Mitochondrial Drugs for Alzheimer Disease

Abstract
:
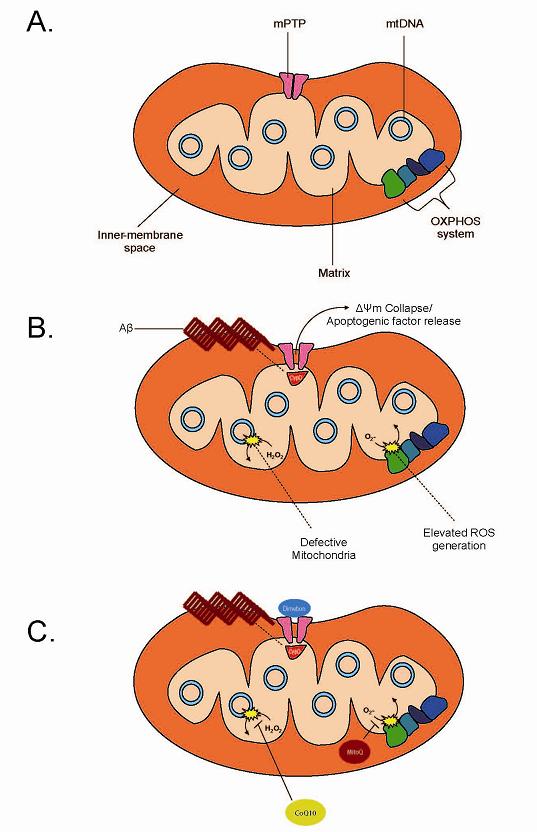{kind=link}
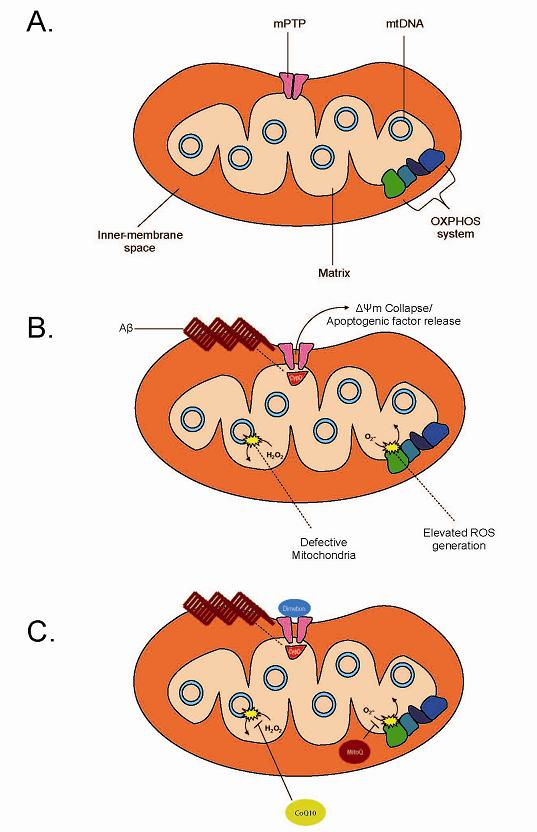{kind=link}
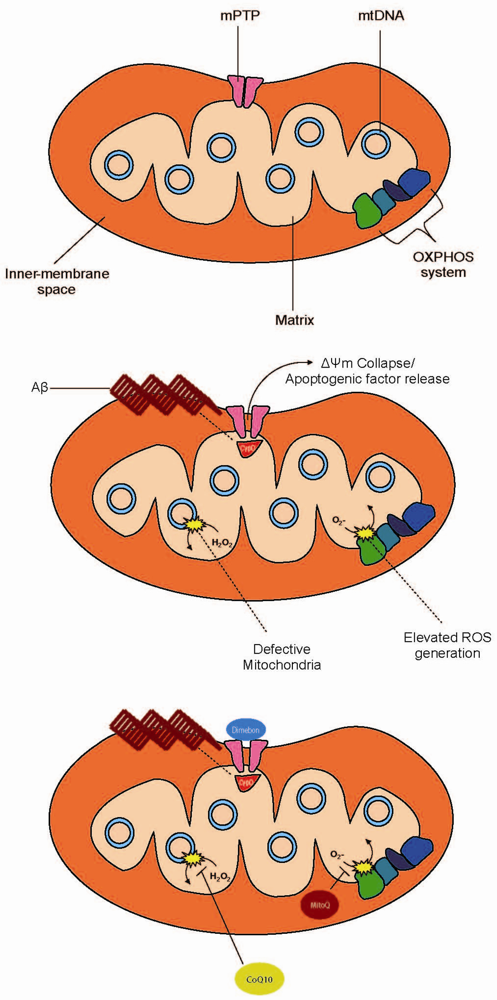{kind=link}
Introduction
Mitochondrial ROS Generation and Oxidative Stress in Alzheimer Disease: An Opportunity for Intervention
Mitochondrial Permeability Transition Pore: A Gateway to Stress Relief
Mitochondrial Dynamics: Fission, Fusion, and Neurodegeneration
Current Treatment Perspectives
Acknowledgments
References
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-betaoverproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar]
- Lee, J.; Boo, J.H.; Ryu, H. The failure of mitochondria leads to neurodegeneration: Do mitochondria need a jump start? Adv. Drug Deliv. Rev. 2009, 61, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar]
- Su, B.; Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 135–142. [Google Scholar]
- Russell, R.L.; Siedlak, S.L.; Raina, A.K.; Bautista, J.M.; Smith, M.A.; Perry, G. Increased neuronal glucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch. Biochem. Biophys. 1999, 370, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, O.; Zhu, X.; Perry, G.; Smith, M.A. Mitochondrial abnormalities and oxidative imbalance in neurodegenerative disease. Sci. Aging Knowledge Environ. 2002, 2002, pe16. [Google Scholar] [PubMed]
- Zhu, X.; Lee, H.G.; Perry, G.; Smith, M.A. Alzheimer disease, the two-hit hypothesis: an update. Biochim. Biophys. Acta 2007, 1772, 494–502. [Google Scholar]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; Shimohama, S.; Cash, A.D.; Siedlak, S.L.; Harris, P.L.; Jones, P.K.; Petersen, R.B.; Perry, G.; Smith, M.A. Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J. Neurochem. 2009, 109 (Suppl. 1), 153–159. [Google Scholar]
- Smith, M.A. Alzheimer disease. Int. Rev. Neurobiol. 1998, 42, 1–54. [Google Scholar]
- Marlatt, M.W.; Lucassen, P.J.; Perry, G.; Smith, M.A.; Zhu, X. Alzheimer's disease: cerebrovascular dysfunction, oxidative stress, and advanced clinical therapies. J. Alzheimers Dis. 2008, 15, 199–210. [Google Scholar] [PubMed]
- Petersen, R.B.; Nunomura, A.; Lee, H.G.; Casadesus, G.; Perry, G.; Smith, M.A.; Zhu, X. Signal transduction cascades associated with oxidative stress in Alzheimer's disease. J. Alzheimers Dis. 2007, 11, 143–152. [Google Scholar]
- Zhu, X.; Castellani, R.J.; Takeda, A.; Nunomura, A.; Atwood, C.S.; Perry, G.; Smith, M.A. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the 'two hit' hypothesis. Mech. Ageing Dev. 2001, 123, 39–46. [Google Scholar]
- Zhu, X.; Raina, A.K.; Perry, G.; Smith, M.A. Alzheimer's disease: the two-hit hypothesis. Lancet Neurol. 2004, 3, 219–226. [Google Scholar]
- Lee, H.G.; Perry, G.; Moreira, P.I.; Garrett, M.R.; Liu, Q.; Zhu, X.; Takeda, A.; Nunomura, A.; Smith, M.A. Tau phosphorylation in Alzheimer's disease: pathogen or protector? Trends Mol. Med. 2005, 11, 164–169. [Google Scholar] [PubMed]
- Lee, H.G.; Zhu, X.; Nunomura, A.; Perry, G.; Smith, M.A. Amyloid beta: the alternate hypothesis. Curr. Alzheimer Res. 2006, 3, 75–80. [Google Scholar]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. 1998, 105, 855–870. [Google Scholar]
- Maurer, I.; Zierz, S.; Moller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar]
- Parker, W.D., Jr.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer's disease brain. Neurology 1994, 44, 1090–1096. [Google Scholar]
- Nagy, Z.; Esiri, M.M.; LeGris, M.; Matthews, P.M. Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathol. (Berl.) 1999, 97, 346–354. [Google Scholar] [CrossRef]
- Smith, M.A.; Perry, G.; Richey, P.L.; Sayre, L.M.; Anderson, V.E.; Beal, M.F.; Kowall, N. Oxidative damage in Alzheimer's. Nature 1996, 382, 120–121. [Google Scholar]
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641. [Google Scholar]
- Castellani, R.J.; Harris, P.L.; Sayre, L.M.; Fujii, J.; Taniguchi, N.; Vitek, M.P.; Founds, H.; Atwood, C.S.; Perry, G.; Smith, M.A. Active glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-(carboxymethyl) lysine and hexitol-lysine. Free Radic. Biol. Med. 2001, 31, 175–180. [Google Scholar]
- Smith, M.A.; Perry, G. Free radical damage, iron, and Alzheimer's disease. J. Neurol. Sci. 1995, 134 (Suppl.), 92–94. [Google Scholar]
- Wataya, T.; Nunomura, A.; Smith, M.A.; Siedlak, S.L.; Harris, P.L.; Shimohama, S.; Szweda, L.I.; Kaminski, M.A.; Avila, J.; Price, D.L.; Cleveland, D.W.; Sayre, L.M.; Perry, G. High molecular weight neurofilament proteins are physiological substrates of adduction by the lipid peroxidation product hydroxynonenal. J. Biol. Chem. 2002, 277, 4644–4648. [Google Scholar]
- Keller, J.N.; Guo, Q.; Holtsberg, F.W.; Bruce-Keller, A.J.; Mattson, M.P. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J. Neurosci. 1998, 18, 4439–4450. [Google Scholar]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar]
- Liu, Q.; Xie, F.; Rolston, R.; Moreira, P.I.; Nunomura, A.; Zhu, X.; Smith, M.A.; Perry, G. Prevention and treatment of Alzheimer disease and aging: antioxidants. Mini Rev. Med. Chem. 2007, 7, 171–180. [Google Scholar]
- Siedlak, S.L.; Casadesus, G.; Webber, K.M.; Pappolla, M.A.; Atwood, C.S.; Smith, M.A.; Perry, G. Chronic antioxidant therapy reduces oxidative stress in a mouse model of Alzheimer's disease. Free Radic. Res. 2009, 43, 156–164. [Google Scholar]
- Pallas, M.; Casadesus, G.; Smith, M.A.; Coto-Montes, A.; Pelegri, C.; Vilaplana, J.; Camins, A. Resveratrol and neurodegenerative diseases: activation of SIRT1 as the potential pathway towards neuroprotection. Curr. Neurovasc. Res. 2009, 6, 70–81. [Google Scholar]
- Beal, M.F. Mitochondrial dysfunction and oxidative damage in Alzheimer's and Parkinson's diseases and coenzyme Q10 as a potential treatment. J. Bioenerg. Biomembr. 2004, 36, 381–386. [Google Scholar]
- Beyer, R.E.; Segura-Aguilar, J.; Di Bernardo, S.; Cavazzoni, M.; Fato, R.; Fiorentini, D.; Galli, M.C.; Setti, M.; Landi, L.; Lenaz, G. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. USA 1996, 93, 2528–2532. [Google Scholar]
- Genova, M.L.; Pich, M.M.; Biondi, A.; Bernacchia, A.; Falasca, A.; Bovina, C.; Formiggini, G.; Parenti Castelli, G.; Lenaz, G. Mitochondrial production of oxygen radical species and the role of Coenzyme Q as an antioxidant. Exp. Biol. Med. (Maywood) 2003, 228, 506–513. [Google Scholar] [PubMed]
- Wadsworth, T.L.; Bishop, J.A.; Pappu, A.S.; Woltjer, R.L.; Quinn, J.F. Evaluation of coenzyme Q as an antioxidant strategy for Alzheimer's disease. J. Alzheimers. Dis. 2008, 14, 225–234. [Google Scholar]
- Kwong, L.K.; Kamzalov, S.; Rebrin, I.; Bayne, A.C.; Jana, C.K.; Morris, P.; Forster, M.J.; Sohal, R.S. Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic. Biol. Med. 2002, 33, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Forster, M.J.; Sohal, R.S. Effects of coenzyme Q10 and alpha-tocopherol administration on their tissue levels in the mouse: elevation of mitochondrial alpha-tocopherol by coenzyme Q10. Free Radic. Biol. Med. 1999, 26, 1375–1382. [Google Scholar]
- Sohal, R.S.; Kamzalov, S.; Sumien, N.; Ferguson, M.; Rebrin, I.; Heinrich, K.R.; Forster, M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 2006, 40, 480–487. [Google Scholar] [PubMed]
- Lu, C.; Zhang, D.; Whiteman, M.; Armstrong, J.S. Is antioxidant potential of the mitochondrial targeted ubiquinone derivative MitoQ conserved in cells lacking mtDNA? Antioxid. Redox Signal. 2008, 10, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Geromel, V.; Darin, N.; Chretien, D.; Benit, P.; DeLonlay, P.; Rotig, A.; Munnich, A.; Rustin, P. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: rationale and comparative benefits. Mol. Genet. Metab. 2002, 77, 21–30. [Google Scholar]
- Murphy, M.P. Development of lipophilic cations as therapies for disorders due to mitochondrial dysfunction. Expert Opin. Biol. Ther. 2001, 1, 753–764. [Google Scholar]
- Smith, R.A.; Kelso, G.F.; James, A.M.; Murphy, M.P. Targeting coenzyme Q derivatives to mitochondria. Methods Enzymol. 2004, 382, 45–67. [Google Scholar]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar]
- Tauskela, J.S. MitoQ--a mitochondria-targeted antioxidant. IDrugs 2007, 10, 399–412. [Google Scholar]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; Gunn-Moore, F.J.; Vonsattel, J.P.; Arancio, O.; Chen, J.X.; Yan, S.D. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar]
- Crompton, M. Mitochondria and aging: a role for the permeability transition? Aging Cell 2004, 3, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; McStay, G.P.; Clarke, S.J. The permeability transition pore complex: another view. Biochimie 2002, 84, 153–166. [Google Scholar]
- Halestrap, A. Biochemistry: a pore way to die. Nature 2005, 434, 578–579. [Google Scholar]
- Zamzami, N.; Larochette, N.; Kroemer, G. Mitochondrial permeability transition in apoptosis and necrosis. Cell Death Differ. 2005, 12 (Suppl. 2), 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Barksby, E.; Johnson, N.; Capano, M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 2002, 84, 143–152. [Google Scholar]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar]
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998, 258, 729–735. [Google Scholar]
- Halestrap, A.P.; Woodfield, K.Y.; Connern, C.P. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 1997, 272, 3346–3354. [Google Scholar] [PubMed]
- Connern, C.P.; Halestrap, A.P. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem. J. 1994, 302 (Pt. 2), 321–324. [Google Scholar] [PubMed]
- Andreeva, L.; Heads, R.; Green, C.J. Cyclophilins and their possible role in the stress response. Int. J. Exp. Pathol. 1999, 80, 305–315. [Google Scholar]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; Robbins, J.; Molkentin, J.D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar]
- Pastorino, J.G.; Chen, S.T.; Tafani, M.; Snyder, J.W.; Farber, J.L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 1998, 273, 7770–7775. [Google Scholar]
- Serrano, F.; Klann, E. Reactive oxygen species and synaptic plasticity in the aging hippocampus. Ageing Res. Rev. 2004, 3, 431–443. [Google Scholar]
- Liu, R.; Liu, I.Y.; Bi, X.; Thompson, R.F.; Doctrow, S.R.; Malfroy, B.; Baudry, M. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc. Natl. Acad. Sci. USA 2003, 100, 8526–8531. [Google Scholar]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puolivali, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar]
- Bachurin, S.; Bukatina, E.; Lermontova, N.; Tkachenko, S.; Afanasiev, A.; Grigoriev, V.; Grigorieva, I.; Ivanov, Y.; Sablin, S.; Zefirov, N. Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann. N. Y. Acad. Sci. 2001, 939, 425–435. [Google Scholar]
- Lermontova, N.N.; Redkozubov, A.E.; Shevtsova, E.F.; Serkova, T.P.; Kireeva, E.G.; Bachurin, S.O. Dimebon and tacrine inhibit neurotoxic action of beta-amyloid in culture and block L-type Ca(2+) channels. Bull. Exp. Biol. Med. 2001, 132, 1079–1083. [Google Scholar]
- Grigorev, V.V.; Dranyi, O.A.; Bachurin, S.O. Comparative study of action mechanisms of dimebon and memantine on AMPA- and NMDA-subtypes glutamate receptors in rat cerebral neurons. Bull. Exp. Biol. Med. 2003, 136, 474–477. [Google Scholar]
- Bachurin, S.O.; Shevtsova, E.P.; Kireeva, E.G.; Oxenkrug, G.F.; Sablin, S.O. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann. NY Acad. Sci. 2003, 993, 334–344, discussion 345–339. [Google Scholar]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer's disease: a randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [PubMed]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell. Dev. Biol. 2006, 22, 79–99. [Google Scholar]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [PubMed]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [PubMed]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: more than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar]
- Parone, P.A.; James, D.I.; Da Cruz, S.; Mattenberger, Y.; Donze, O.; Barja, F.; Martinou, J.C. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol. Cell. Biol. 2006, 26, 7397–7408. [Google Scholar]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2653–2658. [Google Scholar]
- Bleazard, W.; McCaffery, J.M.; King, E.J.; Bale, S.; Mozdy, A.; Tieu, Q.; Nunnari, J.; Shaw, J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999, 1, 298–304. [Google Scholar]
- Sesaki, H.; Jensen, R.E. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J. Cell Biol. 1999, 147, 699–706. [Google Scholar]
- Cheng, X.; Kanki, T.; Fukuoh, A.; Ohgaki, K.; Takeya, R.; Aoki, Y.; Hamasaki, N.; Kang, D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J. Biochem. 2005, 138, 673–678. [Google Scholar]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; Alroy, J.; Wu, M.; Py, B.F.; Yuan, J.; Deeney, J.T.; Corkey, B.E.; Shirihai, O.S. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bonda, D.J.; Wang, X.; Gustaw-Rothenberg, K.A.; Perry, G.; Smith, M.A.; Zhu, X. Mitochondrial Drugs for Alzheimer Disease. Pharmaceuticals 2009, 2, 287-298. https://doi.org/10.3390/ph2030287
Bonda DJ, Wang X, Gustaw-Rothenberg KA, Perry G, Smith MA, Zhu X. Mitochondrial Drugs for Alzheimer Disease. Pharmaceuticals. 2009; 2(3):287-298. https://doi.org/10.3390/ph2030287
Chicago/Turabian StyleBonda, David J., Xinglong Wang, Katarzyna A. Gustaw-Rothenberg, George Perry, Mark A. Smith, and Xiongwei Zhu. 2009. "Mitochondrial Drugs for Alzheimer Disease" Pharmaceuticals 2, no. 3: 287-298. https://doi.org/10.3390/ph2030287
APA StyleBonda, D. J., Wang, X., Gustaw-Rothenberg, K. A., Perry, G., Smith, M. A., & Zhu, X. (2009). Mitochondrial Drugs for Alzheimer Disease. Pharmaceuticals, 2(3), 287-298. https://doi.org/10.3390/ph2030287