Multiple Routes to Oestrogen Antagonism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Chemicals and general molecular methods
2.2. Cell culture, transfection and luciferase assay
2.3. RNA isolation and real-time RT-PCR (qRT-PCR)

3. Results and Discussion
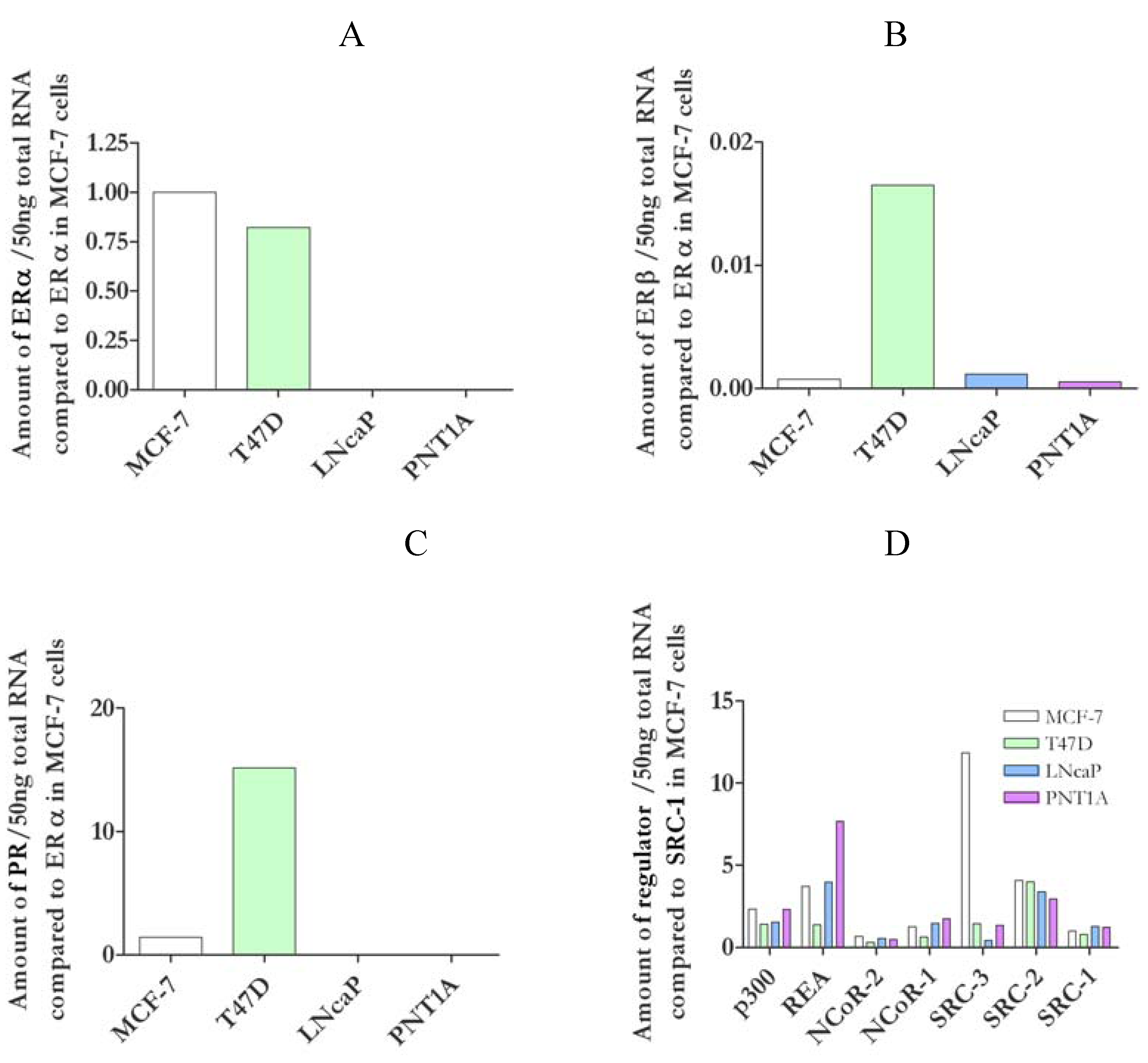
3.1. Characterisation of nuclear receptors and coregulators in cell lines
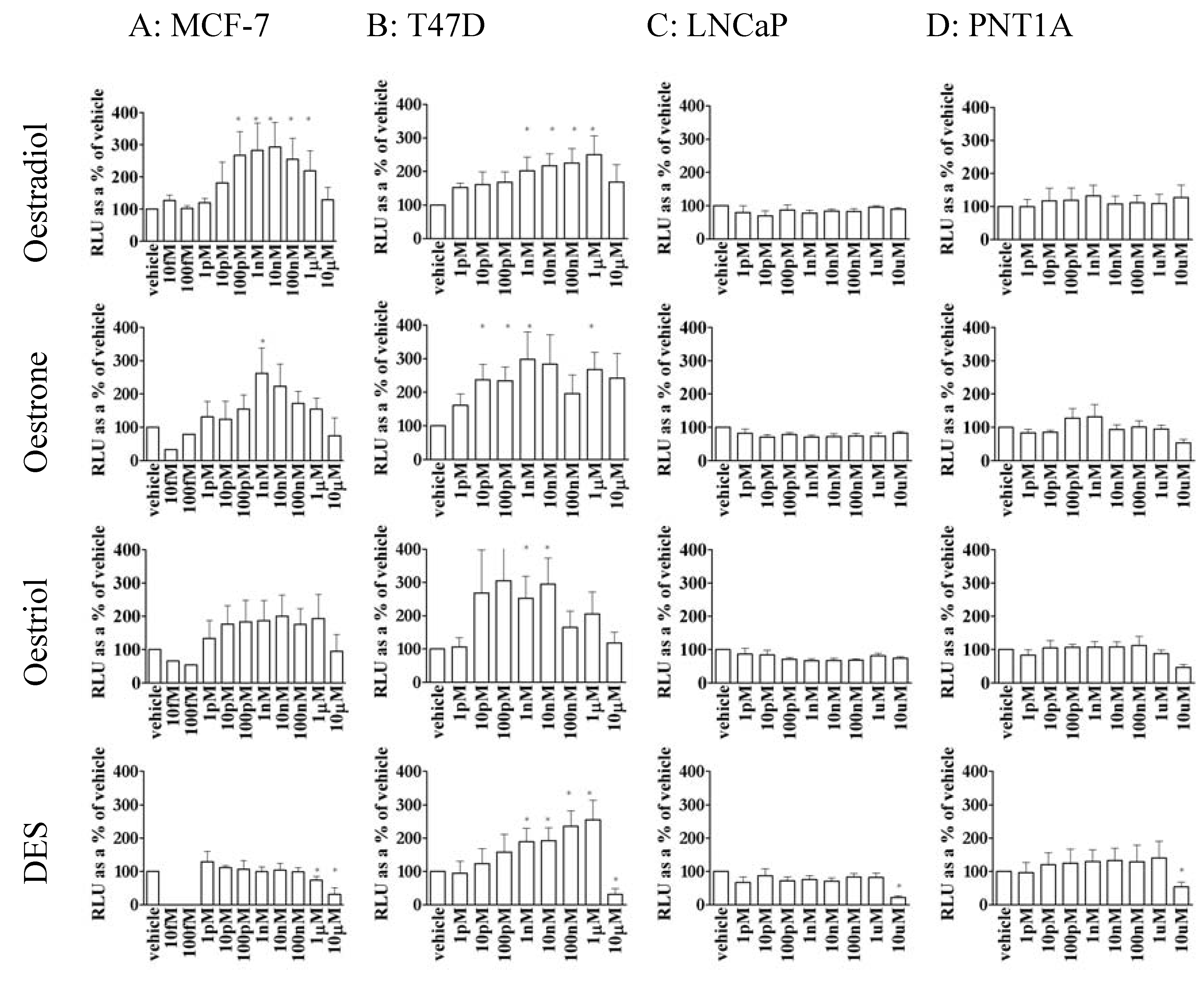
3.2. Oestrogen stimulated transcription was dependent upon cell type
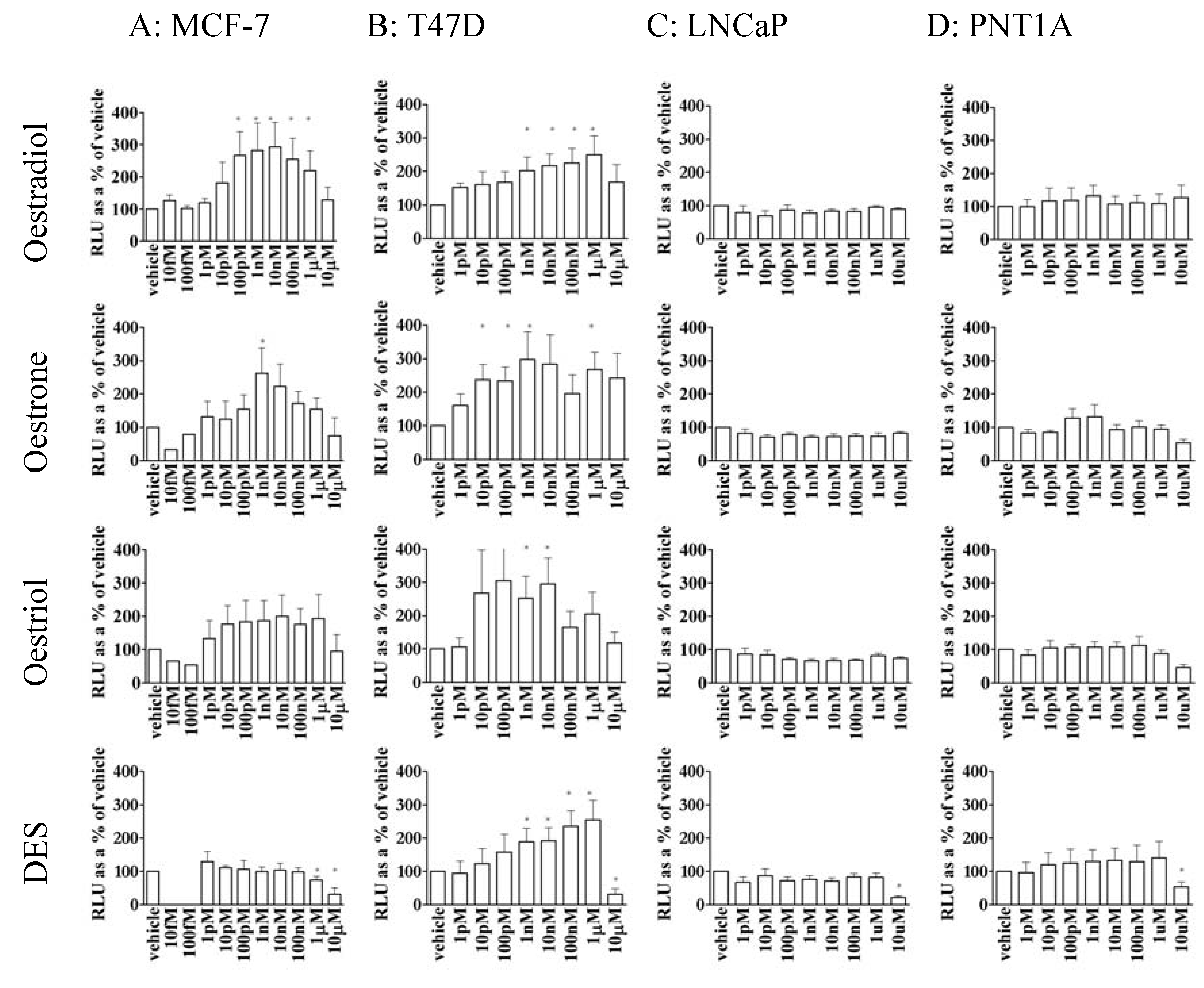
3.3. Oestrone, oestriol and DES were able to stimulate gene transcription in MCF-7 and T47D cells but not prostate cell lines
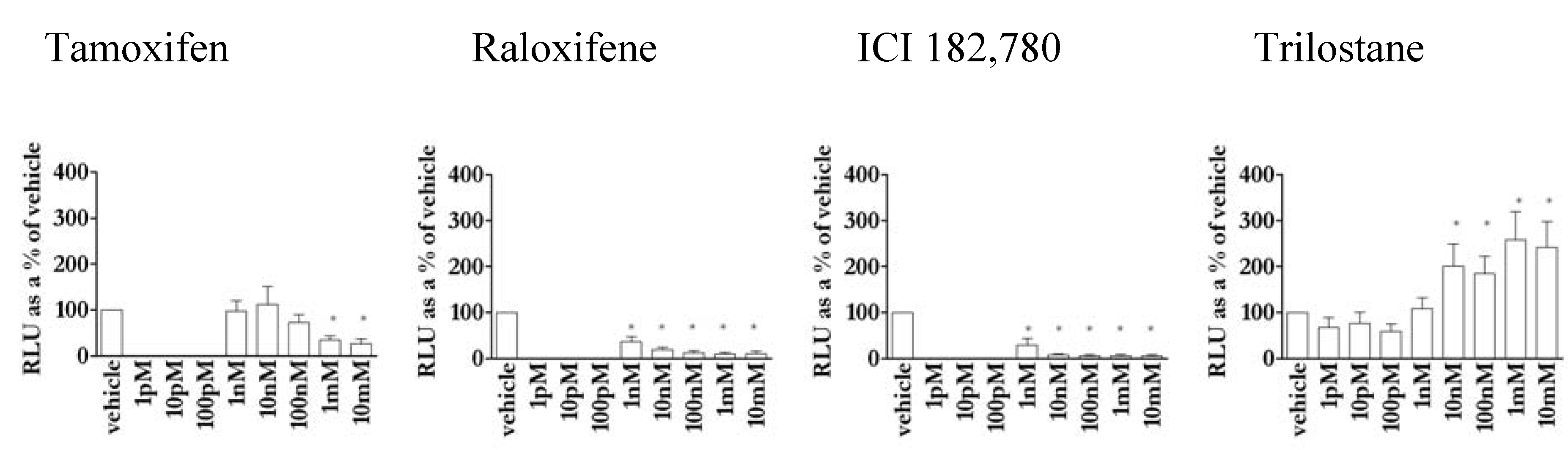
3.4. Except for trilostane, antioestrogens did not stimulate gene transcription in the absence of E2
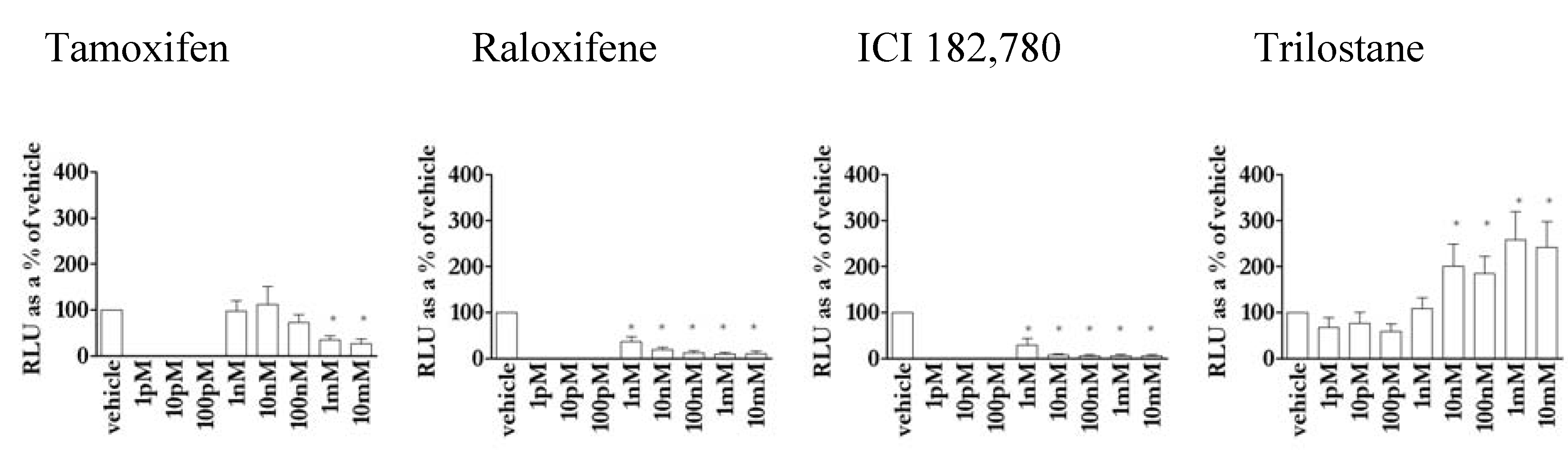
3.5. The action of antioestrogens was dependent on agonist in MCF-7 cells
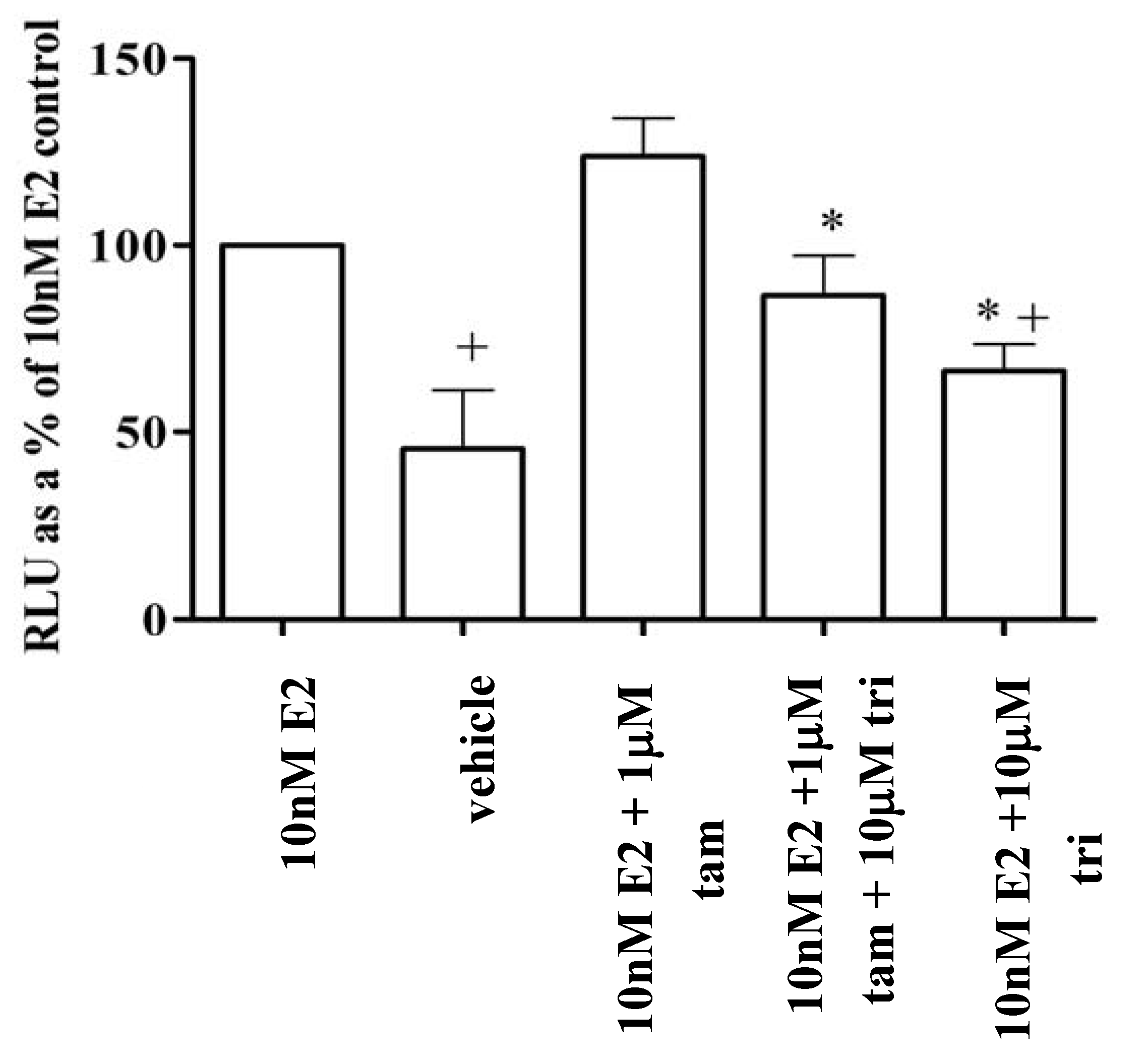
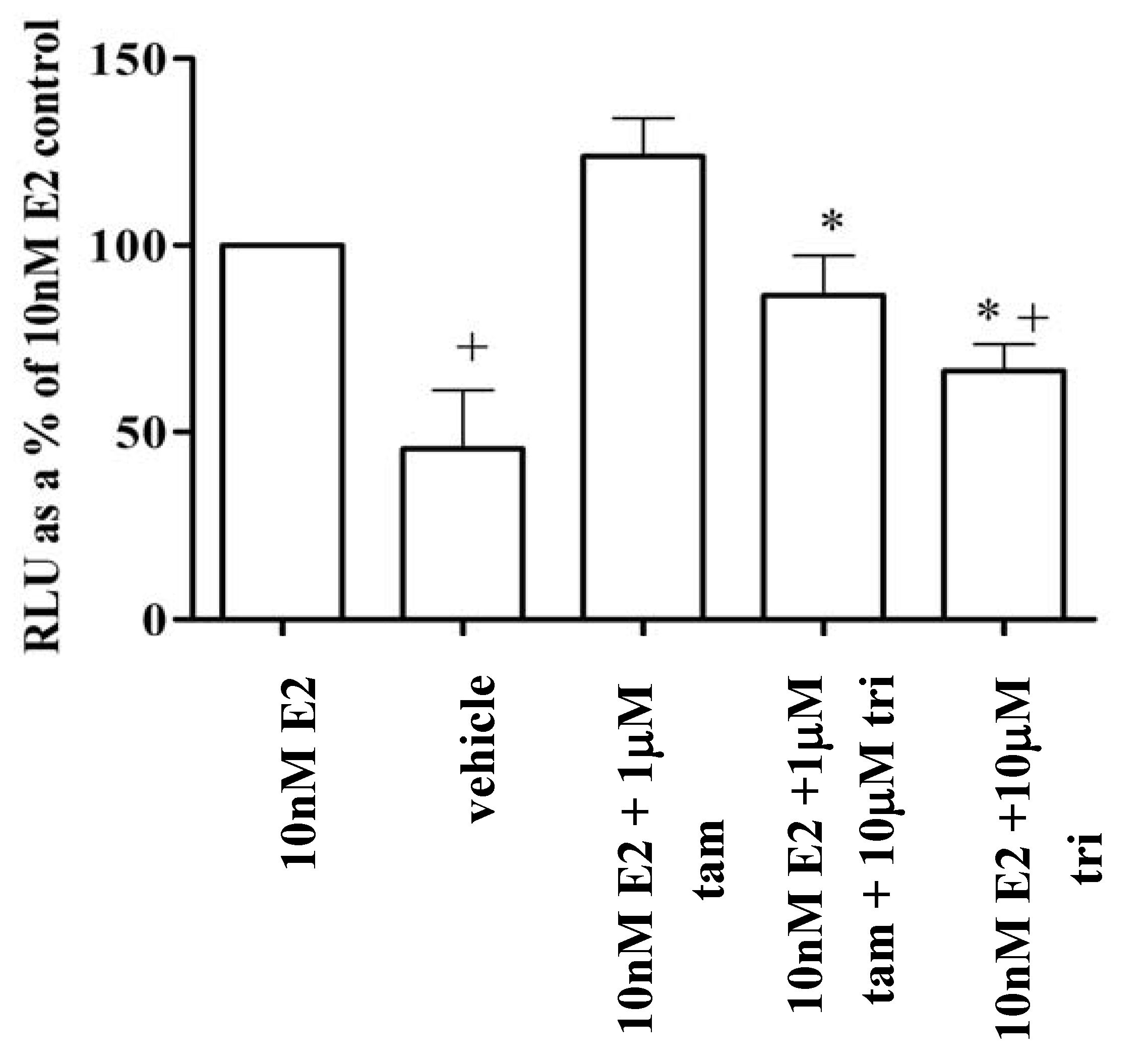
3.6. Trilostane reduced the partial agonist properties of tamoxifen in MCF-7 cells
3.7. Oestrogenic activity is related to ERα, not ERβ

3.8. Novel activity of the antioestrogen trilostane
3.9. Model of a regulatory allosteric ligand binding site on ER
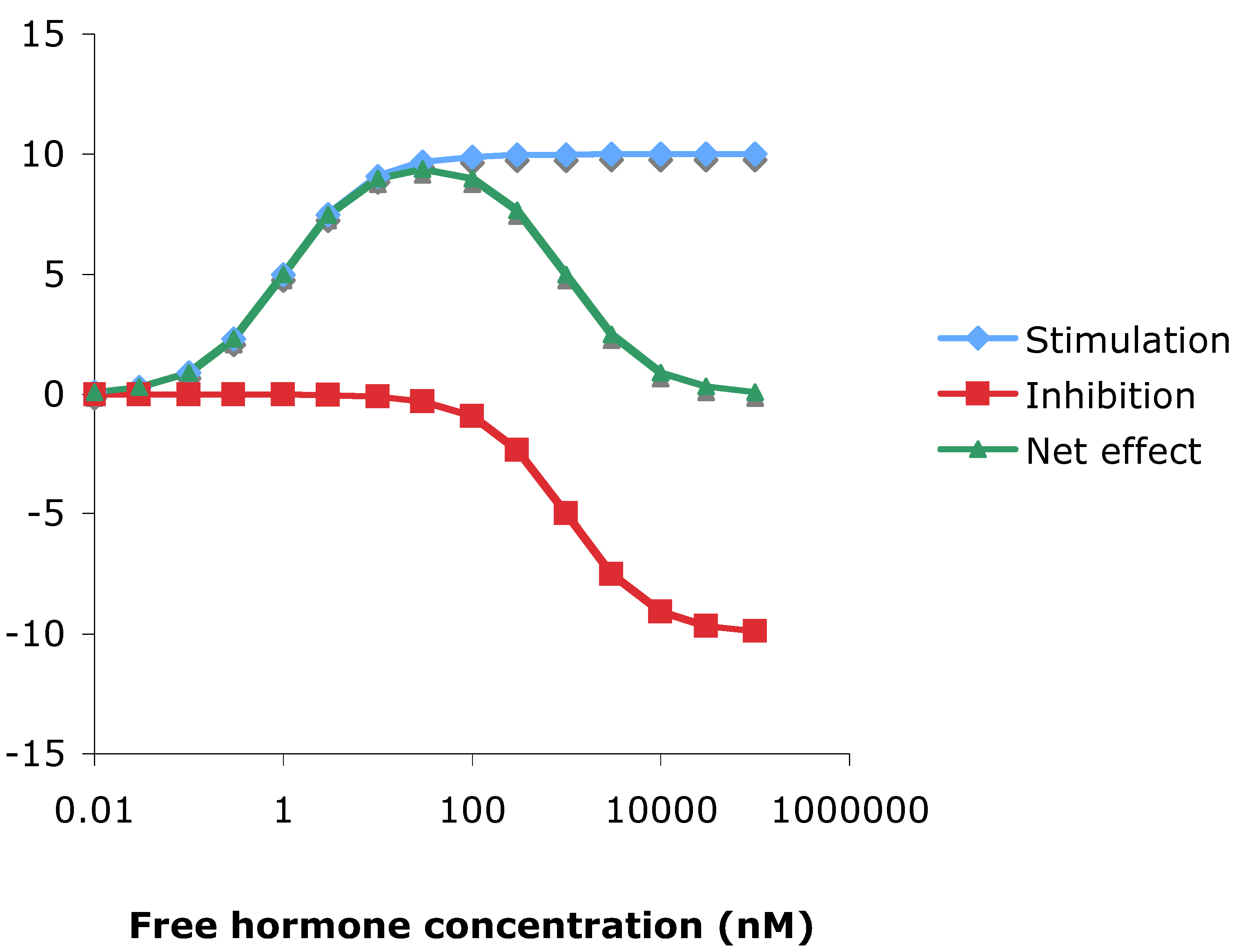
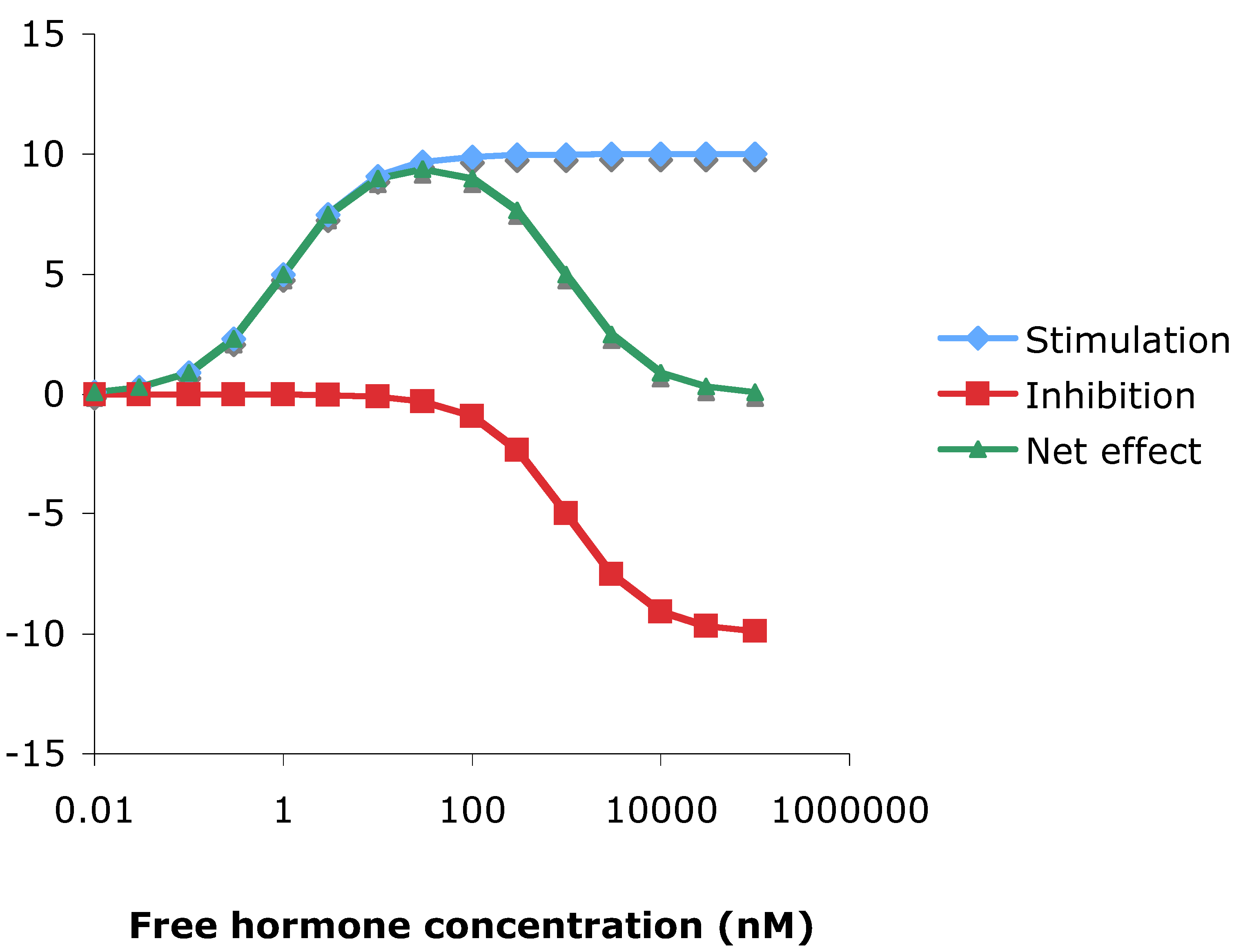
3.10. Theoretical consideration of hormesis
4. Conclusions
Acknowledgements
References and Notes
- Greenspan, F.S.; Strewler, G.J. Basic and Clinical Endocrinology, 5th ed.; Prentice Hall International (UK) Ltd: London, UK, 1997. [Google Scholar]
- Katzenellenbogen, B.S.; Choi, I.; Delage-Mourroux, R.; Ediger, T.R.; Martini, P.G.; Montano, M.; Sun, J.; Weis, K.; Katzenellenbogen, J.A. Molecular mechanisms of estrogen action: selective ligands and receptor pharmacology. J. Steroid Biochem. Mol. Biol. 2000, 74, 279–285. [Google Scholar]
- Jordan, V.C. A century of deciphering the control mechanisms of sex steroid action in breast and prostate cancer: the origins of targeted therapy and chemoprevention. Cancer Res. 2009, 69, 1243–1254. [Google Scholar]
- Brzozowski, A.M.; Pike, A.C.W.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustsfsson, J.-A.; Carlquist, M. Molecular basis of agonism and antagonism in the hER. Nature 1997, 389, 753–758. [Google Scholar]
- Pike, A.C.W.; Brzozowski, A.M.; Thorsell, A.-G.; Engstrom, O.; Ljunggren, J.; Gustafsson, J.-A.; Carlquist, M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO 1999, 18, 4608–4618. [Google Scholar]
- Pike, A.C.W.; Brzozowski, A.M.; Walton, J.; Hubbard, R.E.; Thorsell, A.-G.; Li, Y.-L.; Gustafsson, J.-A.; Carlquist, M. Structural insights into the mode of action of a pure antiestrogen. Structure 2001, 9, 145–153. [Google Scholar]
- Renaud, J.P.; Moras, D. Structural studies on nuclear receptors. Cellular and Molecular Life Sciences 2000, 57, 1748–1769. [Google Scholar]
- Smith, C.L.; O'Malley, B.W. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endo. Revi. 2004, 25, 45–71. [Google Scholar]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; Gustafsson, J.A. Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar]
- Enmark, E.; Pelto-Hukko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjold, M.; Gustafsson, J.-A. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metabol. 1997, 82, 4258–4265. [Google Scholar]
- Jones, P.S.; Parrott, E.; White, I.N.H. Activation of transcription by estrogen receptor α and β is cell type- and promoter-dependent. J. Biol. Chem. 1999, 274, 32008–32014. [Google Scholar]
- DeNardo, D.G.; Kim, H.-T.; Hilsenbeck, S.; Cuba, V.; Tsimelzon, A.; Brown, P.H. Global gene expression analysis of estrogen receptor transcription factor cross talk in breast cancer: identification of estrogen-induced/activator protein-1-dependent genes. Mol. Endocrinol. 2005, 19, 362–378. [Google Scholar]
- Safe, S.; Kim, K. Nuclear receptor-mediated transactivation through interaction with Sp proteins. Prog. Nucl. Acid Res. Mol. Biol. 2004, 77, 1–6. [Google Scholar]
- Kelly, M.J.; Levin, E.R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metabol. 2001, 12, 152–156. [Google Scholar]
- Beatson, G.T. On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new methods of treatment, with illustrative cases. Lancet 1896, 2, 104–107. [Google Scholar]
- Carter, A.C.; Sedransk, N.; Kelley, R.M.; Ansfield, F.J.; Ravdin, R.G.; Talley, R.W.; Potter, N.R. Diethylstilbestrol: recommended dosages for different categories of breast cancer patients. Report of the Cooperative Breast Cancer Group. J. Am. Med. Assoc. 1977, 19, 2079–2085. [Google Scholar]
- Black, L.J.; Jones, C.D.; Falcone, J.F. Antagonism of estrogen action with a new benzothiophene derived antiestrogen. Life Sci. 1983, 32, 1031–1036. [Google Scholar]
- Martino, S.; Cauley, J.A.; Barrett-Connor, E.; Powles, T.J.; Mershon, J.; Disch, D.; Secrest, R.J.; Cummings, S.R. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J. Nat. Cancer Inst. 2004, 96, 1751–1761. [Google Scholar]
- Jordan, V.C. Antioestrogenic action of raloxifene and tamoxifen: today and tomorrow. J. Nat. Cancer Inst. 1998, 90, 967–971. [Google Scholar]
- Vergote, I.; Robertson, J.F.R. Fulvestrant is an effective and well-tolerated endocrine therapy for postmenopausal women with advanced breast cancer: results from clinical trials. Br. J. Cancer 2004, 90, S11–S14. [Google Scholar]
- Peng, J.; Sengupta, S.; Jordan, V.C. Potential of selective estrogen receptor modulators as treatments and preventives of breast cancer. Anticancer Agents Med. Chem. 2009, 9, 481–499. [Google Scholar]
- Clarke, R.; Liu, M.C.; Bouker, K.B.; Gu, Z.; Lee, R.Y.; Zhu, Y.; Skaar, T.C.; Gomez, B.; O’Brien, K.; Wang, Y.; Hilakivi-Clarke, L.A. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003, 22, 7316–7339. [Google Scholar]
- Beardwell, C.G.; Hndley, A.C.; Wilkinsin, P.M.; St John, J.; Bu'lock, D. Hormonal changes in postmenopausal women with breast cancer treated with trilostane and dexamethasone. Clin. Endcrinol. 1985, 23, 413–421. [Google Scholar]
- Williams, C.J.; Barley, V.; Blackheath, G.; Hutcheon, A.; Kaye, S.; Smith, D.; Keen, C.; Webster, D.J.T.; Rowland, C.; Tyrell, C. Multistudy study of trilostane: a new hormonal agent in advanced postmenopausal breast cancer. Cancer Treat. Rep. 1987, 71, 1197–1201. [Google Scholar]
- Chu, P.S.; Buzdar, A.U.; Hortobagyi, G.N. Trilostane with hydrocortisone in treatment of metastatic breast cancer. Breast Cancer Res. Treat. 1989, 13, 117–121. [Google Scholar]
- Williams, C.J.; Barley, V.L.; Blackledge, G.R.; Rowland, C.G.; Tyrell, C.J. Multicentre cross over study of aminoglutethimide and trilostane in advanced postmenopausal breast cancer. Br. J. Cancer 1993, 68, 1210–1215. [Google Scholar]
- Puddefoot, J.R.; Barker, S.; Glover, H.R.; Malouitre, S.; Vinson, G.P. Non-competitive steroid inhibition of oestrogen receptor functions. Int. J. Cancer 2002, 101, 17–22. [Google Scholar]
- Arnold, S.F.; Bergeron, J.M.; Tran, D.Q.; Collins, B.M.; Vonier, P.M.; Crews, D.; Toscano, W.A., Jr.; McLachlan, J.A. Synergistic responses of steroidal estrogens in vitro (yeast) and in in vivo (turtles). Biochem. Biophys. Res. Commun. 1997, 235, 336–342. [Google Scholar]
- Jensen, E.V.; Khan, S.A. A two-site model for antiestrogen action. Mech. Ageing Dev. 2004, 125, 679–682. [Google Scholar]
- Wang, Y.; Chirgadze, N.Y.; Briggs, S.L.; Khan, S.; Jensen, E.V.; Burris, T.P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2006, 103, 9908–9911. [Google Scholar]
- Kojetin, D.J.; Burris, T.P.; Jensen, E.V.; Khan, S.A. Implications of the binding of tamoxifen to the coactivator recognition site of the estrogen receptor. Endocr. Relat. Cancer 2008, 15, 851–870. [Google Scholar]
- Meyers, M.J.; Sun, J.; Carlson, K.E.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor subtype-selective ligands: asymmetric synthesis and biological evaluation of cis- and trans-5,11-dialkyl- 5,6,11, 12-tetrahydrochrysenes. J. Med. Chem. 1999, 42, 2456–2468. [Google Scholar]
- Sun, J.; Meyers, M.J.; Fink, B.E.; Rajendran, R.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-alpha or estrogen receptor-beta. Endocrinology 1999, 140, 800–804. [Google Scholar]
- van Hoorn, W.P. Identification of a second binding site in the estrogen receptor. J. Med. Chem. 2002, 45, 584–589. [Google Scholar]
- Shiau, A.K.; Barstad, D.; Radek, J.T.; Meyers, M.J.; Nettles, K.W.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Agard, D.A.; Greene, G.L. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat. Struct. Biol. 2002, 9, 359–364. [Google Scholar]
- Sumbayev, V.V.; Jensen, J.K.; Hansen, J.A.; Andreasen, P.A. Novel modes of oestrogen receptor agonism and antagonism by hydroxylated and chlorinated biphenyls, revealed by conformation-specific peptide recognition patterns. Mol. Cell Endocrinol. 2008, 287, 30–39. [Google Scholar]
- Puddefoot, J.R.; Barker, S.; Glover, H.R.; Malouitre, S.D.; Vinson, G.P. Non-competitive steroid inhibition of oestrogen receptor functions. Int. J. Cancer 2002, 101, 17–22. [Google Scholar]
- Barker, S.; Malouitre, S.D.; Glover, H.R.; Puddefoot, J.R.; Vinson, G.P. Comparison of effects of 4-hydroxy tamoxifen and trilostane on oestrogen-regulated gene expression in MCF-7 cells: up-regulation of oestrogen receptor beta. J. Steroid Biochem. Mol. Biol. 2006, 100, 141–151. [Google Scholar]
- Lippman, M.; Bolan, G.; Huff, K. The effects of estrogens and antiestrogens on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res. 1976, 36, 4595–4601. [Google Scholar]
- Conolly, R.B.; Lutz, W.K. Nonmonotonic dose-response relationships: mechanistic basis, kinetic modeling, and implications for risk assessment. Toxicol. Sci. 2004, 77, 151–157. [Google Scholar]
- Li, L.; Andersen, M.E.; Heber, S.; Zhang, Q. Non-monotonic dose-response relationship in steroid hormone receptor-mediated gene expression. J. Mol. Endocrinol. 2007, 38, 569–585. [Google Scholar]
- Brooks, S.C.; Skafar, D.F. From ligand structure to biological activity: modified estratrienes and their estrogenic and antiestrogenic effects in MCF-7 cells. Steroids 2004, 69, 401–418. [Google Scholar]
- Kuiper, G.G.J.M.; Lemmem, J.G.; Calsson, B.; Corton, J.C.; Safe, S.H.; Van Der Saag, P.T.; Van Der Burg, B.; Gustafsson, J.-A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor b. Endocriology 1998, 139, 4252–4263. [Google Scholar]
- Liao, L.; Kuang, S.-Q.; Yuan, Y.; Gonzalez, S.M.; O’Malley, B.W.; Xu, J. Molecular structure and biological function of the cancer-amplified nuclear receptor coactivator SRC-3/AIB1. J. Steroid Biochem. Mol. Biol. 2002, 83, 3–14. [Google Scholar]
- Suen, C.-S.; Borrodin, T.J.; Mastroeni, R.; Ceskis, B.J.; Lyttle, C.R.; Frail, D.E. A transcriptional coactivator, steroid receptor coactivator-3 selectively augments steroid receptor transcriptional activity. J. Biol. Chem. 1998, 273, 27645–27653. [Google Scholar]
- Vienonen, A.; Miettinen, S.; Manninen, T.; Altucci, L.; Wilhelm, E.; Ylikomi, T. Regulation of nuclear receptor and cofactor expression in breast cancer cell lines. Eur. J. Endocrinol. 2003, 148, 469–479. [Google Scholar]
- Simon, S.L.R.; Parkes, A.; Leygue, E.; Dotzlaw, H.; Snell, L.; Troup, S.; Adeyinka, A.; Watson, P.H.; Murphy, L.C. Expression of a repressor of estrogen receptor activity in human breast tumors: relationship to some known prognostic markers. Cancer Res. 2000, 60, 2796–2799. [Google Scholar]
- Spiers, V.; Kerin, M.J. Prognostic significance of oestrogen receptor β in breast cancer. British J. Surg. 2000, 87, 405–409. [Google Scholar]
- Cowley, S.M.; Hoare, S.; Mosselman, S.; Parker, M.G. Estrogen receptors alpha and beta form heterodimers on DNA. J. Biol. Chem. 1997, 272(32), 19858–19862. [Google Scholar]
- Katzenellenbogen, B.S. Mechanisms of action and cross-talk between estrogen receptor and progesterone receptor pathways. J. Soc. Gynecol. Investig. 2000, 7, S33–S37. [Google Scholar]
- Montano, M.M.; Ekena, K.; Delage-Mourroux, R.; Chang, W.; Martini, P.; Katzenellenbogen, B.S. An estrogen receptor-selective coregulator that potentiates the effectiveness of antiestrogens and represses the activity of estrogens. Proc. Natl. Acad. Sci. USA 1999, 96, 6947–6952. [Google Scholar]
- Poola, I.; Koduri, S.; Chantra, S.; Clarke, R. Identification of twenty alternatively spliced estrogen receptor alpha mRNAs in breast cancer cell lines and tumors using splice targeted primer approach. J. Steroid Biochem. Mol. Biol. 2000, 72, 249–258. [Google Scholar]
- Pfeffer, U.; Fecarotta, E.; Vidali, G. Coexpression of multiple estrogen receptor variant messenger RNAs in normal and neoplastic breast tissues and in MCF-7 cells. Cancer Res. 1995, 55, 2158–2165. [Google Scholar]
- Madsen, M.W.; Reiter, B.E.; Lykkesfeldt, A.E. Differential expression of estrogen receptor mRNA splice variants in the tamoxifen resistant human breast cancer cell line MCF-7/TAMR-1 compared to the parential MCF-7 line. Mol. Cell. Endocrinol. 1995, 109, 197–207. [Google Scholar]
- Tora, L.; Mullick, A.; Metzger, D.; Ponglikitmongkol, M.; Park, I.; Chambon, P. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO 1989, 8, 1981–1986. [Google Scholar]
- Graham, M.L.; Krett, N.L.; Miller, L.A.; Leslie, K.K.; gordon, D.F.; Wood, W.M.; Wei, L.L.; Horwitz, K.B. T47Dco cells, genetically unstable and containing estrogen receptor mutations, are a model for the progression of breast cancers to hormone resistance. Cancer Res. 1990, 50, 6208–6217. [Google Scholar]
- Wang, Y.; Miksicek, R.J. Identification of a dominant negative form of the human estrogen receptor. Mol. Endocrinol. 1991, 5, 1707–1715. [Google Scholar]
- Hall, J.M.; McDonnell, D.P. The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 1999, 140, 5566–5578. [Google Scholar]
- Malouitre, S.D.; Barker, S.; Puddefoot, J.R.; Jalili, J.; Glover, H.R.; Vinson, G.P. Regulation of hepatic steroid receptors and enzymes by the 3beta-hydroxysteroid dehydrogenase inhibitor trilostane. J. Steroid Biochem. Mol Biol. 2006, 101(2-3), 97–105. [Google Scholar]
- Jozan, S.; Kreitmann, B.; Bayard, F. Different effects of oestradiol, oestriol, oestetrol and of oestrone on human breast cancer cells (MCF-7) in long term tissue culture. Acta Endocrinol. 1981, 98, 73–80. [Google Scholar]
- Margeat, E.; Bourdoncel, A.; Margueron, R.; Poujol, N.; Cavailles, V.; Royer, C. Ligands differentially modulate the protein interactions of the human estrogen receptors a and b. J. Mol. Biol. 2003, 326, 77–92. [Google Scholar]
- Ogawa, S.; Inoue, S.; Watanabe, T.; Orimo, A.; Hosoi, T.; Ouchi, Y.; Muramatsu, M. Molecular cloning and characterization of human estrogen receptor βcx: a potential inhibitor of estrogen action in human. Nucleic Acids Res. 1998, 26, 3505–3512. [Google Scholar]
- Yi, P.; Bhagat, S.; Hilf, R.; Bambara, R.A.; Muyan, M. Differences in the abilities of estrogen receptors to integrate activation functions are critical for sub-type selective transcriptional responses. Mol. Endocrinol. 2002, 16, 1819–1827. [Google Scholar]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar]
- Watanabe, T.; Inoue, S.; Ogawa, S.; Ishii, Y.; Hiroi, H.; Ikeda, K.; Orimo, A.; Muramatsu, M. Agonistic effect of tamoxifen is dependent on cell type, ERE-promoter context, and estrogen receptor subtype: functional difference between estrogen receptors a and b. Biochem. Biophys. Res. Commun. 1997, 236, 140–145. [Google Scholar]
- Mérot, Y.; Métivier, R.; Penot, G.; Manu, D.; Saligaut, C.; Gannon, F.; Pakdel, F.; Kah, O.; Flouriot, G. The relative contribution exerted by AF-1 and AF-2 transactivation functions in estrogen receptor alpha transcriptional activity depends upon the differentiation stage of the cell. J. Biol. Chem. 2004, 279, 26184–26191. [Google Scholar]
- Parker, M.G. Transcriptional activation by oestrogen receptors. Biochem. Soc. Symp. 1998, 63, 45–50. [Google Scholar]
- Arnold, S.F.; Klotz, D.M.; Collins, B.M.; Vonier, P.M.; Guillette, L.J., Jr.; McLachlan, J.A. Synergistic activation of estrogen receptor with combinations of environmental chemicals. Science 1996, 272, 1489–1492. [Google Scholar]
- Stahl, S.; Chun, T.-Y.; Gray, W.G. Phytoestrogens act as estrogen agonist in an estrogen-responsive pituitary cell line. Toxicol. Appl. Pharmacol. 1998, 152, 41–48. [Google Scholar]
- Collins, B.; McLachlan, J.A.; Arnold, S.F. The estrogenic and antiestrogenic activities of phytochemicals with the human estrogen receptor expressed in yeast. Steroids 1997, 62, 365–372. [Google Scholar]
- Tran, D.Q.; Ide, C.F.; McLachlan, J.A.; Arnold, S.F. The anti-estrogenic activity of selected polynuclear aromatic hydrocarbons in yeast expressing human estrogen receptor. Biochem. Biophys. Res. Commun. 1996, 229, 102–108. [Google Scholar]
- Galigniana, M.D.; Vicent, G.P.; Piwien-Pilipuk, G.; Burton, G.; Lantos, C.P. Mechanism of action of the potent sodium-retaining steroid 11, 19-oxidoprogesterone. Mol. Pharmacol. 2000, 58, 58–70. [Google Scholar]
- Christopoulos, A.; El-Fakahany, E.E. Qualitative and quantitative assessment of relative agonist efficacy. Biochem. Pharmacol. 1999, 58, 735–748. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Glover, H.R.; Barker, S.; Malouitre, S.D.M.; Puddefoot, J.R.; Vinson, G.P. Multiple Routes to Oestrogen Antagonism. Pharmaceuticals 2010, 3, 3417-3434. https://doi.org/10.3390/ph3113417
Glover HR, Barker S, Malouitre SDM, Puddefoot JR, Vinson GP. Multiple Routes to Oestrogen Antagonism. Pharmaceuticals. 2010; 3(11):3417-3434. https://doi.org/10.3390/ph3113417
Chicago/Turabian StyleGlover, Hilary R., Stewart Barker, Sylvanie D. M. Malouitre, John R. Puddefoot, and Gavin P. Vinson. 2010. "Multiple Routes to Oestrogen Antagonism" Pharmaceuticals 3, no. 11: 3417-3434. https://doi.org/10.3390/ph3113417
APA StyleGlover, H. R., Barker, S., Malouitre, S. D. M., Puddefoot, J. R., & Vinson, G. P. (2010). Multiple Routes to Oestrogen Antagonism. Pharmaceuticals, 3(11), 3417-3434. https://doi.org/10.3390/ph3113417