Obesity Drug Update: The Lost Decade?

Abstract
:1. Introduction
2. Anti-Obesity Drug Targets in the 1990s

{kind=link}
| Drugs available in 2000 | New anti-obesity drug targets since the discovery of leptin | Drugs available in 2010 |
|---|---|---|
| Orlistat | Leptin | Orlistat |
| Sibutramine | Leptin receptors | |
| MC4 receptor | ||
| NPY receptors | ||
| PYY receptor | ||
| 5-HT2c receptor | ||
| AgRP binding | ||
| MCH | ||
| Orexins | ||
| Incretins | ||
| Ghrelin | ||
| Endocannabinoid receptors |
3. The Discovery of Leptin and the Hypothalamic Circuit for Energy Homeostasis
3.1. Mouse Genetics Lead the Way
3.2. The Melanocortinergic System
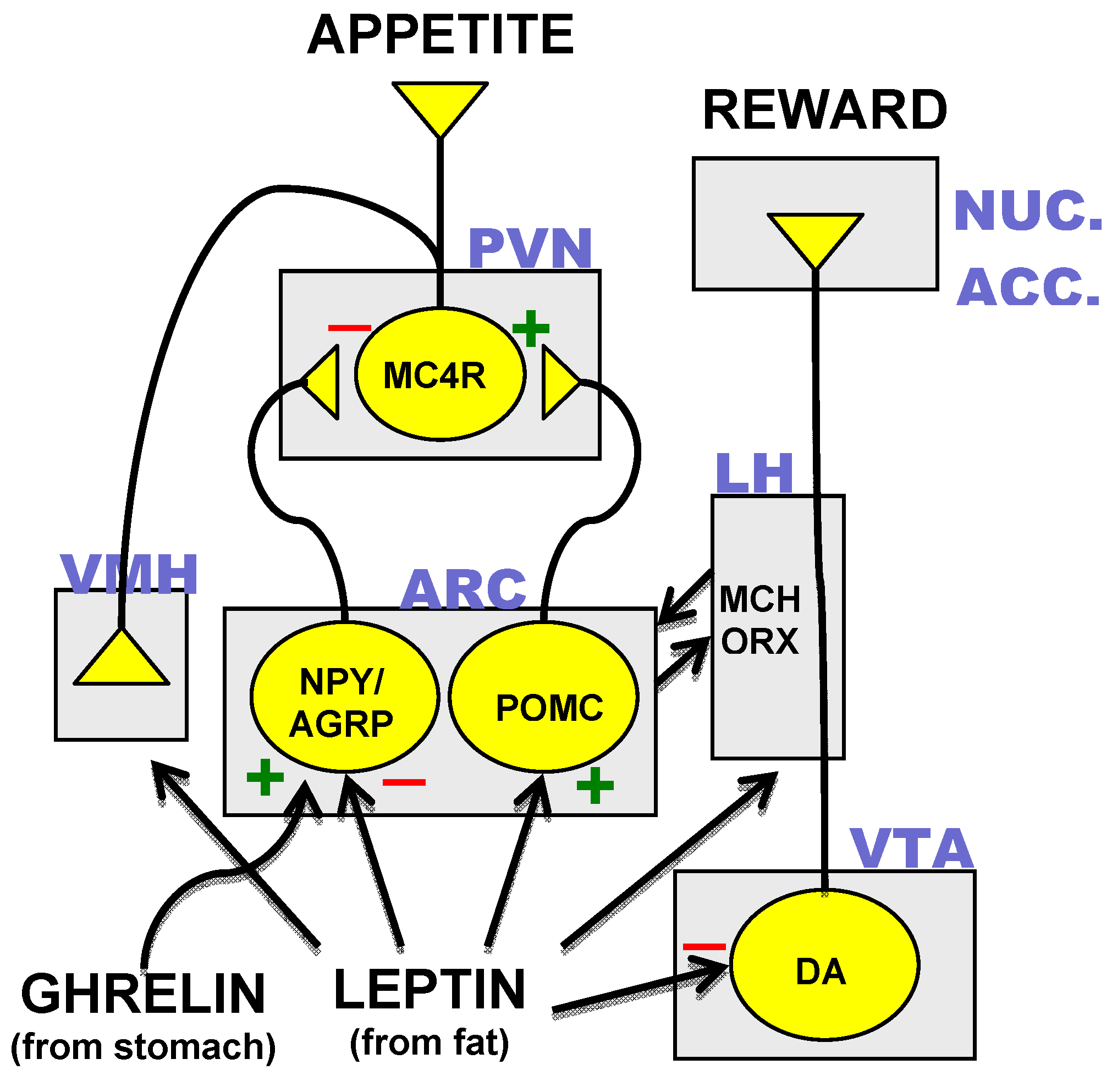
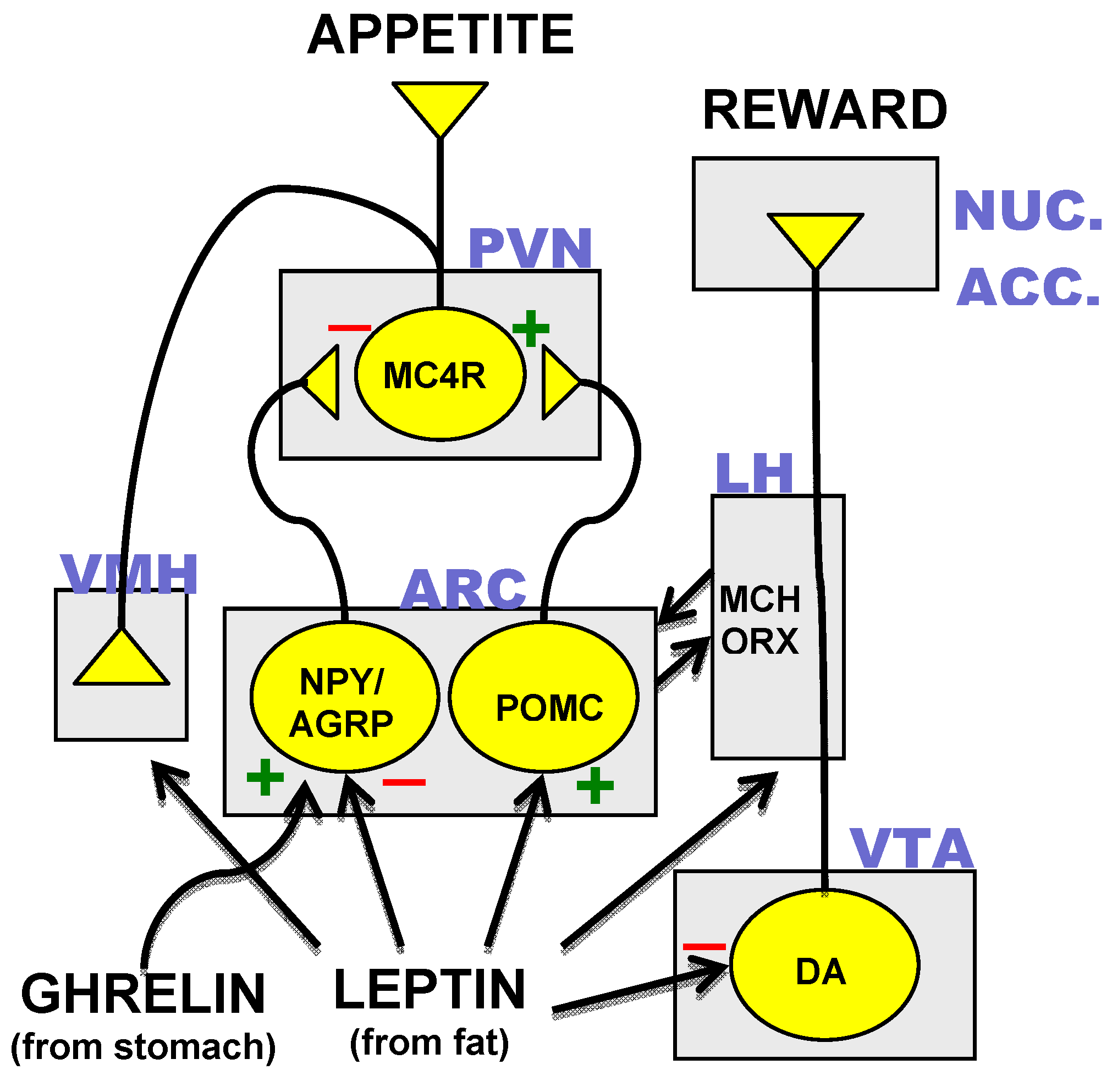
3.3. Hormonal Inputs to the Hypothalamic Circuit
4. CNS Anti-Obesity Drug Targets Since the Discovery of Leptin
4.1. Leptin
4.2. NPY and AgRP
4.3. MC4R
4.4. 5-HT
4.5. Melanin Concentrating Hormone (MCH) and Orexins
4.6. Ghrelin
4.7. Incretins
4.8. Endocannabinoids
4.9. New Combination Therapies
5. Peripheral Anti-Obesity Drug Targets Since the Discovery of Leptin
5.1. Pancreatic Lipase
5.2. 11β-Hydroxysteroid Type 1
5.3. Human Brown Adipose Tissue (BAT)
5.4. Targets That Could Limit Consequences of Obesity
6. Concluding Remarks
Acknowledgements
References
- Puska, P.; Nishida, C.; Porter, D. World Health Organization Report: Obesity and Overweight. Available online: http://www.who.int/dietphysicalactivity/media/en/gsfs_obesity.pdf/ accessed on 24 November 2010.
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell. Biol. 2008, 9, 367–377. [Google Scholar]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar]
- Karmali, S.; Johnson, S.C.; Sharma, A.; Stadnyk, J.; Christiansen, S.; Cottreau, D.; Birch, D.W. Bariatric surgery: a primer. Can. Fam. Physici. 2010, 56, 873–879. [Google Scholar]
- McEwen, L.N.; Coelho, R.B.; Baumann, L.M.; Bilik, D.; Nota-Kirby, B.; Herman, W.H. The cost, quality of life impact, and cost-utility of bariatric surgery in a managed care population. Obes. Surg. 2010, 20, 919–928. [Google Scholar]
- FDA Draft Guidance for Industry Developing Products for Weight Management. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071612.pdf/ accessed date 24 November 2010.
- EMA Guideline on Clinical Evaluation of Medicinal Products Used in Weight Control. 2007. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003264.pdf/ accessed on 24 November 2010.
- Heal, D.J.; Gosden, J.; Smith, S.L. Regulatory challenges for new drugs to treat obesity and comorbid metabolic disorders. Br. J. Clin. Pharmacol. 2009, 68, 861–874. [Google Scholar]
- Kennett, G.A.; Clifton, P.G. New approaches to the pharmacological treatment of obesity: Can they break through the efficacy barrier? Pharmacol. Biochem. Behav. 2010, 97, 63–83. [Google Scholar]
- Weintraub, M.; Sundaresan, P.R.; Schuster, B.; Averbuch, M.; Stein, E.C.; Cox, C.; Byrne, L. Long-term weight control study. IV (weeks 156 to 190). The second double-blind phase. Clin. Pharmacol. Ther. 1992, 51, 608–614. [Google Scholar]
- Greenway, F.L.; Bray, G.A. Combination drugs for treating obesity. Curr. Diab. Rep. 2010, 10, 108–115. [Google Scholar]
- Connolly, H.M.; Crary, J.L.; McGoon, M.D.; Hensrud, D.D.; Edwards, B.S.; Edwards, W.D.; Schaff, H.V. Valvular heart disease associated with fenfluramine-phentermine. N. Engl. J. Med. 1997, 337, 581–588. [Google Scholar]
- Lipworth, B.J. Clinical pharmacology of beta 3-adrenoceptors. Br. J. Clin. Pharmacol. 1996, 42, 291–300. [Google Scholar]
- de Souza, C.J.; Burkey, B.F. Beta 3-adrenoceptor agonists as anti-diabetic and anti-obesity drugs in humans. Curr. Pharm. Des. 2001, 7, 1433–1449. [Google Scholar]
- Ursino, M.G.; Vasina, V.; Raschi, E.; Crema, F.; De Ponti, F. The beta3-adrenoceptor as a therapeutic target: current perspectives. Pharmacol. Res. 2009, 59, 221–234. [Google Scholar]
- Frankish, H.M.; Dryden, S.; Hopkins, D.; Wang, Q.; Williams, G. Neuropeptide Y, the hypothalamus, and diabetes: insights into the central control of metabolism. Peptides 1995, 16, 757–771. [Google Scholar]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; Muir, C.; Sanker, S.; Moriarty, A.; Moore, K.J.; Smutko, J.S.; Mays, G.G.; Wool, E.A.; Monroe, C.A.; Tepper, R.I. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar]
- Barsh, G.S.; Schwartz, M.W. Genetic approaches to studying energy balance: perception and integration. Nat. Rev. Genet. 2002, 3, 589–600. [Google Scholar]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar]
- Sanchez-Lasheras, C.; Konner, A.C.; Bruning, J.C. Integrative neurobiology of energy homeostasis-neurocircuits, signals and mediators. Front. Neuroendocrinol. 2010, 31, 4–15. [Google Scholar]
- Echwald, S.M. Genetics of human obesity: lessons from mouse models and candidate genes. J. Intern. Med. 1999, 245, 653–666. [Google Scholar]
- O'Rahilly, S.; Farooqi, I.S.; Yeo, G.S.; Challis, B.G. Minireview: human obesity-lessons from monogenic disorders. Endocrinology 2003, 144, 3757–3764. [Google Scholar]
- Seeley, R.J.; Drazen, D.L.; Clegg, D.J. The critical role of the melanocortin system in the control of energy balance. Annu. Rev. Nutr. 2004, 24, 133–149. [Google Scholar]
- Barsh, G. From Agouti to Pomc--100 years of fat blonde mice. Nat. Med. 1999, 5, 984–985. [Google Scholar]
- Gantz, I.; Miwa, H.; Konda, Y.; Shimoto, Y.; Tashiro, T.; Watson, S.J.; DelValle, J.; Yamada, T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J. Biol. Chem. 1993, 268, 15174–15179. [Google Scholar]
- Gantz, I.; Konda, Y.; Tashiro, T.; Shimoto, Y.; Miwa, H.; Munzert, G.; Watson, S.J.; DelValle, J.; Yamada, T. Molecular cloning of a novel melanocortin receptor. J. Biol. Chem. 1993, 268, 8246–8250. [Google Scholar]
- Yang, Y.K.; Thompson, D.A.; Dickinson, C.J.; Wilken, J.; Barsh, G.S.; Kent, S.B.; Gantz, I. Characterization of Agouti-related protein binding to melanocortin receptors. Mol. Endocrinol. 1999, 13, 148–155. [Google Scholar]
- Huszar, D.; Lynch, C.A.; Fairchild-Huntress, V.; Dunmore, J.H.; Fang, Q.; Berkemeier, L.R.; Gu, W.; Kesterson, R.A.; Boston, B.A.; Cone, R.D.; Smith, F.J.; Campfield, L.A.; Burn, P.; Lee, F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997, 88, 131–141. [Google Scholar]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in virto and in vivo by agouti-related protein. Science 1997, 278, 135–138. [Google Scholar]
- Shutter, J.R.; Graham, M.; Kinsey, A.C.; Scully, S.; Luthy, R.; Stark, K.L. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes Dev. 1997, 11, 593–602. [Google Scholar]
- Reizes, O.; Lincecum, J.; Wang, Z.; Goldberger, O.; Huang, L.; Kaksonen, M.; Ahima, R.; Hinkes, M.T.; Barsh, G.S.; Rauvala, H.; Bernfield, M. Transgenic expression of syndecan-1 uncovers a physiological control of feeding behavior by syndecan-3. Cell 2001, 106, 105–116. [Google Scholar]
- Creemers, J.W.; Pritchard, L.E.; Gyte, A.; Le Rouzic, P.; Meulemans, S.; Wardlaw, S.L.; Zhu, X.; Steiner, D.F.; Davies, N.; Armstrong, D.; Lawrence, C.B.; Luckman, S.M.; Schmitz, C.A.; Davies, R.A.; Brennand, J.C.; White, A. Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83-132: interaction between AGRP83-132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology 2006, 147, 1621–1631. [Google Scholar]
- Hahn, T.M.; Breininger, J.F.; Baskin, D.G.; Schwartz, M.W. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat. Neurosci. 1998, 1, 271–272. [Google Scholar]
- Fan, W.; Dinulescu, D.M.; Butler, A.A.; Zhou, J.; Marks, D.L.; Cone, R.D. The central melanocortin system can directly regulate serum insulin levels. Endocrinology 2000, 141, 3072–3079. [Google Scholar]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Gruters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar]
- Yaswen, L.; Diehl, N.; Brennan, M.B.; Hochgeschwender, U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med. 1999, 5, 1066–1070. [Google Scholar]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; Christiansen, L.M.; Edelstein, E.; Choi, B.; Boss, O.; Aschkenasi, C.; Zhang, C.Y.; Mountjoy, K.; Kishi, T.; Elmquist, J.K.; Lowell, B.B. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar]
- Baskin, D.G.; Hahn, T.M.; Schwartz, M.W. Leptin sensitive neurons in the hypothalamus. Horm. Metab. Res. 1999, 31, 345–350. [Google Scholar]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar]
- Cowley, M.A.; Cone, R.D.; Enriori, P.; Louiselle, I.; Williams, S.M.; Evans, A.E. Electrophysiological actions of peripheral hormones on melanocortin neurons. Ann. NY Acad. Sci. 2003, 994, 175–186. [Google Scholar]
- Marsh, D.J.; Hollopeter, G.; Huszar, D.; Laufer, R.; Yagaloff, K.A.; Fisher, S.L.; Burn, P.; Palmiter, R.D. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat. Genet. 1999, 21, 119–122. [Google Scholar]
- van de Wall, E.; Leshan, R.; Xu, A.W.; Balthasar, N.; Coppari, R.; Liu, S.M.; Jo, Y.H.; MacKenzie, R.G.; Allison, D.B.; Dun, N.J.; Elmquist, J.; Lowell, B.B.; Barsh, G.S.; de Luca, C.; Myers, M.G., Jr.; Schwartz, G.J.; Chua, S.C., Jr. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 2008, 149, 1773–1785. [Google Scholar]
- Davis, J.F.; Choi, D.L.; Benoit, S.C. Insulin, leptin and reward. Trends Endocrinol. Metab. 2010, 21, 68–74. [Google Scholar]
- Fry, M.; Ferguson, A.V. Ghrelin: central nervous system sites of action in regulation of energy balance. Int. J. Pept. 2010, 2010. [Google Scholar]
- Cowley, M.A.; Grove, K.L. Ghrelin--satisfying a hunger for the mechanism. Endocrinology 2004, 145, 2604–2606. [Google Scholar]
- Chen, H.Y.; Trumbauer, M.E.; Chen, A.S.; Weingarth, D.T.; Adams, J.R.; Frazier, E.G.; Shen, Z.; Marsh, D.J.; Feighner, S.D.; Guan, X.M.; Ye, Z.; Nargund, R.P.; Smith, R.G.; Van der Ploeg, L.H.; Howard, A.D.; MacNeil, D.J.; Qian, S. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology 2004, 145, 2607–2612. [Google Scholar]
- Feldkircher, K.M.; Mistry, A.M.; Romsos, D.R. Adrenalectomy reverses pre-existing obesity in adult genetically obese (ob/ob) mice. Int. J. Obes. Relat. Metab. Disord. 1996, 20, 232–235. [Google Scholar]
- Gyengesi, E.; Liu, Z.W.; D'Agostino, G.; Gan, G.; Horvath, T.L.; Gao, X.B.; Diano, S. Corticosterone regulates synaptic input organization of POMC and NPY/AgRP neurons in adult mice. Endocrinology 2010, 151, 5395–5402. [Google Scholar]
- Uchoa, E.T.; Mendes da Silva, L.E.; de Castro, M.; Antunes-Rodrigues, J.; Elias, L.L. Hypothalamic oxytocin neurons modulate hypophagic effect induced by adrenalectomy. Horm. Behav. 2009, 56, 532–538. [Google Scholar]
- Uchoa, E.T.; Sabino, H.A.; Ruginsk, S.G.; Antunes-Rodrigues, J.; Elias, L.L. Hypophagia induced by glucocorticoid deficiency is associated with an increased activation of satiety-related responses. J. Appl. Physiol. 2009, 106, 596–604. [Google Scholar]
- Dallman, M.F. Stress-induced obesity and the emotional nervous system. Trends Endocrinol. Metab. 2010, 21, 159–165. [Google Scholar]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjorbaek, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Invest. 2000, 105, 1827–1832. [Google Scholar]
- Thaler, J.P.; Choi, S.J.; Schwartz, M.W.; Wisse, B.E. Hypothalamic inflammation and energy homeostasis: resolving the paradox. Front. Neuroendocrinol. 2010, 31, 79–84. [Google Scholar]
- Kleinridders, A.; Schenten, D.; Konner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Bruning, J.C. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009, 10, 249–259. [Google Scholar]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS One 2010, 5, e11376. [Google Scholar]
- Trevaskis, J.L.; Parkes, D.G.; Roth, J.D. Insights into amylin-leptin synergy. Trends Endocrinol. Metab. 2010, 21, 473–479. [Google Scholar]
- Ravussin, E.; Smith, S.R.; Mitchell, J.A.; Shringarpure, R.; Shan, K.; Maier, H.; Koda, J.E.; Weyer, C. Enhanced weight loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity (Silver Spring) 2009, 17, 1736–1743. [Google Scholar]
- Takeda press release. Amylin and Takeda Announce Decision to Advance Development of Pramlintide/Metreleptin Combination Treatment for Obesity. Available online: http://www.takeda.com/press/article_35851.html/ accecced on 24 November 2010.
- Lantz, K.A.; Hart, S.G.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity (Silver Spring) 2010, 18, 1516–1523. [Google Scholar]
- Wortley, K.E.; Anderson, K.D.; Yasenchak, J.; Murphy, A.; Valenzuela, D.; Diano, S.; Yancopoulos, G.D.; Wiegand, S.J.; Sleeman, M.W. Agouti-related protein-deficient mice display an age-related lean phenotype. Cell Metab. 2005, 2, 421–427. [Google Scholar]
- Palmiter, R.D.; Erickson, J.C.; Hollopeter, G.; Baraban, S.C.; Schwartz, M.W. Life without neuropeptide Y. Recent Prog. Horm. Res. 1998, 53, 163–199. [Google Scholar]
- Flier, J.S. AgRP in energy balance: Will the real AgRP please stand up? Cell Metab 2006, 3, 83–85. [Google Scholar]
- Dumont, Y.; Martel, J.C.; Fournier, A.; St-Pierre, S.; Quirion, R. Neuropeptide Y and neuropeptide Y receptor subtypes in brain and peripheral tissues. Prog. Neurobiol. 1992, 38, 125–167. [Google Scholar]
- Sato, N.; Ogino, Y.; Mashiko, S.; Ando, M. Modulation of neuropeptide Y receptors for the treatment of obesity. Expert Opin. Ther. Pat. 2009, 19, 1401–1415. [Google Scholar]
- MacNeil, D.J. NPY Y1 and Y5 receptor selective antagonists as anti-obesity drugs. Curr. Top. Med. Chem. 2007, 7, 1721–1733. [Google Scholar]
- Kamiji, M.M.; Inui, A. Neuropeptide y receptor selective ligands in the treatment of obesity. Endocr. Rev. 2007, 28, 664–684. [Google Scholar]
- Kuo, L.E.; Kitlinska, J.B.; Tilan, J.U.; Li, L.; Baker, S.B.; Johnson, M.D.; Lee, E.W.; Burnett, M.S.; Fricke, S.T.; Kvetnansky, R.; Herzog, H.; Zukowska, Z. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 2007, 13, 803–811. [Google Scholar]
- Erondu, N.; Wadden, T.; Gantz, I.; Musser, B.; Nguyen, A.M.; Bays, H.; Bray, G.; O'Neil, P.M.; Basdevant, A.; Kaufman, K.D.; Heymsfield, S.B.; Amatruda, J.M. Effect of NPY5R antagonist MK-0557 on weight regain after very-low-calorie diet-induced weight loss. Obesity (Silver Spring) 2007, 15, 895–905. [Google Scholar]
- Matsuyama, K. Obesity Becomes Shionogi’s Gamble for Another Crestor (Update3). Bloomberg online 2009. [Google Scholar]
- Shionogi press release. Shionogi announces positive top-line efficacy results from year-long studies of velneperit, a novel NPY Y5 receptor antagonist being investigated for the treatment of obesity. 2009. Available online: http://www.shionogi.co.jp/ir_en/news/detail/e_090217-2.pdf/ Accessed on 24 November 2010.
- Safety Study of the Inhibition of Agouti-related Protein (AgRP) for the Management of Obesity and Weight Loss. Available online: http://clinicaltrials.gov/ct2/show/record/NCT00779519/ accessed on 24 November 2010.
- Tao, Y.X. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr. Rev. 2010, 31, 506–543. [Google Scholar]
- MacKenzie, R.G. Obesity-associated mutations in the human melanocortin-4 receptor gene. Peptides 2006, 27, 395–403. [Google Scholar]
- Farooqi, I.S.; Keogh, J.M.; Yeo, G.S.; Lank, E.J.; Cheetham, T.; O'Rahilly, S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 2003, 348, 1085–1095. [Google Scholar]
- Santini, F.; Maffei, M.; Pelosini, C.; Salvetti, G.; Scartabelli, G.; Pinchera, A. Melanocortin-4 receptor mutations in obesity. Adv. Clin. Chem. 2009, 48, 95–109. [Google Scholar]
- Aslan, I.R.; Ranadive, S.A.; Ersoy, B.A.; Rogers, S.J.; Lustig, R.H.; Vaisse, C. Bariatric surgery in a patient with complete MC4R deficiency. Int. J. Obes. (Lond) 2010. [Google Scholar]
- Raposinho, P.D.; Castillo, E.; d'Alleves, V.; Broqua, P.; Pralong, F.P.; Aubert, M.L. Chronic blockade of the melanocortin 4 receptor subtype leads to obesity independently of neuropeptide Y action, with no adverse effects on the gonadotropic and somatotropic axes. Endocrinology 2000, 141, 4419–4427. [Google Scholar]
- Lu, X.Y.; Nicholson, J.R.; Akil, H.; Watson, S.J. Time course of short-term and long-term orexigenic effects of Agouti-related protein (86-132). Neuroreport 2001, 12, 1281–1284. [Google Scholar]
- Chen, A.S.; Marsh, D.J.; Trumbauer, M.E.; Frazier, E.G.; Guan, X.M.; Yu, H.; Rosenblum, C.I.; Vongs, A.; Feng, Y.; Cao, L.; Metzger, J.M.; Strack, A.M.; Camacho, R.E.; Mellin, T.N.; Nunes, C.N.; Min, W.; Fisher, J.; Gopal-Truter, S.; MacIntyre, D.E.; Chen, H.Y.; Van der Ploeg, L.H. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat. Genet. 2000, 26, 97–102. [Google Scholar]
- Calton, M.A.; Ersoy, B.A.; Zhang, S.; Kane, J.P.; Malloy, M.J.; Pullinger, C.R.; Bromberg, Y.; Pennacchio, L.A.; Dent, R.; McPherson, R.; Ahituv, N.; Vaisse, C. Association of functionally significant Melanocortin-4 but not Melanocortin-3 receptor mutations with severe adult obesity in a large North American case-control study. Hum. Mol. Genet. 2009, 18, 1140–1147. [Google Scholar]
- Tao, Y.X. Chapter 6 mutations in Melanocortin-4 receptor and human obesity. Prog. Mol. Biol. Transl. Sci. 2009, 88C, 173–204. [Google Scholar]
- Tan, K.; Pogozheva, I.D.; Yeo, G.S.; Hadaschik, D.; Keogh, J.M.; Haskell-Leuvano, C.; O'Rahilly, S.; Mosberg, H.I.; Farooqi, I.S. Functional characterization and structural modeling of obesity associated mutations in the melanocortin 4 receptor. Endocrinology 2009, 150, 114–125. [Google Scholar]
- Xiang, Z.; Proneth, B.; Dirain, M.L.; Litherland, S.A.; Haskell-Luevano, C. Pharmacological characterization of 30 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists, synthetic agonists, and the endogenous agouti-related protein antagonist. Biochemistry 2010, 49, 4583–4600. [Google Scholar]
- Xiang, Z.; Pogozheva, I.D.; Sorenson, N.B.; Wilczynski, A.M.; Holder, J.R.; Litherland, S.A.; Millard, W.J.; Mosberg, H.I.; Haskell-Luevano, C. Peptide and small molecules rescue the functional activity and agonist potency of dysfunctional human melanocortin-4 receptor polymorphisms. Biochemistry 2007, 46, 8273–8287. [Google Scholar]
- Granell, S.; Mohammad, S.; Ramanagoudr-Bhojappa, R.; Baldini, G. Obesity-linked variants of melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can be rescued to the cell surface by a chemical chaperone. Mol. Endocrinol. 2010, 24, 1805–1821. [Google Scholar]
- Fan, Z.C.; Tao, Y.X. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J. Cell. Mol. Med. 2009, 13, 3268–3282. [Google Scholar]
- Sayk, F.; Heutling, D.; Dodt, C.; Iwen, K.A.; Wellhoner, J.P.; Scherag, S.; Hinney, A.; Hebebrand, J.; Lehnert, H. Sympathetic function in human carriers of melanocortin-4 receptor gene mutations. J. Clin. Endocrinol. Metab. 2010, 95, 1998–2002. [Google Scholar]
- Greenfield, J.R.; Miller, J.W.; Keogh, J.M.; Henning, E.; Satterwhite, J.H.; Cameron, G.S.; Astruc, B.; Mayer, J.P.; Brage, S.; See, T.C.; Lomas, D.J.; O'Rahilly, S.; Farooqi, I.S. Modulation of blood pressure by central melanocortinergic pathways. N. Engl. J. Med. 2009, 360, 44–52. [Google Scholar]
- Corander, M.P.; Fenech, M.; Coll, A.P. Science of self-preservation: how melanocortin action in the brain modulates body weight, blood pressure, and ischemic damage. Circulation 2009, 120, 2260–2268. [Google Scholar]
- Maier, T.; Hoyer, J. Modulation of blood pressure by central melanocortinergic pathways. Nephrol. Dial. Transplant. 2010, 25, 674–677. [Google Scholar]
- King, S.H.; Mayorov, A.V.; Balse-Srinivasan, P.; Hruby, V.J.; Vanderah, T.W.; Wessells, H. Melanocortin receptors, melanotropic peptides and penile erection. Curr. Top. Med. Chem. 2007, 7, 1098–1106. [Google Scholar]
- Palatin Technologies, Inc. Press Release. Palatin Technologies, Inc. Announces Positive Safety Results in Subcutaneous Bremelanotide Tiarl in Men. 2010. Available online: http://www.palatin.com/news/news.asp?param=248.
- Walker, W.P.; Gunn, T.M. Piecing together the pigment-type switching puzzle. Pigment Cell Melanoma Res. 2010, 23, 4–6. [Google Scholar]
- Perez-Oliva, A.B.; Olivares, C.; Jimenez-Cervantes, C.; Garcia-Borron, J.C. Mahogunin ring finger-1 (MGRN1) E3 ubiquitin ligase inhibits signaling from melanocortin receptor by competition with Galphas. J. Biol. Chem. 2009, 284, 31714–31725. [Google Scholar]
- Kaelin, C.B.; Candille, S.I.; Yu, B.; Jackson, P.; Thompson, D.A.; Nix, M.A.; Binkley, J.; Millhauser, G.L.; Barsh, G.S. New ligands for melanocortin receptors. Int. J. Obes. (Lond) 2008, 32 Suppl. 7, S19–S27. [Google Scholar]
- MacKenzie, R.G. Signaling in obesity neurons. Curr. Med. Chem. 2004, 4, 113–117. [Google Scholar]
- Ryan, D.H. Use of sibutramine and other noradrenergic and serotonergic drugs in the management of obesity. Endocrine 2000, 13, 193–199. [Google Scholar]
- Williams, G. Withdrawal of sibutramine in Europe. BMJ 2010, 340, c824. [Google Scholar]
- FDA Safety Alert. Meridia (sibutramine hydrochloride): Follow-Up to an Early Communication about an Ongoing Safety Review. Available online: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm198221.htm/ accessed on 24 November 2010.
- James, W.P.T.; Caterson, I.D.; Coutinho, W.; Finer, N.; Van Gaal, L.F.; Maggioni, A.P.; Torp-Pedersen, C.; Sharma, A.M.; Shepherd, G.M.; Rode, R.A.; Renz, C.L. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. NEJM 2010, 363, 905–917. [Google Scholar]
- Abbott Press Release. Abbott to Voluntarily Withdraw Meridia® (Sibutramine) in the U.S. 2010. Available online: http://www.abbott.com/global/url/pressRelease/en_US/60.5:5/Press_Release_0908.htm.
- Astrup, A.; Madsbad, S.; Breum, L.; Jensen, T.J.; Kroustrup, J.P.; Larsen, T.M. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1906–1913. [Google Scholar]
- Xu, Y.; Jones, J.E.; Kohno, D.; Williams, K.W.; Lee, C.E.; Choi, M.J.; Anderson, J.G.; Heisler, L.K.; Zigman, J.M.; Lowell, B.B.; Elmquist, J.K. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron 2008, 60, 582–589. [Google Scholar]
- Heisler, L.K.; Jobst, E.E.; Sutton, G.M.; Zhou, L.; Borok, E.; Thornton-Jones, Z.; Liu, H.Y.; Zigman, J.M.; Balthasar, N.; Kishi, T.; Lee, C.E.; Aschkenasi, C.J.; Zhang, C.Y.; Yu, J.; Boss, O.; Mountjoy, K.G.; Clifton, P.G.; Lowell, B.B.; Friedman, J.M.; Horvath, T.; Butler, A.A.; Elmquist, J.K.; Cowley, M.A. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron 2006, 51, 239–249. [Google Scholar]
- Smith, S.R.; Weissman, N.J.; Anderson, C.M.; Sanchez, M.; Chuang, E.; Stubbe, S.; Bays, H.; Shanahan, W.R. Multicenter, placebo-controlled trial of lorcaserin for weight management. N. Engl. J. Med. 2010, 363, 245–256. [Google Scholar]
- Pollack, A. Panel urges denial of diet drug. New York Times 2010. [Google Scholar]
- Griffond, B.; Risold, P.Y. MCH and feeding behavior-interaction with peptidic network. Peptides 2009, 30, 2045–2051. [Google Scholar]
- Tsujino, N.; Sakurai, T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol. Rev. 2009, 61, 162–176. [Google Scholar]
- Ito, M.; Ishihara, A.; Gomori, A.; Matsushita, H.; Metzger, J.M.; Marsh, D.J.; Haga, Y.; Iwaasa, H.; Tokita, S.; Takenaga, N.; Sato, N.; MacNeil, D.J.; Moriya, M.; Kanatani, A. Mechanism of the anti-obesity effects induced by a novel melanin-concentrating hormone 1-receptor antagonist in mice. Br. J. Pharmacol. 2010, 159, 374–383. [Google Scholar]
- AMRI press release. AMRI Announces Phase I Study of Novel Drug for Obesity Treatment. Available online: http://www.amriglobal.com/img/document_files/np072110.pdf/ accessed on 24 November 2010.
- Actelion press release. Almorexant in development for insomnia. Available online: http://www1.actelion.com/en/scientists/development-pipeline/phase-3/almorexant.page/ accessed on 24 November 2010.
- Cox, C.D.; Breslin, M.J.; Whitman, D.B.; Schreier, J.D.; McGaughey, G.B.; Bogusky, M.J.; Roecker, A.J.; Mercer, S.P.; Bednar, R.A.; Lemaire, W.; Bruno, J.G.; Reiss, D.R.; Harrell, C.M.; Murphy, K.L.; Garson, S.L.; Doran, S.M.; Prueksaritanont, T.; Anderson, W.B.; Tang, C.; Roller, S.; Cabalu, T.D.; Cui, D.; Hartman, G.D.; Young, S.D.; Koblan, K.S.; Winrow, C.J.; Renger, J.J.; Coleman, P.J. Discovery of the dual orexin receptor antagonist [(7R)-4-(5-chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]-methanone (MK-4305) for the treatment of insomnia. J. Med. Chem. 2010, 53, 5320–5332. [Google Scholar]
- Coleman, P.J.; Renger, J.J. Orexin receptor antagonists: a review of promising compounds patented since 2006. Expert Opin. Ther. Pat. 2010, 20, 307–324. [Google Scholar]
- Bingham, M.J.; Cai, J.; Deehan, M.R. Eating, sleeping and rewarding: orexin receptors and their antagonists. Curr. Opin. Drug Discov. Devel. 2006, 9, 551–559. [Google Scholar]
- Chung, S.; Hopf, F.W.; Nagasaki, H.; Li, C.Y.; Belluzzi, J.D.; Bonci, A.; Civelli, O. The melanin-concentrating hormone system modulates cocaine reward. Proc. Natl. Acad. Sci. USA 2009, 106, 6772–6777. [Google Scholar]
- Sears, R.M.; Liu, R.J.; Narayanan, N.S.; Sharf, R.; Yeckel, M.F.; Laubach, M.; Aghajanian, G.K.; DiLeone, R.J. Regulation of nucleus accumbens activity by the hypothalamic neuropeptide melanin-concentrating hormone. J. Neurosci. 2010, 30, 8263–8273. [Google Scholar]
- Sharf, R.; Sarhan, M.; Dileone, R.J. Role of orexin/hypocretin in dependence and addiction. Brain Res. 2010, 1314, 130–138. [Google Scholar]
- Winrow, C.J.; Tanis, K.Q.; Reiss, D.R.; Rigby, A.M.; Uslaner, J.M.; Uebele, V.N.; Doran, S.M.; Fox, S.V.; Garson, S.L.; Gotter, A.L.; Levine, D.M.; Roecker, A.J.; Coleman, P.J.; Koblan, K.S.; Renger, J.J. Orexin receptor antagonism prevents transcriptional and behavioral plasticity resulting from stimulant exposure. Neuropharmacology 2010, 58, 185–194. [Google Scholar]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar]
- Tschop, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar]
- Dickson, S.L.; Luckman, S.M. Induction of c-fos messenger ribonucleic acid in neuropeptide Y and growth hormone (GH)-releasing factor neurons in the rat arcuate nucleus following systemic injection of the GH secretagogue, GH-releasing peptide-6. Endocrinology 1997, 138, 771–777. [Google Scholar]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschop, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; Garcia-Segura, L.M.; Nillni, E.A.; Mendez, P.; Low, M.J.; Sotonyi, P.; Friedman, J.M.; Liu, H.; Pinto, S.; Colmers, W.F.; Cone, R.D.; Horvath, T.L. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661. [Google Scholar]
- Willesen, M.G.; Kristensen, P.; Romer, J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 1999, 70, 306–316. [Google Scholar]
- Cummings, D.E.; Purnell, J.Q.; Frayo, R.S.; Schmidova, K.; Wisse, B.E.; Weigle, D.S. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 2001, 50, 1714–1719. [Google Scholar]
- Kojima, M.; Kangawa, K. Ghrelin: more than endogenous growth hormone secretagogue. Ann. NY Acad. Sci. 2010, 1200, 140–148. [Google Scholar]
- Pournaras, D.J.; le Roux, C.W. Ghrelin and metabolic surgery. Int. J. Pept. 2010, 2010. [Google Scholar]
- Sun, Y.; Ahmed, S.; Smith, R.G. Deletion of ghrelin impairs neither growth nor appetite. Mol. Cell. Biol. 2003, 23, 7973–7981. [Google Scholar]
- Wortley, K.E.; Anderson, K.D.; Garcia, K.; Murray, J.D.; Malinova, L.; Liu, R.; Moncrieffe, M.; Thabet, K.; Cox, H.J.; Yancopoulos, G.D.; Wiegand, S.J.; Sleeman, M.W. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc. Natl. Acad. Sci. USA 2004, 101, 8227–8232. [Google Scholar]
- Longo, K.A.; Charoenthongtrakul, S.; Giuliana, D.J.; Govek, E.K.; McDonagh, T.; Qi, Y.; DiStefano, P.S.; Geddes, B.J. Improved insulin sensitivity and metabolic flexibility in ghrelin receptor knockout mice. Regul. Pept. 2008, 150, 55–61. [Google Scholar]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar]
- Hayes, M.R.; De Jonghe, B.C.; Kanoski, S.E. Role of the glucagon-like-peptide-1 receptor in the control of energy balance. Physiol. Behav. 2010, 100, 503–510. [Google Scholar]
- Edholm, T.; Degerblad, M.; Gryback, P.; Hilsted, L.; Holst, J.J.; Jacobsson, H.; Efendic, S.; Schmidt, P.T.; Hellstrom, P.M. Differential incretin effects of GIP and GLP-1 on gastric emptying, appetite, and insulin-glucose homeostasis. Neurogastroenterol Motil 2010.
- Ma, X.; Bruning, J.; Ashcroft, F.M. Glucagon-like peptide 1 stimulates hypothalamic proopiomelanocortin neurons. J. Neurosci. 2007, 27, 7125–7129. [Google Scholar]
- Sandoval, D.A.; Bagnol, D.; Woods, S.C.; D'Alessio, D.A.; Seeley, R.J. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 2008, 57, 2046–2054. [Google Scholar]
- Williams, D.L.; Baskin, D.G.; Schwartz, M.W. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 2009, 150, 1680–1687. [Google Scholar]
- Holst, J.J.; Vilsboll, T.; Deacon, C.F. The incretin system and its role in type 2 diabetes mellitus. Mol. Cell. Endocrinol. 2009, 297, 127–136. [Google Scholar]
- Kruger, D.F.; Bode, B.; Spollett, G.R. Understanding GLP-1 analogs and enhancing patients success. Diabetes Educ. 2010, 36 Suppl. 3, 44S–72S, quiz 73S-74S.. [Google Scholar]
- Irwin, N.; Flatt, P.R. Therapeutic potential for GIP receptor agonists and antagonists. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 499–512. [Google Scholar]
- Kim, S.J.; Nian, C.; McIntosh, C.H. Glucose-dependent insulinotropic polypeptide (GIP) increases human adipocyte lipoprotein lipase (LPL) expression through cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2 (TORC2) mediated trans-activation of the LPL gene. J. Lipid. Res. 2010. [Google Scholar]
- Beckman, L.M.; Beckman, T.R.; Earthman, C.P. Changes in gastrointestinal hormones and leptin after Roux-en-Y gastric bypass procedure: a review. J. Am. Diet. Assoc. 2010, 110, 571–584. [Google Scholar]
- Thomas, S.; Schauer, P. Bariatric surgery and the gut hormone response. Nutr. Clin. Pract. 2010, 25, 175–182. [Google Scholar]
- Bose, M.; Teixeira, J.; Olivan, B.; Bawa, B.; Arias, S.; Machineni, S.; Pi-Sunyer, F.X.; Scherer, P.E.; Laferrere, B. Weight loss and incretin responsiveness improve glucose control independently after gastric bypass surgery. J. Diabetes 2009, 2, 47–55. [Google Scholar]
- Laferrere, B.; Swerdlow, N.; Bawa, B.; Arias, S.; Bose, M.; Olivan, B.; Teixeira, J.; McGinty, J.; Rother, K.I. Rise of oxyntomodulin in response to oral glucose after gastric bypass surgery in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2010, 95, 4072–4076. [Google Scholar]
- Suzuki, K.; Simpson, K.A.; Minnion, J.S.; Shillito, J.C.; Bloom, S.R. The role of gut hormones and the hypothalamus in appetite regulation. Endocr. J. 2010, 57, 359–372. [Google Scholar]
- Andre, A.; Gonthier, M.P. The endocannabinoid system: Its roles in energy balance and potential as a target for obesity treatment. Int. J. Biochem. Cell. Biol. 2010, 42, 1788–1801. [Google Scholar]
- Maccarrone, M.; Gasperi, V.; Catani, M.V.; Diep, T.A.; Dainese, E.; Hansen, H.S.; Avigliano, L. The endocannabinoid system and its relevance for nutrition. Annu. Rev. Nutr. 2010, 30, 423–440. [Google Scholar]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Batkai, S.; Jarai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; Kunos, G. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar]
- Jamshidi, N.; Taylor, D.A. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br. J. Pharmacol. 2001, 134, 1151–1154. [Google Scholar]
- Kirkham, T.C.; Williams, C.M.; Fezza, F.; Di Marzo, V. Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br. J. Pharmacol. 2002, 136, 550–557. [Google Scholar]
- Verty, A.N.; McGregor, I.S.; Mallet, P.E. Paraventricular hypothalamic CB(1) cannabinoid receptors are involved in the feeding stimulatory effects of Delta(9)-tetrahydrocannabinol. Neuropharmacology 2005, 49, 1101–1109. [Google Scholar]
- Jelsing, J.; Larsen, P.J.; Vrang, N. Identification of cannabinoid type 1 receptor expressing cocaine amphetamine-regulated transcript neurons in the rat hypothalamus and brainstem using in situ hybridization and immunohistochemistry. Neuroscience 2008, 154, 641–652. [Google Scholar]
- Ravinet, T.C.; Delgorge, C.; Menet, C.; Arnone, M.; Soubrie, P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 640–648. [Google Scholar]
- Cota, D.; Sandoval, D.A.; Olivieri, M.; Prodi, E.; D'Alessio, D.A.; Woods, S.C.; Seeley, R.J.; Obici, S. Food intake-independent effects of CB1 antagonism on glucose and lipid metabolism. Obesity (Silver Spring) 2009, 17, 1641–1645. [Google Scholar]
- Nogueiras, R.; Veyrat-Durebex, C.; Suchanek, P.M.; Klein, M.; Tschop, J.; Caldwell, C.; Woods, S.C.; Wittmann, G.; Watanabe, M.; Liposits, Z.; Fekete, C.; Reizes, O.; Rohner-Jeanrenaud, F.; Tschop, M.H. Peripheral, but not central, CB1 antagonism provides food intake-independent metabolic benefits in diet-induced obese rats. Diabetes 2008, 57, 2977–2991. [Google Scholar]
- Bermudez-Silva, F.J.; Viveros, M.P.; McPartland, J.M.; Rodriguez de Fonseca, F. The endocannabinoid system, eating behavior and energy homeostasis: the end or a new beginning? Pharmacol. Biochem. Behav. 2010, 95, 375–382. [Google Scholar]
- FDA Briefing Document. NDA 21-888. 2007. Available online: http://www.fda.gov/ohrms/dockets/ac/07/briefing/2007-4306b1-fda-backgrounder.pdf/ Accessed on 24 November 2010.
- European Medicines Agency Press Release. The European Medicines Agency recommends suspension of the marketing authorisation of Acomplia. 2008. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2009/11/WC500014774.pdf/ Accessed on 24 November 2010.
- Di Marzo, V.; Despres, J.P. CB1 antagonists for obesity--what lessons have we learned from rimonabant? Nat. Rev. Endocrinol. 2009, 5, 633–638. [Google Scholar]
- Di Marzo, V.; Ligresti, A.; Cristino, L. The endocannabinoid system as a link between homoeostatic and hedonic pathways involved in energy balance regulation. Int. J. Obes. (Lond) 2009, 33 Suppl. 2, S18–S24. [Google Scholar]
- Dodd, G.T.; Stark, J.A.; McKie, S.; Williams, S.R.; Luckman, S.M. Central cannabinoid signaling mediating food intake: a pharmacological-challenge magnetic resonance imaging and functional histology study in rat. Neuroscience 2009, 163, 1192–1200. [Google Scholar]
- Fattore, L.; Melis, M.; Fadda, P.; Pistis, M.; Fratta, W. The endocannabinoid system and nondrug rewarding behaviours. Exp. Neurol. 2010, 224, 23–36. [Google Scholar]
- Receveur, J.M.; Murray, A.; Linget, J.M.; Norregaard, P.K.; Cooper, M.; Bjurling, E.; Nielsen, P.A.; Hogberg, T. Conversion of 4-cyanomethyl-pyrazole-3-carboxamides into CB1 antagonists with lowered propensity to pass the blood-brain-barrier. Bioorg. Med. Chem. Lett. 2010, 20, 453–457. [Google Scholar]
- Tam, J.; Vemuri, V.K.; Liu, J.; Batkai, S.; Mukhopadhyay, B.; Godlewski, G.; Osei-Hyiaman, D.; Ohnuma, S.; Ambudkar, S.V.; Pickel, J.; Makriyannis, A.; Kunos, G. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Invest. 2010, 120, 2953–2966. [Google Scholar]
- Berridge, K.C.; Ho, C.Y.; Richard, J.M.; DiFeliceantonio, A.G. The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res. 2010, 1350, 43–64. [Google Scholar]
- Cota, D.; Tschop, M.H.; Horvath, T.L.; Levine, A.S. Cannabinoids, opioids and eating behavior: the molecular face of hedonism? Brain Res. Rev. 2006, 51, 85–107. [Google Scholar]
- Kirkham, T. Endocannabinoids and the neurochemistry of gluttony. J. Neuroendocrinol. 2008, 20, 1099–1100. [Google Scholar]
- Pollack, A. Panel votes against obesity drug. New York Times 2010. [Google Scholar]
- Greenway, F.L.; Whitehouse, M.J.; Guttadauria, M.; Anderson, J.W.; Atkinson, R.L.; Fujioka, K.; Gadde, K.M.; Gupta, A.K.; O'Neil, P.; Schumacher, D.; Smith, D.; Dunayevich, E.; Tollefson, G.D.; Weber, E.; Cowley, M.A. Rational design of a combination medication for the treatment of obesity. Obesity (Silver Spring) 2009, 17, 30–39. [Google Scholar]
- Sinnayah, P.; Wallingford, N.M.; Evans, A.E.; Cowley, M.A. Bupropion and naltrexone interact synergistically to decrease food intake in mice. In North American Association for the Study of Obesity. Annual Scientific Meeting, New Orleans, LA, USA, 2007.
- Orexigen press release. Orexigen(R) Therapeutics Phase 2b Trial for Empatic(TM) Meets Primary Efficacy Endpoint Demonstrating Significantly Greater Weight Loss Versus Comparators in Obese Patients. 2010. Available online: http://ir.orexigen.com/phoenix.zhtml?c=207034&p=irol-newsArticle&ID=1336796&highlight=.
- Fujioka, K.; Rubino, D.; Wyatt, H.; Apovian, C.; Greenway, F.; Hu, J.; Kim, D.; Dunayevich, E.; Landbloom, R. A Phase 2B, 24-Week Study Evaluating the Efficacy and Safety of Two Doses of Zonisamide SR/Bupropion SR Combination Therapy in Overweight and Obese Subjects. In ADA Annual Meeting. Abstract. # 1841-P; 2010. [Google Scholar]
- Enna, S.J.; Williams, M. Challenges in the search for drugs to treat central nervous system disorders. J. Pharmacol. Exp. Ther. 2009, 329, 404–411. [Google Scholar]
- Reichel, A. Addressing central nervous system (CNS) penetration in drug discovery: basics and implications of the evolving new concept. Chem. Biodivers. 2009, 6, 2030–2049. [Google Scholar]
- Powell, T.M.; Khera, A. Therapeutic approaches to obesity. Curr. Treat. Options. Cardiovasc. Med. 2010, 12, 381–395. [Google Scholar]
- Johansson, K.; Neovius, K.; DeSantis, S.M.; Rossner, S.; Neovius, M. Discontinuation due to adverse events in randomized trials of orlistat, sibutramine and rimonabant: a meta-analysis. Obes. Rev. 2009, 10, 564–575. [Google Scholar]
- FDA Drug Safety Communication: Completed Safety Review of Xenical/Alli (orlistat) and Severe Liver Injury. Available online: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationfor-PatientsandProviders/ucm213038.htm/ accessed on 24 November 2010.
- Norgine BV Website. Product Pipeline. 2010. Available online: http://www.norgine.com/pages/norgine_development/product_pipeline_products.html.
- Morton, N.M.; Seckl, J.R. 11beta-hydroxysteroid dehydrogenase type 1 and obesity. Front.Horm. Res. 2008, 36, 146–164. [Google Scholar]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar]
- Hughes, K.A.; Webster, S.P.; Walker, B.R. 11-Beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitors in type 2 diabetes mellitus and obesity. Expert Opin. Investig. Drugs 2008, 17, 481–496. [Google Scholar]
- Tseng, Y.H.; Cypess, A.M.; Kahn, C.R. Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug Discov. 2010, 9, 465–482. [Google Scholar]
- Barbe, P.; Millet, L.; Galitzky, J.; Lafontan, M.; Berlan, M. In situ assessment of the role of the beta 1-, beta 2- and beta 3-adrenoceptors in the control of lipolysis and nutritive blood flow in human subcutaneous adipose tissue. Br. J. Pharmacol. 1996, 117, 907–913. [Google Scholar]
- Deng, C.; Paoloni-Giacobino, A.; Kuehne, F.; Boss, O.; Revelli, J.P.; Moinat, M.; Cawthorne, M.A.; Muzzin, P.; Giacobino, J.P. Respective degree of expression of beta 1-, beta 2- and beta 3-adrenoceptors in human brown and white adipose tissues. Br. J. Pharmacol. 1996, 118, 929–934. [Google Scholar]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; Kolodny, G.M.; Kahn, C.R. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar]
- Enerback, S. Human brown adipose tissue. Cell. Metab. 2010, 11, 248–252. [Google Scholar]
- Lidell, M.E.; Enerback, S. Brown adipose tissue--a new role in humans? Nat. Rev. Endocrinol. 2010, 6, 319–325. [Google Scholar]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; Nuutila, P. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar]
- Phillips, S.A.; Kung, J.T. Mechanisms of adiponectin regulation and use as a pharmacological target. Curr. Opin. Pharmacol. 2010, 10, 676–683. [Google Scholar]
- Ziemke, F.; Mantzoros, C.S. Adiponectin in insulin resistance: lessons from translational research. Am. J. Clin. Nutr. 2010, 91, 258S–261S. [Google Scholar]
- Canto, C.; Auwerx, J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell. Mol. Life Sci. 2010, 67, 3407–3423. [Google Scholar]
- Goodyear, L.J. The exercise pill--too good to be true? N. Engl. J. Med. 2008, 359, 1842–1844. [Google Scholar]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; Kang, H.; Shaw, R.J.; Evans, R.M. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar]
- Yessoufou, A.; Wahli, W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels. Swiss Med. Wkly 2010, 140, w13071. [Google Scholar]
- Silva, J.P.; Wahlestedt, C. Role of Sirtuin 1 in metabolic regulation. Drug Discov. Today 2010, 15, 781–791. [Google Scholar]
- Oh da, Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar]
- Kepez, A.; Oto, A.; Dagdelen, S. Peroxisome proliferator-activated receptor-gamma: novel therapeutic target linking adiposity, insulin resistance, and atherosclerosis. BioDrugs 2006, 20, 121–135. [Google Scholar]
- Knouff, C.; Auwerx, J. Peroxisome proliferator-activated receptor-gamma calls for activation in moderation: lessons from genetics and pharmacology. Endocr. Rev. 2004, 25, 899–918. [Google Scholar]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell. Metab. 2005, 1, 133–144. [Google Scholar]
- FDA News Release. FDA Significantly Restricts Access to the Diabetes Drug AVANDIA. Available online: http://www.fda.gov/newsevents/newsroom/pressannouncements/UCM226975.htm/ accessed on 24 November 2010.
- EMA press release. European Medicines Agency Recommends Suspension of Avandia, Avandamet and Avaglim. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2010/09/news_detail_001119.jsp&murl=menus/news_and_events/news_and_events.jsp&mid=WC0b01ac058004d5c1/ accessed on 24 November 2010.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yao, F.; MacKenzie, R.G. Obesity Drug Update: The Lost Decade? Pharmaceuticals 2010, 3, 3494-3521. https://doi.org/10.3390/ph3123494
Yao F, MacKenzie RG. Obesity Drug Update: The Lost Decade? Pharmaceuticals. 2010; 3(12):3494-3521. https://doi.org/10.3390/ph3123494
Chicago/Turabian StyleYao, Fayi, and Robert George MacKenzie. 2010. "Obesity Drug Update: The Lost Decade?" Pharmaceuticals 3, no. 12: 3494-3521. https://doi.org/10.3390/ph3123494
APA StyleYao, F., & MacKenzie, R. G. (2010). Obesity Drug Update: The Lost Decade? Pharmaceuticals, 3(12), 3494-3521. https://doi.org/10.3390/ph3123494