Nanomedicine Faces Barriers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Targeting Challenge
2.1. Clinically Relevant Targets
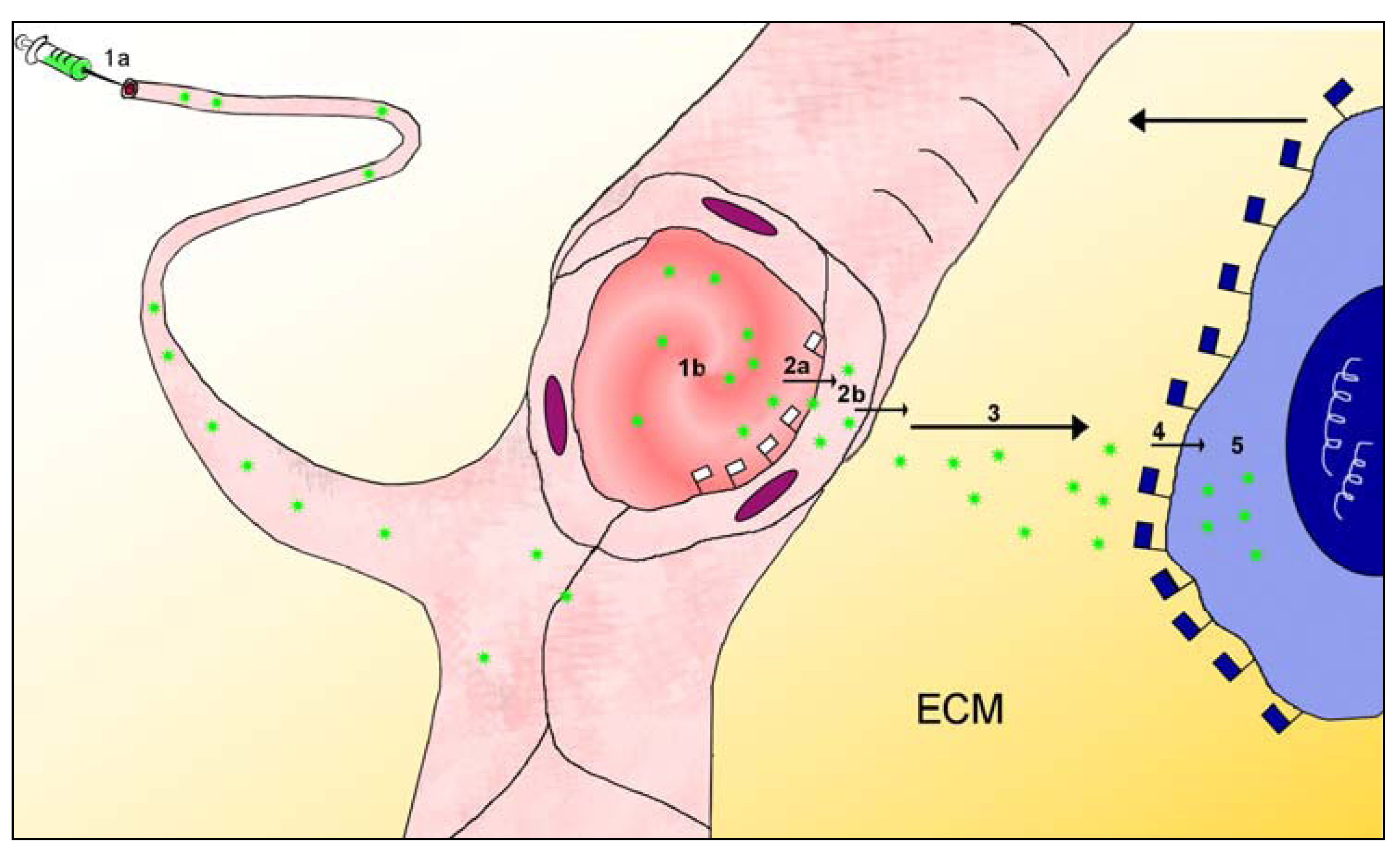
2.2. The Multistage Path to the Target
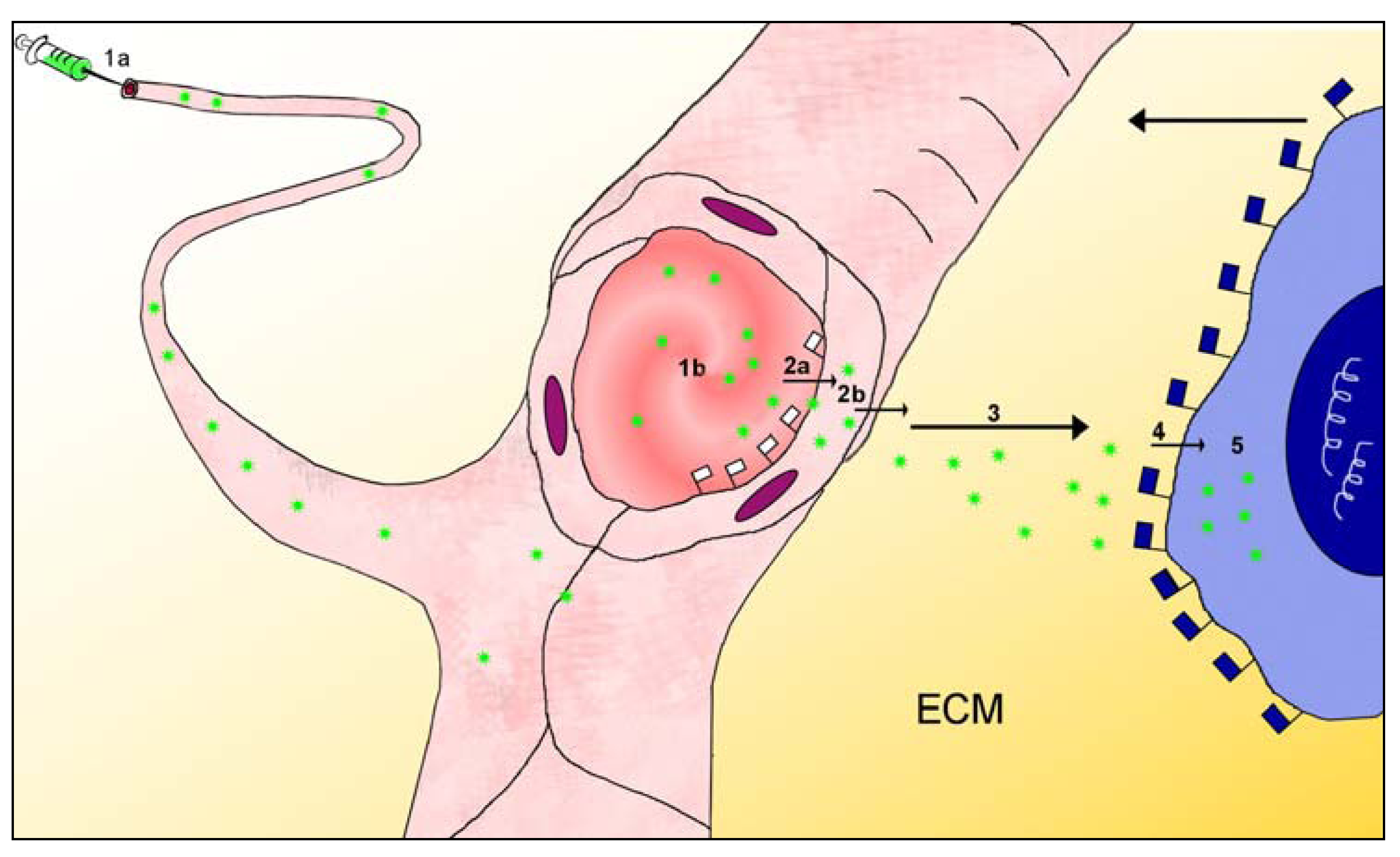
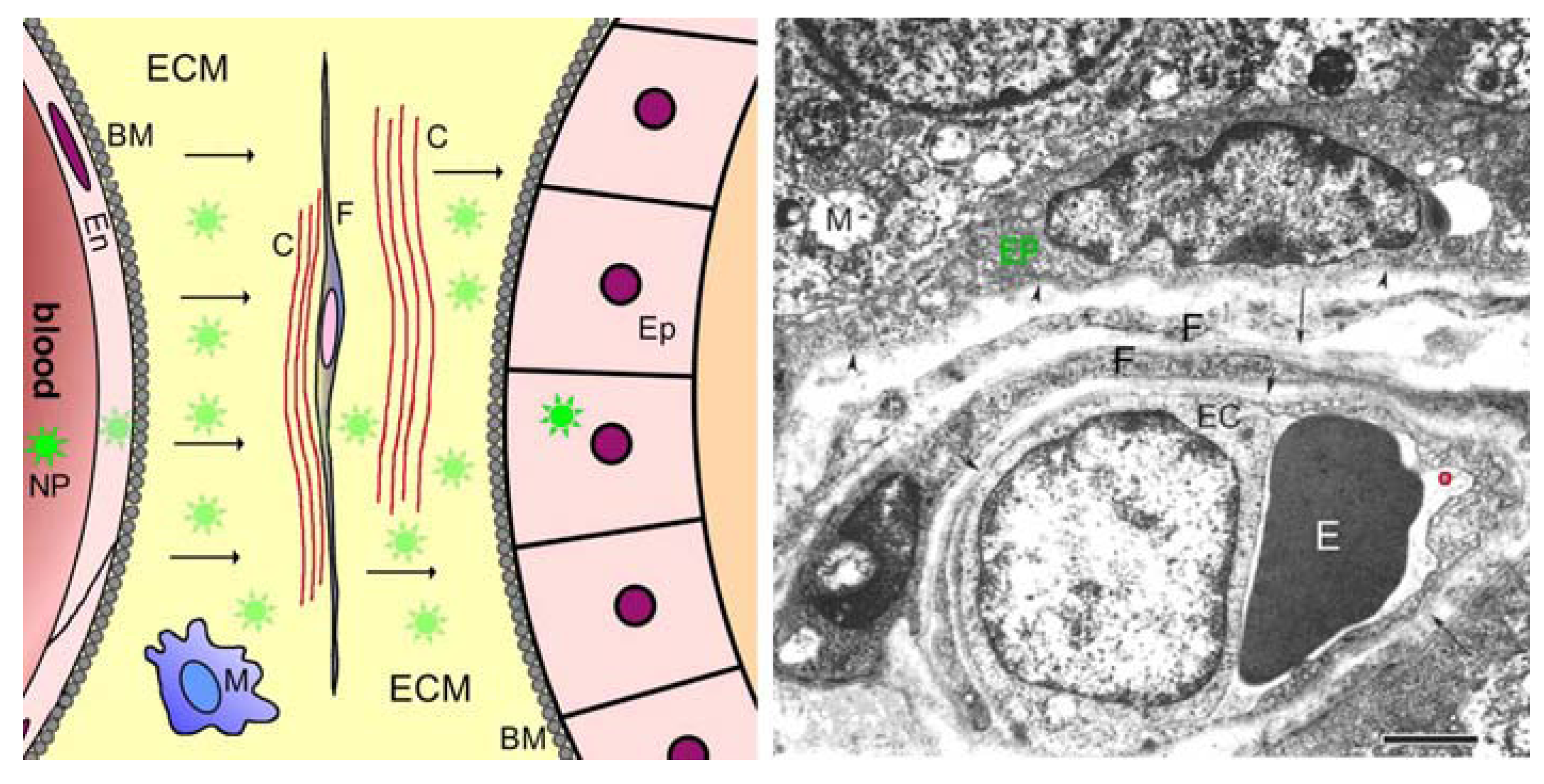
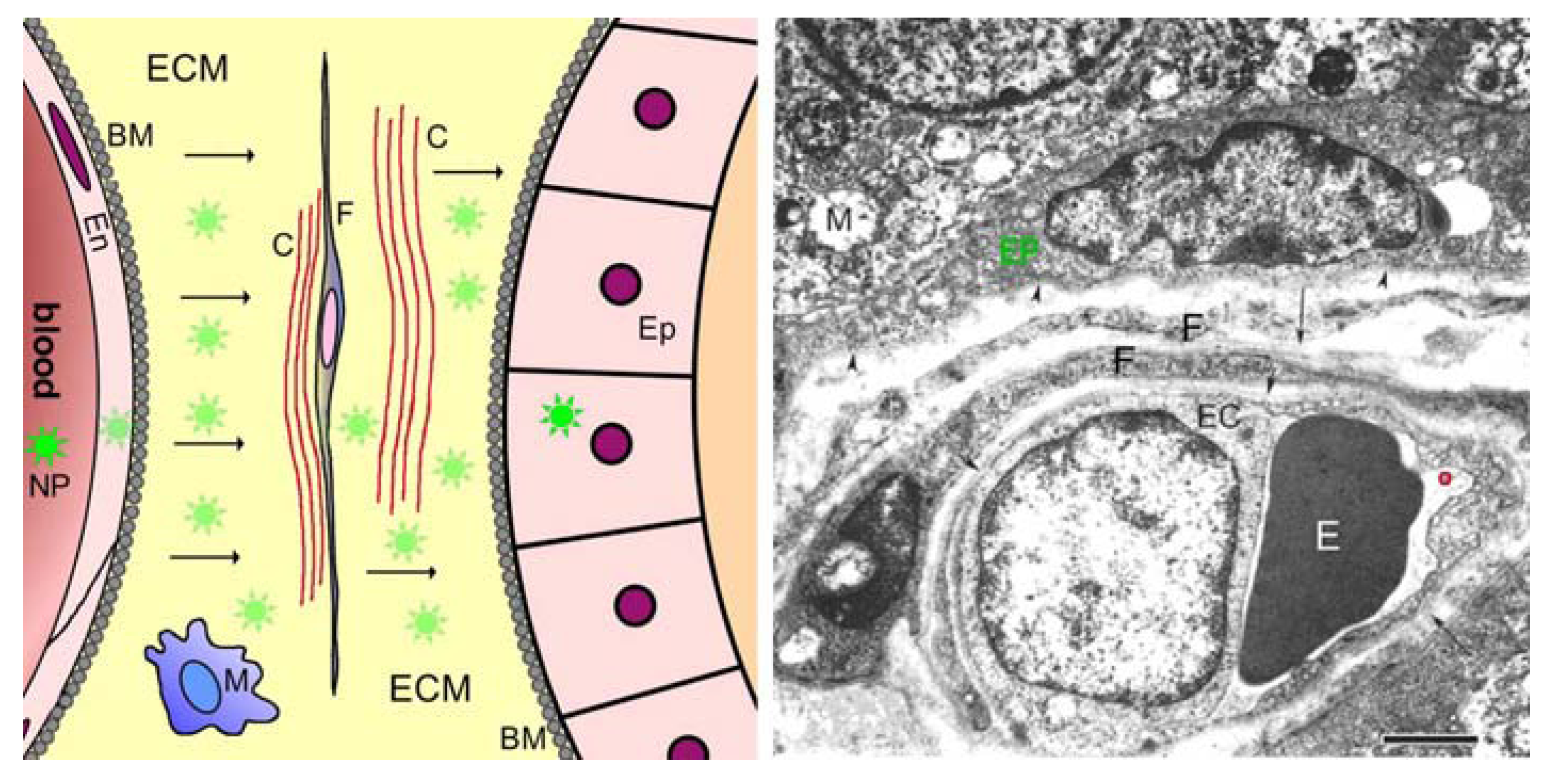
2.3. Barriers between Blood and Tissues
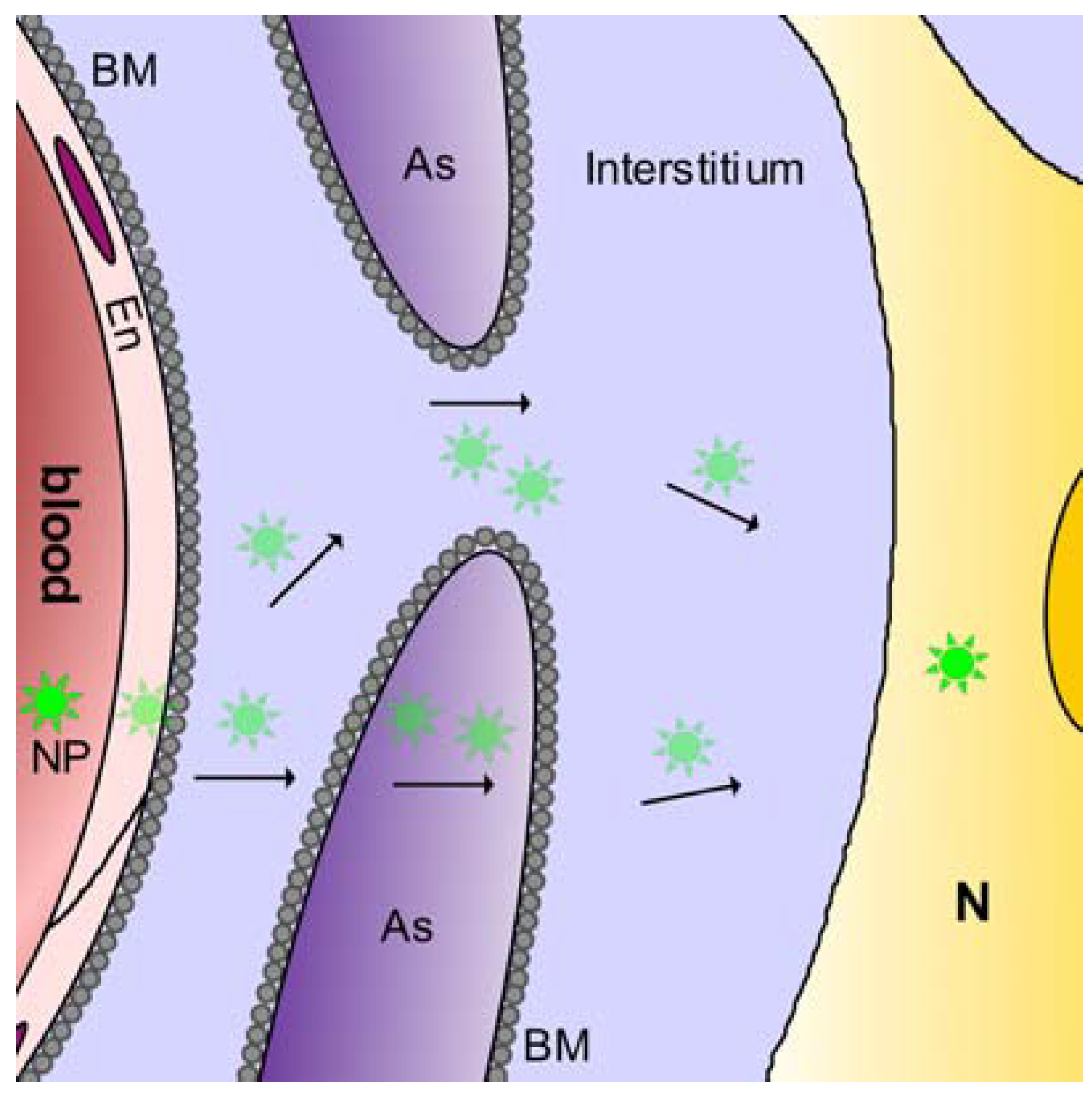
2.3.1. Blood-brain barrier
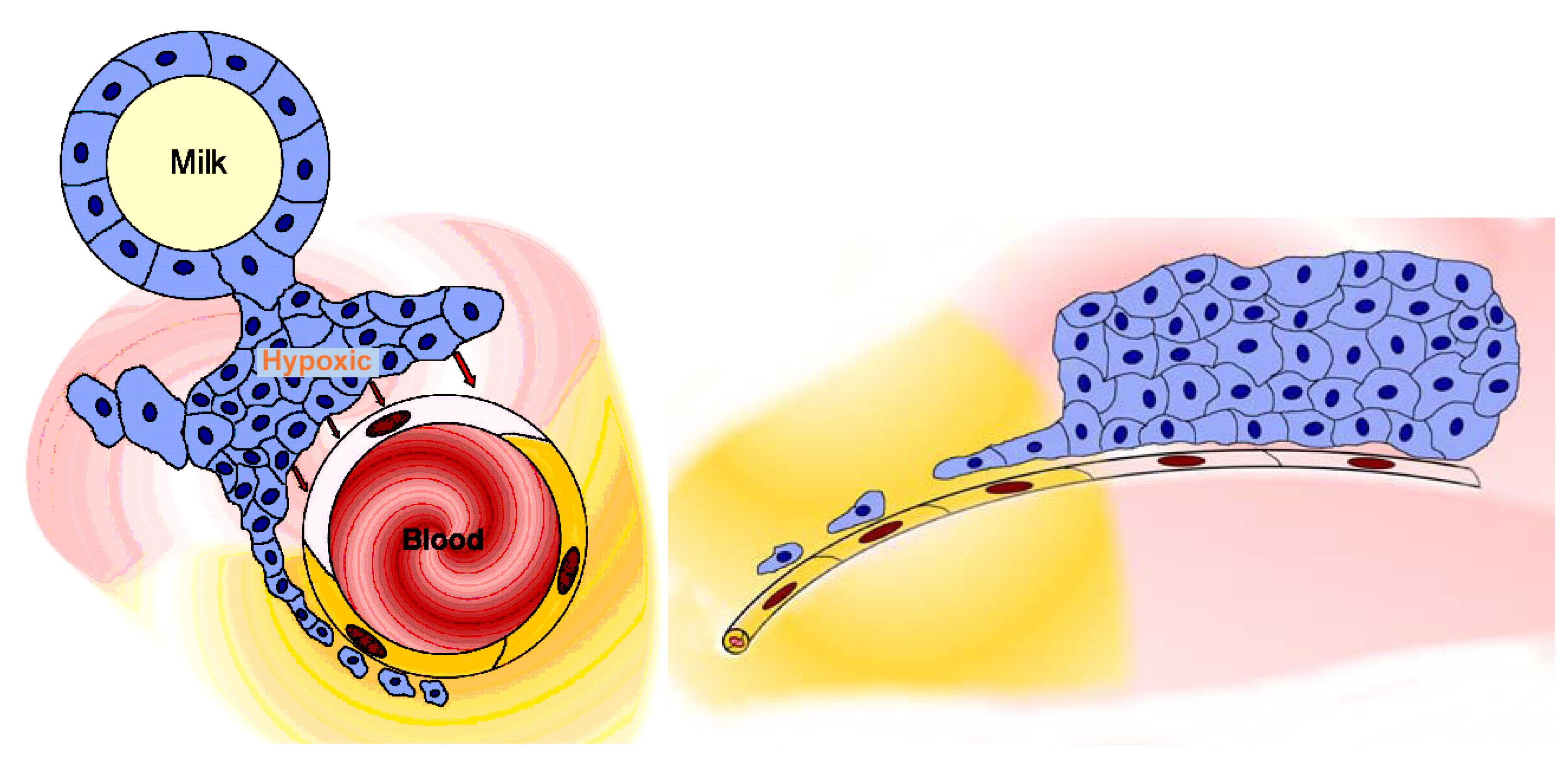
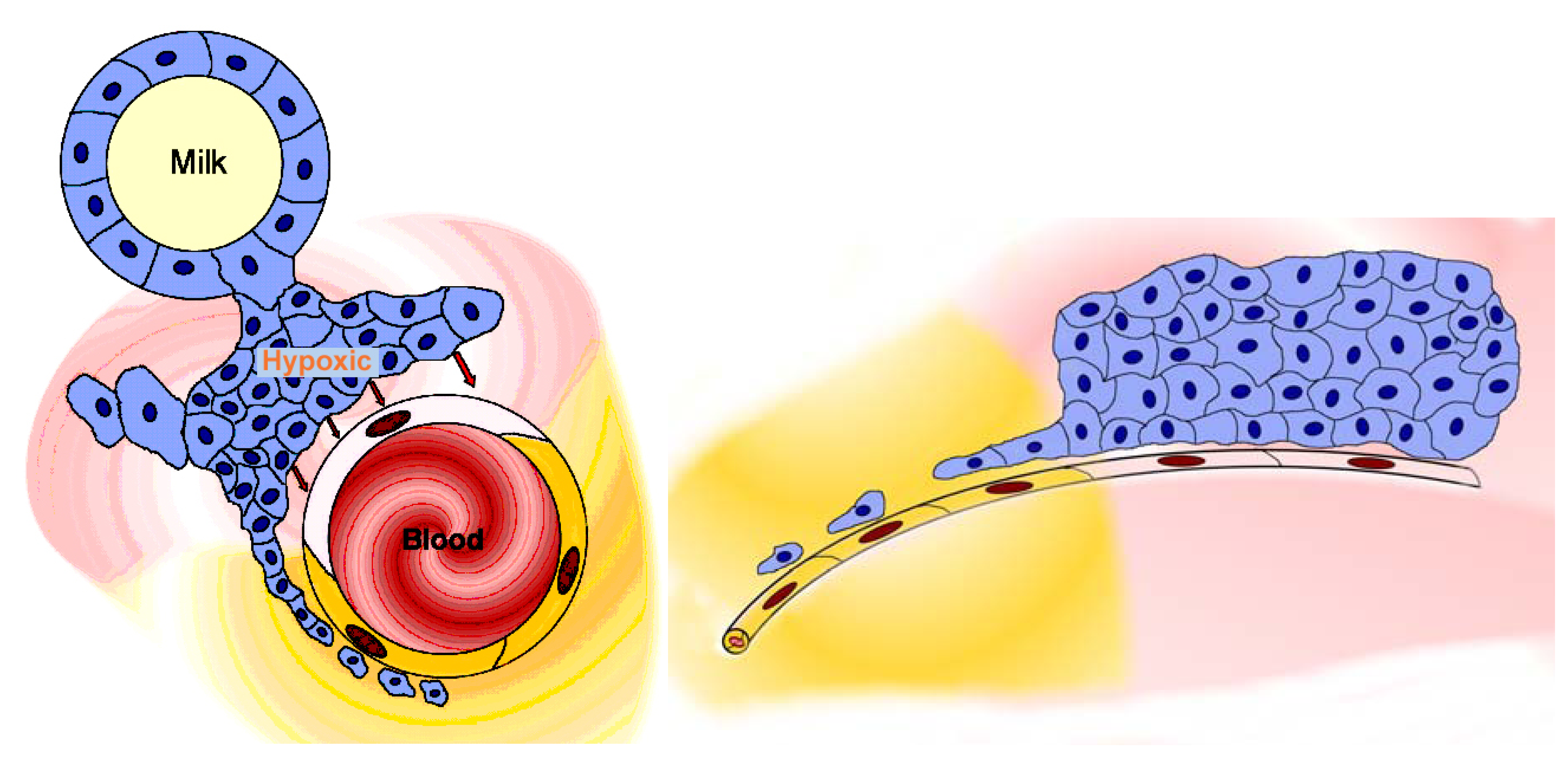
2.3.2. Blood-milk barrier
2.3.3. Blood tissue barriers in muscles
2.3.4. Blood-tissue barriers in other organs
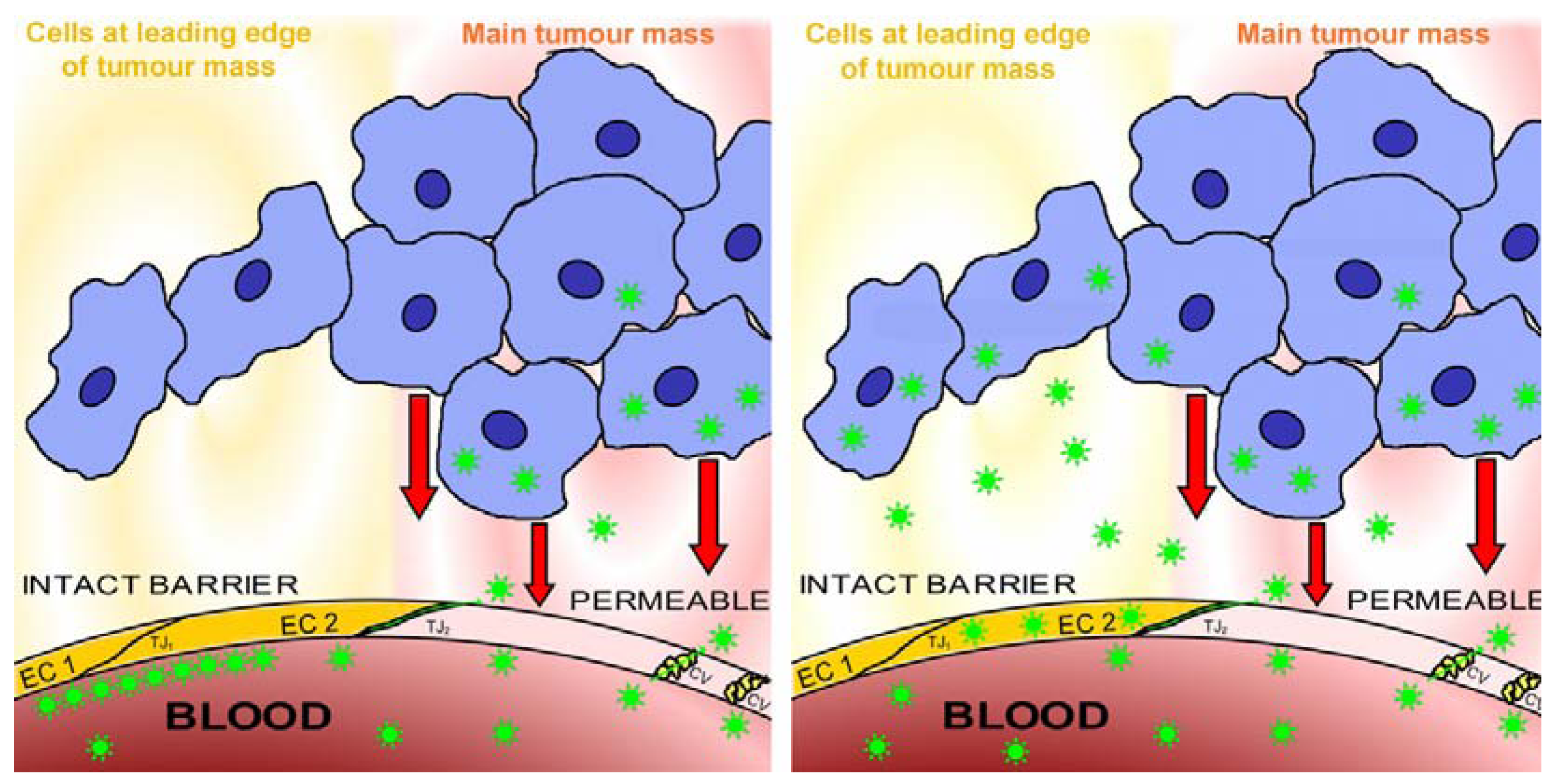
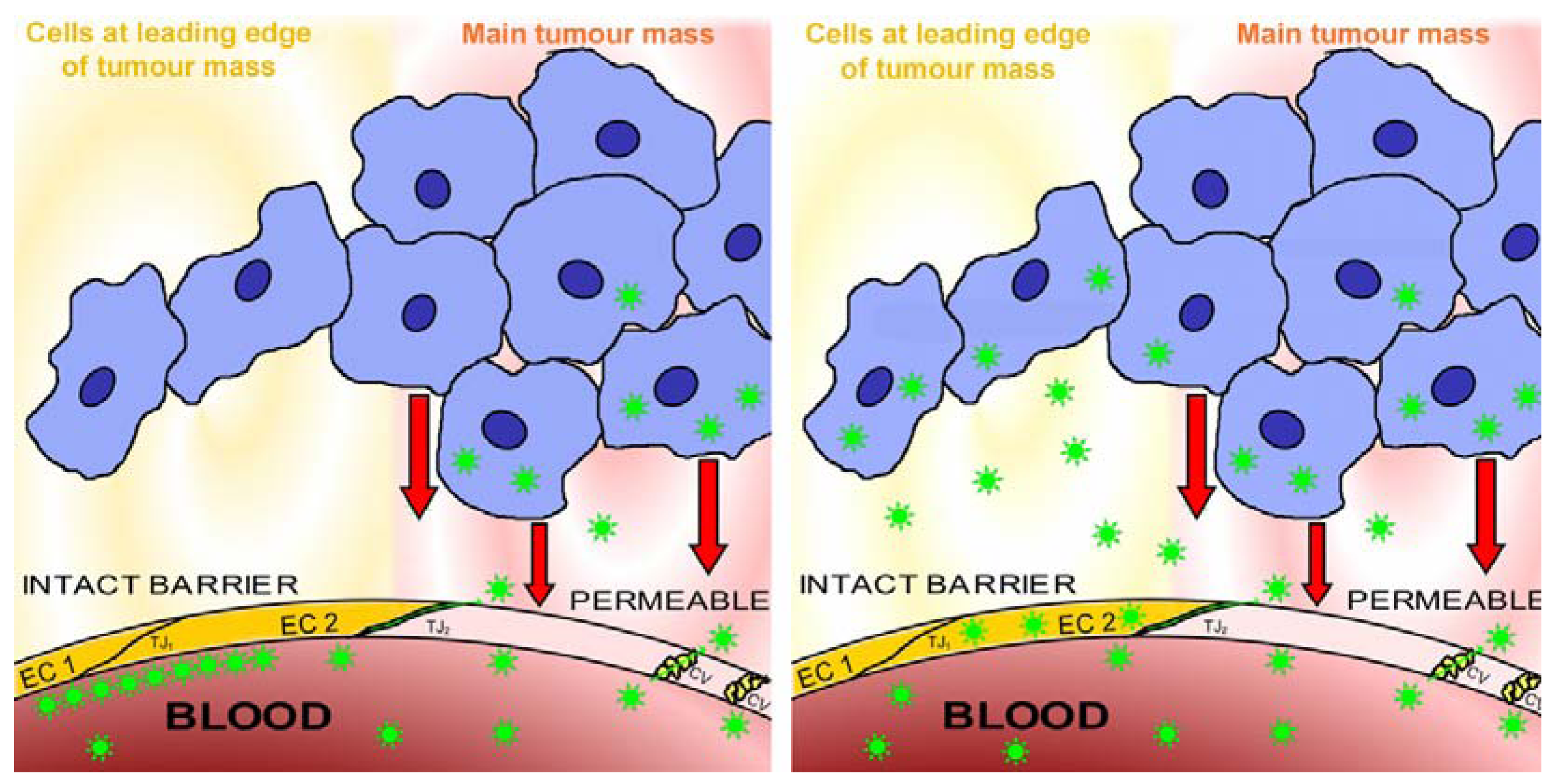
2.4. Blood-Tissue Barriers in Disease States, EPR
2.5. Lesions behind Barriers
3. Particles Face Barriers
3.1. Small Drugs Pass Barriers by Diffusion
3.2. Differentiated Delivery Strategies Optimise the Use of Nanoparticles
3.3. Evading Blood-Tissue Barriers
3.3.1. Opening the barrier
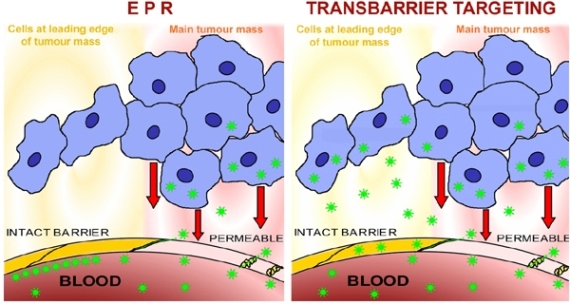
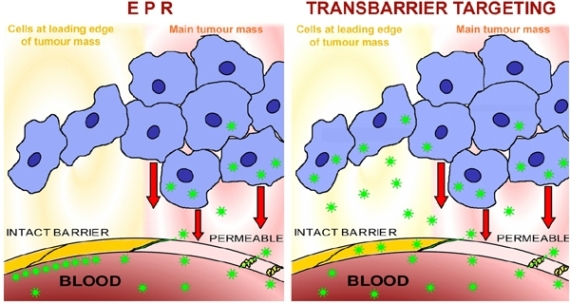
3.3.2. EPR
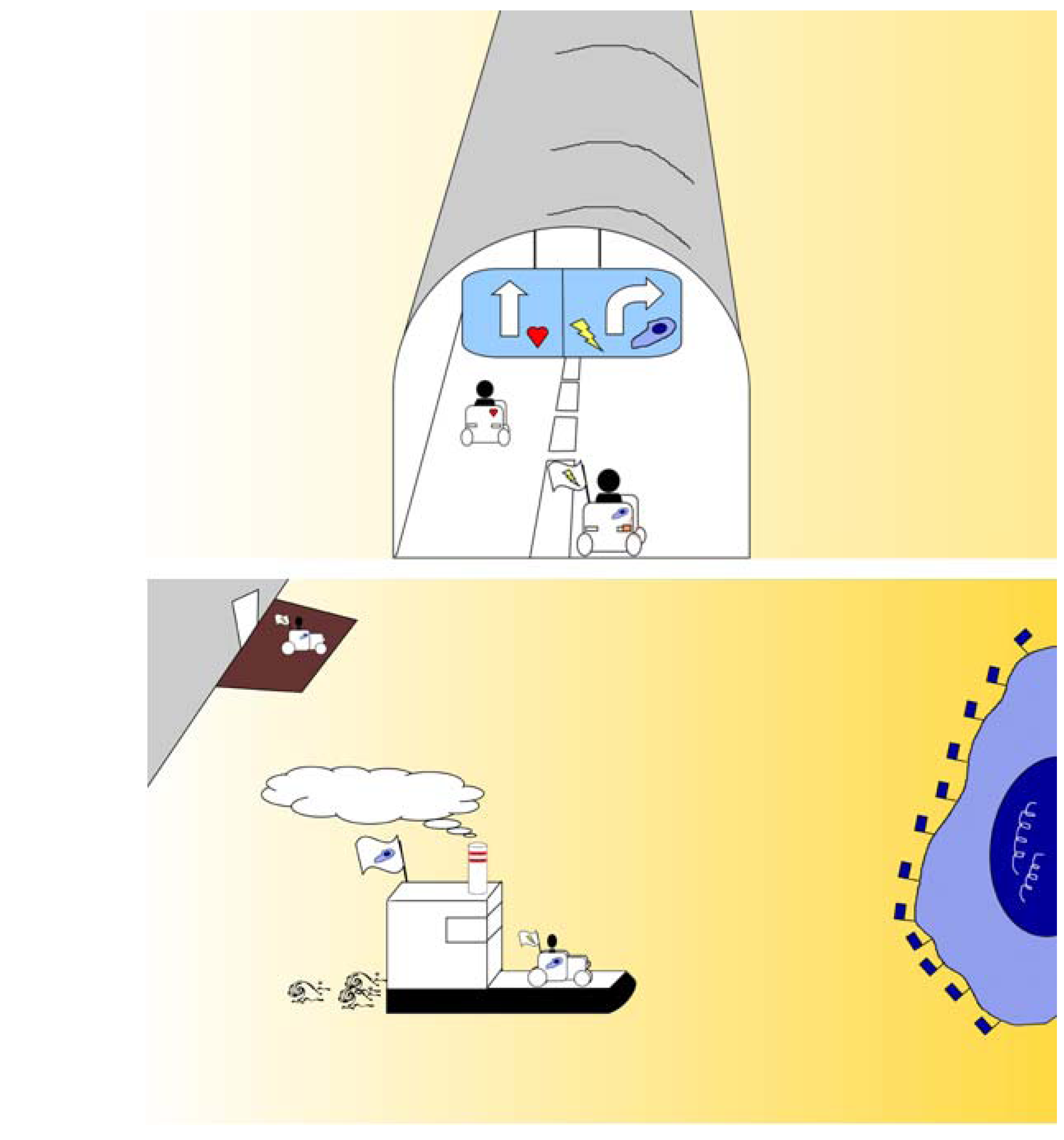
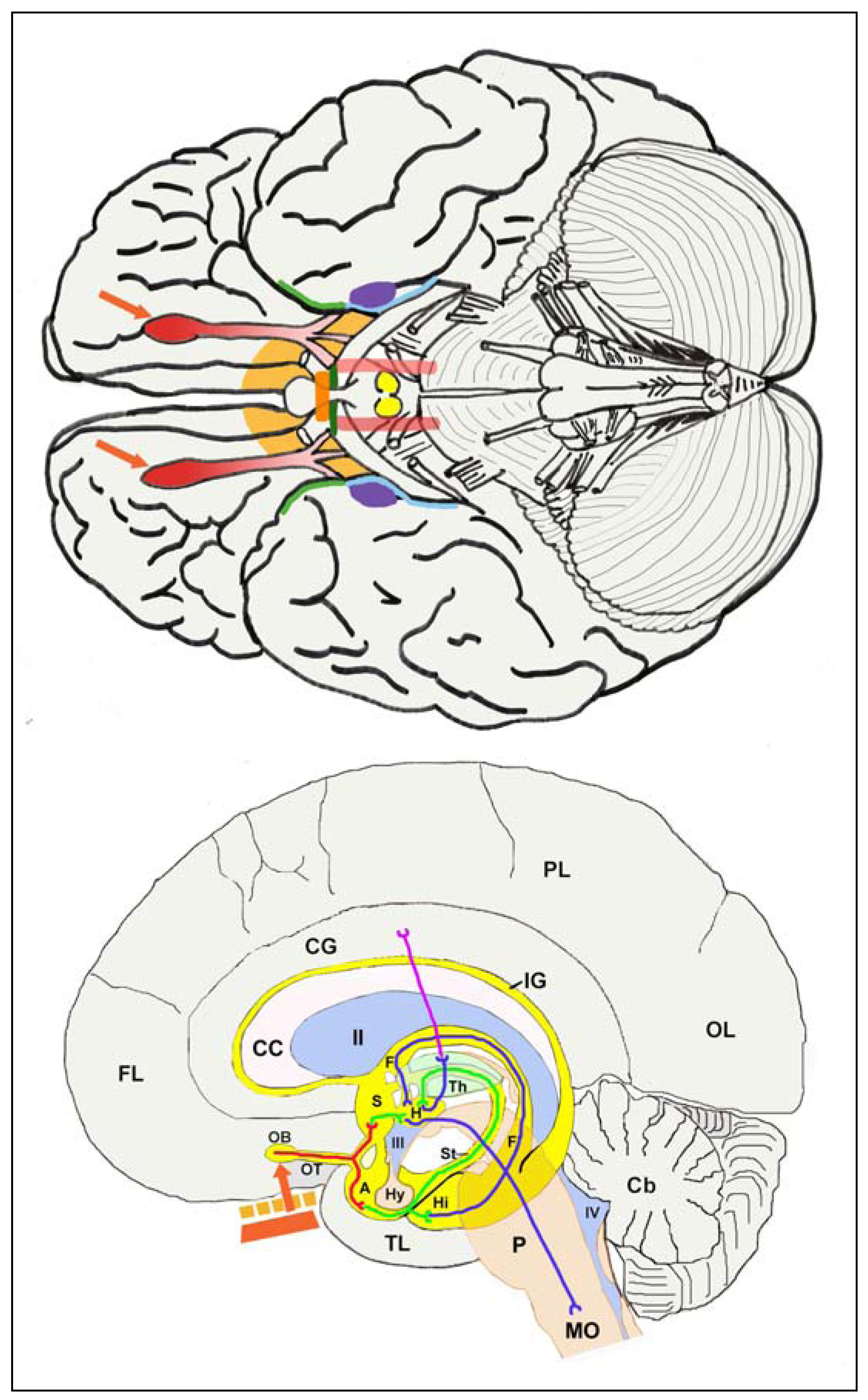
3.3.3. Olfactory route to evade the blood-brain barrier
3.3.4. Immune therapy
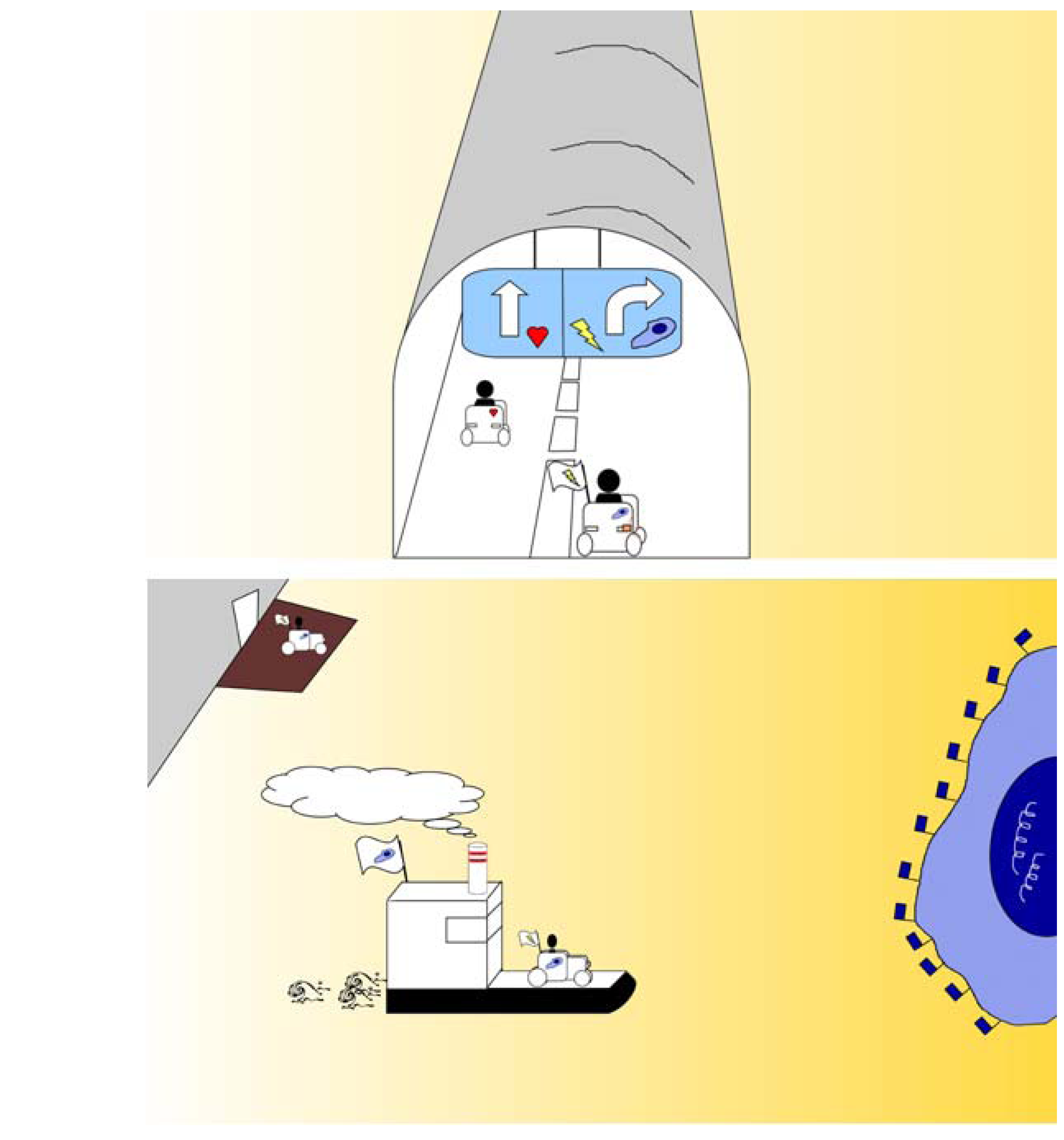
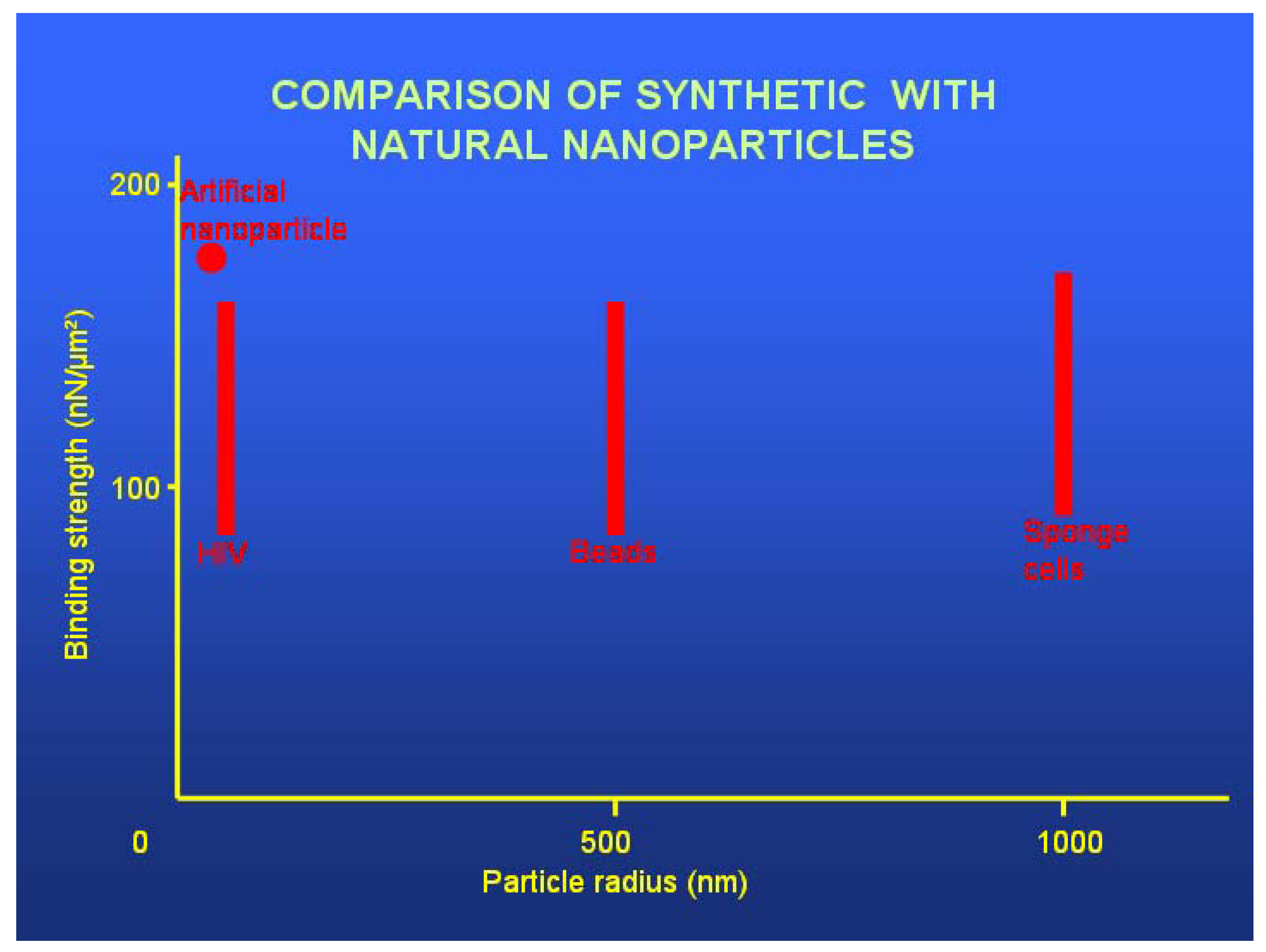
3.4. Confronting Intact Blood-Tissue Barriers: Penetrating Barriers by Flagging and Ferrying
3.4.1. Flagging
3.4.2. Ferrying
4. Design of Barrier-Passing Nanoparticles
4.1. Transbarrier Targeting as Essential Requirement
4.2. The Numbers of Targeting Groups per Nanoparticle
4.3. Targeting Chemistry
4.4. Nanoparticle Chemistry
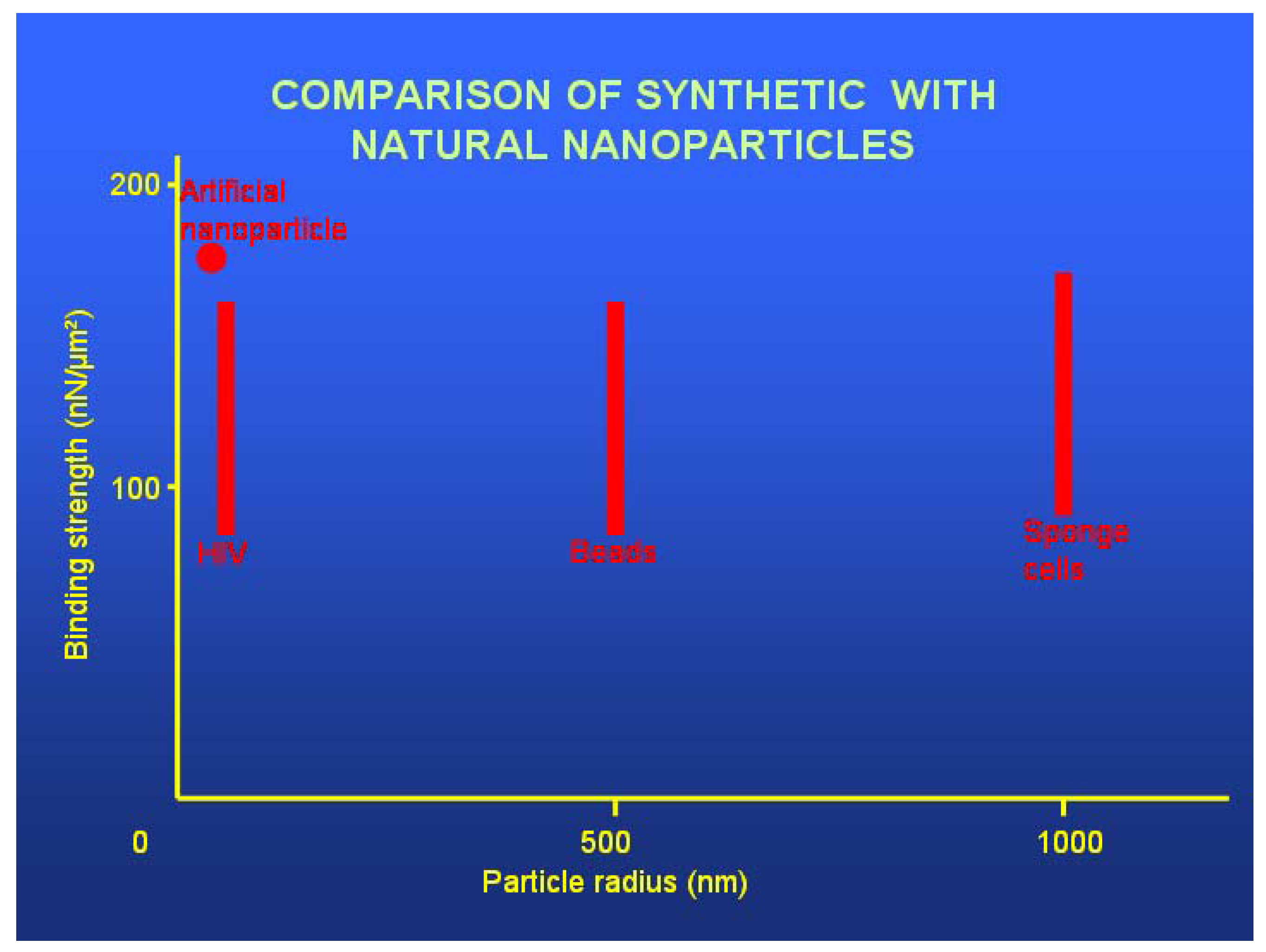
4.5. Nanoparticle Size
4.6. Nanoparticle Stability and Flexibility
4.7. Control of Nanoparticle Clearance
4.8. Market and Regulatory Issues
5. Future Directions
Acknowledgements
References
- Ehrlich, P. On immunity with special reference to cell life. Proc. R. Soc. 1900, 66, 424–448. [Google Scholar]
- Stumpf, W.E. The dose makes the medicine. Drug Dis. Today 2006, 11, 550–555. [Google Scholar]
- Evans, W.E.; Pratt, C.B.; Taylor, R.H.; Barker, L.F.; Crom, W.R. Pharmacokinetic monitoring of high-dose methotrexate. Cancer Chemother. Pharmacol. 1979, 3, 161–166. [Google Scholar]
- Evans, W.E.; Crom, W.R.; Abromowitch, M.; Dodge, R.; Look, A.T.; Bowman, W.P.; George, S.L.; Pui, C.H. Clinical pharmacodynamics of high-dose methotrexate in acute lymphocytic leukemia. Identification of a relation between concentration and effect. New Engl. J. Med. 1986, 314, 471–477. [Google Scholar]
- Evans, W.E.; Relling, M.V.; Rodman, J.H.; Crom, W.R.; Boyett, J.M.; Pui, C.H. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. New Engl. J. Med. 1998, 338, 499–505. [Google Scholar]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. New Engl. J. Med. 2006, 354, 166–178. [Google Scholar]
- Gamelin, E.; Delva, R.; Jacob, J.; Merrouche, Y.; Raoul, J.L.; Pezet, D.; Dorval, E.; Piot, G.; Morel, A.; Boisdron-Celle, M. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 2099–2105. [Google Scholar]
- Saif, M.W.; Choma, A.; Salamone, S.J.; Chu, E. Pharmacokinetically guided dose adjustment of 5-fluorouracil: a rational approach to improving therapeutic outcomes. J. Nat. Cancer Inst. 2009, 101, 1–10. [Google Scholar]
- de Jonge, M.E.; Huitema, A.D.R.; Schellens, J.H.M.; Rodenhuis, S.; Beijnen, J.H. Individualised cancer chemotherapy: Strategies and performance of prospective studies on therapeutic drug monitoring with dose adaptation: a review. Clin. Pharmacokinet. 2005, 44, 147–173. [Google Scholar]
- Galpin, A.J.; Evans, W.E. Therapeutic drug monitoring in cancer management. Clin. Chem. 1993, 39, 2419–2430. [Google Scholar]
- Krynetski, E.Y.; Evans, W.E. Pharmacogenetics of cancer therapy: getting personal. Am. J. Hum. Genet. 1998, 63, 11–16. [Google Scholar]
- Rousseau, A.; Marquet, P. Application of pharmacokinetic modelling to the routine therapeutic drug monitoring of anticancer drugs. Fundam. Clin. Pharmacol. 2002, 16, 253–262. [Google Scholar]
- Holzer, A.K.; Katano, K.; Klomp, L.W.J.; Howell, S.B. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 6744–6749. [Google Scholar]
- Schiffer, R.; Neis, M.; Höller, D.; Rodríguez, F.; Geier, A.; Gartung, C.; Lammert, F.; Dreuw, A.; Zwadlo-Klarwasser, G.; Merk, H.; Jugert, F.; Baron, J.M. Active influx transport is mediated by members of the organic anion transporting polypeptide family in human epidermal keratinocytes. J. Invest. Dermatol. 2003, 120, 285–291. [Google Scholar]
- Suzuki, H.; Sugiyama, Y. Transport of drugs across the hepatic sinusoidal membrane: sinusoidal drug influx and efflux in the liver. Semin. Liver Dis. 2000, 20, 251–263. [Google Scholar]
- Kato, Y.; Miyazaki, T.; Kano, T.; Sugiura, T.; Kubo, Y.; Tsuji, A. Involvement of influx and efflux transport systems in gastrointestinal absorption of celiprolol. J. Pharma. Sci. 2009, 98, 2529–2539. [Google Scholar]
- Tomlinson, E. Theory and practice of site-specific drug delivery. Adv. Drug Deliver. Rev. 1987, 1, 87–198. [Google Scholar]
- Schnitzer, J.E. Update on the cellular and molecular basis of capillary permeability. Trends Cardiovasc. Med. 1993, 3, 124–130. [Google Scholar]
- Schnitzer, J.E. Vascular targeting as a strategy for cancer therapy. N. Engl. J. Med. 1998, 339, 472–474. [Google Scholar]
- Denekamp, J. Vasculature as a target for tumour therapy. Prog. Appl. Microcirc. 1984, 4, 28–38. [Google Scholar]
- Burrows, F.J.; Thorpe, P.E. Vascular targeting - a new approach to the therapy of solid tumors. Pharmacol. Ther. 1994, 64, 155–174. [Google Scholar]
- Dykes, P.W.; Bradwell, A.R.; Chapman, C.E.; Vaughan, A.T.M. Radioimmunotherapy of cancer, clinical studies and limiting factors. Cancer Treat. Rev. 1987, 14, 87–106. [Google Scholar]
- Jain, R.K. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer Res. 1990, 50, 814s–819s. [Google Scholar]
- Sands, H.; Jones, P.L. Physiology of monoclonal antibody accretion by tumors. Cancer Treat. Res. 1990, 51, 97–122. [Google Scholar]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar]
- Christofidou-Solomidou, M.; Pietra, G.G.; Solomides, C.C.; Arguiris, E.; Harshaw, D.; Fitzgerald, G.A.; Albelda, S.M.; Muzykantov, V.R. Immunotargeting of glucose oxidase to endothelium in vivo causes oxidative vascular injury in the lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L794–L805. [Google Scholar]
- Holton, O.D.; Black, C.D.; Parker, R.J.; Covell, D.G.; Barbet, J.; Sieber, S.M.; Talley, M.J.; Weinstein, J.N. Biodistribution of monoclonal IgG1, F(ab´)2, and Fab´ in mice after intravenous injection. Comparison between anti-B cell (anti-Lyb8.2) and irrelevant (MOPC-21) antibodies. J. Immunol. 1987, 139, 3041–3049. [Google Scholar]
- Hughes, B.J.; Kennel, S.; Lee, R.; Huang, L. Monoclonal antibody targeting of liposomes to mouse lung in vivo. Cancer Res. 1989, 49, 6214–6220. [Google Scholar]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar]
- Pimm, M.V.; Baldwin, R.W. Quantitative evaluation of the localization of a monoclonal antibody (791T/36) in human osteogenic sarcoma xenografts. Eur. J. Cancer Clin. Oncol. 1984, 20, 515–524. [Google Scholar]
- Weinstein, J.N.; van Osdol, W. The macroscopic and microscopic pharmacology of monoclonal antibodies. Int. J. Immunopharmacol. 1992, 14, 457–463. [Google Scholar]
- Ferrari, M. Cancer nanotechnology, opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar]
- Dvorak, H.F.; Nagy, J.A.; Dvorak, A.M. Structure of solid tumors and their vasculature: implications for therapy with monoclonal antibodies. Cancer Cells 1991, 3, 77–85. [Google Scholar]
- Baker, M. Upping the ante on antibodies. Nat. Biotechnol. 2005, 23, 1065–1072. [Google Scholar]
- Glennie, M.J.; Johnson, P.W.M. Clinical trials of antibody therapy. Trends Immunol. 2000, 21, 403–410. [Google Scholar]
- Fair, W.R.; Israeli, R.S.; Heston, W.D.W. Prostate-specific membrane antigen. Prostate 1997, 32, 140–148. [Google Scholar]
- Gao, X.; Porter, A.T.; Grignon, D.J.; Pontes, J.E.; Honn, K.V. Diagnostic and prognostic markers for human prostate cancer. Prostate 1997, 31, 264–281. [Google Scholar]
- Murphy, G.P.; Barren, R.J.; Erickson, S.J.; Bowes, V.A.; Wolfert, R.L.; Bartsch, G.; Klocker, H.; Pointner, J.; Reissigl, A.; McLeod, D.G.; Douglas, T.; Morgan, T.; Kenny, G.M.; Ragde, H.; Boynton, A.L.; Holmes, E.H. Evaluation and comparison of two new prostate carcinoma markers: Free-prostate specific antigen and prostate specific membrane antigen. Cancer 1998, 78, 809–818. [Google Scholar]
- Jain, R.K. Understanding barriers to drug delivery: high resolution in vivo imaging is key. Clin. Cancer Res. 1999, 5, 1605–1606. [Google Scholar]
- Torchilin, V.P. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. Nanotechnol. Drug Deliv. 2007, 9, E128–E147. [Google Scholar]
- Dimotakis, P.E. The mixing transition in turbulent flows. J. Fluid Mechan. 2000, 409, 69–98. [Google Scholar]
- Presley, J.F.; Mayor, S.; McGraw, T.E.; Dunn, K.W.; Maxfield, F.R. Bafilomycin A1 treatment retards transferrin receptor recycling more than bulk membrane recycling. J. Biol. Chem. 1997, 272, 13929–13936. [Google Scholar]
- Paulos, C.M.; Reddy, J.A.; Leamon, C.P.; Turk, M.J.; Low, P.S. Ligand binding and kinetics of folate receptor recycling in vivo: impact on receptor-mediated drug delivery. Mol. Pharmacol. 2004, 66, 1406–1414. [Google Scholar]
- Benacerraf, B.; Sebestyen, M.; Cooper, N.S. The clearance of antigen-antibody complexes from the blood by the reticulo-endothelial system. J. Immunol. 1959, 82, 131–137. [Google Scholar]
- Jain, R.K. The next frontier of molecular medicine: delivery of therapeutics. Nat. Med. 1998, 4, 655–657. [Google Scholar]
- Thrush, G.R.; Lark, L.R.; Clinchy, B.C.; Vitetta, E.S. Immunotoxins: an update. Annu. Rev. Immunol. 1996, 14, 49–71. [Google Scholar]
- Harvey, S.J.; Miner, J.H. Revisiting the glomerular charge barrier in the molecular era. Curr. Opin. Nephrol. Hypertension 2008, 17, 393–398. [Google Scholar]
- Kossmann, C.E.; Palade, G.E. Blood capillaries of the heart and other organs. Circulation 1961, 24, 368–384. [Google Scholar]
- Lum, H.; Malik, A.B. Regulation of vascular endothelial barrier function. Am. J. Physiol. Lung Cell Mol. Physiol. 1994, 267, L223–L241. [Google Scholar]
- Zhou, H.; Ohno, N.; Terada, N.; Saitoh, S.; Fujii, Y.; Ohno, S. Involvement of follicular basement membrane and vascular endothelium in blood-follicle barrier formation of mice revealed by ‘in vivo cryotechnique’. Reproduction 2007, 134, 307–317. [Google Scholar]
- Wieser, E.; Strohmeyer, D.; Rogatsch, H.; Horninger, W.; Bartsch, G; Debbage, P. Access of tumor-derived macromolecules and cells to the blood: an electron microscopical study of structural barriers in microvessel clusters in highly malignant primary prostate carcinomas. Prostate 2004, 62, 123–132. [Google Scholar]
- Bucior, I.; Scheuring, S.; Engel, A.; Burger, M.M. Carbohydrate-carbohydrate interaction provides adhesion force and specificity for cellular recognition. J. Cell Biol. 2004, 165, 529–537. [Google Scholar]
- Santacroce, M.; Orsini, F.; Mari, S.A.; Marinone, M.; Lenardi, C.; Bettè, S.; Sacchi, V.F.; Poletti, G. Atomic force microscopy imaging of Xenopus laevis oocyte plasma membrane purified by ultracentrifugation. Microsc. Res. Tech. 2008, 71, 397–402. [Google Scholar]
- Tate, R.L.; Holmes, J.M.; Kohn, L. Characteristics of a solubilized thyrotropin receptor from bovine thyroid plasma membranes. J. Biol. Chem. 1975, 250, 6527–6533. [Google Scholar]
- Ando, T.; Davies, T.F. Monoclonal antibodies to the thyrotropin receptor. Clin. Devel. Immunol. 2005, 12, 137–143. [Google Scholar]
- Camarillo, I.G.; Thordarson, G.; Moffat, J.G.; van Horn, K.M.; Binart, N.; Kelly, P.A.; Talamantes, F. Prolactin receptor expression in the epithelia and stroma of the rat mammary gland. J. Endocrinol. 2001, 171, 85–95. [Google Scholar]
- Barker, S.; Laird, S.M.; Ho, M.M.; Vinson, G.P.; Hinson, J.P. Characterization of a rat adrenocortical inner zone-specific antigen and identification of its putative precursor. J. Mol. Endocrinol. 1992, 9, 95–102. [Google Scholar]
- Molineux, I.J. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol. Microbiol. 2001, 40, 1–8. [Google Scholar]
- Förster, C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008, 130, 55–70. [Google Scholar]
- Minn, A.; Ghersi-Egea, J.F.; Perrin, R.; Leininger, B.; Siest, G. Drug metabolizing enzymes in the brain and cerebral microvessels. Brain Res. Rev. 1991, 16, 65–82. [Google Scholar]
- Brownlees, J.; Williams, C.H. Peptidases, peptides and the mammalian blood-brain barrier. J. Neurochem. 1993, 60, 793–803. [Google Scholar]
- Abbott, N.J.; Romero, I.A. Transporting therapeutics across the blood-brain barrier. Mol. Med. Today 1996, 2, 106–113. [Google Scholar]
- Beigneux, A.P.; Davies, B.S.J.; Gin, P.; Weinstein, M.M.; Farber, E.; Qiao, X.; Peale, F.; Bunting, S.; Walzem, R.L.; Wong, J.S.; Blaner, W.S.; Ding, Z.M.; Melford, K.; Wongsiriroj, N.; Shu, X.; de Sauvage, F.; Ryan, R.O.; Fong, L.G.; Bensadoun, A.; Young, S.G. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007, 5, 279–291. [Google Scholar]
- van Haaren, P.M.A.; van Bavel, E.; Vink, H.; Spaan, J.A.E. Charge modification of the endothelial surface layer modulates the permeability barrier of isolated rat mesenteric small arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2503–H2507. [Google Scholar]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar]
- Brunskill, N.J.; Stuart, J.; Tobin, A.B.; Walls, J.; Nahorski, S. Receptor-mediated endocytosis of albumin by kidney proximal tubule cells is regulated by phosphatidylinositide 3-kinase. J. Clin. Invest. 1998, 101, 2140–2150. [Google Scholar]
- Joó, F. The role of second messenger molecules in the regulation of permeability in the cerebral endothelial cells. Adv. Exp. Med. Biol. 1993, 331, 155–164. [Google Scholar]
- Keys, J.L.; King, G.J. Morphology of pig uterine subepithelial capillaries after topical and systemic oestrogen treatment. J. Reprod. Fertil. 1995, 105, 287–294. [Google Scholar]
- Schnitzer, J.E.; McIntosh, D.P.; Dvorak, A.M.; Liu, J.; Oh, P. Separation of caveolae from associated microdomains of GPI-anchored proteins. Science 1995, 269, 1435–1439. [Google Scholar]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar]
- Bannerman, D.D.; Paape, M.J.; Hare, W.R.; Sohn, E.J. Increased levels of LPS-binding protein in bovine blood and milk following bacterial lipopolysaccharide challenge. J. Dairy Sci. 2003, 86, 3128–3137. [Google Scholar]
- Disson, O.; Grayo, S.; Huillet, E.; Nikitas, G.; Langa-Vives, F.; Dussurget, O.; Ragon, M.; Le Monnier, A.; Babinet, C.; Cossart, P.; Lecuit, M. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 2008, 455, 1114–1118. [Google Scholar]
- Le Monnier, A.; Autret, N.; Join-Lambert, O.F.; Jaubert, F.; Charbit, A.; Berche, P.; Kalyal, S. ActA is required for crossing of the fetoplacental barrier by Listeria monocytogenes. Infect. Immun. 2007, 75, 950–957. [Google Scholar]
- Chiovato, L.; Latrofa, F.; Braverman, L.E.; Pacini, F.; Capezzone, M.; Masserini, L.; Grasso, L.; Pinchera, A. Disappearance of humoral thyroid autoimmunity after complete removal of thyroid antigens. Ann. Intern. Med. 2003, 139, 346–351. [Google Scholar]
- Fulmer, B.R.; Turner, T.T. A blood-prostate barrier restricts cell and molecular movement across the rat ventral prostate epithelium. J. Urol. 2000, 163, 1591–1594. [Google Scholar]
- Schadewinkel-Scherkl, A.M.; Rasmussen, F.; Merck, C.C.; Nielsen, P.; Frey, H.H. Active transport of benzyl-penicillin across the blood-milk barrier. Pharmacol. Toxicol. 2009, 73, 14–19. [Google Scholar]
- Banerjee, D.K.; Ornberg, R.L.; Youdim, M.B.H.; Heldman, E.; Pollard, H.B. Endothelial cells from bovine adrenal medulla develop capillary-like growth patterns in culture. Proc. Natl. Acad. Sci. USA 1985, 82, 4702–4706. [Google Scholar]
- Bennett, H.S.; Luft, J.H.; Hampton, J.C. Morphological classifications of vertebrate blood capillaries. Am. J. Physiol. 1959, 196, 381–390. [Google Scholar]
- Friend, D.S.; Gilula, N.B. Variations in tight and gap junctions in mammalian tissues. J. Cell Biol. 1972, 53, 758–776. [Google Scholar]
- Lewandowsky, M. Zur Lehre der Zerebrospinalflüssigkiet. Z. Klin. Med. 1890, 40, 480–494. [Google Scholar]
- Biedl, A.; Kraus, R. Über eine bisher unbekannte toxische Wirkung der Gallensäure auf das Zentralnervensystem. Zentralbl. Inn. Med. 1898, 19, 1185–1200. [Google Scholar]
- Ehrlich, P. Über die Beziehung chemischer Constitution, Vertheilung, und pharmakologischer Wirkung; Gesammelte Arbeiten zur Immunitätsforschung: Berlin, Germany, 1904. [Google Scholar]
- Goldmann, E.E. Vitalfärbung am Zentralnervensystem. Abh. Preuss. Wissensch. Phys. Math. 1913, 1, 1–60. [Google Scholar]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar]
- Brightman, M.W.; Reese, T.S. Junctions between intimately apposed cell membranes in the vertebrate brain. J. Cell Biol. 1969, 40, 648–677. [Google Scholar]
- Simionescu, M.; Popov, D.; Sima, A. Endothelial transcytosis in health and disease. Cell Tissue Res. 2009, 335, 27–40. [Google Scholar]
- Dempsey, E.W.; Wislocki, G.B. An electron microscopic study of the blood-brain barrier in the rat, employing silver nitrate as a vital stain. J. Biophys. Biochem. Cytol. 1955, 1, 245–256. [Google Scholar]
- Dermietzel, R.; Krause, D. Molecular anatomy of the blood-brain barrier as defined by immunocytochemistry. Int. Rev. Cytol. 1991, 127, 57–109. [Google Scholar]
- Revest, P.A.; Jones, H.C.; Abbott, N.J. The transendothelial DC potential of rat blood-brain barrier vessels in situ. Adv. Exp. Med. Biol. 1993, 331, 71–74. [Google Scholar]
- Pardridge, W.M.; Boado, R.J.; Farrell, C.R. Brain-type glucose transporter (GLUT-1) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridization. J. Biol. Chem. 1990, 265, 18035–18040. [Google Scholar]
- Duport, S.; Robert, E.; Muller, D.; Grau, G.; Parisi, L.; Stoppini, L. An in vitro blood-brain barrier model: cocultures between endothelial cells and organotypic brain slice cultures. Proc. Natl. Acad. Sci. USA 1998, 95, 1840–1845. [Google Scholar]
- Green, E. Blood-brain barrier: Size matters at the blood-brain barrier. Nat. Rev. Neurosci. 2003, 4, 525. [Google Scholar]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar]
- Bradbury, M.W.B. The blood-brain barrier. Exp. Physiol. 1993, 78, 453–472. [Google Scholar]
- Bonetta, L. Endothelial tight junctions form the blood-brain barrier. J. Cell Biol. 2005, 169, 378–379. [Google Scholar]
- Engelhardt, B. Development of the blood-brain barrier. Cell Tissue Res. 2003, 314, 119–129. [Google Scholar]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood-brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010. [Google Scholar]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar]
- Rubin, L.L.; Staddon, J.M. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 1999, 22, 11–28. [Google Scholar]
- Ueno, M. Molecular anatomy of the brain endothelial barrier: an overview of the distributional features. Curr. Med. Chem. 2007, 14, 1199–1206. [Google Scholar]
- Holash, J.A.; Harik, S.I.; Perry, G.; Stewart, P.A. Barrier properties of testis microvessels. Proc. Natl. Acad. Sci. USA 1993, 90, 11069–11073. [Google Scholar]
- Wong, C.H.; Cheng, C.Y. The blood-testis barrier: its biology, regulation, and physiological role in spermatogenesis. Curr. Top. Dev. Biol. 2005, 71, 263–296. [Google Scholar]
- Debbage, P.L.; Sölder, E.; Seidl, S.; Hutzler, P.; Hugl, B.; Öfner, D.; Kreczy, A. Intravital lectin perfusion analysis of vascular permeability in human micro- and macro- blood vessels. Histochem. Cell Biol. 2001, 116, 349–359. [Google Scholar]
- Enders, A.C.; Blankenship, T.N. Comparative placental structure. Adv. Drug Deliv. Rev. 1999, 38, 3–15. [Google Scholar]
- Soelder, E.; Hutzler, P.; Debbage, P. Permeabilitäten im Plazentaren Gefässbaum am Termin. 23. Jahrestagung der Österreichischen Gesellschaft für Reproduktionsmedizin und Endokrinologie, Innsbruck, Austria, 20-22 September 2007.
- Sölder, E.; Kremser, C.; Rohr, I.; Hutzler, P.; Debbage, P. Molecular mapping deep within a living human organ: analysis of microvessel function on the timescale of seconds and with sub-micrometre spatial resolution. Histochem. Cell Biol. 2009, 131, 537–551. [Google Scholar]
- Soelder, E.; Rohr, I.; Hutzler, P.; Debbage, P. Imaging of placental transport mechanisms: a review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 144 (Suppl. 1), 114–120. [Google Scholar]
- Vähäkangas, K.; Myllynen, P. Drug transporters in the human blood-placental barrier. Brit. J. Pharm. 2009, 158, 665–678. [Google Scholar]
- Tzelepi, V.N.; Tsamandas, A.C.; Vlotinou, H.D.; Vagianos, C.E.; Scopa, C.D. Tight junctions in thyroid carcinogenesis: diverse expression of claudin-1, claudin-4, claudin-7 and occludin in thyroid neoplasms. Mod. Pathol. 2008, 21, 22–30. [Google Scholar]
- Shennan, D.B.; Peaker, M. Transport of milk constituents by the mammary gland. Physiol. Rev. 2000, 80, 925–951. [Google Scholar]
- McManaman, J.L.; Neville, M.C. Mammary physiology and milk secretion. Adv. Drug Delivery Rev. 2003, 55, 629–641. [Google Scholar]
- Nguyen, D.D.; Neville, M.C. Tight junction regulation in the mammary gland. J. Mammary Gland Biol. Neopl. 1998, 3, 233–246. [Google Scholar]
- Monks, J.; Neville, M.C. Albumin transcytosis across the epithelium of the lactating mouse mammary gland. J. Physiol. 2004, 560, 267–280. [Google Scholar]
- Linzell, J.L.; Peaker, M. Changes in colostrum composition and in the permeability of the mammary epithelium at about the time of parturition in the goat. J. Physiol. 1974, 243, 129–151. [Google Scholar]
- Berga, S.E. Electrical potentials and cell-to-cell dye movement in mouse mammary gland during lactation. Am. J. Physiol. Cell Physiol. 1984, 247, C20–C25. [Google Scholar]
- Nguyen, D.D.; Parlow, A.F.; Neville, M.C. Hormonal regulation of tight junction closure in the mouse mammary epithelium during the transition from pregnancy to lactation. J. Endocrinol. 2001, 170, 347–356. [Google Scholar]
- Halsey, J.F.; Mitchell, C.; Meyer, R.; Cebra, J.J. Metabolism of immunoglobulin A in lactating mice: origins of immunoglobulin A in milk. European J. Immunol. 1982, 12, 107–112. [Google Scholar]
- Geursen, A.; Grigor, M.R. Serum albumin secretion in rat milk. J. Physiol. 1987, 391, 419–427. [Google Scholar]
- Swaisgood, H.E. Protein and amino acid composition of bovine milk. In Handbook of Milk Composition; Jensen, R.G., Ed.; Academic Press: San Diego, Washington DC, USA, 1995; pp. 464–467. [Google Scholar]
- Lonnerdal, B.; Atkinson, S. Nitrogenous components of milk: A. Human milk proteins. In Handbook of Milk Composition; Jensen, R.G., Ed.; Academic Press: Washington DC, USA, 1995; pp. 352–368. [Google Scholar]
- Jain, N.C.; Schalm, O.W.; Carroll, E.J.; Lasmanis, J. Experimental mastitis in leukopenic cows: immunologically induced neutropenia and response to intra-mammary inoculation of Aerobacter aerogenes. Am. J. Vet. Res. 1968, 37, 2089–2097. [Google Scholar]
- Carlos, T.M.; Harlan, J.M. Membrane proteins involved in phagocyte adherence to endothelium. Immunol. Rev. 1990, 114, 5–28. [Google Scholar]
- Guidry, A.J.; O'Brien, C.N.; Douglass, L.W. A bovine mammary endothelial/epithelial cell culture model of the blood/milk barrier. Can. J. Vet. Res. 1998, 62, 117–121. [Google Scholar]
- Landis, E.M.; Pappenheimer, J.R. Exchange of substances through the capillary walls. Handb. Physiol. 1963, 2, 961–1034. [Google Scholar]
- Palade, G.E. Blood capillaries of the heart and other organs. Circulation 1961, 24, 368–384. [Google Scholar]
- Jennings, M.A.; Marchesi, V.T.; Florey, H. The transport of particles across the walls of small blood vessels. Proc. R. Soc. Lond. B. Biol. Sci. 1962, 156, 14–19. [Google Scholar]
- Weihe, E.; Kalmbach, P. Ultrastructure of capillaries in the conduction system of the heart in various mammals. Cell Tissue Res. 1978, 192, 77–87. [Google Scholar]
- Anversa, P.; Giacomelli, F.; Wiener, J. Intercellular junctions of rat endocardium. Anat. Rec. 1975, 183, 477–483. [Google Scholar]
- Bruns, R.R.; Palade, G.E. Studies on blood capillaries: II. The transport of ferritin molecules across the wall of muscle capillaries. J. Cell Biol. 1968, 37, 277–299. [Google Scholar]
- Karnovsky, M.J. Vesicular transport of exogenous peroxidase across capillary endothelium into the T system of muscle. J. Cell Biol. 1965, 27, 49A–50A. [Google Scholar]
- Palade, G.E. Fine structure of blood capillaries. J. Appl. Phys. 1953, 24, 1424. [Google Scholar]
- Simionescu, N.; Simionescu, M.; Palade, G.E. Permeability of muscle capillaries to exogenous myoglobin. J. Cell Biol. 1973, 57, 2424–2452. [Google Scholar]
- Karnovsky, M.J. The ultrastructural basis of capillary permeability studied with peroxidase as a tracer. J. Cell Biol. 1967, 35, 213–236. [Google Scholar]
- Williams, M.C.; Wissig, S.L. The permeability of muscle capillaries to horseradish peroxidase. J. Cell Biol. 1975, 66, 531–555. [Google Scholar]
- Schlingemann, R.O.; Hofman, P.; Klooster, J.; Blaauwgeers, H.G.T.; Van der Gaag, R.; Vrensen, G.F.J.M. Ciliary muscle capillaries have blood-tissue barrier characteristics. Exp. Eye Res. 1998, 66, 747–754. [Google Scholar]
- Pietra, G.G.; Szidon, J.P.; Leventhal, M.M.; Fishman, A.P. Hemoglobin as a tracer in hemodynamic pulmonary edema. Science 1969, 166, 1643–1646. [Google Scholar]
- Schneeberger, E.E.; Karnovsky, M.J. The influence of intravascular fluid volume on the permeability of newborn and adult mouse lungs to ultrastructural protein tracers. J. Cell Biol. 1971, 49, 319–334. [Google Scholar]
- Schneeberger, E.E.; Karnovsky, M.J. The ultrastructural basis of alveolar-capillary membrane permeability to peroxidase used as a tracer. J. Cell Biol. 1975, 37, 781–793. [Google Scholar]
- Schneeberger, E.E.; Karnovsky, M.J. Substructure of intercellular junctions in freeze-fractured alveolar-capillary membranes of mouse lung. Circ. Res. 1976, 38, 404–411. [Google Scholar]
- Kim, K.-J.; Malik, A.B. Protein transport across the lung epithelial barrier. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L247–L259. [Google Scholar]
- Chapman, A.L.; Bopp, W.J. Electron microscopy of vascular barrier in thymus, tonsil, and lymph node of beagle pups. Am. J. Vet. Res. 1970, 31, 1255–1268. [Google Scholar]
- Abe, K.; Ito, T. Vascular permeability in the thymus of the mouse. Arch. Histol. Jpn. 1974, 36, 251–264. [Google Scholar]
- Mikata, A.; Niki, R. Permeability of postcapillary venules of the lymph node. An electron microscopic study. Exp. Mol. Pathol. 1971, 14, 289–305. [Google Scholar]
- Raviola, E.; Karnovsky, M.J. Evidence for a blood-thymus barrier using electron-opaque tracers. J. Exp. Medicine 1972, 136, 466–498. [Google Scholar]
- Kimm, M.H.; Hardin, J.A.; Gall, D.G. Transport of albumin into the intestinal lumen of the rat. Can. J. Physiol. Pharmacol. 1997, 75, 193–198. [Google Scholar]
- Simionescu, M.; Simionescu, N.; Palade, G.E. Permeability of intestinal capillaries. Pathway followed by dextrans and glycogen. J. Cell Biol. 1972, 53, 365–392. [Google Scholar]
- Sztul, E.S.; Howell, K.E.; Palade, G.E. Intracellular transport of secretory component and albumin in rat hepatocytes. J. Cell Biol. 1983, 97, 1582–1591. [Google Scholar]
- Maltby, B.; Lemon, G.J.; Bassingthwaighte, J.B.; Kelly, P.J. Exchange of potassium and strontium in adult bone. Am. J. Physiol. Heart Circ. Physiol. 1982, 242, H705–H712. [Google Scholar]
- Sörensson, J.; Matejka, G.L.; Ohlson, M.; Haraldsson, B. Human endothelial cells produce orosomucoid, an important component of the capillary barrier. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H530–H534. [Google Scholar]
- Dvorak, H.F. How tumors make bad blood vessels and stroma. Am. J. Pathol. 2003, 162, 1747–1757. [Google Scholar]
- Shlosberg, D.; Benifla, M.; Kaufer, D.; Friedman, A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 2010, 6, 393–403. [Google Scholar]
- Petrovski, K.R.; Stefanov, E. Milk composition changes during mastitis. Available online: www.milkproduction.com.
- Antonetti, D.A.; Barber, A.J.; Khin, S.; Lieth, E.; Tarbell, J.M.; Gardner, T.W. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content. Vascular endothelial growth factor decreases occludin in retinal endothelial cells. Diabetes 1998, 47, 1953–1959. [Google Scholar]
- Greenwood, J. Mechanisms of blood-brain barrier breakdown. Neuroradiology 2004, 33, 95–100. [Google Scholar]
- Schneider, S.W.; Ludwig, T.; Tatenhorst, L.; Braune, S.; Oberleithner, H.; Senner, V.; Paulus, W. Glioblastoma cells release factors that disrupt blood-brain barrier feature. Acta Neuropathol. 2004, 107, 272–276. [Google Scholar]
- Roberts, H.C.; Roberts, T.P.L.; Brasch, R.C.; Dillon, W.P. Quantitative measurement of microvascular permeability in human brain tumors achieved using dynamic contrast-enhanced MR imaging: correlation with histologic grade. Am. J. Neuroradiol. 2000, 21, 891–899. [Google Scholar]
- Cohen, F.M.; Kuwatsuru, R.; Shames, D.M.; Neuder, M.; Mann, J.S.; Vexler, V.; Rosenau, W.; Brasch, R.C. Contrast-enhanced magnetic resonance imaging estimation of altered capillary permeability in experimental mammary carcinomas after X-irradiation. Investig. Radiol. 1994, 29, 970–977. [Google Scholar]
- Baillie, C.T.; Winslet, M.C.; Bradley, N.J. Tumour vasculature - a potential therapeutic target. Brit. J. Cancer 1995, 72, 257–267. [Google Scholar]
- Denekamp, J.; Hobson, B. Endothelial cell proliferation in experimental tumours. Brit. J. Cancer 1982, 46, 711–720. [Google Scholar]
- Warren, B.A. The vascular morphology of tumors. In Tumor Blood Circulation: Angiogenesis, Vascular Morphology and Blood Flow of Experimental and Human Tumors; Peterson, H., Ed.; CRC Press: Boca Raton, FL, 1979; pp. 1–47. [Google Scholar]
- Mattsson, J.; Appelgren, L.; Hamberger, B.; Peterson, H. Tumor vessel innervation and influence of vasoactive drugs on tumor blood flow. In Tumor Blood Circulation: Angiogenesis, Vascular Morphology and Blood Flow of Experimental and Human Tumors; Peterson, H., Ed.; CRC Press: Boca Raton, FL, 1979; pp. 129–135. [Google Scholar]
- Dvorak, H.F.; Nagy, J.A.; Dvorak, J.T.; Dvorak, A.M. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am. J. Pathol. 1988, 133, 95–109. [Google Scholar]
- Furman-Haran, E.; Margalit, R.; Grobgeld, D.; Degani, H. Dynamic contrast-enhanced magnetic resonance imaging reveals stress-induced angiogenesis in MCF7 human breast tumors. Proc. Natl. Acad. Sci. USA 1996, 93, 6247–6251. [Google Scholar]
- van Dijke, C.F.; Brasch, R.C.; Roberts, T.P.; Weidner, N.; Mathur, A.; Shames, D.M.; Mann, J.S.; Demsar, F.; Lang, P.; Schwickert, H.C. Mammary carcinoma model: correlation of macromolecular contrast-enhanced MR imaging characterizations of tumor microvasculature and histologic capillary density. Radiology 1996, 198, 813–818. [Google Scholar]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release 2000, 65, 271–284. [Google Scholar]
- Greish, K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J. Drug Targeting 2007, 15, 457–464. [Google Scholar]
- Iyer, A.K.; Greish, K.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar]
- Tanaka, T.; Shiramoto, S.; Miyashita, M.; Fujishima, Y.; Kaneo, Y. Tumor targeting based on the effect of enhanced permeability and retention (EPR) and the mechanism of receptor-mediated endocytosis (RME). Int. J. Pharm. 2004, 277, 39–61. [Google Scholar]
- Lamprecht, A. IBD: Selective nanoparticle adhesion can enhance colitis therapy. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 311–312. [Google Scholar]
- Wang, D.; Miller, S.C.; Liu, X-M.; Anderson, B.; Wang, X.S.; Goldring, S.R. Novel dexamethasone-HPMA copolymer conjugate and its potential application in treatment of rheumatoid arthritis. Arthr. Res. Therap. 2007, 9, R2. [Google Scholar]
- Tarner, I.H.; Müller-Ladner, U. Drug delivery systems for the treatment of rheumatoid arthritis. Expert Opin. Drug Deliv. 2008, 5, 1027–1037. [Google Scholar]
- Lukyanov, A.N.; Hartner, W.C.; Torchilin, V.P. Increased accumulation of PEG-PE micelles in the area of experimental myocardial infarction in rabbits. J. Control. Release 2004, 94, 187–193. [Google Scholar]
- Schiffelers, R.M.; Banciu, M.; Metselaar, J.M.; Storm, G. Therapeutic application of long-circulating liposomal glucocorticoids in auto-immune diseases and cancer. J. Liposome Res. 2006, 16, 185–194. [Google Scholar]
- Mulder, W.J.M.; Strijkers, G.J.; van Tilborg, G.A.F.; Griffioen, A.W.; Nicolay, K. Lipid-based nanoparticles for contrast-enhanced MRI and molecular imaging. NMR Biomed. 2006, 19, 142–164. [Google Scholar]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer's and Parkinson's disease: implications for drug therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar]
- Kalaria, R.N. The blood-brain barrier and cerebrovascular pathology in Alzheimer's Disease. Ann. N. York Acad. Sci. 2006, 893, 113–125. [Google Scholar]
- Lien, W.M.; Ackerman, N.B. The blood supply of experimental liver metastases. II. A microcirculatory study of normal and tumor vessels of the liver with the use of perfused silicone rubber. Surgery 1970, 68, 334–340. [Google Scholar]
- Molema, G.; de Leij, L.F.; Meijer, D.K. Tumor vascular endothelium: barrier or target in tumor directed drug delivery and immunotherapy. Pharm. Res. 1997, 14, 2–10. [Google Scholar]
- Béduneau, A.; Saulnier, P.; Benoit, J.-P. Active targeting of brain tumors using nanocarriers. Biomaterials 2007, 28, 4947–4967. [Google Scholar]
- Debbage, P.; Jaschke, W. Molecular imaging with nanoparticles: giant roles for dwarf actors. Histochem. Cell Biol. 2008, 130, 845–875. [Google Scholar]
- Pardridge, W.M. CNS drug design based on principles of blood-brain barrier transport. J. Neurochem. 1998, 70, 1781–1792. [Google Scholar]
- Vaughan, A.T.M.; Anderson, P.; Dykes, P.W.; Chapman, C.E.; Bradwell, A.R. Limitations to the killing of tumours using radiolabelled antibodies. Brit. J. Radiol. 1987, 60, 567–572. [Google Scholar]
- Mathijssen, R.H.J.; de Jong, F.A.; Loos, W.J.; van der Bol, J.M.; Verweij, J.; Sparreboom, A. Flat-fixed dosing versus body surface area-based dosing of anticancer drugs in adults: Does it make a difference? Oncologist 2007, 12, 913–923. [Google Scholar]
- Walko, C.M.; McCloud, H.L. Will we ever be ready for blood level-guided therapy? J. Clin. Oncol. 2008, 26, 2078–2079. [Google Scholar]
- Debbage, P. Targeted drugs and nanomedicine: present and future. Curr. Pharm. Design 2009, 15, 153–172. [Google Scholar]
- McIntosh, D.P.; Tan, X-Y.; Oh, P.; Schnitzer, J.E. Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: a pathway to overcome cell barriers to drug and gene delivery. Proc. Nat. Acad. Sci. USA 2002, 99, 1996–2001. [Google Scholar]
- Siegal, T.; Rubinstein, R.; Bokstein, F.; Schwartz, A.; Lossos, A.; Shalom, E.; Chisin, R.; Gomori, J.M. In vivo assessment of the window of barrier opening after osmotic blood-brain barrier disruption in humans. J. Neurosurg. 2000, 92, 599–605. [Google Scholar]
- Greenwood, J.; Mason, J.C. Statins and the vascular endothelial inflammatory response. Trends Immunol. 2007, 28, 88–98. [Google Scholar]
- Kimura, H.; Weisz, A.; Kurashima, Y.; Hashimoto, K.; Ogura, T.; D'Acquisto, F.; Addeo, R.; Makuuchi, M.; Esumi, H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood 2000, 95, 189–197. [Google Scholar]
- Kreuter, J.; Alyautdin, R.N.; Kharkevich, D.A.; Ivanov, A.A. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). Brain Res. 1995, 674, 171–174. [Google Scholar]
- Sun, W.; Xie, C.; Wang, H.; Hu, Y. Specific role of polysorbate 80 coating on the targeting of nanoparticles to the brain. Biomaterials 2004, 25, 3065–3071. [Google Scholar]
- Flacke, S.; Fischer, S.; Scott, M.J.; Fuhrhop, R.J.; Allen, J.S.; McLean, M.; Winter, P.; Sicard, G.A.; Gaffney, P.J.; Wickline, S.A.; Lanza, G.M. Novel MRI contrast agent for molecular imaging of fibrin: implications for detecting vulnerable plaques. Circulation 2001, 104, 1280–1285. [Google Scholar]
- Winter, P.M.; Cai, K.; Chen, J.; Adair, C.R.; Kiefer, G.E.; Athey, P.S.; Gaffney, P.J.; Buff, C.E.; Robertson, J.D.; Caruthers, S.D.; Wickline, S.A.; Lanza, G.M. Targeted PARACEST nanoparticle contrast agent for the detection of fibrin. Magn. Reson. Med. 2006, 56, 1384–1388. [Google Scholar]
- Miele, E.; Spinelli, G.P.; Miele, E.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast cancer. Int. J. Nanomed. 2009, 4, 99–105. [Google Scholar]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol. Sci. 1999, 30, 592–599. [Google Scholar]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Nanomedicine: current status and future prospects. FASEB J. 2005, 19, 311–330. [Google Scholar]
- Sinha, R.; Kim, G.J.; Nie, S.; Shin, D.M. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol. Cancer Ther. 2006, 5, 1909–1917. [Google Scholar]
- Smith, T.D.; Bhatnagar, K.P.; Tuladhar, P.; Burrows, A.M. Distribution of olfactory epithelium in the primate nasal cavity: Are “microsmia” and “macrosmia” valid morphological concepts? Anatom. Record 2004, 281A, 1173–1181. [Google Scholar]
- Nedelec, S.; Dubacq, C.; Trembleau, A. Morphological and molecular features of the mammalian olfactory sensory neuron axons: What makes these axons so special? J. Neurocytol. 2005, 34, 49–64. [Google Scholar]
- Dahan, M.; Lévi, S.; Luccardini, C.; Rostaing, P.; Riveau, B.; Triller, A. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science 2003, 302, 442–445. [Google Scholar]
- Zhang, Q.; Li, Y.; Tsien, R.W. The dynamic control of kiss-and-run and vesicular reuse probed with single nanoparticles. Science 2009, 323, 1448–1453. [Google Scholar]
- Rajan, S.S.; Vu, T.Q. Quantum dots monitor TrkA receptor dynamics in the interior of neural PC12 cells. Nanoletters 2006, 6, 2049–2059. [Google Scholar]
- Wang, Y-C.; Wu, Y-T.; Huang, H.-Y.; Lin, H-I.; Lo, L.-W.; Tzeng, S.-F.; Yang, C.-S. Sustained intraspinal delivery of neurotrophic factor encapsulated in biodegradable nanoparticles following contusive spinal cord injury. Biomaterials 2008, 29, 4546–4553. [Google Scholar]
- Oberdörster, G.; Elder, A.; Rinderknecht, A. Nanoparticles and the brain: cause for concern? J. Nanosci. Nanotechnol. 2009, 9, 4996–5007. [Google Scholar]
- Zhan, J.; Brys, M.; Glodzik, L.; Tsui, W.; Javier, E.; Wegie, J.; Kuchna, I.; Pirraglia, E.; Li, Y.; Mosconi, L.; Saint Louis, L.A.; Switalski, R.; De Santi, S.; Kim, B.C.; Wisniewski, T.; Reisberg, B.; Bobinski, M.; de Leon, M.J. An entorhinal cortex sulcal pattern is associated with Alzheimer's disease. Hum. Brain Mapp. 2009, 3, 874–882. [Google Scholar]
- Bergot, A.-S.; Durgeau, A.; Levacher, B.; Colombo, B.M.; Cohenand, J.L.; Klatzmann, D. Antigen quality determines the efficiency of antitumor immune responses generated in the absence of regulatory T cells. Cancer Gene Therap. 2010, 17, 645–654. [Google Scholar]
- Kaptzan, T.; Skutelsky, E.; Itzhaki, O.; Sinai, J.; Michowitz, M.; Yossipov, Y.; Schiby, G.; Leibovici, J. Age-dependent differences in the efficacy of cancer immunotherapy in C57BL and AKR mouse strains. Exp. Gerontol. 2004, 39, 1035–1048. [Google Scholar]
- Trefzer, U.; Weingart, G.; Chen, Y.; Herberth, G.; Adrian, K.; Winter, H.; Audring, H.; Guo, Y.; Sterry, W.; Walden, P. Hybrid cell vaccination for cancer immune therapy: first clinical trial with metastatic melanoma. Int. J. Cancer 2000, 85, 618–626. [Google Scholar]
- Chapman, P.T.; Jamar, F.; Keelan, E.T.; Peters, A.M.; Haskard, D.O. Use of a radiolabeled monoclonal antibody against E-selectin for imaging of endothelial activation in rheumatoid arthritis. Arthritis Rheum. 1996, 39, 1371–1375. [Google Scholar]
- Jones, S.P.; Trocha, S.D.; Strange, M.B.; Granger, D.N.; Kevil, C.G.; Bullard, D.C.; Lefer, D.J. Leukocyte and endothelial cell adhesion molecules in a chronic murine model of myocardial reperfusion injury. Am. J. Physiol. 2000, 279, H2196–H2201. [Google Scholar]
- Schurmann, G.M.; Bishop, A.E.; Facer, P.; Vecchio, M.; Lee, J.C.; Rampton, D.S.; Polak, J.M. Increased expression of cell adhesion molecule P-selectin in active inflammatory bowel disease. Gut 1995, 36, 411–418. [Google Scholar]
- Shieh, C.-C.; Sadasivan, B.K.; Russell, G.J.; Schön, M.P.; Parker, C.M.; Brenner, M.B. Lymphocyte adhesion to epithelia and endothelia mediated by the lymphocyte endothelial-epithelial cell adhesion molecule glycoprotein. J. Immunol. 1999, 163, 1592–1601. [Google Scholar]
- Cybulsky, M.I.; Gimbrone, M.A., Jr. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science 1991, 251, 788–791. [Google Scholar]
- van der Wal, A.C.; Das, P.K.; Tigges, A.J.; Becker, A.E. Adhesion molecules on the endothelium and mononuclear cells in human atherosclerotic lesions. Am. J. Pathol. 1992, 141, 1427–1433. [Google Scholar]
- Clarke, M.S.F.; West, D.C. The identification of proliferation and tumour-induced proteins in human endothelial cells: a possible target for tumour therapy. Electrophoresis 1991, 12, 500–508. [Google Scholar]
- Davies, G.; Rmali, K.A.; Watkins, G.; Mansel, R.E.; Mason, M.D.; Jiang, W.G. Elevated levels of tumour endothelial marker-8 in human breast cancer and its clinical significance. Int. J. Oncol. 2006, 29, 1311–1317. [Google Scholar]
- Magnussen, A.; Kasman, I.M.; Norberg, S.; Baluk, P.; Murray, R.; McDonald, D.M. Rapid access of antibodies to α5ß1 integrin overexpressed on the luminal surface of tumor blood vessels. Cancer Res. 2005, 65, 2712–2721. [Google Scholar]
- Rmali, K.A.; Watkins, G.; Harrison, G.; Parr, C.; Puntis, M.C.A.; Jiang, W.G. Tumour endothelial marker 8 (TEM-8) in human colon cancer and its association with tumour progression. Eur. J. Surg. Oncol. 2004, 30, 948–953. [Google Scholar]
- Strickland, L.A.; Jubb, A.M.; Hongo, J.-A.; Zhong, F.; Burwick, J.; Fu, L.; Frantz, G.D.; Koeppen, H. Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-A (VEGF). J. Pathol. 2005, 206, 466–475. [Google Scholar]
- Sakhalkar, H.S.; Dalal, M.K.; Salem, A.K.; Ansari, R.; Fu, J.; Kiani, M.F.; Kurjiaka, D.T.; Hanes, J.; Shakesheff, K.M.; Goetz, D.J. Leukocyte-inspired biodegradable particles that selectively and avidly adhere to inflamed endothelium in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 15895–15900. [Google Scholar]
- Oh, P.; Li, Y.; Yu, J.; Durr, E.; Krasinska, K.M.; Carver, L.A.; Testa, J.E.; Schnitzer, J.E. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature 2004, 429, 629–635. [Google Scholar]
- Valadon, P.; Garnett, J.D.; Testa, J.E.; Bauerle, M.; Oh, P.; Schnitzer, J.E. Screening phage display libraries for organ-specific vascular immunotargeting in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 407–412. [Google Scholar]
- Brekken, R.A.; Sage, E.H. SPARC, a matricellular protein: at the crossroads of cell-matrix. Matrix Biol. 2000, 19, 569–580. [Google Scholar]
- Broadwell, R.D.; Baker-Cairns, B.J.; Friden, P.M.; Oliver, C.; Villegas, J.C. Transcytosis of protein through the mammalian cerebral epithelium and endothelium: III. Receptor-mediated transcytosis through the blood-brain barrier of blood-borne transferrin and antibody against the transferrin receptor. Exp. Neurol. 1996, 142, 47–65. [Google Scholar]
- Fishman, J.B.; Rubin, J.B.; Handrahan, J.V.; Connor, J.R.; Fine, R.E. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J. Neurosci. Res. 1987, 18, 299–304. [Google Scholar]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312, 162–163. [Google Scholar]
- Megias, L.; Guerri, C.; Fornas, E.; Azorin, I.; Bendala, E.; Sancho-Tello, M.; Durán, J.M.; Tomás, M.; Gomez-Lechon, M.J.; Renau-Piqueras, J. Endocytosis and transcytosis in growing astrocytes in primary culture. Possible implications in neural development. Int. J. Dev. Biol. 2000, 44, 209–221. [Google Scholar]
- Lu, W.; Zhang, Y.; Tan, Y.-Z.; Hu, K.-L; Jiang, X.-G.; Fu, S.-K. Cationic albumin-conjugated pegylated nanoparticles as novel drug carrier for brain delivery. J. Control. Release 2005, 107, 428–448. [Google Scholar]
- Zensi, A.; Begley, D.; Pontikis, C.; Legros, C.; Mihoreanu, L.; Wagner, S.; Büchel, C.; von Briesen, H.; Kreuter, J. Albumin nanoparticles targeted with ApoE enter the CNS by transcytosis and are delivered to neurones. J. Control. Release 2009, 137, 78–86. [Google Scholar]
- Michaelis, K.; Hoffmann, M.M.; Dreis, S.; Herbert, E.; Alkyautdin, R.N.; Michaelis, M.M.; Kreuter, J.; Langer, K. Covalent linkage of apolipoprotein E to albumin nanoparticles strongly enhances drug transport into the brain. J. Pharmacol. Exp. Ther. 2006, 317, 1246–1253. [Google Scholar]
- Cormode, D.P.; Briley-Saebo, K.C.; Mulder, W.J.; Aguinaldo, J.G.; Barazza, A.; Ma, Y.; Fisher, E.A.; Fayad, Z.A. An ApoA-I mimetic peptide high-density-lipoprotein-based MRI contrast agent for atherosclerotic plaque composition detection. Small 2008, 4, 1437–1444. [Google Scholar]
- Cormode, D.P.; Chandrasekar, R.; Delshad, A.; Briley-Saebo, K.C.; Calcagno, C.; Barazza, A.; Mulder, W.J.M.; Fisher, E.A.; Fayad, Z.A. Comparison of synthetic high density lipoprotein (HDL) contrast agents for MR imaging of atherosclerosis. Bioconjug. Chem. 2009, 20, 937–943. [Google Scholar]
- Frias, J.C.; Ma, Y.; Williams, K.J.; Fayad, Z.A.; Fisher, E.A. Properties of a versatile nanoparticle platform contrast agent to image and characterize atherosclerotic plaques by magnetic resonance imaging. Nano Lett. 2006, 6, 2220–2224. [Google Scholar]
- Steinman, R.M.; Brodie, S.E.; Cohn, Z.A. Membrane flow during pinocytosis. J. Cell Biol. 1976, 68, 665–687. [Google Scholar]
- Oh, P.; Borgström, P.; Witkiewicz, H.; Li, Y.; Borgström, B.J.; Chrastina, A.; Iwata, K.; Zinn, K.R.; Baldwin, R.; Testa, J.E.; Schnitzer, J.E. Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat. Biotechnol. 2007, 25, 327–337. [Google Scholar]
- Schnitzer, J.E. Caveolae: from basic trafficking mechanisms to targeting transcytosis for tissue-specific drug and gene delivery in vivo. Adv. Drug Deliv. Rev. 2001, 49, 265–280. [Google Scholar]
- Tuma, P.L.; Hubbard, A.L. Transcytosis: crossing cellular barriers. Physiol. Rev. 2003, 83, 871–932. [Google Scholar] [PubMed]
- Le Roy, C.; Wrana, J.L. Clathrin- and non-clathrin mediated endocytic regulation of cell signaling. Nat. Rev. 2005, 6, 112–126. [Google Scholar]
- Miaczynska, M.; Stenmark, H. Mechanisms and functions of endocytosis. J. Cell Biol. 2008, 180, 7–11. [Google Scholar]
- Qian, Z.M.; Li, H.; Sun, H.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar]
- Roth, M.G. Clathrin-mediated endocytosis before fluorescent proteins. Nat. Rev. Mol. Cell Biology 2006, 7, 63–68. [Google Scholar]
- Rippe, B.; Rosengren, B.-I.; Carlsson, O.; Venturoli, D. Transendothelial transport: the vesicle controversy. J. Vasc. Res. 2002, 39, 375–390. [Google Scholar]
- Juurlink, B.H.J.; Devon, R.M. Macromolecular translocation - a possible function of astrocytes. Brain Res. 1990, 533, 73–77. [Google Scholar]
- Brightman, M.W. The distribution within the brain of ferritin injected into cerebrospinal fluid compartments. I. Ependymal distribution. J. Cell Biol. 1965, 26, 99–123. [Google Scholar] [PubMed]
- Phillipe, J.M.; Dubois, J.M.; Rouzaire-Dubois, B.; Cartron, P.F.; Vallette, F.; Morel, N. Functional expression of V-ATPases in the plasma membrane of glial cells. Glia 2002, 37, 365–373. [Google Scholar]
- Tabernero, A.; Velasco, A.; Granda, B.; Lavado, E.M.; Medina, J.M. Transcytosis of albumin in astrocytes activates the sterol regulatory element-binding protein-1, which promotes the synthesis of the neurotrophic factor oleic acid. J. Biol. Chem. 2002, 277, 4240–4246. [Google Scholar]
- Brasnjevic, I.; Steinbusch, H.W.M.; Schmitz, C.; Martinez-Martinez, P.; the European NanoBioPharmaceutics Research Initiative. Delivery of peptide and protein drugs over the blood-brain barrier. Progr. Neurobiol. 2009, 87, 212–251. [Google Scholar]
- Curry, S.; Brick, P.; Franks, N.P. Fatty acid binding to human serum albumin: new insights from crystallographic studies. Biochem. Biophys. Acta 1999, 1441, 131–140. [Google Scholar]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar]
- Carver, L.A.; Schnitzer, J.E. Caveolae: Mining little caves for new cancer targets. Nat. Rev. 2003, 3, 571–581. [Google Scholar]
- Milici, A.J.; Watrous, N.E.; Stukenbrok, H.; Palade, G.E. Transcytosis of albumin in capillary endothelium. J. Cell Biol. 1987, 105, 2603–2612. [Google Scholar]
- Ghitescu, L.; Fixman, A.; Simionescu, M.; Simionescu, N. Specific binding sites for albumin restricted to plasmalemmal vesicles of continuous capillary endothelium: receptor-mediated transcytosis. J. Cell Biol. 1986, 102, 1304–1311. [Google Scholar]
- Oh, P.; McIntosh, D.P.; Schnitzer, J.E. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J. Cell Biol. 1998, 141, 101–114. [Google Scholar]
- Schnitzer, J.E.; Oh, P.; McIntosh, D.P. Role of GTP hydrolysis in fission of caveolae directly from plasma membranes. Science 1996, 274, 239–242. [Google Scholar]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 2002, 109, 1541–1550. [Google Scholar]
- Conrad, P.A.; Smart, E.J.; Ying, Y.S.; Anderson, R.G.; Bloom, G.S. Caveolin cycles between plasma membrane caveolae and the Golgi complex by microtubule-dependent and microtubule-independent steps. J. Cell Biol. 1995, 131, 1421–1433. [Google Scholar]
- Smart, E.J.; Ying, Y.S.; Conrad, P.A.; Anderson, R.G.W. Caveolin moves from caveolae to the Golgi apparatus in response to cholesterol oxidation. J. Cell Biol. 1994, 127, 1185–1197. [Google Scholar]
- Le, P.U.; Nabi, I.R. Distinct caveolae-mediated endocytic pathways target the Golgi apparatus and the endoplasmic reticulum. J. Cell Biol. 2003, 116, 1059–1071. [Google Scholar]
- Puri, V.; Watanabe, R.; Singh, R.D.; Dominguez, M.; Brown, J.C.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Clathrin dependent and -independent internalization of plasma membrane sphingolipids initiates two Golgi targeting pathways. J. Cell Biol. 2001, 154, 535–547. [Google Scholar]
- Kartenbeck, J.; Stukenbrok, H.; Helenius, A. Endocytosis of simian virus 40 into the endoplasmic reticulum. J. Cell Biol. 1989, 109, 2721–2729. [Google Scholar]
- Pelkmans, L.; Kartenback, J.; Helenius, A. Caveolar endocytosis of Simian virus 40 reveals a novel two-step vesicular transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar]
- Benlimame, N.; Le, P.U.; Nabi, I.R. Localization of autocrine motility factor receptor to caveolae and clathrin independent internalization of its ligand to smooth endoplasmic reticulum. Mol. Biol. Cell 1998, 9, 1773–1786. [Google Scholar]
- Schnitzer, J.E.; Oh, P.; Pinney, E.; Allard, J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecule. J. Cell Biol. 1994, 127, 1217–1232. [Google Scholar]
- Tran, D.; Carpentier, J.L.; Sawano, F.; Gorden, P.; Orci, L. Ligands internalized through coated or noncoated invaginations follow a common intracellular pathway. Proc. Natl. Acad. Sci. USA 1987, 84, 7957–7961. [Google Scholar]
- Schnitzer, J.E. The endothelial cell surface and caveolae in health and disease. In Vascular Endothelium: Physiology, Pathology and Therapeutic Opportunities; Born, G.V.R., Schwartz, C.J., Eds.; Schattauer: Stuttgart, Germany, 1997; pp. 77–95. [Google Scholar]
- Simionescu, M.; Simionescu, N. Endothelial transport of macromolecules: transcytosis and endocytosis. A look from cell biology. Cell Biol. Rev. 1991, 25, 1–78. [Google Scholar]
- Zhu, P.; Chertova, E.; Bess, J., Jr.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar]
- Huang, C.-C.; Tang, M.; Zhang, M.-Y.; Majeed, S.; Montabana, E.; Stanfield, R.L.; Dimitrov, D.S.; Korber, B.; Sodroski, J.; Wilson, I.A.; Wyatt, R.; Kwong, P.D. Structure of a V3-containing HIV-1 gp120 core. Science 2005, 310, 1025–1028. [Google Scholar]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 2008, 455, 109–113. [Google Scholar]
- Park, E.Y.; Smith, M.J.; Stropp, E.S.; Snapp, K.R.; DiVietro, J.A.; Walker, W.F.; Schmidtke, D.W.; Diamond, S.L.; Lawrence, M.B. Comparison of PSGL-1 microbead and neutrophil rolling: microvillus elongation stabilizes P-selectin bond clusters. Biophys. J. 2002, 82, 1835–1847. [Google Scholar]
- Kienberger, F.; Kada, G.; Mueller, H.; Hinterdorfer, P. Single molecule studies of antibody-antigen interaction strength versus intra-molecular antigen stability. J. Mol. Biol. 2005, 347, 597–606. [Google Scholar]
- Hinterdorfer, P.; Baumgartner, W.; Gruber, H.J.; Schilcher, K.; Schindler, H. Detection and localization of individual antibody-antigen recognition events by atomic force microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 3477–3481. [Google Scholar]
- Saleh, O.A.; Sohn, L.L. Direct detection of antibody-antigen binding using an on-chip artificial pore. Proc. Natl. Acad. Sci. USA 2003, 100, 820–824. [Google Scholar]
- Fritz, J.; Katopodis, A.G.; Kolbinger, F.; Anselmetti, D. Force-mediated kinetics of single P-selectin/ligand complexes observed by atomic force microscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 12283–12288. [Google Scholar]
- Hanley, W.; McCarty, O.; Jadhav, S.; Tseng, Y.; Wirtz, D.; Konstantopoulos, K. Single molecule characterization of P-selectin/ligand binding. J. Biol. Chem. 2003, 278, 10556–10561. [Google Scholar]
- Dammer, U.; Popescu, O.; Wagner, P.; Anselmetti, D.; Guntherodt, H.J.; Misevic, G.N. Binding strength between cell adhesion proteoglycans measured by atomic force microscopy. Science 1995, 267, 1173–1175. [Google Scholar]
- Popescu, O.; Checiu, I.; Ghergel, P.; Simon, Z.; Misevic, G.N. Quantitative and qualitative approach of glycan-glycan interactions in marine sponges. Biochimie 2003, 85, 181–188. [Google Scholar]
- Shao, J.-Y.; Hochmuth, R.M. Micropipette suction for measuring piconewton forces of adhesion and tether formation from neutrophil membranes. Biophys. J. 1996, 71, 2892–2901. [Google Scholar]
- Waugh, R.E.; Bauserman, R.G. Physical measurements of bilayer-skeletal separation forces. Ann. Biomed. Eng. 1995, 23, 308–321. [Google Scholar]
- Dai, J.; Sheetz, M.P. Mechanical properties of neuronal growth cone membrane studied by tether formation with laser optical tweezers. Biophys. J. 1995, 68, 988–996. [Google Scholar]
- Hochmuth, R.M.; Shao, J.-Y.; Dai, J.; Sheetz, M.P. Deformation and flow of membrane into tethers extracted from neuronal growth cones. Biophys. J. 1996, 70, 358–369. [Google Scholar]
- Alon, R.; Hammer, D.A.; Springer, T.A. Lifetime of the P-selectin-carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature 1995, 374, 539–542. [Google Scholar]
- Veronese, F.M. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar]
- Berry, C.C.; Curtis, A.S.G. Functionalisation of magnetic nanoparticles for applications in biomedicine. J. Phys. D. Appl. Phys. 2003, 36, R198–R206. [Google Scholar]
- Geiser, M.; Kreyling, W.G. Deposition and biokinetics of inhaled nanoparticles. Par. Fibre Toxicol. 2010, 7, 2. [Google Scholar]
- Chen, W.; Yang, X. Researches on PEG-modified copolymer nanoparticle. Article in Chinese. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi 2003, 20, 143–147. [Google Scholar]
- Brunner, R.; Jensen-Jarolim, E.; Pali-Schöll, I. The ABC of clinical and experimental adjuvants - A brief overview. Immunol. Lett. 2010, 128, 29–35. [Google Scholar]
- Frey, A.; Mantis, N.; Kozlowski, P.A.; Quayle, A.J.; Bajardi, A.; Perdomo, J.J.; Robey, F.A.; Neutra, M.R. Immunization of mice with peptomers covalently coupled to aluminum oxide nanoparticles. Vaccine 1998, 17, 3007–3019. [Google Scholar]
- Frey, A.; Neutra, M.R.; Robey, F.A. Spatially aligned conjugated composition having a thioether bond linkage. US Patent 6086881, 2000. [Google Scholar]
- Schöll, I.; Weissenböck, A.; Förster-Waldl, E.; Untersmayr, E.; Walter, F.; Willheim, M.; Boltz-Nitulescu, G.; Scheiner, O.; Gabor, F.; Jensen-Jarolim, E. Allergen-loaded biodegradable poly(D,L-lactic-co-glycolic) acid nanoparticles down-regulate an ongoing Th2 response in the BALB/c mouse model. Clin. Exp. Aller. 2004, 34, 315–321. [Google Scholar]
- Baumeister, H. A novel human expression system for production of higher active biotherapeutics with optimised glycosylation. Pharm. Chem. Biopharm. 2006, 2, 21–24. [Google Scholar]
- Baumeister, H.; Goletz, S. Novel glycosylation technologies for the development of biosimilars and biobetters. Innova. Pharma. Technol. 2009, 52–58. [Google Scholar]
- Brooks, S.A. Appropriate glycosylation of recombinant proteins for human use: implications of choice of expression system. Mol. Biotechnol. 2004, 28, 241–255. [Google Scholar]
- Kawasaki, N.; Itoh, S.; Hashii, N.; Takakura, D.; Qin, Y.; Huang, X.; Yamaguchi, T. The significance of glycosylation analysis in development of biopharmaceuticals. Biol. Pharm. Bull. 2009, 32, 796–800. [Google Scholar]
- Stollenwerk, M.M.; Pashkunova-Martic, I.; Kremser, C.; Talasz, H.; Thurner, G.C.; Abdelmoez, A.A.; Wallnöfer, E.A.; Helbok, A.; Neuhauser, E.; Klammsteiner, N.; Klimaschewski, L.; von Guggenberg, E.; Fröhlich, E.; Keppler, B.; Jaschke, W.; Debbage, P. Albumin-based nanoparticles as Magnetic Resonance contrast agents: I. Concept, first syntheses and characterisation. Histochem. Cell Biol. 2010, 133, 375–404. [Google Scholar]
- Abdelmoez, A.A.; Thurner, G.C.; Wallnöfer, E.A.; Klammsteiner, N.; Kremser, C.; Talasz, H.; Mrakovcic, M.; Froehlich, E.; Jaschke, W.; Debbage, P. Albumin-based nanoparticles as Magnetic Resonance contrast agents: II. Physicochemical characterization of purified and standardised nanoparticles. Histochem. Cell Biol. 2010, 134, 171–196. [Google Scholar]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar]
- Peters, T., Jr. All about Albumin. Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, USA, 1996; p. 432. [Google Scholar]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar]
- Greco, F.; Vicent, M.J. Polymer-drug conjugates: current status and future trends. Front. Biosci. 2008, 13, 2744–2756. [Google Scholar]
- Bharali, D.J.; Khalil, M.; Gurbuz, M.; Simone, T.M.; Mousa, S.A. Nanoparticles and cancer therapy: A concise review with emphasis on dendrimers. Int. J. Nanomed. 2009, 4, 1–7. [Google Scholar]
- Duncan, R.; Izzo, L. Dendrimer biocompatibility and toxicity. Adv. Drug Deliv. Rev. 2005, 57, 2215–2237. [Google Scholar]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar]
- Moghimi, S.M.; Patel, H.M. Serum-mediated recognition of liposomes by phagocytic cells of the reticuloendothelial system - The concept of tissue specificity. Adv. Drug Deliv. Rev. 1998, 32, 45–60. [Google Scholar]
- Senior, J.H. Fate and behaviour of liposomes in vivo: a review of controlling factors. CRC Crit. Rev. Ther. Drug Carrier System 1987, 3, 123–193. [Google Scholar]
- Bonté, F.; Hsu, M.J.; Papp, A.; Wu, K.; Regen, S.L.; Juliano, R.L. Interactions of polymerizable phosphatidylcholine vesicles with blood components: relevance to biocompatibility. Biochim. Biophys. Acta 1987, 900, 1–9. [Google Scholar]
- Juliano, R.L. Factors controlling the kinetics and tissue distribution of liposomes, microspheres, and emulsion. Adv. Drug Delivery Rev. 1988, 2, 31–54. [Google Scholar]
- Chonn, A.; Cullis, P.R.; Devine, D.V. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J. Immunol. 1991, 146, 4234–4241. [Google Scholar]
- Chonn, A.; Semple, S.C.; Cullis, P.R. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J. Biol. Chem. 1992, 267, 18759–18765. [Google Scholar]
- Patel, H.M. Serum opsonins and liposomes: Their interaction and opsonophagocytosis. Crit. Rev. Ther. Drug Carrier Syst. 1992, 9, 39–90. [Google Scholar]
- Dave, J.; Patel, H.M. Differentiation in hepatic and splenic phagocytic activity during reticuloendothelial blockade with cholesterol free and cholesterol rich vesicles. Biochim. Biophys. Acta 1986, 888, 184–190. [Google Scholar]
- Liu, D.; Hu, Q.; Song, Y.K. Liposome clearance from blood: different animal species have different mechanisms. Biochim. Biophys. Acta 1995, 1240, 277–284. [Google Scholar]
- Torchilin, V.; Trubetskoy, V. Which polymers can make nanoparticulate drug carriers long-circulating? Adv. Drug Deliv. Rev. 1995, 16, 141–155. [Google Scholar]
- Wattendorf, U.; Merkle, H.P. PEGylation as a tool for the biomedical engineering of surface modified microparticles. J. Pharm. Sci. 2008, 97, 4655–4669. [Google Scholar]
- Lemarchand, C.; Gref, R.; Couvreur, P. Polysaccharide decorated nanoparticles. Eur. J. Pharm. Biopharm. 2004, 58, 327–341. [Google Scholar]
- OECD Joint Meeting of the Chemicals Committee. No. 6. List of manufactured nanomaterials and list of endpoints for phase one of the OECD testing programme. Working Party on Manufactured Nanomaterials (2008) Series on the Safety of Manufactured Nanomaterials. ENV/JM/MONO(2008)13/REV. Available online: www.oecd.org/ehs For updates see www.oecd.org/env/nanosafety/.
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Debbage, P.; Thurner, G.C. Nanomedicine Faces Barriers. Pharmaceuticals 2010, 3, 3371-3416. https://doi.org/10.3390/ph3113371
Debbage P, Thurner GC. Nanomedicine Faces Barriers. Pharmaceuticals. 2010; 3(11):3371-3416. https://doi.org/10.3390/ph3113371
Chicago/Turabian StyleDebbage, Paul, and Gudrun C. Thurner. 2010. "Nanomedicine Faces Barriers" Pharmaceuticals 3, no. 11: 3371-3416. https://doi.org/10.3390/ph3113371
APA StyleDebbage, P., & Thurner, G. C. (2010). Nanomedicine Faces Barriers. Pharmaceuticals, 3(11), 3371-3416. https://doi.org/10.3390/ph3113371