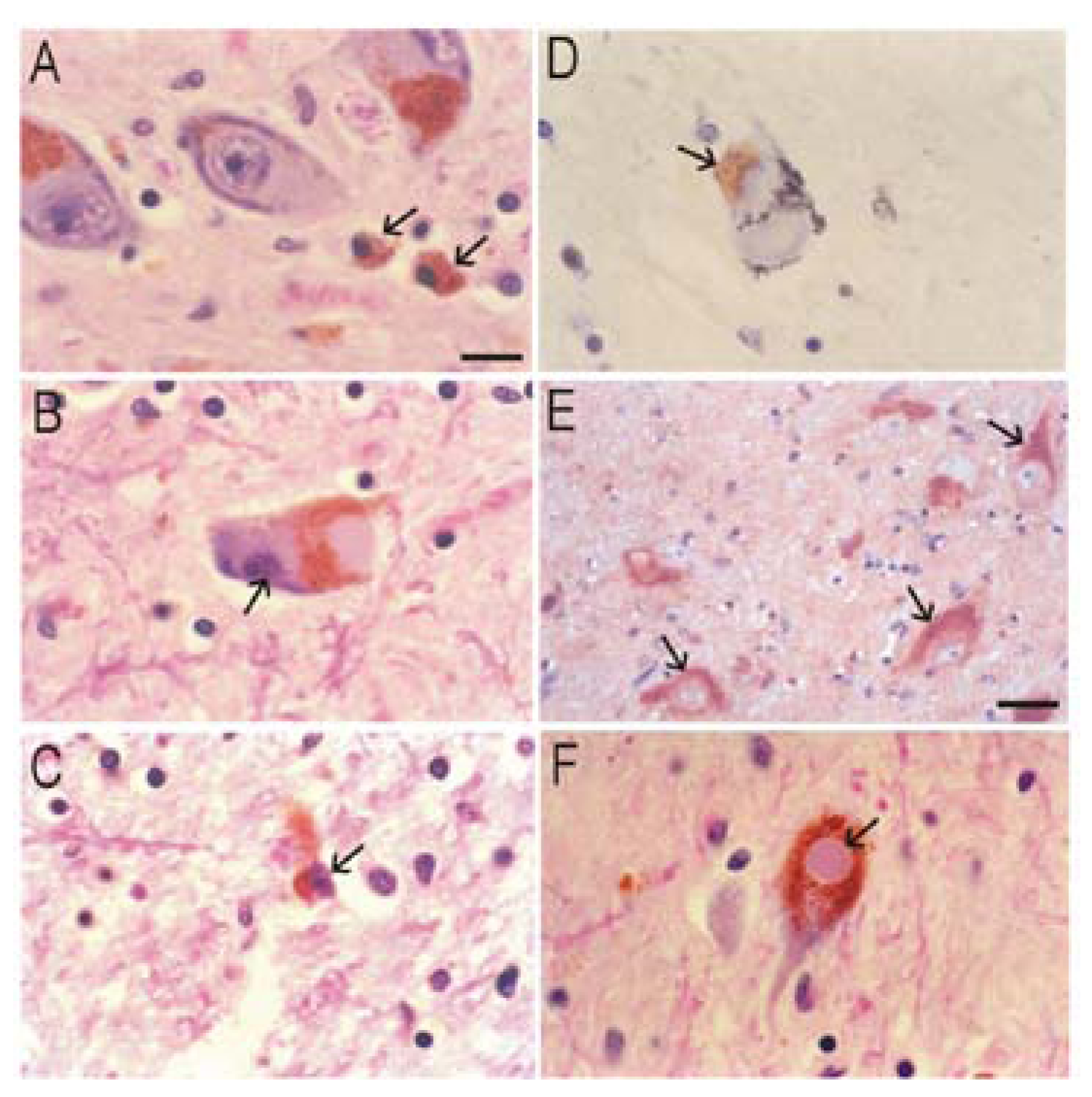
Parkinson’s Disease (PD)
| Locus | Inheritance | Gene | Protein Name/ Function |
|---|---|---|---|
| PARK1/4q21 | autosomal dominant | α-syn | α-Syn/presynaptic maintenance? |
| PARK2/6q25.2-27 | autosomal recessive | parkin | Parkin/ubiquitin E3 ligase |
| PARK3/2p13 | autosomal dominant | ? | ? |
| PARK4/4p15 | autosomal dominant | α-syn | α-Syn/presynaptic maintenance? |
| PARK5/4p14 | autosomal dominant | UCHL1 | UCHL1/polyubiquitin hydrolase |
| PARK6/1p36 | autosomal recessive | PINK1 | PTEN-induced putative kinase-1/mitochondrial protein kinase |
| PARK7/1p36.33-36-12 | autosomal recessive | DJ-1 | DJ-1/mitochondrial antioxidant, chaperone |
| PARK8/12q12 | autosomal dominant | LRRK2 | Dardarin/multifunctional kinase/GTPase |
| PARK9/1p36 | autosomal recessive | ATP13A2 | Lysosomal type 5 P-ATPase |
| PARK10/1p32 | ? | ? | |
| PARK11/2q36-37 | autosomal dominant | GIGYF2? | Grb10-interacting GYP protein 2, modulates tyrosine kinase receptor signaling, including IGF-1 |
| PARK12/Xq21-q25 | X-linked | ? | ? |
| PARK13/2p12 | autosomal recessive susceptibility factor | Omi/HtrA2 | Omi/HtrA2, mitochondrial serine peptidase, inhibitor of IAPs |
| PARK14/22q13.1 | autosomal recessive | PLA2G6 | Phospholipase A2 group VI |
| PARK15/22q12-q13 | autosomal recessive | FBXO7 | F-box protein 7 |
Mutant Genes that Cause Some Forms of PD
α-Syn
UCH-L1 and Parkin
PINK1
DJ-1
LRRK2
Neuronal Cell Death in Human PD
PD α-Syn Tg Mice Develop Neuronal Mitochondrial Degeneration and Cell Death
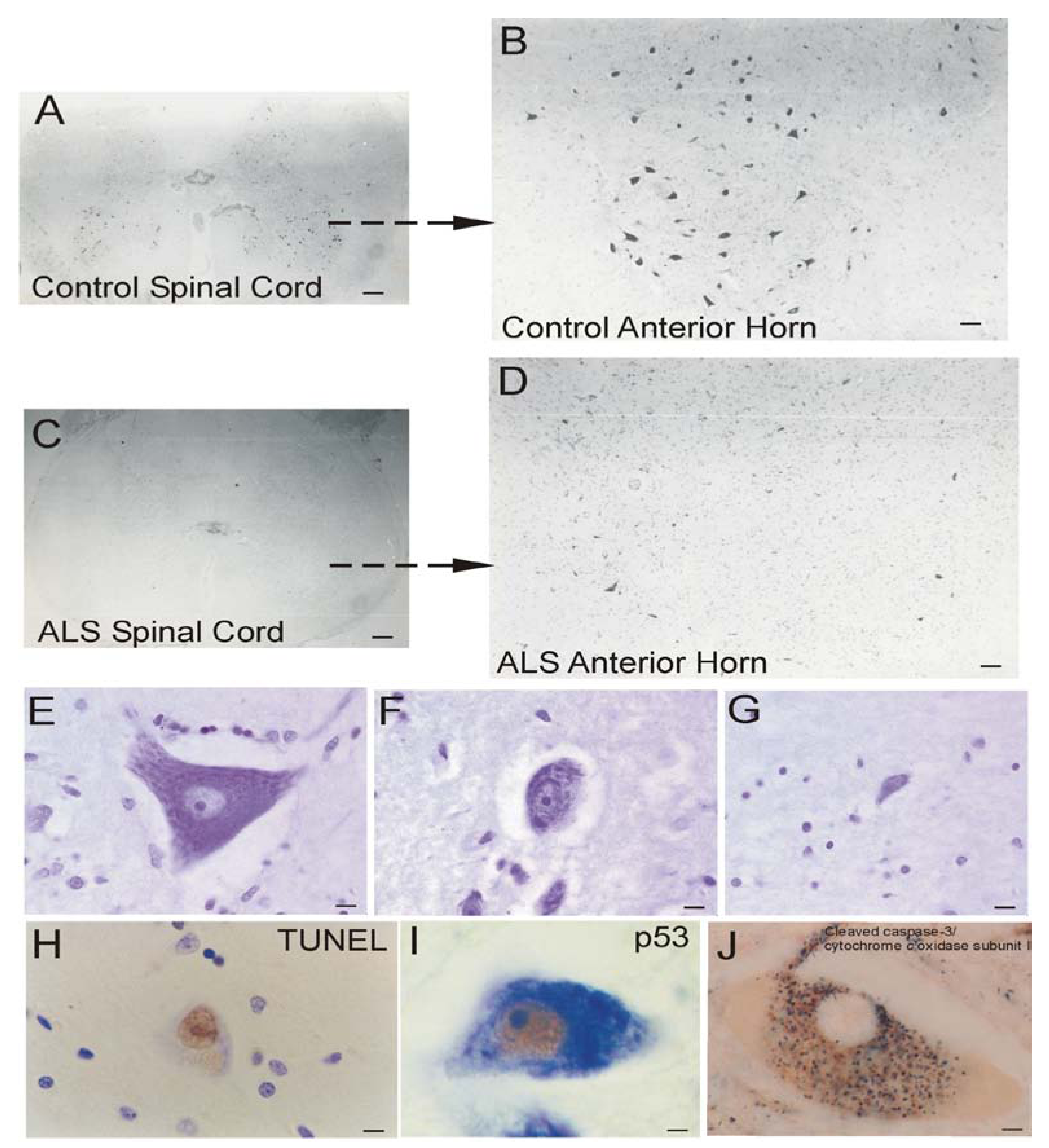
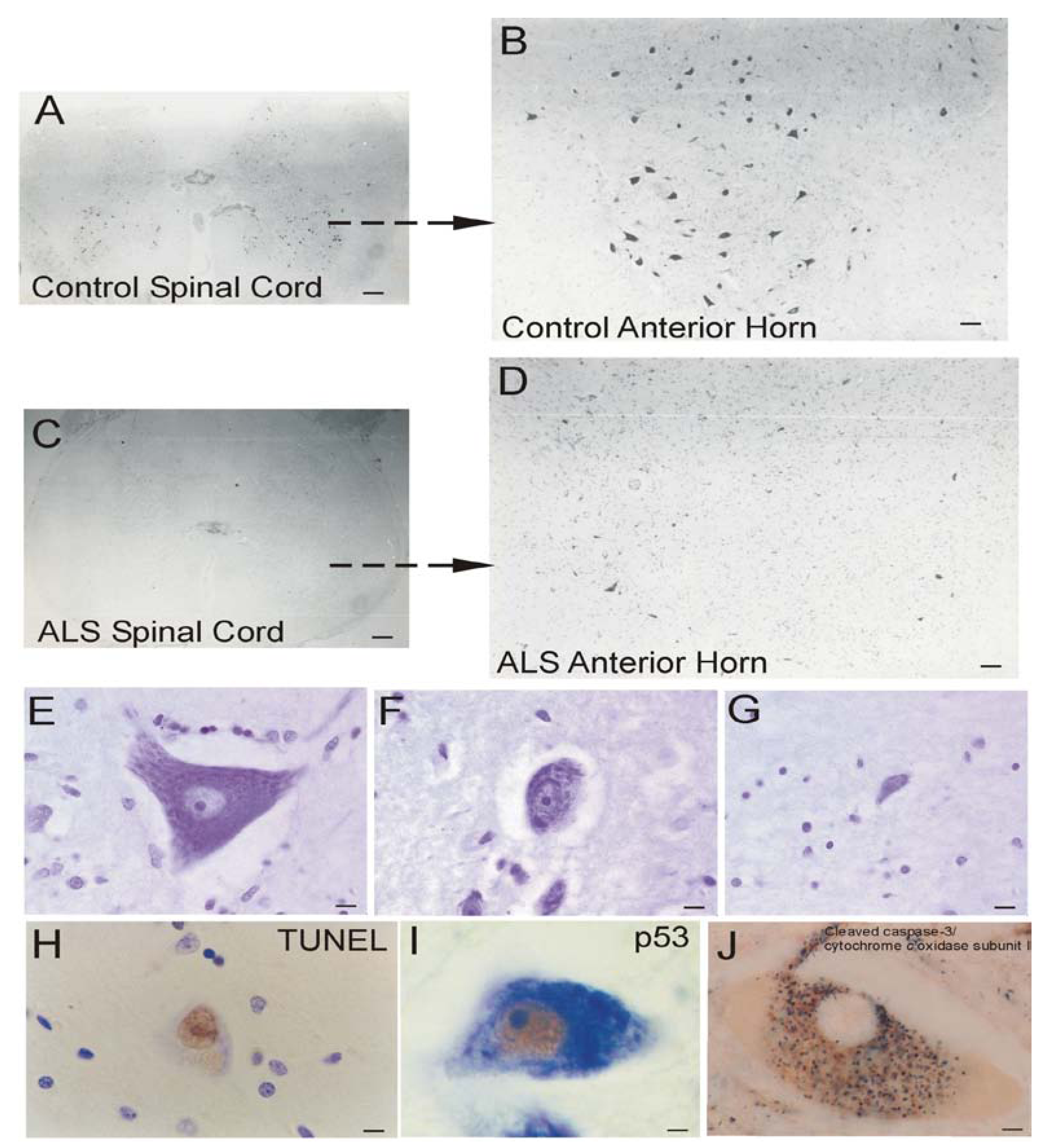
ALS
| Locus | Inheritance | Gene | Protein Name/ Function |
|---|---|---|---|
| ALS1/21q22 | autosomal dominant (adult onset) | SOD1 | Cu/Zn superoxide dismutase/ dismutation of superoxide |
| ALS2/2q33.2 | autosomal recessive (juvenile onset primary lateral sclerosis) | Alsin | Alsin/guanine exchange factor for RAB5A and Rac1 |
| ALS4/9q34 | autosomal dominant (adult onset) | SETX | Senataxin/helicase, RNA processing |
| ALS6/16q12 | autosomal recessive (adult onset) | FUS | Fused in sarcoma, component of heterogeneous nuclear ribonuclear protein complex; RNA/DNA binding protein |
| ALS8/20q13.33 | autosomal dominant | VAPB | VAMP-associated protein B/part of SNARE complex |
| 2q13 | autosomal dominant (adult onset, atypical ALS) | DCTN1 | Dynactin p150glued/axonal transport, link between dynein and microtubule network |
| ALS10/1p36.22 | autosomal dominant | TARDBP | TAR DNA binding protein, DNA and RNA binding protein, regulates RNA splicing |
| ALS11/6q21 | autosomal recessive | FIG4 | FIG4 homolog, SAC1 lipid phosphatase domain containing protein; regulates phosphotidylinositol turnover |
| 14q11.1-q11.2 | susceptibility factor | ANG | Angiogenin; angiogenesis; stimulates production of rRNA |
| 22q12.2 | susceptibility factor | NEFH | Neurofilament, heavy polypeptide; neurofilament subunit |
| 12q12-q13 | susceptibility factor | PRPH | Peripherin; intermediate filament formation |
| 5q13 | susceptibility factor | SMN | Survival motor neuron; RNA processing |
| 7q36.6 | susceptibility factor? | DPP6 | Dipeptidyl-peptidase 6; S9B serine protease, binds voltage-gated potassium channels |
Mitochondrial Dysfunction in Human ALS
Human ALS and Mitochondrial-Orchestrated PCD Involving p53
Mitochondrial Pathobiology in Cell and Mouse Models of ALS
The mPTP Contributes to the Disease Mechanisms of ALS in Mice
Summary and Outlook
Abbreviations
| Aβ | amyloid beta protein |
| AD | Alzheimer’s disease |
| AIF | apoptosis-inducing factor |
| ALS | amyotrophic lateral sclerosis |
| ANT | adenine nucleotide translocator |
| Apaf | apoptotic protease activating factor |
| APP | amyloid precursor protein |
| CNS | central nervous system |
| Cu/ZnSOD | copper/zinc superoxide dismutase (also SOD1) |
| CyPD | cyclophilin D |
| DISC | death-inducing signaling complex |
| EM | electron microscopy |
| ER | endoplasmic reticulum |
| GPe | globus pallidus external |
| GPi | globus pallidus internal |
| HtrA2 | high temperature requirement protein A2 |
| IAP | inhibitor of apoptosis protein |
| IMM | inner mitochondrial membrane |
| KA | kainic acid |
| LB | Lewy body |
| LGN | lateral geniculate nucleus |
| LRRK2 | leucine-rich repeat kinase 2 |
| mnSOD | manganese SOD (also SOD2) |
| mPT | mitochondrial permeability transition |
| mPTP | mitochondrial permeability transition pore |
| mSOD1 | mutant SOD1 |
| mtDNA | mitochondrial DNA |
| NAIP | neuronal apoptosis inhibitory protein |
| NFT | neurofibrillary tangle |
| NMDA | N-methy-D-aspartate |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| O2•- | superoxide radical |
| OMM | outer mitochondrial membrane |
| ONOO- | peroxynitrite |
| PCD | programmed cell death |
| PD | Parkinson’s disease |
| PINK1 | phosphatase and tensin homolog-induced putative kinase-1 |
| ROS | reactive oxygen species |
| SNc | substantia nigra compacta |
| Syn | α-synuclein |
| Tg | transgenic |
| TIMM | translocase of inner mitochondrial membrane |
| TOMM | translocase of outer mitochondrial membrane |
| TSPO | translocator protein 18 kDa (peripheral benzodiazepine receptor) |
| TNF | tumor necrosis factor |
| TUNEL | terminal transferase-mediated biotin-dUTP nick-end labeling |
| UCH-L1 | ubiquitin carboxy-terminal hydrolyase-L1 |
| VDAC | voltage-dependent anion channel |
Acknowledgments
References
- Virchow, R. Cellular Pathology as Based Upon Physiological and Pathological Histology; John Churchill: London (Translation), UK, 1860. [Google Scholar]
- Martin, L.J. Neurodegenerative disorders of the human brain and spinal cord. In Encyclopedia of the Human Brain; Ramachandran, V.S., Ed.; Elsevier Science Academic Press: San Diego, USA, 2002; Volume 3, pp. 441–463. [Google Scholar]
- Rich, T.; Allen, R.L.; Wyllie, A.H. Defying death after DNA damage. Nature 2000, 407, 777–783. [Google Scholar]
- Zheng, T.S. Death by design: the big debut of small molecules. Nat. Cell Biol. 2001, 3, E1–E3. [Google Scholar]
- Martin, L.J. Mitochondriopathy in Parkinson’s disease and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2006, 65, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Isave, N.K.; Plotnikov, E.Y.; Zorova, L.D.; Stelmashook, E.V.; Vasileva, A.K.; Arkhagelskaya, A.A.; Khrjapenkova, T.G. The mitochondrion as Janus Bifrons. Biochemistry (Moscow) 2007, 72, 1115–1126. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Intl. J. Biochem. Cell Biol. 2002, 34, 1372–1381. [Google Scholar]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn of evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar]
- Halliwell, B. Role of free radicals in the neurodegenerative diseases. Drugs Aging 2001, 18, 685–716. [Google Scholar]
- Mungrue, I.N.; Bredt, D.S.; Stewart, D.J.; Husain, M. From molecules to mammals: what’s NOS got to do with it? Acta Physiol. Scand. 2003, 179, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Carson, M.; Smith, C.D.; Koppenol, W.H. ALS, SOD and peroxynitrite. Nature 1993, 364, 548. [Google Scholar]
- Martin, L.J.; Liu, Z. DNA damage profiling in motor neurons: a single-cell analysis by comet assay. Neurochem. Res. 2002, 27, 1089–1100. [Google Scholar]
- Giulini, C. Characterization and function of mitochondrial nitric-oxide synthase. Free Radic. Biol. Med. 2003, 34, 397–408. [Google Scholar]
- Brown, G.C.; Borutaite, V. Nitric oxide, cytochrome c, and mitochondria. Biochem. Soc. Sym. 1999, 66, 17–25. [Google Scholar]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer type) dominant optic atrophy: a novel mitochondrial disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar]
- Martin, L.J.; Al-Abdulla, N.A.; Brambrink, A.M.; Kirsch, J.R.; Sieber, F.E.; Portera-Cailliau, C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res. Bull. 1998, 46, 281–309. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J.; Graham, E.M.; Martin, L.J. Apoptosis in perinatal hypoxic-ischemic brain injury: how important is it and should it be inhibited? Brain Res. Rev. 2005, 50, 244–257. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 186–197. [Google Scholar] [PubMed]
- Waldmeier, P.C.; Zimmermann, K.; Qian, T.; Tintelnot-Blomley, M.; Lemasters, J.J. Cyclophilin D as a drug target. Curr. Med. Chem. 2003, 10, 1485–1506. [Google Scholar]
- Crompton, M. Mitochondria and aging: a role for the permeability transition? Aging Cell 2004, 3, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Krauskopf, A; Basso, E.; Petronilli, V.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [PubMed]
- Martin, L.J. Neuronal cell death in nervous system development, disease, and injury. Int. J. Mol. Med. 2001, 7, 455–478. [Google Scholar] [PubMed]
- Lockshin, R.A.; Zakeri, Z. Caspase-independent cell deaths. Curr. Opin. Cell Biol. 2002, 14, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S. Developmental Biology; Sinauer Associates: Sunderland, MA, USA, 2006. [Google Scholar]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Portera-Cailliau, C.; Price, D.L.; Martin, L.J. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J. Comp. Neurol. 1997, 378, 70–87. [Google Scholar]
- Portera-Cailliau, C.; Price, D.L.; Martin, L.J. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J. Comp. Neurol. 1997, 378, 88–104. [Google Scholar]
- Formigli, L.; Papucci, L.; Tani, N.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Zecchi Orlandini, S. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell. Physiol. 2000, 182, 41–49. [Google Scholar]
- Lennon, S.V.; Martin, S.J.; Cotter, T.G. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif. 1991, 24, 203–214. [Google Scholar]
- Fernandes, R.S.; Cotter, T.G. Apoptosis or necrosis: intracellular levels of glutathione influence mode of cell death. Biochem. Pharmacol. 1994, 48, 675–681. [Google Scholar]
- Bonfoco, E.; Krainc, D.; Ankarcrona, M.; Nicotera, P.; Lipton, S.A. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell culture. Proc. Natl. Acad. Sci. USA 1995, 92, 7162–7166. [Google Scholar]
- Raffray, M.; Cohen, G.M. Apoptosis and necrosis in toxicology: a continuum or distinct modes of cell death? Pharmacol. Ther. 1997, 75, 153–177. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lipinski, M.; Degterev, A. Diversity in the mechanisms of neuronal cell death. Neuron 2003, 40, 401–413. [Google Scholar]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The Bcl-2 protein family: opposing activities that mediate cell death. Nat. Rev. 2008, 9, 47–59. [Google Scholar]
- Merry, D.E.; Korsmeyer, S.J. Bcl-2 gene family in the nervous system. Ann. Rev. Neurosci. 1997, 20, 245–267. [Google Scholar]
- Trump, B.F.; Berezesky, I.K. The role of altered [Ca2+]i regulation in apoptosis, oncosis, and necrosis. Biochim. Biophys. Acta 1996, 1313, 173–178. [Google Scholar] [CrossRef]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Trump, B.J.; Goldblatt, P.J.; Stowell, R.E. Studies on necrosis of mouse liver in vitro. Ultrastructural alterations in the mitochondria of hepatic parenchymal cells. Lab. Invest. 1964, 14, 343–371. [Google Scholar]
- Leist, M.; Single, B.; Castoldi, A.F.; Kuhnles, S.; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar]
- Martin, L.J.; Brambrink, A.M.; Price, A.C.; Kaiser, A.; Agnew, D.M.; Ichord, R.N.; Traystman, R.J. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol. Dis. 2000, 7, 169–191. [Google Scholar]
- Golden, W.C.; Brambrink, A.M.; Traystman, R.J.; Martin, L.J. Failure to sustain recovery of Na,K ATPase function is a possible mechanism for striatal neurodegeneration in hypoxic-ischemic newborn piglets. Mol. Brain Res. 2001, 88, 94–102. [Google Scholar] [CrossRef]
- Castro, J.; Ruminot, I.; Porras, O.H.; Flores, C.M.; Hermosilla, T.; Verdugo, E.; Venegas, F.; Hartel, S.; Michea, L.; Barros, L.F. ATP steal between cation pumps: a mechanism linking Na+ influx to the onset or necrotic Ca2+ overload. Cell Death Diff. 2006, 13, 1675–1685. [Google Scholar] [CrossRef]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: a specific form of programmed cell death. Exp. Cell Res. 2003, 283, 1–16. [Google Scholar]
- Kim, Y-S.; Morgan, M.J.; Choksi, S.; Lu, Z-G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 2007, 26, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol. Dis. 2000, 7, 225–239. [Google Scholar]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochem. Biophys. Acta 1995, 1241, 139–176. [Google Scholar]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar]
- Van Gurp, M.; Festjens, N.; van Loo, G.; Saelens, X.; Vandenabeele, P. Mitochondrial intermembrane proteins in cell death. Biochem. Biophys. Res. Comm. 2003, 304, 487–497. [Google Scholar]
- Leung, A.W.C.; Halestrap, A.P. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2008, 1777, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The voltage-dependent anion channel (VDAC): function in intracellular signaling, cell life and cell death. Curr. Pharm. Des. 2006, 12, 2249–2270. [Google Scholar]
- Rostovtseva, T.K.; Tan, W.; Colombini, M. On the role of VDAC in apoptosis: fact and fiction. J. Bioenerget. Biomembr. 2005, 37, 129–142. [Google Scholar]
- Granville, D.J.; Gottlieb, R.A. The mitochondrial voltage-dependent anion channel (VDAC) as a therapeutic target for initiating cell death. Curr. Med. Chem. 2003, 10, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Ruitenbeek, W.; van den Heuvel, L.P.; Dolce, V.; Iacobazzi, V.; Smeitink, J.A.M.; Palmieri, F.; Trijbels, J.M.F. Human mitochondrial transmembrane metabolite carriers: tissue distribution and its implication for mitochondrial disorders. J. Bioenerg. Biomembr. 1998, 30, 277–284. [Google Scholar]
- Wu, S.; Sampson, M.J.; Decker, W.K.; Craigen, W.J. Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochem. Biophys. Acta 1999, 1452, 68–78. [Google Scholar] [CrossRef]
- Anflous, K.; Armstrong, D.D.; Craigen, W.J. Altered sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J. Biol. Chem. 2001, 276, 1954–1960. [Google Scholar] [PubMed]
- Sampson, M.J.; Decker, W.K.; Beaudet, A.L.; Ruitenbeek, W.; Armstrong, D.; Hicks, M.J.; Craigen, W.J. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J. Biol. Chem. 2001, 276, 39206–39212. [Google Scholar]
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsemeyer, S.J. VDAC2 inhibits Bak activation and mitochondrial apoptosis. Science 2003, 301, 513–517. [Google Scholar]
- Chandra, D.; Choy, G.; Daniel, P.T.; Tang, D.G. Bax-dependent regulation of Bak by voltage-dependent anion channel 2. J. Biol. Chem. 2005, 280, 19051–19061. [Google Scholar]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar]
- Karachitos, A.; Galganska, H.; Wojtkowska, M.; Budzinska, M.; Stobienia, O.; Bartosz, G.; Kimita, H. Cu,Zn-superoxide dismutase is necessary for proper function of VDAC in Saccharomyces cerevisiae cells. FEBS Lett. 2009, 583, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.H.; Waymire, K.G.; Cottrell, B.; Trounce, I.A.; MacGregor, G.R.; Wallace, D.C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997, 16, 226–234. [Google Scholar]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallace, D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Bio. Chem. 1992, 267, 14592–14597. [Google Scholar]
- Vyssokikh, M.Y.; Katz, A.; Rueck, A.; Wuensch, C.; Dorner, A.; Zorov, D.B.; Brdiczka, D. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem. J. 2001, 358, 349–358. [Google Scholar]
- Kikoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [PubMed]
- Machida, K.; Hayashi, Y.; Osada, H. A novel adenine nucleotide translocase inhibitor, MT-21, induces cytochrome c release through a mitochondrial permeability transition-independent mechanisms. J. Biol. Chem. 2002, 277, 31243–31248. [Google Scholar] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, H.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn II, G.W.; Robbins, J.; Molkentin, J.D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar]
- Woodfield, K.; Rück, A.; Brdiczka, D.; Halestrap, A.P. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998, 336, 287–290. [Google Scholar]
- Johnson, N.; Khan, A.; Virji, S.; Ward, J.M.; Crompton, M. Import and processing of heart mitochondrial cyclophilin D. Eur. J. Biochem. 1999, 263, 353–359. [Google Scholar] [CrossRef] [PubMed]
- McEnery, M.W.; Dawson, T.M.; Verma, A.; Gurley, D.; Colombini, M.; Snyder, S.H. Mitochondrial voltage-dependent anion channel. J. Biol. Chem. 1993, 268, 23289–23296. [Google Scholar]
- Shoshan-Barmatz, V.; Zalk, R.; Gincel, D.; Vardi, N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochem. Biophys. Acta 2004, 1657, 105–114. [Google Scholar] [CrossRef]
- Akanda, N.; Tofight, R.; Brask, J.; Tamm, C.; Elinder, F.; Ceccatelli, S. Voltage-dependent anion channels (VDAC) in the plasma membrane play a critical role in apoptosis in differentiated hippocampal neurons but not in neural stem cells. Cell Cycle 2008, 7, 3225–3234. [Google Scholar]
- Yu, W.H.; Wolfgang, W.; Forte, M. Subcellular localization of human voltage-dependent anion channel isoforms. J. Biol. Chem. 1995, 270, 13998–14006. [Google Scholar]
- Buck, C.R.; Jurynec, M.J.; Upta, D.E.; Law, A.K.T.; Bilger, J.; Wallace, D.C.; McKeon, R.J. Increased adenine nucleotide translocator 1 in reactive astrocytes facilitates glutamate transport. Exp. Neurol. 2003, 181, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Hazelton, J.L.; Petrasheuskaya, M.; Fiskum, G.; Kristian, T. Cyclophilin D is expressed predominantly in mitochondria of γ-aminobutyric acidergic interneurons. J. Neurosci. Res. 2009, 87, 1250–1259. [Google Scholar]
- Naga, K.K.; Sullivan, P.G.; Geddes, J.W. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J. Neurosci. 2007, 27, 7469–7475. [Google Scholar]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Freedman, R.B. Peptidyl prolyl cis-trans-isomerase activity associated with the lumen of the endoplasmic reticulum. Biochem. J. 1994, 300, 865–870. [Google Scholar] [PubMed]
- Sullivan, P.G.; Rabchevsky, A.G.; Keller, J.N.; Lovell, M.; Sodhi, A.; Hart, R.P.; Scheff, S.W. Intrinsic differences in brain and spinal cord mitochondria: implications for therapeutic interventions. J. Comp. Neurol. 2004, 474, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Morota, S.; Hansson, M.J.; Ishii, N.; Kudo, Y.; Elmer, E.; Uchino, H. Spinal cord mitochondria display lower calcium retention capacity compared with brain mitochondria without inherent differences in sensitivity to cyclophilin D inhibition. J. Neurochem. 2007, 103, 2066–2076. [Google Scholar]
- Collins, T.J.; Bootman, M.D. Mitochondria are morphologically heterogeneous within cells. J. Exp. Biol. 2003, 206, 1993–2000. [Google Scholar]
- Jensen, R.E. Control of mitochondrial shape. Curr. Opin. Cell Biol. 2005, 17, 384–388. [Google Scholar]
- Hamberger, A.; Blomstrand, C.; Lehninger, A.L. Comparative studies of mitochondria isolated from neuron-enriched and glia-enriched fractions of rabbit and beef brain. J. Cell Biol. 1970, 45, 221–234. [Google Scholar]
- Tata, J.R. Requirement for RNA and protein synthesis for induced regression of tadpole tail in organ culture. Dev. Biol. 1966, 13, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. p53’s believe it or not: lessons on transcription-independent death. J. Clin. Immunol. 2003, 23, 355–361. [Google Scholar]
- Martin, L.J.; Liu, Z.; Pipino, J.; Chestnut, B.; Landek, M.A. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb. Cortex 2009, 19, 1273–1293. [Google Scholar]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar]
- Schwartz, L.M.; Smith, S.W.; Jones, M.E.; Osborne, B.A. Do all programmed cell deaths occur via apoptosis? Proc. Natl. Acad. Sci. USA 1993, 90, 980–984. [Google Scholar]
- Amin, F.; Bowen, I.D.; Szegedi, Z.; Mihalik, R.; Szende, B. Apoptotic and non-apoptotic modes of programmed cell death in MCF-7 human breast carcinoma cells. Cell Biol. Intl. 2000, 24, 253–260. [Google Scholar]
- Jacobson, M. Developmental Neurobiology; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Glücksmann, A. Cell deaths in normal vertebrate ontogeny. Biol. Rev. 1951, 26, 59–86. [Google Scholar]
- Lockshin, R.A.; Williams, C.M. Programmed cell death: II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J. Insect Physiol. 1964, 10, 643–649. [Google Scholar] [CrossRef]
- Saunders, J.W. Death in embryonic systems. Science 1966, 154, 604–612. [Google Scholar]
- Bursch, W.; Paffe, S.; Putz, B.; Barthel, G.; Schulte-Hermann, R. Determination of the length of the histological stages of apoptosis in normal liver and in altered hepatic foci of rats. Carcinogenesis 1990, 11, 847–853. [Google Scholar]
- Wyllie, A.H.; Kerr, J.F.R.; Currie, A.R. Cell death: the significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar]
- Nagata, S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 1999, 33, 29–55. [Google Scholar]
- Clarke, P.G.H. Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar]
- Wyllie, A.H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 1980, 284, 555–556. [Google Scholar]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. [Google Scholar]
- Sakahira, H.; Enari, M.; Ohsawa, Y.; Uchiyama, Y.; Nagata, S. Apoptotic nuclear morphological change without DNA fragmentation. Curr. Biol. 1999, 9, 543–546. [Google Scholar]
- Pilar, G.; Landmesser, L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. J. Cell Biol. 1976, 68, 339–356. [Google Scholar]
- Nakajima, W.; Ishida, A.; Lange, M.S.; Gabrielson, K.L.; Wilson, M.A.; Martin, L.J.; Blue, M.E.; Johnston, M.V. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J. Neurosci. 2000, 20, 7994–8004. [Google Scholar]
- Northington, F.J.; Ferriero, D.M.; Graham, E.M.; Traystman, R.J.; Martin, L.J. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 2001, 8, 207–219. [Google Scholar]
- Northington, F.J.; Zelaya, M.E.; O’Riordan, D.P.; Blomgren, K.; Flock, D.L.; Hagberg, H.; Ferriero, D.M.; Martin, L.J. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 2007, 149, 822–833. [Google Scholar]
- Schweichel, J.U.; Merker, H.J. The morphology of various types of cell death in prenatal tissues. Teratology 1973, 7, 253–266. [Google Scholar]
- Xue, L. Z.; Fletcher, G. C.; Tolkovsky, A. M. Autophagy is activated by apoptotic signaling in sympathetic neurons: an alternative mechanism of death execution. Mol. Cell. Neurosci. 1999, 14, 180–198. [Google Scholar]
- Yue, Z.; Horton, A.; Bravin, M.; DeJager, P.L.; Selimi, F.; Heintz, N. A novel protein complex linking the δ2 glutamate receptor and autophagy: implications for neurodegeneration in Lurcher mice. Neuron 2002, 35, 921–933. [Google Scholar] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; Misushima, N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 44, 885–889. [Google Scholar]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; Tanaka, K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 44, 880–884. [Google Scholar]
- Nakendra, D.; Tanaka, A.; Suen, D-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Todde, V.; Veenhuis, M.; van der Klei, I.J. Autophagy: principles and significance in health and disease. Biochim. Biophys. Acta 2009, 1792, 3–13. [Google Scholar] [PubMed]
- Bursch, W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Diff. 2001, 8, 569–581. [Google Scholar]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar]
- Liange, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar]
- Ogier-Denis, E.; Codogno, P. Autophagy: a barrier or an adaptive response to cancer. Biochim. Biophys. Acta 2003, 1603, 113–128. [Google Scholar] [PubMed]
- Ameisen, J.C. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Diff. 2002, 9, 367–393. [Google Scholar] [CrossRef]
- Metzstein, M.M.; Stanfield, G.M.; Horvitz, H.R. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 1998, 14, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The bcl-2 family: regulators of the cellular life-or-death switch. Nat. Rev. 2002, 2, 647–656. [Google Scholar]
- Wolf, B.B.; Green, D.R. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J. Biol. Chem. 1999, 274, 20049–20052. [Google Scholar]
- Levrero, M.; De Laurenzi, V.; Costanzo, A.; Sabatini, S.; Gong, J.; Wang, J.Y.J.; Melino, G. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J. Cell Sci. 2000, 113, 1661–1670. [Google Scholar]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar]
- Klein, J.A.; Longo-Guess, C.M.; Rossmann, M.P.; Seburn, R.E.; Hurd, R.E.; Frankel, W.N.; Bronson, R.T.; Ackerman, S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002, 419, 367–374. [Google Scholar]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilentei, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 4432–4538. [Google Scholar]
- Liston, P.; Roy, N.; Tamai, K.; Lefebvre, C.; Baird, S.; Cherton-Horvat, G.; Farahani, R.; McLean, M.; Ikeda, J-E.; MacKenzie, A.; Korneluk, R.G. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 1996, 379, 349–353. [Google Scholar] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, T.J.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. Bax and Bak regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.L.; Fesik, S.W. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1999, 381, 335–341. [Google Scholar]
- Martin, L.J.; Price, A.C.; McClendon, K.B.; Al-Abdulla, N.A.; Subramaniam, J.R.; Wong, P.C.; Liu, Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J. Neurochem. 2003, 85, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Wolter, K.G.; Hsu, Y-T.; Smith, C.L.; Nechushtan, A.; Xi, X-G.; Youle, R.L. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar] [PubMed]
- Nechushtan, A.; Smith, C.L.; Lamensdorf, I.; Yoon, S-H.; Youle, R.J. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 2001, 153, 1265–1276. [Google Scholar] [PubMed]
- Antonsson, B.; Conti, F.; Ciavatta, A.; Montessuit, S.; Lewis, S.; Martinou, I.; Bernasconi, L.; Bernard, A.; Mermod, J-J.; Mazzei, G.; Maundrell, K.; Gambale, F.; Sadoul, R.; Martinou, J-C. Inhibition of bax channel-forming activity by bcl-2. Science 1997, 277, 370–372. [Google Scholar] [PubMed]
- Shimizu, S.; Ide, T.; Yanagida, T.; Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000, 275, 12321–12325. [Google Scholar]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: a primary site for bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T-I.; Jones, D.P.; Wang, X. Prevention of apoptosis by bcl-2: release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar]
- Vander Heiden, M.G.; Chandel, N.S.; Williamson, E.K.; Schumacker, P.T.; Thompson, C.B. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91, 627–637. [Google Scholar]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar]
- Wei, M.C.; Zong, W-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic Bax and Bak: a requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [PubMed]
- Hu, Y.; Benedict, M.A.; Wu, D.; Inohara, N.; Núñez, G. Bcl-xL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl. Acad. Sci. USA 1998, 95, 4386–4391. [Google Scholar]
- Song, Q.; Kuang, Y.; Dixit, V.M.; Vincenz, C. Boo, a negative regulator of cell death, interacts with Apaf-1. EMBO J. 1999, 18, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Jena, N.; Croce, C.M. Inactivation of Bcl-2 by phosphorylation. Proc. Natl. Acad. Sci. USA 1995, 92, 4507–4511. [Google Scholar]
- Wang, H-G.; Rapp, U.R.; Reed, J.C. Bcl-2 targets the protein kinase raf-1 to mitochondria. Cell 1996, 87, 629–638. [Google Scholar] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar]
- del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-induced phosphorylation of bad through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvensen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14-3-3 not Bcl-xL. Cell 1996, 87, 619–628. [Google Scholar]
- Wang, H-G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [PubMed]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [PubMed]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C-Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; Gygi, S.P.; Korsmeyer, S.J. Bad and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952–956. [Google Scholar] [PubMed]
- Chowdhury, I.; Tharakan, B.; Bhat, G.H. Caspases- an update. Comp. Biochem. Physiol. 2008, 151, 10–27. [Google Scholar]
- Mancini, M.; Nicholson, D.W.; Roy, S.; Thornberry, N.A.; Peterson, E.P.; Casciola-Rosen, L.A.; Rosen, A. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J. Cell Biol. 1998, 140, 1485–1495. [Google Scholar]
- Krajewski, S.; Krajewska, M.; Ellerby, L.M.; Welsh, K.; Xie, Z.; Deveraus, Q.L.; Salvesen, G.S.; Bredesen, D.E.; Rosenthal, R.E.; Fiskum, G.; Reed, J.C. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 5752–5757. [Google Scholar]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An Apaf-1-cytochrome c multimeric complex is functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar]
- Li, H.; Zhu, H.; Xu, C-J.; Yuan, J. Cleavage of Bid by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [PubMed]
- Robertson, J. D.; Enoksson, M.; Suomela, M.; Zhivotovsky, B.; Orrenius, S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J. Biol. Chem. 2002, 277, 29803–29809. [Google Scholar] [PubMed]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar]
- Holcik, M. The IAP proteins. Trends Gen. 2002, 18, 537–538. [Google Scholar]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [PubMed]
- Hao, Y.; Sekine, K.; Kawabata, A; Nakamura, H.; Ishioka, T.; Ohata, H.; Katayama, R.; Hashimoto, C.; Zhang, X.; Noda, T.; Tsuruo, T.; Naito, M. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 2004, 6, 849–860. [Google Scholar] [PubMed]
- Jiang, Y.; de Bruin, A.; Caldas, H.; Fangusaro, J.; Hayes, J.; Conway, E.M.; Robinson, M.L.; Altura, A. Essential role for survivin in early brain development. J. Neurosci. 2005, 25, 6962–6970. [Google Scholar] [PubMed]
- Xu, D.G.; Korneluk, R.G.; Tamai, K.; Wigle, N.; Hakim, A.; MacKenzie, A.; Robertson, G.S. Distribution of neuronal apoptosis inhibitory protein-like immunoreactivity in the rat central nervous system. J. Comp. Neurol. 1997, 382, 247–259. [Google Scholar]
- McPhail, L.T.; Vanderluit, J.L.; McBride, C.B.; Oschipok, L.W.; crocker, S.J.; Xu, D.; Thompson, C.S.; Liston, P.; Holcik, M.; Robertson, G.S.; Tetzlaff, W. Endogenous expression of inhibitor of apoptosis proteins in facial motoneurons of neonatal and adult rats following axotomy. Neuroscience 2003, 117, 567–575. [Google Scholar]
- Roy, N.; Mahadevan, M.S.; McLean, M.; Shutler, G.; Yaraghi, Z.; Farahani, R.; Baird, S.; Besner-Johnston, A.; Lefebvre, C.; Kang, X.; Salih, M.; Aubry, H.; Tamai, K.; Guan, X.; Ioannou, P.; Crawford, T.O.; de Jong, P.J.; Surh, L.; Ikeda, J.-E.; Korneluk, R.G.; Mackenzie, A. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80, 167–178. [Google Scholar]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K; Takio, K; Takahashi, R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Bogaerts, V.; Nuytemans, K.; Reumers, J.; Pals, R.; Engelborghs, S.; Pickut, B.; Corsmit, E.; Peeters, K.; Schymkowizt, J.; De Deyn, P.P.; Cras, P.; Rousseau, F.; Theuns, J.; Van Broeckhoven, C. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson’s disease. Hum. Mut. 2008, 29, 832–840. [Google Scholar]
- Simon-Sanchez, J.; Singleton, A.B. Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum. Mol. Gen. 2008, 17, 1988–1993. [Google Scholar]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Constantini, P.; Loeffler, M.; Larochette, N.; Goodlett, D.R.; Aebersold, R.; Siderovski, D.P.; Penninger, J.M.; Kroemer, G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar]
- Mate, M.J.; Ortiz-Lombardia, M.; Boitel, B.; Haouz, A.; Tello, D.; Susin, S.A.; Penninger, J.; Kroemer, G.; Alzari, P.M. The crystal structure of the mouse apoptosis-inducing factor AIF. Nature Struct. Biol. 2002, 9, 442–446. [Google Scholar]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Develop. 1998, 12, 2973–2983. [Google Scholar]
- Maki, C.G.; Huibregtse, J.M.; Howley, P.M. In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996, 56, 2649–2654. [Google Scholar] [PubMed]
- Shieh, S-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [PubMed]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science 2003, 302, 1036–1038. [Google Scholar]
- Erster, S.; Moll, U.M. Stress-induced p53 runs a transcription-independent death program. Biochem Biophys. Res. Comm. 2005, 331, 843–850. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar]
- Leu, J. I-J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Aloyz, R.S.; Bamji, S.X.; Pozniak, C.D.; Toma, J.G.; Atwal, J.; Kaplan, D.R.; Miller, F.D. p53 is essential for developmental neuron death regulated by the TrkA and p75 neurotrophin receptors. J. Cell. Biol. 1998, 143, 1691–1703. [Google Scholar]
- Martin, L.J.; Kaiser, A.; Yu, J.W.; Natale, J.E.; Al-Abdulla, N.A. Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J. Comp. Neurol. 2001, 433, 299–311. [Google Scholar] [PubMed]
- Morrison, R.S.; Kinoshita, Y.; Johnson, M.D.; Guo, W.; Garden, G.A. p53-dependent cell death signaling in neurons. Neurochem. Res. 2003, 28, 15–27. [Google Scholar]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar] [PubMed]
- Li, L.; Liu, F.; Salmonsen, R.A.; Turner, T.K.; Litofsky, N.S.; Di Cristofano, A.; Pandolfi, P.P.; Jones, S.N.; Recht, L.D.; Ross, A.H. PTEN in neural precursor cells: regulation of migration, apoptosis, and proliferation. Mol. Cell. Neurosci. 2002, 20, 21–29. [Google Scholar] [PubMed]
- Desagher, S.; Osen-Sand, A.; Nichols, A.; Eskes, R.; Montessuit, S.; Lauper, S.; Maundrell, K.; Antonsson, B.; Martinou, J-C. Bid-induced conformational change of bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell. Biol. 1999, 144, 891–901. [Google Scholar]
- Troy, C.M.; Friedman, J. E.; Friedman, W. J. Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J. Biol. Chem. 2002, 277, 34295–34302. [Google Scholar] [PubMed]
- Martin, L.J.; Chen, K.; Liu, Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J. Neurosci. 2005, 25, 6449–6459. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.R.; Newhouse, J.P. The toxic effect of sodium L-glutamate on the inner layers of the retina. Arch. Ophthal. 1957, 58, 193–201. [Google Scholar]
- Olney, J.W. Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J. Neuropathol. Exp. Neurol. 1971, 30, 75–90. [Google Scholar]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar]
- Martin, L.J. The apoptosis-necrosis cell death continuum in CNS development, injury and disease: contributions and mechanisms. In Neuroprotection; Lo, E.H., Marwah, J., Eds.; Prominent Press: Scottsdale, AR, USA, 2002; pp. 379–412. [Google Scholar]
- Martin, L.J.; Sieber, F.E.; Traystman, R.J. Apoptosis and necrosis occur in separate neuronal populations in hippocampus and cerebellum after ischemia and are associated with alterations in metabotropic glutamate receptor signaling pathways. J. Cereb. Blood Flow. Metab. 2000, 20, 153–167. [Google Scholar]
- Martin, L.J. Excitotoxicity. In Encyclopedia of Neuroscience; Adelman, G., Smith, B.H., Eds.; Elsevier Science: Amsterdam, The Netherland, 2004; CD-ROM. [Google Scholar]
- Reynolds, I.J. Mitochondrial membrane potential and the permeability transition in excitotoxicity. Ann. N.Y. Acad. Sci. 1999, 893, 33–41. [Google Scholar]
- Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. Dementia of Alzheimer’s disease and other neurodegenerative disorders- memantine, a new hope. Pharma. Res. 2005, 51, 1–17. [Google Scholar]
- Gwag, B.J.; Koh, J.Y.; DeMaro, J.A.; Ying, H.S.; Jacquin, M.; Choi, D.W. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience 1997, 77, 393–401. [Google Scholar]
- Kure, S.; Tominaga, T.; Yoshimoto, T.; Tada, K.; Narisawa, K. Glutamate triggers internucleosomal DNA cleavage in neuronal cells. Biochem. Biophys. Res. Commun. 1991, 179, 39–45. [Google Scholar]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar]
- Simonian, N.A.; Getz, R.L.; Leveque, J.C.; Konradi, C.; Coyle, J. T. Kainate induces apoptosis in neurons. Neuroscience 1996, 74, 675–683. [Google Scholar]
- Dessi, F.; Charriaut-Marlangue, C.; Khrestchatisky, M.; Ben-Ari, Y. Glutamate-induced neuronal death is not a programmed cell death in cerebellar culture. J. Neurochem. 1993, 60, 1953–1955. [Google Scholar]
- Xiang, H.; Kinoshita, Y.; Knudson, C.M.; Korsmeyer, S.J.; Schwartzkroin, P.A.; Morrison, R.S. Bax involvement in p53-mediated neuronal cell death. J. Neurosci. 1998, 18, 1363–1373. [Google Scholar]
- Miller, T.M.; Moulder, K.L.; Knudson, C.M.; Creedon, D.J.; Deshmukh, M.; Korsmeyer, S.J.; Johnson, E.M., Jr. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell. Biol. 1997, 139, 205–217. [Google Scholar]
- Dargusch, R.; Piasecki, D.; Tan, S.; Liu, Y.; Schubert, D. The role of Bax in glutamate-induced nerve cell death. J. Neurochem. 2001, 76, 295–301. [Google Scholar]
- Johnson, M.D.; Kinoshita, Y.; Xiang, H.; Ghatan, S.; Morrison, R.S. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J. Neurosci. 1999, 19, 2996–3006. [Google Scholar]
- Tenneti, L.; Lipton, S.A. Involvement of activated caspase-3-like proteases in N-methyl-D-aspartate-induced apoptosis in cerebrocortical neurons. J. Neurochem. 2001, 74, 134–142. [Google Scholar] [CrossRef]
- Simons, M.; Beinroth, S.; Gleichmann, M.; Liston, P.; Korneluk, R.G.; MacKenzie, A.E.; Bahr, M.; Klockgether, T.; Robertson, G.S.; Weller, M.; Schulz, J.B. Adenovirus-mediated gene transfer of inhibitors of apoptosis proteins delays apoptosis in cerebellar granule neurons. J. Neurochem. 1999, 72, 292–301. [Google Scholar]
- van Lookeren Campagne, M.; Lucassen, P.J.; Vermeulen, J.P.; Balázs, R. NMDA and kainate induced internucleosomal DNA cleavage associated with both apoptotic and necrotic cell death in the neonatal rat brain. Eur. J. Neurosci. 1995, 7, 1627–1640. [Google Scholar]
- Holcik, M.; Thompson, C.S.; Yaraghi, Z.; Lefebvre, C.A.; MacKenzie, A.E.; Korneluk, R.G. The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury. Proc. Natl. Acad. Sci. USA 1999, 97, 2286–2290. [Google Scholar]
- Schreiber, S.S.; Tocco, G.; Najm, I.; Thompson, R.F.; Baudry, M. Cycloheximide prevents kainate-induced neuronal death and c-fos expression in adult rat brain. J. Mol. Neurosci. 1993, 4, 149–159. [Google Scholar]
- Leppin, C.; Finiels-Marlier, F.; Crawley, J.N.; Montpied, P.; Paul, S.M. Failure of a protein synthesis inhibitor to modify glutamate receptor-mediated neurotoxicity in vivo. Brain Res. 1992, 581, 168–170. [Google Scholar]
- Lok, J.; Martin, L.J. Rapid subcellular redistribution of Bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. J. Neurotrauma 2002, 19, 815–828. [Google Scholar]
- Mueller, D.; Shamblott, M.J.; Fox, H.F.; Gearhart, J.D.; Martin, L.J. Transplanted human embryonic germ cell-derived neural stem cells replace neurons and oligodendrocytes in the forebrain of neonatal mice with excitotoxic brain damage. J. Neurosci. Res. 2005, 82, 592–608. [Google Scholar]
- Al-Abdulla, N.A.; Portera-Cailliau, C.; Martin, L.J. Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience 1998, 86, 191–209. [Google Scholar]
- Yang, Y.; Xie, Y.; Chai, H.; Fan, M.; Liu, S.; Liu, H.; Bruce, I.; Wu, W. Microarray analysis of gene expression patterns in adult spinal motoneurons after different types of axonal injuries. Brain Res. 2006, 1075, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdulla, N.A.; Martin, L.J. Apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of perikaryal mitochondria and oxidative damage to the nucleus. Am. J. Pathol. 1998, 153, 447–456. [Google Scholar]
- Fujikawa, D.G. Confusion between neuronal apoptosis and activation of programmed cell death mechanisms in acute necrotic insults. Trends Neurosci. 2000, 23, 410–411. [Google Scholar]
- Ishimaru, M.J.; Ikonomidou, C.; Tenkova, T.I.; Der, T.C.; Dikranian, K; Sesma, M.A.; Olney, J.W. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J. Comp. Neurol. 1999, 408, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S. Apoptosis: a guide for the perplexed. Trends Pharmacol. Sci. 2002, 23, 19–24. [Google Scholar]
- Baille, V.; Clarke, P.G.; Brochier, G.; Dorander, F.; Verna, J.M.; Four, E.; Lallement, G.; Carpentier, P. Soman-induced convulsions: the neuropathology revisited. Toxicology 2005, 215, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Hall, J.J.; Noble, L.J.; Ferriero, D.M. Delayed cell death in neonatal mouse hippocampus from hypoxia-ischemia is neither apoptotic nor necrotic. Neurosci. Lett. 2001, 304, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ying, D.J.; Cui, L.; Lungsdorf, J.; Yu, S.P. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004, 1022, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J. Neuropathol. Exp. Neurol. 1999, 58, 459–471. [Google Scholar]
- Zoccolella, S.; Santamato, A.; Lamberti, P. Current and emerging treatments for amyotrophic lateral sclerosis. Neuropsychiatr. Dis. Treat. 2009, 5, 577–595. [Google Scholar]
- Katzman, R. Education and the prevalence of dementia and Alzheimer’s disease. Neurology 1993, 43, 13–20. [Google Scholar]
- Evans, D.A.; Funkenstein, H.H.; Albert, M.S.; Scherr, P.A.; Cook, N.R.; Chown, M.J.; Hebert, L.E.; Hennekens, C.H.; Taylor, J.O. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. JAMA 1989, 262, 2551–2556. [Google Scholar] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under the auspices of the Department of Health and Human Services task force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar]
- Olshansky, S.J.; Carnes, B.A.; Cassel, C.K. The aging of the human species. Sci. Am. 1993, 268, 46–52. [Google Scholar]
- Minati, L.; Edginton, T.; Bruzzone, M.G.; Giaccone, G. Current concepts in Alzheimer's disease: A multidisciplinary review. Am. J. Alz. Dis. Other Demen. 2009, 24, 95–121. [Google Scholar]
- Chartier-Harlin, M.-C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.; Mullan, M. Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 1991, 353, 844–846. [Google Scholar]
- Tilley, L.; Morgan, K.; Kalsheker, N. Genetic risk factors for Alzheimer’s disease. J. Clin. Pathol. Mol. Pathol. 1998, 51, 293–304. [Google Scholar]
- Goate, A.; Chartier-Harlin, M.-.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; Mant, R.; Newton, P.; Rooke, K.; Roques, P.; Talbot, C.; Pericak-Vance, M.; Roses, A.; Williamson, R.; Rossor, M.; Owen, M.; Hardy, J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991, 349, 704–706. [Google Scholar] [PubMed]
- Naruse, S.; Igarashi, S.; Kobayashi, H.; Aoki, K.; Inuzuka, T.; Kaneko, K.; Shimizu, T.; Iihara, K.; Kojima, T.; Miyatake, T.; Tsuji, S. Mis-sense mutation Val->Ile in exon 17 of amyloid precursor protein gene in Japanese familial Alzheimer's disease. Lancet 1991, 337, 978–979. [Google Scholar]
- Campion, D.; Flaman, J.M.; Brice, A.; Hannequin, D.; Dubois, B.; Martin, C.; Moreau, V.; Charbonnier, F.; Didierjean, O.; Tardieu, S.; Penet, C.; Puel, M.; Pasquier, F.; Ledoze, F.; Bellis, G.; Calenda, A.; Heilig, R.; Martinez, M.; Mallet, J.; Bellis, M.; Clergetdarpoux, F.; Agid, Y.; Frebourg, T. Mutations of the presenilin 1 gene in families with early-onset Alzheimer's disease. Hum. Mol. Genet. 1995, 4, 2373–2377. [Google Scholar]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; Tsuda, T.; Mar, L.; Foncin, J.-F.; Bruni, A.C.; Montesi, M.P.; Sorbi, S.; Rainero, I.; Pinessi, L.; Nee, L.; Chumakov, I.; Pollen, D.; Brookes, A.; Sanseau, P.; Polinsky, R.J.; Wasco, W.; Da Silva, H.A.R.; Haines, J.L.; Pericak-Vance, M.A.; Tanzi, R.E.; Roses, A.D.; Fraser, P.E.; Rommens, J.M.; St George-Hyslop, P.H. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995, 375, 754–760. [Google Scholar]
- Kalaria, R.N. Dementia comes of age in the developing world. Lancet 2003, 361, 888–889. [Google Scholar]
- Roses, A.D. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar]
- Gomez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar]
- Mouton, P.R.; Martin, L.J.; Calhoun, M.E.; Dal Forno, G.; Price, D.L. Cognitive decline strongly correlates with cortical atrophy in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 371–377. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Kawas, C.H.; Martin, L.J.; Troncoso, J.C. The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann. N.Y. Acad. Sci. 2000, 908, 255–259. [Google Scholar] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Regeur, L.; Oster, S.; Pakkenberg, B. Neocortical glial cell numbers in Alzheimer's disease. A stereological study. Dement. Geriatr. Cogn. Disord. 2003, 16, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer’s disease-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar]
- Kermer, P.; Liman, J; Weishaupt, J.H.; Bahr, M. Neuronal apoptosis in neurodegenerative diseases: from basic research to clinical application. Neurodeg. Dis. 2004, 1, 9–19. [Google Scholar]
- Smale, G.; Nichols, N.R.; Brady, D.R.; Finch, C.E.; Horten, W.E., Jr. Evidence for apoptotic cell death in Alzheimer's disease. Exp. Neurol. 1995, 133, 225–230. [Google Scholar]
- Anderson, A.J.; Su, J.H.; Cotman, C.W. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J. Neurosci. 1996, 16, 1710–1719. [Google Scholar]
- Adamec, E.; Vonsattel, J.P.; Nixon, R.A. DNA strand breaks in Alzheimer's disease. Brain Res. 1999, 849, 67–77. [Google Scholar]
- Stadelmann, C.; Deckwerth, T.L.; Srinivasan, A.; Bancher, C.; Brock, W.; Lassmann, H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am. J. Pathol. 1999, 155, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Lucassen, P.J.; Chung, W.C.J.; Kamphorst, W.; Swaab, D.F. DNA damage distribution in the human brain as shown by in situ end labeling; area-specific differences in aging and Alzheimer's disease in the absence of apoptotic morphology. J. Neuropathol. Exp. Neurol. 1997, 56, 887–900. [Google Scholar]
- Su, J.H.; Zhao, M.; Anderson, A.J.; Srinivasan, A.; Cotman, C.W. Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res. 2001, 898, 350–357. [Google Scholar]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Ota, T.; Matsuoka, Y.; Nomura, Y.; Smith, M.A.; Perry, G.; Whitehouse, P.J.; Taniguchi, T. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer's disease. Brain Res. 1998, 780, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Gastard, M.C.; Troncoso, J.C.; Koliatsos, V.E. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann. Neurol. 2003, 54, 393–398. [Google Scholar]
- Pompl, P.N.; Yemul, S.; Xiang, Z.; Ho, L.; Haroutunian, V.; Purohit, D.; Mohs, R.; Pasinetti, G.M. Caspase gene expression in the brain as a function of the clinical progression of Alzheimer's disease. Arch. Neurol. 2003, 60, 369–376. [Google Scholar]
- Rohn, T.T.; Rissman, R.A.; Davis, M.C.; Kim, Y.E.; Cotman, C.W.; Head, E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Neurobiol. Dis. 2002, 11, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Albrecht, S.; Bourdeau, M.; Petzke, T.; Bergeron, C.; LeBlanc, A.C. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol. 2004, 165, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Selznick, L.A.; Holtzman, D.M.; Han, B.H.; Gokden, M.; Srinivasan, A.N.; Johnson, E.M., Jr.; Roth, K.A. In situ immunodetection of neuronal caspase-3 activation in Alzheimer's disease. J. Neuropath. Exp. Neurol. 1999, 58, 1020–1026. [Google Scholar] [CrossRef]
- Lesuisse, C.; Martin, L.J. Immature and mature neurons engage different apoptotic mechanisms involving caspase-3 and the mitogen-activated protein kinase pathway. J. Cereb. Blood Flow Metabol. 2002, 22, 935–950. [Google Scholar]
- Shimohama, S.; Tanino, H.; Fujimoto, S. Differential subcellular localization of caspase family protein in the adult rat brain. Neurosci. Lett. 2001, 315, 125–128. [Google Scholar]
- Yan, X.X.; Najbauer, J.; Woo, C.C.; Dashtipour, K.; Ribak, C.E.; Leon, M. Expression of active caspase-3 in mitotic and postmitotic cells or rat forebrain. J. Comp. Neurol. 2001, 433, 4–22. [Google Scholar]
- Troncoso, J.C.; Cataldo, A.M.; Nixon, R.A.; Barnett, J.L.; Lee, M.K.; Checler, F.; Fowler, D.R.; Smialek, J.E.; Crain, B.; Martin, L.J.; Kawas, C.H. Neuropathology of preclinical and clinical late-onset Alzheimer’s disease. Ann. Neurol. 1998, 43, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau is a major antigenic component of paired helical filaments in Alzheimer's disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar]
- Sze, C.-I.; Bi, H.; Kleinschmidt-DeMasters, B.K.; Filley, C.M.; Martin, L.J. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer’s disease. J. Neurol. Sci. 2001, 182, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.A.; McKernan, R.M. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002, 5, 1039–1042, (suppl). [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1993, 12, 376–389. [Google Scholar]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katsman, R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar]
- Martin, L.J.; Pardo, C.A.; Cork, L.C.; Price, D.L. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am. J. Pathol. 1994, 145, 1358–1381. [Google Scholar]
- Sze, C.-I.; Troncoso, J.C.; Kawas, C.; Mouton, P.; Price, D.L.; Martin, L.J. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1997, 56, 933–994. [Google Scholar]
- Selkoe, D.J. Alzheimer's disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar]
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science 1989, 245, 417–420. [Google Scholar]
- Younkin, S.G. Evidence that Abeta 42 is the real culprit in Alzheimer's disease. Ann. Neurol. 1995, 37, 287–288. [Google Scholar]
- Fath, T.; Eidenmuller, J.; Brandt, R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J. Neurosci. 2002, 22, 9733–9741. [Google Scholar]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Gen. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; Trinchese, F.; Liu, S.; Gunn-Moore, F.; Lue, L.-F.; Walker, D.G.; Kuppsamy, P.; Zewier, Z.L.; Arancio, O.; Stern, D.; Yan, S.S.; Wu, H. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; Gunn-Moore, F.J.; Vonsattel, J.P.; Aranico, O.; Chen, J.X.; Yan, S.D. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar]
- Duyckaerts, C.; Potier, M.C.; Delatour, B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008, 115, 5–38. [Google Scholar] [PubMed]
- Irizarry, M.C.; McNamara, M.; Fedorchak, K.; Hsiao, K.; Hyman, B.T. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol. 1997, 56, 965–973. [Google Scholar]
- Irizarry, M.C.; Soriano, F.; McNamara, M.; page, K.J.; Schenk, D.; Games, D.; Hyman, B.T. Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyoid precursor protein V717F (PDAPP) transgenic mouse. J. Neurosci. 1997, 17, 7053–7059. [Google Scholar] [PubMed]
- Takeuchi, A.; Irizarry, M.C.; Duff, K.; Saido, T.C.; Hsiao-Ashe, K.; Hasegawa, M.; Mann, D.M.A.; Hyman, B.T.; Iwatsubo, T. Age-related amyloid β deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid β precursor protein Swedish mutant is not associated with global neuronal loss. Am. J. Pathol. 2000, 157, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, M.E.; Wiederhold, K.H.; Abramowski, D.; Phinney, A.L.; Probst, A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Jucker, M. Neuron loss in APP transgenic mice. Nature 1998, 395, 755–756. [Google Scholar]
- Selznick, L.A.; Holtzman, D.M.; Han, B.H.; Gokden, M.; Srinivasan, A.N.; Johnson, E.M., Jr.; Roth, K.A. In situ immunodetection of neuronal caspase-3 activation in Alzheimer's disease. J. Neuropath. Exp. Neurol. 1999, 58, 1020–1026. [Google Scholar]
- Yang, D.-S.; Kumar, A.; Stavrides, P.; Peterson, J.; Peterhoff, C.M.; Pawlik, M.; Levy, E.; Cataldo, A.M.; Nixon, R.A. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am. J. Pathol. 2008, 173, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar]
- LaFerla, F.M.; Hall, C.K.; Ngo, L.; Jay, G. Extracellular deposition of β-amyloid upon p53-dependent neuronal cell death in transgenic mice. J. Clin. Invest. 1996, 98, 1626–1632. [Google Scholar]
- Zhang, Y.; McLaughlin, R.; Goodyer, C.; LeBlanc, A. Selective cytotoxicity of intracellular amyloid β peptide1-42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002, 156, 519–529. [Google Scholar]
- Loo, D.T.; Copani, A.; Pike, C.J.; Whittemore, E.R.; Walencewicz, A.J.; Cotman, C.W. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc. Natl. Acad. Sci. USA 1993, 90, 7951–7955. [Google Scholar]
- Behl, C.; Davis, J.B.; Klier, F.G.; Schubert, D. Amyloid beta peptide induces necrosis rather than apoptosis. Brain Res. 1994, 645, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Land, J.M.; Sharpe, M.A.; Clark, J.B.; Duchen, M.R.; Canevari, L. β-amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol. Dis. 2002, 10, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Duker, N.J.; Sperling, J.; Soprano, K.J.; Druin, D.P.; Davis, A.; Ashworth, R. β-Amyloid protein induces the formation of purine dimers in cellular DNA. J. Cell Biochem. 2001, 81, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Kuperstein, F; Yavin, E. ERK activation and nuclear translocation in amyloid-beta peptide- and iron-stressed neuronal cell cultures. Eur. J. Neurosci. 2002, 16, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. β-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. [Google Scholar] [PubMed]
- Yaar, M.; Zhai, S.; Fine, R.E.; Eisenhauer, P.B.; Arble, B.L.; Stewart, K.B.; Gilchrest, B.A. Amyloid β binds trimers as well as monomers of the 75-kDa neurotrophin receptor and activates receptor signaling. J. Biol. Chem. 2002, 277, 7720–7725. [Google Scholar]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar]
- Ethell, D.W.; Kinloch, R.; Green, D.R. Metalloproteinase shedding of Fas ligand regulates β-amyloid neurotoxicity. Curr. Biol. 2002, 12, 1595–1600. [Google Scholar]
- Culmsee, C.; Zhu, X.; Yu, Q.S.; Chan, S.L.; Camandola, S.; Guo, Z.; Greig, N.H.; Mattson, M.P. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 2001, 77, 220–228. [Google Scholar]
- Ma, L.; Ohyagi, Y.; Miyoshi, K.; Sakae, N.; Motomaura, K.; Taniwaki, T.; Furuya, H.; Takeda, K.; Tabira, T.; Kira, J. Increase in p53 protein levels by presenilin 1 gene mutations and its inhibition by secretase inhibitors. J. Alz. Dis. 2009, 16, 565–575. [Google Scholar]
- Giovanni, A.; Keramaris, E.; Morris, E.J.; Hou, S.T.; O'Hare, M.; Dyson, N.; Robertson, G.S.; Slack, R.S.; Park, D.S. E2F1 mediates death of β-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J. Biol. Chem. 2000, 275, 11553–11560. [Google Scholar]
- Paradis, E.; Douillard, H.; Koutroumanis, M.; Goodyer, C.; LeBlanc, A. Amyloid β peptide of Alzheimer's disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J. Neurosci. 1997, 16, 7533–7539. [Google Scholar]
- Weidemann, A.; Paliga, K.; Durrwang, U.; Reinhard, F.B.M.; Schuckert, O.; Evin, G.; Masters, C.L. Proteolytic processing of the Alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J. Biol. Chem. 1999, 274, 5823–5829. [Google Scholar] [PubMed]
- LeBlanc, A.; Liu, H.; Goodyer, C.; Bergeron, C.; Hammond, J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 1999, 274, 23426–23436. [Google Scholar] [PubMed]
- Gervais, F.G.; Xu, D.; Robertson, G.S.; Vaillancourt, J.P.; Zhu, Y.; Huang, J.; LeBlanc, A.; Smith, D.; Rigby, M.; Shearman, M.S.; Clarke, E.E.; Zheng, H.; van der Ploeg, L.H.; Ruffolo, S.C.; Thornberry, N.A.; Xanthoudakis, S.; Zamboni, R.J.; Roy, S.; Nicholson, D.W. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 97, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavign, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar]
- Passer, B.J.; Pellegrini, L.; Vito, P.; Ganjei, J.K.; D'Adamio, L. Interaction of Alzheimer’s presenilin-1 and presenilin-2 with Bcl-X(L). A potential role in modulating the threshold of cell death. J. Biol. Chem. 1999, 274, 24007–24013. [Google Scholar] [PubMed]
- Kim, T.-W.; Pettingell, W.H.; Jung, Y.-K.; Kovacs, D.M.; Tazi, R.E. Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protein. Science 1997, 277, 373–376. [Google Scholar] [PubMed]
- Bursztajn, S.; DeSouza, R.; McPhie, D.L.; Berman, S.A.; Shioi, J.; Robakis, N.K.; Neve, R.L. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J. Neurosci. 1998, 18, 9790–9799. [Google Scholar]
- Gamliel, A.; Teicher, C.; Hartmann, T.; Beyreuther, K.; Stein, R. Overexpression of wild-type presenilin 2 or its familial Alzheimer's disease-associated mutant does not induce or increase susceptibility to apoptosis in different cells. Neuroscience 2003, 117, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Culmsee, C.; Haughey, N.; Klapper, W.; Mattson, M.P. Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism involving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol. Dis. 2002, 11, 2–19. [Google Scholar]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Indicence of Parkinson's disease: variations by age, gender and race ethnicity. Am. J. Epidemol. 2003, 157, 1015–1022. [Google Scholar]
- Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar]
- Lowe, J.; Lennox, G.; Leigh, P.N. Disorders of movement and system degeneration. In Greenfields Neuropathology; Graham, D.I., Lantos, P.L., Eds.; London: Arnold, 1997; pp. 281–366. [Google Scholar]
- Braak, H.; Del Tredici, K.; Rüb, U.; d Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Etiology of Parkinson's disease. Neurology 2006, 66 (Suppl. 4), S10–S23. [Google Scholar] [PubMed]
- Ascherio, A.; Chen, H.; Weisskopf, M.G.; O’Reilly, E.; McCullough, M.L.; Calle, E.E.; Schwarzchild, M.A.; Thun, M.J. Pesticide exposure and risk for Parkinson’s disease. Ann. Neurol. 2006, 60, 197–203. [Google Scholar]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Reiss, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; Stenroos, E.S.; Chandrasekharappa, S.; Athanassiadou, A.; Papapetropoulos, T.; Johnson, W.G.; Lazzarini, A.M.; Duvoisin, R.C.; Di Iorio, G.; Golbe, L.I.; Nussbaum, R.L. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; Lincoln, S.; Crawley, A.; Hanson, M.; Maraganore, D.; Adler, C.; Cookson, M.R.; Muenter, M.; Baptista, M.; Miller, D.; Blancato, J.; Hardy, J.; Gwinn-Hardy, K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [PubMed]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Browstein, M.J.; Jonnalagada, S.; Chernova, T.; Dehejia, A.; Lavedan, C.; Gasser, T.; Steinbach, P.J.; Wilkinson, K.D.; Polymeropoulos, M.H. The ubiquitin pathway in Parkinson's disease. Nature 1998, 395, 451–452. [Google Scholar]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; van Dongen, J.W.; Vanacore, N.; van Swieten, J.C.; Brice, A.; Meco, G.; van Duijn, C.M.; Oostra, B.A.; Heutink, P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar]
- Hatano, Y.; Li, Y.; Sato, K.; Asakawa, S.; Yamamura, Y.; Tomiyama, H.; Yoshino, H.; Asahina, M.; Kobayashi, S.; Hassin-Baer, S.; Lu, C.S.; Ng, A.R.; Rosales, P.L.; Shimizu, N.; Toda, T.; Mizuno, Y.; Hattori, N. Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol. 2004, 56, 424–427. [Google Scholar]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, A.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; Albanese, A.; Nussbaum, R.; Gonzalez-Maldonado, R.; Deller, T.; Salvi, S.; Cortelli, P.; Gilks, W.P.; Latchman, D.S.; Harvey, R.J.; Dallapiccola, B.; Auburger, G.; Wood, N.W. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar]
- Paisán-Ruίz, C.; Jain, S.; Whitney Evans, E.; Gilks, W.P.; Simón, J.; van der Brug, M.; López de Munain, A.; Aparicio, S.; Martίnez-Gil, A.; Khan, N.; Johnson, J.; Ruiz Martinez, J.; Nicholl, D.; Marti Carrera, I.; Saénz Peňa, A.; de Silva, R.; Lees, A.; Martί-Massó, J.F.; Pérez-Tur, J.; Wood, N.W.; Singleton, A.B. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 2004, 44, 595–600. [Google Scholar] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; Stoessi, J.; Pfeiffer, R.F.; Patenge, N.; Carballo Carbajal, I.; Vieregge, P.; Asmus, F.; Müller-Myhsok, B.; Dickson, D.W.; Meitinger, T.; Storm, T.M.; Wszolek, Z.K.; Gasser, T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar]
- Ramirez, A.; Heimbach, A.; grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A,F.; Wriekat, A.-L.; Roeper, J.; Al-Din, A.; Hillmer, A.M.; Karsak, M.; Liss, B.; Woods, C.G.; Behrens, M.I. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Gen. 2006, 38, 1184–1191. [Google Scholar]
- Lesuisse, C.; Martin, L.J. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J. Neurobiol. 2002, 51, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminals. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.Y. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar]
- Nakamura, K.; Nemani, V.M.; Wallender, E.K.; Kaehlcke, K.; Ott, M.; Edwards, R.H. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 2008, 28, 12305–12317. [Google Scholar] [PubMed]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; Sudhof, T.C. Double knockout mice for α- and β-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar]
- Gurevicine, I.; Gurevicius, K.; Tanila, H. Role of α-synuclein in synaptic glutamate release. Neurobiol. Dis. 2007, 28, 83–89. [Google Scholar]
- Liu, S.; Fa, M.; Ninan, I.; Trinchese, F.; Dauer, W.; Aranico, O. α-Synuclein involvement in hippocampla synaptic plasticity: role of NO, cGMP, cGK and CAMKII. Eur. J. Neurosci. 2007, 25, 3583–3596. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.L.; Nemani, V.M.; Voglmaier, S.M.; Anthony, M.D.; Ryan, T.A.; Edwards, R.H. Neural activity control the synaptic accumulation of α-synuclein. J. Neurosci. 2005, 25, 10913–10921. [Google Scholar] [PubMed]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudholf, T.C. α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Schluter, O.M.; Sudhof, T.C. A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, M.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrilization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibril, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar]
- Junn, E.; Mouradian, M.M. Human α-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 2002, 320, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Orth, M.; Wilkinson, J.M.; Taanman, J.W.; Warner, T.T.; Cooper, J.M.; Schapira, A.H. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum. Mol. Genet. 2000, 9, 2683–2689. [Google Scholar]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, W.M-Y. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, JQ.; Lee, V.M-Y. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [PubMed]
- Ischiropoulos, H. Oxidative modification of alpha-synuclein. Ann. N.Y. Acad. Sci. 2003, 991, 93–100. [Google Scholar]
- Wilkinson, K.D.; Lee, K.M.; Deshpande, S.; Duerken-Hughes, P.; Boss, J.M.; Pohl, J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl terminal hydrolase. Science 1989, 246, 670–673. [Google Scholar] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar]
- Lansbury, P.T., Jr.; Brice, A. Genetics of Parkinson's disease and biochemical studies of implicated gene products. Curr. Opin. Cell Biol. 2002, 14, 653–660. [Google Scholar]
- McNaught, K.S.; Mytilineou, C.; Jnobaptiste, R.; Yabut, J.; Shahidharan, P.; Jennert, P.; Olanow, C.W. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J. Neurochem. 2002, 81, 301–306. [Google Scholar]
- Imai, Y.; Takahashi, R. How do Parkin mutations result in neurodegeneration? Curr. Opin. Neurobiol. 2004, 14, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Matsumine, H.; Asakawa, S.; Kitada, T.; Yoshino, H.; Elibol, B; brookes, A.J.; Yamamura, Y.; Kobayashi, T.; Wang, M.; Yoritaka, A.; Minoshima, S.; Shimizu, N.; Mizuno, Y. Point mutations (Thr240Arg and Gln311Stop) in the Parkin gene. Biochem. Biophys. Res. Commun. 1998, 249, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Van Der Brug, M.; Ahmad, R.; Kesavapany, S.; Miller, D.W.; Petsko, G.A.; Cookson, M.R. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA 2005, 102, 5703–5708. [Google Scholar]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and RGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [PubMed]
- Taymans, J-M.; Van den Haute, C.; Baekelandt, V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J. Neurochem. 2006, 98, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Thomas, K.J.; Ostazewski, B.L.; Cooksen, M.R.; Selkoe, D.J. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009, 48, 2045–2052. [Google Scholar] [PubMed]
- Deng, H.; Jankovic, J.; Guo, Y.; Xie, W.; Le, W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem. Biophys. Res. Commun. 2005, 337, 1133–1138. [Google Scholar]
- Marongiu, R.; Spencer, B.; Crews, L.; Adame, A.; Patrick, C.; Trejo, M.; dallapiccola, B.; Valente, E.M.; Masliah, E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem. 2009, 108, 1561–1574. [Google Scholar]
- Wilson, M.A.; Collins, J.L.; Hod, Y.; Ringe, D.; Petsko, G.A. The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc. Natl. Acad. Sci. USA 2003, 100, 9256–9261. [Google Scholar]
- Shang, H.; Lang, D.; Jean-Marc, B.; Kaelin-Lang, A. Localization of DJ-1 mRNA in the mouse brain. Neurosci. Lett. 2004, 367, 273–277. [Google Scholar]
- Canet-Aviles, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar]
- Miller, D.W.; Ahmad, R.; Hague, S.; Baptista, M.J.; Canet-Aviles, R.; McLendon, C.; Carter, D.M.; Zhu, P.P.; Stadler, J.; Chandran, J.; Klinefelter, G.R.; Blackstone, C.; Cookson, M.R. L166P mutant DJ-1, causative for recessive Parkinson's disease, is degraded through the ubiquitin-proteasome system. J. Biol. Chem. 2003, 278, 36588–36595. [Google Scholar] [PubMed]
- Takahashi-Niki, K.; Niki, T.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson's disease patients. Biochem. Biophys. Res. Commun. 2004, 320, 389–397. [Google Scholar]
- Galter, D.; Westerlund, M.; Carmine, A.; Lindqvist, E.; Sydow, O.; Olson, L. LRRK2 expression linked to dopamine-innervated areas. Ann. Neurol. 2006, 59, 714–719. [Google Scholar]
- Melrose, H.; Lincoln, S.; Tyndall, G.; Dickson, D.; Farrer, M. Anatomical localization of leucine-rich repeat kinase 2 mouse brain. Neuroscience 2006, 139, 791–794. [Google Scholar]
- Iaccarino, C.; Crosio, C.; Vitale, C.; Sanna, G.; Carri, M.T.; Barone, P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum. Mol. Gen. 2007, 16, 1319–1326. [Google Scholar]
- Ho, C. C-Y.; Rideout, H.J.; Ribe, E.; Troy, C.M.; Dauer, W.T. The Parkinson’s disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J. Neurosci. 2009, 29, 1011–1016. [Google Scholar] [PubMed]
- Tatton, N.A.; MaClean-Fraser, A.; Tatton, W.G.; Perl, D.P.; Olnanow, C.W. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson’s disease. Ann. Neurol. 1998, 44, S142–S148. [Google Scholar]
- Jellinger, K.A. Is there apoptosis in Lewy body disease? Acta Neuropathol. 1999, 97, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Wüllner, U.; Kornhuber, J.; Weller, M.; Schulz, J.B.; Loschmann, P.A.; Riederer, P.; Klockgether, T. Cell death and apoptosis regulating proteins in Parkinson’s disease- a cautionary note. Acta Neuropathol. 1999, 97, 408–412. [Google Scholar]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marguez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Martin, L.J.; Liu, Z.; Troncoso, J.C.; Price, D.L. Neuronal cell death in human neurodegenerative diseases and their animal/cell models. In Apoptosis in Health and Disease; Holcik, M., LaCasse, E., Korneluk, R., MacKenzie, A., Eds.; Cambridge University Press: Cambridge, UK, 2005; pp. 242–315. [Google Scholar]
- Tompkins, M.M.; Basgall, E.J.; Zamrini, E.; Hill, W.D. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigra neurons. Am. J. Pathol. 1997, 150, 119–131. [Google Scholar]
- Ginsberg, S.D.; Hemby, S.E.; Mufson, E.J.; Martin, L.J. Cell and tissue microdissection in combination with genomic and proteomic profiling. In Neuroanatomical Tract-Tracing 3, Molecules, Neurons, and Systems; Zaborszky, L., Wouterlood, F.G., Lanciego, J.L., Eds.; Springer: New York, NY, USA, 2006; pp. 109–141. [Google Scholar]
- Martin, L.J; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Capedam, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; Gajendiram, M.; Roth, B.L.; Chesselet, M.F.; Maidment, N.T.; Levine, M.S.; Shen, J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [PubMed]
- Perez, F.A.; Palmiter, R.D. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. USA 2005, 102, 2174–2179. [Google Scholar]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar]
- Lu, X-H.; Fleming, S.M.; Meurers, B.; Ackerson, L.C.; Mortazavi, F.; Lo, V.; Hernandez, D,; Sulzer, D.; Jackson, G.R.; Maidment, N.T.; Chesselet, M-F.; Yang, X.W. Bacterial artificial chromosome transgenic mice expressing a truncated mutant Parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant α-synuclein. J. Neurosci. 2009, 29, 1962–1976. [Google Scholar] [PubMed]
- Chen, L.; Cagniard, B.; Mathews, T.; Jones, S.; Koh, H.C.; Ding, Y.; Carvey, P.M.; Ling, Z.; Kang, U.J.; Zhuang, X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J. Biol. Chem. 2005, 280, 21418–21426. [Google Scholar]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, Y.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; Bernardi, G.; Roth, B.L.; Pothos, E.N.; Calabresi, P.; Shen, J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar]
- Gispert, S.; Del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.W.; Deller, T.; Braak, H.; Auburger, G.; Nussbaum, R.L. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence or aggregate formation. Mol. Cell. Neurosci. 2003, 24, 419–429. [Google Scholar]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van der Putten, H.; Probst, A.; Kremmer, E.; Kretzschmar, H.A.; Haassm, C. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [PubMed]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar]
- Richfield, E.K.; Thiruchelvam, M.J.; Cory-Slechta, D.A.; Wuetzer, C.; Gainetdinov, R.R.; Caron, M.G.; Di Monte, D.A.; Federoff, H.J. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp. Neurol. 2002, 175, 35–48. [Google Scholar]
- van der Putten, H.; Wiederhold, K-H.; Probst, A.; Barbieri, S.; Mistl, C.; Danner, S.; Kauffmann, S.; Hofele, K.; Spooren, W.P.; Ruegg, M.A.; Lin, S.; Caroni, P.; Sommer, B.; Tolnay, M.; Bilbe, G. Neuropathology in mice expressing human α-synuclein. J. Neurosci. 2000, 20, 6021–6029. [Google Scholar] [PubMed]
- Wakamatsu, M.; Ishii, A.; Iwata, S.; Sakagami, J.; Ukai, Y.; Ono, M.; Kanbe, D.; Muramatsu, S-i.; Kabayashi, K.; Iwatsubo, T.; Yoshimoto, M. Selective loss of nigral dopamine neurons induced by overexpression of truncated human α-synuclein. Neurobiol. Aging 2008, 29, 547–585. [Google Scholar]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar]
- Lieberman, A.R. The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int. Rev. Neurobiol. 1971, 14, 49–124. [Google Scholar]
- Poon, H.F.; Frasier, M.; Shreve, N.; Calabrese, V.; Wolozin, B.; Butterfield, D.A. Mitochondrial associated metabolic proteins are selectively oxidized in A30P α-synuclein transgenic mice- a model of familial Parkinson’s disease. Neurobiol. Dis. 2005, 18, 492–498. [Google Scholar]
- Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.; Moore, S.; Davies, Y.; Allsop, D. Alpha-synuclein implicated in Parkinson’s disease catalyses the formation of hydrogen peroxide in vitro. Free Radic. Biol. Med. 2001, 30, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Orth, M.; Wilkinson, J.M.; Taanman, J.W.; Warner, T.T.; Cooper, J.M.; Schapira, A.H. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum. Mol. Genet. 2000, 9, 2683–2689. [Google Scholar]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, VM-Y.; Ischiropoulos, H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, VM-Y.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers. J. Biol. Chem. 2000, 365, 18344–18349. [Google Scholar]
- Chen, K.; Northington, F.J.; Martin, L.J. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 2010, in press.. [Google Scholar]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar]
- Sathasivam, S.; Ince, P.G.; Shaw, P.J. Apoptosis in amyotrophic lateral sclerosis: a review of the evidence. Neuropathol. Appl. Neurobiol. 2001, 27, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Stephens, B.; Guiloff, R.J.; Navarrete, R.; Newman, P.; Nikhar, N.; Lewis, P. Widespread loss of neuronal populations in spinal ventral horn in sporadic motor neuron disease. A morphometric study. J. Neurol. Sci. 2006, 244, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, S.; Al-Sarraj, S.; Kibble, M.; Landau, S.; Parnavelas, J.; Cotter, D.; Everall, I.; Leigh, P.N. Cortical selective vulnerability in motor neurons disease: a morphometric study. Brain 2004, 127, 1237–1251. [Google Scholar]
- Kabashi, E.; Valdmains, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; Pradat, P-F.; Camu, W.; Meininger, V.; Dupre, N.; Rouleau, G.A. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [PubMed]
- Schymick, J.C.; Talbot, K.; Traynor, G.J. Genetics of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2007, 16, R233–R242. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Lander, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; Figlewicz, D.; Brown, R.H.; Meisler, M.H. Deleterious variants of FIG4, a phosphoinositade phosphatase, in patients with ALS. Am. J. Human Gen. 2009, 84, 85–88. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Ultrastructural changes of synapses of Betz cell in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 268, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Ince, P.G.; Shaw, P.J. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem. Intl. 2002, 40, 543–551. [Google Scholar] [CrossRef]
- Comi, G.P.; Bordoni, A.; Salani, S.; Franeschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N.; Moggio, M.; Ausenda, C.D.; Taanman, J.W.; Scarlato, G. Cytochrome c oxidase subunit I microdeletion in a paitent with motor neuron disease. Ann. Neurol. 1998, 43, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, G.M.; Taylo, R.W.; Walls, T.J.; Tonska, K.; Taylor, G.A.; Shaw, P.J.; Ince, P.G.; Turnbull, D.M. Motor neuron disease in a patient with a mitochondrial tRNAIle mutation. Ann. Neurol. 2006, 59, 570–574. [Google Scholar]
- Soong, N.W.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar]
- Mawrin, C.; Kirches, E.; Krause, G.; Wiedemann, F.R.; Vorwerk, C.K.; Bogerts, B.; Schildhaus, H.U.; Dietzmann, K.; Schneider-Stock, R. Single-cell analysis of mtDNA levels in sporadic amyotrophic lateral sclerosis. NeuroReport 2004, 15, 939–943. [Google Scholar]
- Corti, S.; Donadonu, C.; Ronchi, D.; Bordoni, A.; Fortunato, F.; Santoro, D.; Del Bo, R.; Lucchini, V.; Crugnola, V.; Papadimitriou, D.; Salani, S.; Moggio, M.; Bresolin, N.; Comi, G.P. Amyotrophic lateral sclerosis linked to a novel SOD1 mutation with muscle mitochondrial dysfunction. J. Neurol. Sci. 2009, 276, 170–174. [Google Scholar]
- Babcock, D.; Hille, B. Mitochondrial oversight of cellular Ca2+ signaling. Curr. Opin. Neurobiol. 1998, 8, 398–404. [Google Scholar]
- Siklos, L.; Engelhardt, J.; Harat, Y.; Smith, R.G.; Joo, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar]
- Choi, D.W. Cellular defences destroyed. Nature 2005, 433, 696–698. [Google Scholar]
- Brown, M.R.; Sullivan, P.G.; Geddes, J.W. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J. Biol. Chem. 2006, 281, 11658–11668. [Google Scholar]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar]
- Heath, P.R.; Tomkins, J.; Ince, P.G.; Shaw, P.J. Quantitative assessment of AMPA receptor mRNA in human spinal motor neurons isolated by laser capture microdissection. NeuroReport 2002, 13, 1753–1757. [Google Scholar]
- Kwak, S.; Kawahara, Y. Deficient RNA editing of GluR2 and neuronal death in amyotrophic lateral sclerosis. J. Mol. Med. 2005, 83, 110–120. [Google Scholar]
- Chang, D.T.W.; Reynolds, I.J. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog. Brain Res. 2006, 80, 241–268. [Google Scholar]
- Hansson, M.J.; Mansson, R.; Morota, S.; Uchino, H.; Kallur, T.; Sumi, T.; Ishii, N.; Shimazu, M.; Keep, M.F.; Jegorov, A.; Elmer, E. Calcium-induced generation of reactive oxygen species in brain mitochondria is mediated by permeability transition. Free Radic. Biol. Med. 2008, 45, 284–294. [Google Scholar]
- Bergmann, F.; Keller, B.U. Impact of mitochondrial inhibition on excitability and cytosolic Ca2+ levels in brainstem motoneurones. J. Physiol. 2004, 555, 45–59. [Google Scholar]
- Beal, M.F. Oxidatively modified protein in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar]
- Abe, K.; Pan, L-H.; Watanabe, M.; Kato, T.; Itoyama, Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci. Lett. 1995, 199, 152–154. [Google Scholar] [PubMed]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar]
- Sasaki, S.; Warita, H.; Abe, K.; Iwata, M. Inducible nitric oxide synthase (iNOS) and nitrotyrosine immunoreactivity in the spinal cords of transgenic mice with mutant SOD1 gene. J. Neuropathol. Exp. Neurol. 2001, 60, 839–846. [Google Scholar]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H., Jr; Beal, M.F. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 281–287. [Google Scholar] [PubMed]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbul, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Wiedemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123, 1339–1348. [Google Scholar]
- Soraru, G.; Vergani, L.; Fedrizzi, L.; D’Ascenzo, C.; Polo, A.; Bernazzi, B.; Angelini, C. Activities of mitochondrial complexes correlate with nNOS amount in muscle from ALS patients. Neuropath. Appl. Neurobiol. 2007, 33, 204–211. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N’Guessan, B.; Tranchant, C.; Loeffler, J-P.; Lampert, E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar] [PubMed]
- Martin, L.J.; Liu, Z. Opportunities for neuroprotection in ALS using cell death mechanism rationales. Drug Discov. Today 2004, 1, 135–143. [Google Scholar] [CrossRef]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar]
- Yamazaki, M.; Esumi, E.; Nakani, I. Is motoneuronal death in amyotrophic lateral sclerosis apoptosis? Neuropathology 2005, 25, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Liu, Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and Bax-dependent. J. Neurobiol. 2002, 50, 181–197. [Google Scholar]
- Fornai, F.; Longone, P.; Ferrucci, M.; Lenzi, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A. Autophagy and amyotrophic lateral sclerosis. Autophagy 2008, 4, 527–530. [Google Scholar]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar]
- McCord, J.M.; Fridovich, I. Superoxide dismutase, an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Rakhit, R.; Crow, J.P.; Lepock, J.R.; Kondejewski, L.H.; Cashman, N.R.; Chakrabartty, A. Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic sclerosis. J. Biol. Chem. 2004, 279, 15499–15504. [Google Scholar]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar]
- Estévez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, L. ; Barbeito, M.M.; Beckman, J.S. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 1999, 286, 2498–2500. [Google Scholar] [PubMed]
- Flanagan, S.W.; Anderson, R.D.; Ross, M.A.; Oberley, L.W. Overexpression of manganese superoxide dismutase attenuates neuronal death in human cells expressing mutant (G37R) Cu/Zn-superoxide dismutase. J. Neurochem. 2002, 81, 170–177. [Google Scholar]
- Bilsland, L.G.; Nirmalananthan, N.; Yip, J.; Greensmith, L.; Duhcen, M.R. Expression of mutant SOD1G93A in astrocytes induces functional deficits in motoneuron mitochondria. J. Neurochem. 2008, 107, 1271–1283. [Google Scholar]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; Chen, W.; Zhai, P.; Sufit, R.L.; Siddique, T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar]
- Dal Canto, M.C.; Gurney, M.E. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am. J. Pathol. 1994, 145, 1271–1279. [Google Scholar]
- Chang, Q.; Martin, L.J. Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a confocal quantitative analysis. Am. J. Path. 2009, 174, 574–585. [Google Scholar]
- Bendotti, C.; Calvaresi, N.; Chiveri, L.; Prelle, A.; Moggio, M.; Braga, M.; Silani, V.; De Biasi, S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J. Neurol. Sci. 2001, 191, 25–33. [Google Scholar]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [PubMed]
- Jaarsma, D.; Rognoni, F.; van Duijn, W.; Verspaget, H.W.; Haasdijk, E.D.; Holstege, J.C. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001, 102, 293–305. [Google Scholar]
- Sasaki, S.; Warita, H.; Murakami, T.; Abe, K.; Iwata, M. Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2004, 107, 461–474. [Google Scholar]
- Deng, H-X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W-Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; Warner, C.; Deng, G.; Soriano, E.; Smyth, C.; Parge, H.E.; Ahmed, A.; Roses, A.D.; Hallewell, R.A.; Pericak-Vance, M.A.; Siddique, T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [PubMed]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Guarnieri, M.; Xu, Z-S.; Wong, P.C.; Brown, R.H., Jr.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar]
- Yim, M.B.; Kang, J-H.; Yim, H-S.; Kwak, H-S.; Chock, P.B.; Stadtman, E.R. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1996, 93, 5709–5714. [Google Scholar]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Rouleau, G.A. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann. Neurol. 2007, 62, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Ezzi, S.A.; Urushitani, M.; Julien, J-P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. Mutant Cu,Zn superoxide dismutases and familial amyotrophic lateral sclerosis: evaluation of oxidative hypotheses. Free Radic. Biol. Med. 2003, 34, 1383–1389. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 2041–2048. [Google Scholar]
- Poon, H.F.; Hensley, K.; Thongboonkerd, V.; Merchant, M.L.; Lynn, B.C.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Butterfield, D.A. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice- a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005, 39, 435–462. [Google Scholar]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn Superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215:1–RC215:6. [Google Scholar]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide (SOD) in rat liver. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar]
- Liu, J.; Lillo, C.; Jonsson, A.; Vande Velde, C.; Ward, C.W.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Anderson, P.M.; Brannstrom, T.M.; Gredal, O.; Wong, P.C.; Williams, D.S.; Cleveland, D.W. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–15. [Google Scholar]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant protein bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar]
- Goldsteins, G.; Keksa-Goldsteine, V.; Ahtiniemi, T.; Jaronen, M.; Arens, E.; Akerman, K.; Chan, R.H.; Koistinaho, J. Deleterious role of superoxide dismutase in the mitochondrial intermembrane space. J. Biol. Chem. 2008, 283, 8446–8452. [Google Scholar]
- Higgins, C.M.; Jung, C.; Xu, Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K-F.; Browlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; Shaw, C.E.; Leigh, P.N.; Miller, C.C. J.; Grierson, A.J. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondrial content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [PubMed]
- Siklos, L.; Engelhardt, J.I.; Alexianu, M.E.; Gurney, M.E.; Siddique, T.; Appel, S.H. Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J. Neuropath. Exp. Neurol. 1998, 57, 571–587. [Google Scholar]
- Jaiswal, M.K.; Keller, B.U. Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 2009, 75, 478–489. [Google Scholar]
- Nguyen, K.T.; Garcia-Chacon, L.E.; Barrett, J.N.; Barrett, E.F.; David, G. The ψm depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc. Natl. Acad. Sci. USA 2009, 106, 2007–2011. [Google Scholar]
- Sasaki, S.; Shibata, N.; Komori, T.; Iwata, M. iNOS and nitrotyrosine immunoreactivity in amyotrophic lateral sclerosis. Neurosci. Lett. 2000, 291, 44–48. [Google Scholar]
- Kunz, W.S. Different metabolic properties of mitochondrial oxidative phosphorylation in different cell types- important implications for mitochondrial cytopathies. Exp. Physiol. 2003, 88.1, 149–154. [Google Scholar] [CrossRef]
- Keep, M.; Elmér, E.; Fong, K.S.K.; Csiszar, K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001, 894, 327–331. [Google Scholar]
- Karlsson, J.; Fong, K.S.; Hansson, M.J.; Elmer, E.; Csiszar, K.; Keep, M.F. Life span extension and reduced neuronal death after weekly intraventricular cyclosporine injections in the G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosurg. 2004, 101, 128–137. [Google Scholar]
- Kirkinezos, I.G.; Hernandez, D.; Bradley, W.G.; Moraes, C.T. An ALS mouse model with a permeable blood-brain barrier benefits from systemic cyclosporine A treatment. J. Neurochem. 2004, 88, 821–826. [Google Scholar]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; lacapere, JJ-J.; Massaad, C.; Schmacher, M.; Steidl, E-M.; Maux, D.; Delaage, M.; Henderson, C.E.; Pruss, R.M. Identification and characterization of Cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [PubMed]
- Mills, C.; Makwana, M.; Wallace, A.; Benn, S.; Schmidt, H.; Tegeder, I.; Costigan, M.; Brown, R.H., Jr; Raivich, G.; Woolf, C. Ro5-4864 promotes neonatal motor neuron survival and nerve regeneration in adult rats. Eur. J. Neurosci. 2008, 27, 937–946. [Google Scholar]
- Yan, L-J.; Sohal, R.S. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl. Acad. Sci. USA 1998, 95, 12896–12901. [Google Scholar]
- Prokai, L.; Yan, L-J; Vera-Serrano, J.L.; Stevens, S.M., Jr; Forster, M.J. Mass spectrometry-based survey of age-associated protein carbonylation in rat brain mitochondria. J. Mass Spectrom. 2007, 42, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Vieira, H.L.A.; Belzacq, A-S.; Haouzu, D.; Bernassola, F.; Cohen, I.; Jacotot, E.; Ferri, K.F.; Hamel, C.E.; Bartle, L.M.; Melino, G.; Brenner, C.; Goldmacher, V.; Kroemer, G. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene 2001, 20, 4305–4316. [Google Scholar] [PubMed]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548. [Google Scholar]
- Costantini, P.; Belzacq, A-S.; Vieira, H.L.A.; Larochette, N.; de Pablo, M.A.; Zamzami, N.; Susin, S.A.; Brenner, C.; Kroemer, G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 2000, 19, 307–314. [Google Scholar] [PubMed]
- García, N.; Martínez-Abundis, E.; Pavón, N.; Correa, F.; Chávez, E. Copper induces permeability transition through its interaction with the adenine nucleotide translocase. Cell Biol. Int. 2007, 31, 893–899. [Google Scholar]
- Grimm, S.; Brdiczka, D. The permeability transition pore in cell death. Apoptosis 2007, 12, 841–855. [Google Scholar]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, Z.; Fowlkes, J.; Rahder, M.; Stern, K.; Bernardi, P.; Bourdette, D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hertz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martin, L.J. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals 2010, 3, 839-915. https://doi.org/10.3390/ph3040839
Martin LJ. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals. 2010; 3(4):839-915. https://doi.org/10.3390/ph3040839
Chicago/Turabian StyleMartin, Lee J. 2010. "Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases" Pharmaceuticals 3, no. 4: 839-915. https://doi.org/10.3390/ph3040839
APA StyleMartin, L. J. (2010). Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals, 3(4), 839-915. https://doi.org/10.3390/ph3040839