Insulin and Insulin-Sensitizing Drugs in Neurodegeneration: Mitochondria as Therapeutic Targets

Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Mitochondria and the Brain
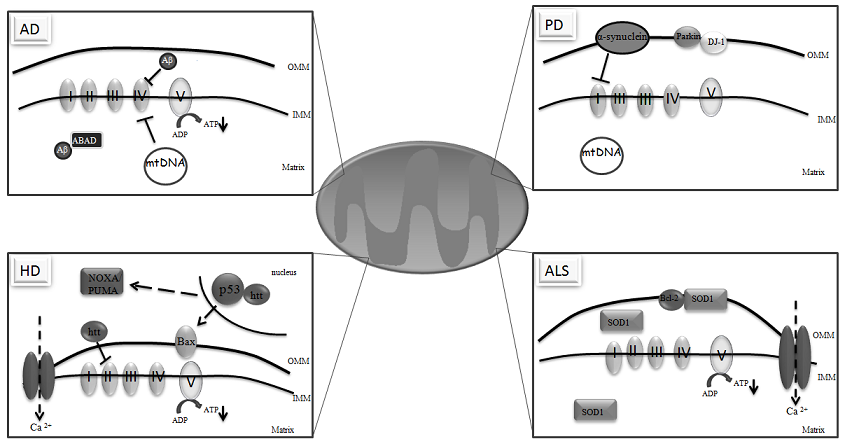
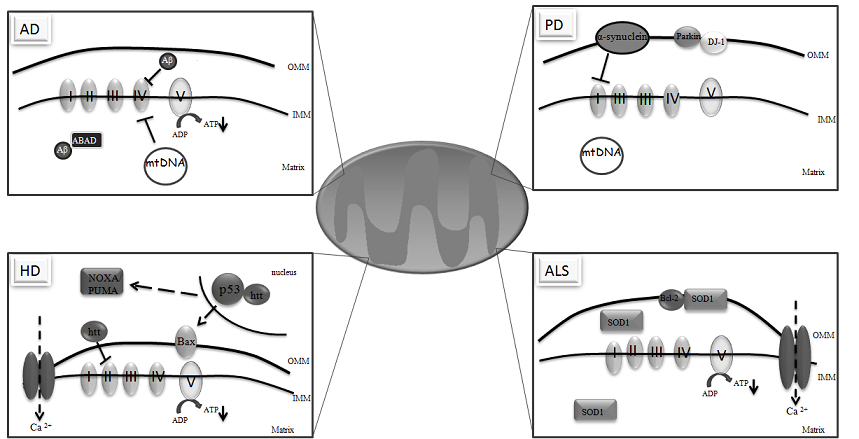
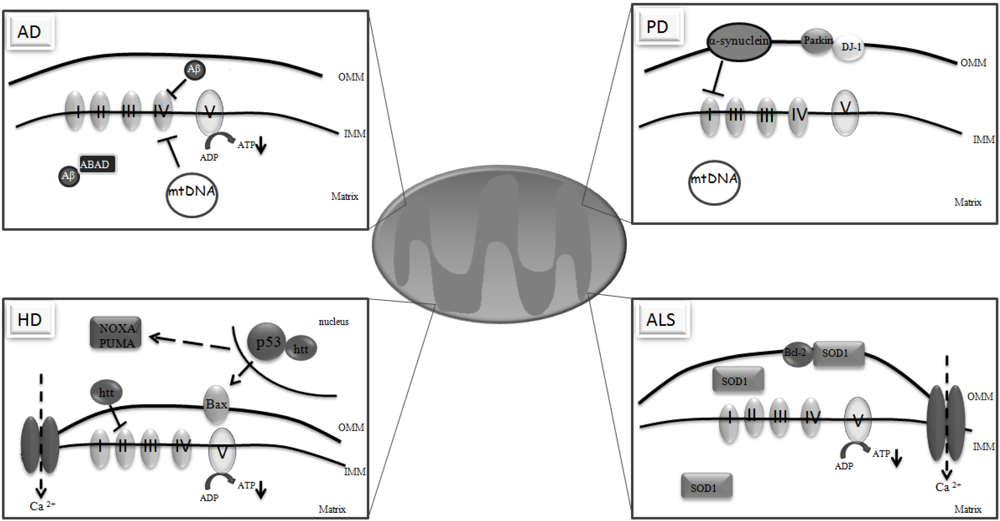
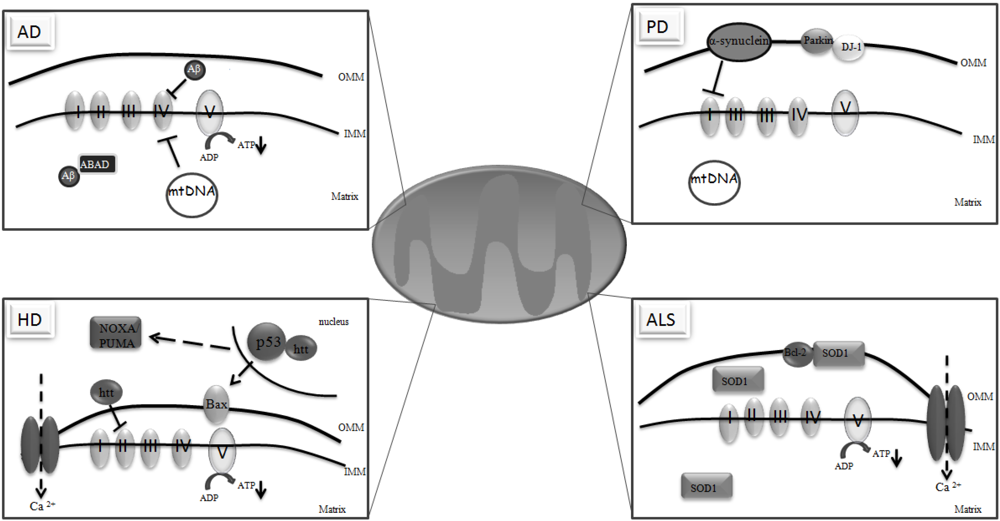
3. Mitochondria and Neurodegeneration
4. Insulin and the Brain
5. Insulin Signaling Dysregulation and Neurodegeneration
6. Role of Insulin and Insulin-Sensitizers in Neurodegeneration: Mitochondria as Potential Therapeutic Targets
7. Conclusions
References
- Van Der Heide, L.P.; Ramakers, G.M.; Smidt, M.P. Insulin signaling in the central nervous system: Learning to survive. Prog. Neurobiol. 2006, 79, 205–221. [Google Scholar]
- White, M.F.; Kahn, C.R. The insulin signaling system. J. Biol. Chem. 1994, 269, 1–4. [Google Scholar]
- Taguchi, A.; White, M.F. Insulin-like signaling, nutrient homeostasis, and life span. Annu. Rev. Physiol. 2008, 70, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [PubMed]
- Giannakou, M.E.; Goss, M.; Jünger, M.A.; Hafen, E.; Leevers, S.J.; Partridge, L. Long-lived drosophila With overexpressed dFOXO in adult fat body. Science 2004, 305, 361. [Google Scholar]
- Rincon, M.; Muzumdar, R.; Atzmon, G.; Barzilai, N. The paradox of the insulin/IGF-1 signaling pathway in longevity. Mech. Ageing Dev. 2004, 125, 397–403. [Google Scholar]
- Cohen, E.; Dillin, A. The insulin paradox: Aging, proteotoxicity and neurodegeneration. Nat. Rev. Neurosci. 2008, 10, 759–767. [Google Scholar]
- Dou, J.T.; Chen, M.; Dufour, F.; Alkon, D.L.; Zhao, W.Q. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn. Mem. 2005, 12, 646–655. [Google Scholar]
- Ding, Q.; Vaynman, S.; Akhavan, M.; Ying, Z.; Gomez-Pinilla, F. Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 2006, 140, 823–833. [Google Scholar]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory improvement following induced hyperinsulinemia in Alzheimer's disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [PubMed]
- Gasparini, L.; Xu, H. Potential roles of insulin and IGF-1 in Alzheimer's disease. Trends Neurosci. 2003, 26, 404–406. [Google Scholar]
- Watson, G.S.; Craft, S. The role of insulin resistance in the pathogenesis of Alzheimer's disease: Implications for treatment. CNS Drugs 2003, 17, 27–45. [Google Scholar]
- Bowling, A.C.; Beal, M.F. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 1995, 56, 1151–1171. [Google Scholar]
- McNay, E.C. The impact of recurrent hypoglycemia on cognitive function in aging. Neurobiol. Aging 2005, 26, 76–79. [Google Scholar]
- Moreira, P.I.; Duarte, A.I.; Santos, M.S.; Rego, A.C.; Oliveira, C.R. An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer's disease. J. Alzheimers Dis. 2009, 741–761. [Google Scholar]
- Turner, N.; Heilbronn, L.K. Is mitochondrial dysfunction a cause of insulin resistance? Trends Endocrinol. Metab. 2008, 19, 324–330. [Google Scholar] [PubMed]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R. Alzheimer's disease: A lesson from mitochondrial dysfunction. Antioxid. Redox Signal 2007, 9, 1621–1630. [Google Scholar]
- Chung, J.H.; Seo, A.Y.; Chung, S.W.; Kim, M.K.; Leeuwenburgh, C.; Yu, B.P.; Chung, H.Y. Molecular mechanism of PPAR in the regulation of age-related inflammation. Ageing Res. Rev. 2008, 7, 126–136. [Google Scholar]
- Chaturvedi, R.K.; Beal, M.F. PPAR: A therapeutic target in Parkinson's disease. J. Neurochem. 2008, 106, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors: New targets for the pharmacological modulation of macrophage gene expression and function. Curr. Opin. Lipidol. 2003, 14, 459–468. [Google Scholar]
- Rangwala, S.M.; Lazar, M.A. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol. Sci. 2004, 25, 331–336. [Google Scholar]
- Patsouris, D.; Müller, M.; Kersten, S. Peroxisome proliferator activated receptor ligands for the treatment of insulin resistance. Curr. Opin. Investig. Drugs 2004, 5, 1045–1050. [Google Scholar] [PubMed]
- Sundararajan, S.; Jiang, Q.; Heneka, M.; Landreth, G. PPARgamma as a therapeutic target in central nervous system diseases. Neurochem. Int. 2006, 49, 136–144. [Google Scholar]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARgamma agonists as therapeutics for the treatment of Alzheimer's disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar]
- Roses, A.D.; Saunders, A.M.; Huang, Y.; Strum, J.; Weisgraber, K.H.; Mahley, R.W. Complex disease-associated pharmacogenetics: Drug efficacy, drug safety, and confirmation of a pathogenetic hypothesis (Alzheimer's disease). Pharmacogenomics J. 2007, 7, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. The mitochondrial impairment, oxidative stress and neurodegeneration connection: Reality or just an attractive hypothesis? Trends Neurosci. 2008, 31, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial dysfunction in Parkinson's disease. Cell Death Differ. 2007, 14, 1261–1266. [Google Scholar]
- Schon, E.A.; Manfredi, G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest 2003, 111, 303–312. [Google Scholar]
- Mancuso, M.; Coppede, F.; Migliore, L.; Siciliano, G.; Murri, L. Mitochondrial dysfunction, oxidative stress and neurodegeneration. J. Alzheimers Dis. 2006, 10, 59–73. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar]
- Torres, M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003, 8, 369–391. [Google Scholar]
- Heffetz, D.; Bushkin, I.; Dror, R.; Zick, Y. The insulin mimetic agents H2O2 and vanadate stimulate protein tyrosine phosphorylation in intact cells. J. Biol. Chem. 1990, 265, 2896–2902. [Google Scholar]
- Konishi, H.; Matsuzaki, H.; Tanaka, M.; Takemura, Y.; Kuroda, S.; Ono, Y.; Kikkawa, U. Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Lett. 1997, 410, 493–498. [Google Scholar]
- Nulton, P.A.C.; Szweda, L.I. Modulation of mitochondrial function by hydrogen peroxide. J. Biol. Chem. 2001, 276, 23357–23361. [Google Scholar]
- Manna, S.K.; Zhang, H.J.; Yan, T.; Oberley, L.W.; Aggarwal, B.B. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappaB and activated protein-1. J. Biol. Chem. 1998, 273, 13245–13254. [Google Scholar]
- Perry, G.; Nunomura, A.; Hirai, K.; Zhu, X.; Perez, M.; Avila, J.; Castellani, R.J.; Atwood, C.S.; Aliev, G.; Sayre, L.M.; Takeda, A.; Smith, M.A. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer’s and other neurodegenerative diseases? Free Radic. Biol. Med. 2002, 33, 1475–1479. [Google Scholar] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar]
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 2008, 21, 172–188. [Google Scholar]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar]
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 2008, 43, 24–33. [Google Scholar]
- Wanrooij, S.; Goffart, S.; Pohjoismäki, J.L.; Yasukawa, T.; Spelbrink, J.N. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 2007, 35, 3238–3251. [Google Scholar]
- Sedensky, M.M.; Morgan, P.G. Mitochondrial respiration and reactive oxygen species in C. elegans. Exp. Gerontol. 2006, 41, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Bussière, F.; Hekimi, S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar]
- Dillin, A.; Hsu, A.L.; Arantes, O.N.; Lehrer, G.J.; Hsin, H.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Rates of behavior and aging specified by mitochondrial function during development. Science 2002, 298, 2398–2401. [Google Scholar] [PubMed]
- Ashford, J.W.; Mortimer, J.A. Non-familial Alzheimer's disease is mainly due to genetic factors. J. Alzheimers Dis. 2002, 4, 169–177. [Google Scholar]
- Petrozzi, L.; Ricci, G.; Giglioli, N.J.; Siciliano, G.; Mancuso, M. Mitochondria and neurodegeneration. Biosci. Rep. 2007, 27, 87–104. [Google Scholar]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. NY Acad. Sci. 2008, 1147, 395–412. [Google Scholar]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer's disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar]
- Rhein, V.; Eckert, A. Effects of Alzheimer's amyloid-beta and tau protein on mitochondrial function—Role of glucose metabolism and insulin signalling. Arch. Physiol. Biochem. 2007, 113, 131–141. [Google Scholar]
- Moreira, P.I.; Cardoso, S.M.; Santos, M.S.; Oliveira, C.R. The key role of mitochondria in Alzheimer's disease. J. Alzheimers. Dis. 2006, 9, 101–110. [Google Scholar]
- Moreira, P.I.; Cardoso, S.M.; Pereira, C.M.; Santos, M.S.; Oliveira, C.R. Mitochondria as a Therapeutic Target in Alzheimer's Disease and Diabetes. CNS Neurol. Disord. Drug Targets. 2009, 8, 492–511. [Google Scholar]
- Fernandez, V.P.; Fernandez, A.P.; Castro, B.S.; Serrano, J.; Bentura, M.L.; Martinez, M.R.; Martinez, A.; Rodrigo, J. Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s disease. Histol. Histopathol. 2004, 19, 823–844. [Google Scholar] [PubMed]
- Cardoso, S.M.; Santos, S.; Swerdlow, R.H.; Oliveira, C.R. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001, 15, 1439–1441. [Google Scholar]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Oliveira, C.R. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci. Rep. 2001, 21, 789–800. [Google Scholar]
- Moreira, P.I.; Santos, M.S.; Moreno, A.; Rego, A.C.; Oliveira, C.R. Effect of amyloid beta-peptide on permeability transition pore: A comparative study. J Neurosci. Res. 2002, 69, 257–267. [Google Scholar]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Seiça, R.; Oliveira, C.R. Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes 2003, 52, 1449–1456. [Google Scholar]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; Trinchese, F.; Liu, S.; Gunn, M.F.; Lue, L.F.; Walker, D.G.; Kuppusamy, P.; Zewier, Z.L.; Arancio, O.; Stern, D.; Yan, S.S.; Wu, H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 2004, 304, 448–452. [Google Scholar] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; Chiba, S.; Atwood, C.S.; Petersen, R.B.; Smith, M.A. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar]
- Praticò, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; Quinn, J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Dröse, S.; Brandt, U.; Müller, W.E.; Eckert, A.; Götz, J. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar]
- Drzezga, A.; Lautenschlager, N.; Siebner, H.; Riemenschneider, M.; Willoch, F.; Minoshima, S.; Schwaiger, M.; Kurz, A. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer's disease: A PET follow-up study. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1104–1113. [Google Scholar]
- Alexander, G.E.; Chen, K.; Pietrini, P.; Rapoport, S.I.; Reiman, E.M. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer's Disease Treatment Studies. Am. J. Psychiatry 2002, 159, 738–745. [Google Scholar]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar]
- Huang, H.M.; Zhang, H.; Xu, H.; Gibson, G.E. Inhibition of the alpha-ketoglutarate dehydrogenase complex alters mitochondrial function and cellular calcium regulation. Biochim. Biophys. Acta 2003, 1637, 119–126. [Google Scholar]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer's disease varies with apolipoprotein E genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar]
- Sorbi, S.; Fani, C.; Piacentini, S.; Giannini, E.; Amaducci, L. Energy metabolism in demented brain. Prog. Neuropsychopharmacol. Biol. Psychiatry. 1986, 10, 591–597. [Google Scholar]
- Moreira, P.I.; Harris, P.L.; Zhu, X.; Santos, M.S.; Oliveira, C.R.; Smith, M.A.; Perry, G. Lipoic acid and N-acetyl cysteine decrease mitochondrial-related oxidative stress in Alzheimer disease patient fibroblasts. J. Alzheimers Dis. 2007, 12, 195–206. [Google Scholar]
- Canevari, L.; Clark, J.B.; Bates, T.E. Beta-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999, 457, 131–134. [Google Scholar]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar]
- Cottrell, D.A.; Blakely, E.L.; Johnson, M.A.; Ince, P.G.; Borthwick, G.M.; Turnbull, D.M. Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age. Neurobiol. Aging 2001, 22, 265–272. [Google Scholar]
- Chandrasekaran, K.; Hatanpää, K.; Rapoport, S.I.; Brady, D.R. Decreased expression of nuclear and mitochondrial DNA-encoded genes of oxidative phosphorylation in association neocortex in Alzheimer disease. Brain Res. Mol. Brain Res. 1997, 44, 99–104. [Google Scholar]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology 1990, 40, 1302–1303. [Google Scholar]
- Curti, D.; Rognoni, F.; Gasparini, L.; Cattaneo, A.; Paolillo, M.; Racchi, M.; Zani, L.; Bianchetti, A.; Trabucchi, M.; Bergamaschi, S.; Govoni, S. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer's disease (AD) patients. Neurosci. Lett. 1997, 236, 13–16. [Google Scholar]
- Valla, J.; Schneider, L.; Niedzielko, T.; Coon, K.D.; Caselli, R.; Sabbagh, M.N.; Ahern, G.L.; Baxter, L.; Alexander, G.; Walker, D.G.; Reiman, E.M. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion 2006, 6, 323–330. [Google Scholar]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Cybrids in Alzheimer's disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [PubMed]
- Trimmer, P.A.; Keeney, P.M.; Borland, M.K.; Simon, F.A.; Almeida, J.; Swerdlow, R.H.; Parks, J.P.; Parker, W.D., Jr.; Bennett, J.P., Jr. Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer's disease worsen with passage in culture. Neurobiol. Dis. 2004, 15, 29–39. [Google Scholar] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar]
- Lin, M.T.; Simon, D.K.; Ahn, C.H.; Kim, L.M.; Beal, M.F. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer's disease brain. Hum. Mol. Genet. 2002, 11, 133–145. [Google Scholar] [PubMed]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer's disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar]
- Pyle, A.; Foltynie, T.; Tiangyou, W.; Lambert, C.; Keers, S.M.; Allcock, L.M.; Davison, J.; Lewis, S.J.; Perry, R.H.; Barker, R.; Burn, D.J.; Chinnery, P.F. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann. Neurol. 2005, 57, 564–567. [Google Scholar]
- Edland, S.D.; Silverman, J.M.; Peskind, E.R.; Tsuang, D.; Wijsman, E.; Morris, J.C. Increased risk of dementia in mothers of Alzheimer's disease cases: Evidence for maternal inheritance. Neurology 1996, 47, 254–256. [Google Scholar] [PubMed]
- Wolf, P.A.; Beiser, A.; Au, R.; Auerbach, S.; DeCarli, C. Neurology 2005, 64, 267–268.
- Davis, R.E.; Miller, S.; Herrnstadt, C.; Ghosh, S.S.; Fahy, E.; Shinobu, L.A.; Galasko, D.; Thal, L.J.; Beal, M.F.; Howell, N.; Parker, W.D., Jr. Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 4526–4531. [Google Scholar]
- Elson, J.L.; Herrnstadt, C.; Preston, G.; Thal, L.; Morris, C.M.; Edwardson, J.A.; Beal, M.F.; Turnbull, D.M.; Howell, N. Does the mitochondrial genome play a role in the etiology of Alzheimer's disease? Hum. Genet. 2006, 119, 241–254. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer's disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; Shimohama, S.; Cash, A.D.; Siedlak, S.L.; Harris, P.L.; Jones, P.K.; Petersen, R.B.; Perry, G.; Smith, M.A. Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar]
- Moreira, P.I.; Siedlak, S.L.; Wang, X.; Santos, M.S.; Oliveira, C.R.; Tabaton, M.; Nunomura, A.; Szweda, L.I.; Aliev, G.; Smith, M.A.; Zhu, X.; Perry, G. Autophagocytosis of mitochondria is prominent in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2007, 66, 525–532. [Google Scholar]
- Moreira, P.I.; Siedlak, S.L.; Wang, X.; Santos, M.S.; Oliveira, C.R.; Tabaton, M.; Nunomura, A.; Szweda, L.I.; Aliev, G.; Smith, M.A.; Zhu, X.; Perry, G. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 2007, 3, 614–615. [Google Scholar]
- Fukui, H.; Diaz, F.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA 2007, 104, 14163–14168. [Google Scholar]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia, O.M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of Parkinson disease: Multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035. [Google Scholar]
- Gu, M.; Cooper, J.M.; Taanman, J.W.; Schapira, A.H. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson's disease. Ann. Neurol. 1998, 44, 177–186. [Google Scholar]
- Esteves, A.R.; Domingues, A.F.; Ferreira, I.L.; Januário, C.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial function in Parkinson's disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008, 8, 219–228. [Google Scholar]
- Valente, E.M.; Abou, S.P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del, D.; Bentivoglio, A.R.; Healy, D.G.; Albanese, A.; Nussbaum, R.; González, M.R.; Deller, T.; Salvi, S.; Cortelli, P.; Gilks, W.P.; Latchman, D.S.; Harvey, R.J.; Dallapiccola, B.; Auburger, G.; Wood, N.W. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar]
- Liu, W.; Vives, B.C.; Acín, P.R.; Yamamoto, A.; Tan, Y.; Li, Y.; Magrané, J.; Stavarache, M.A.; Shaffer, S.; Chang, S.; Kaplitt, M.G.; Huang, X.Y.; Beal, M.F.; Manfredi, G.; Li, C. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson's disease. PLoS One 2009, 4, 1–14. [Google Scholar]
- Bialecka, M.; Hui, S.; Klodowska, D.G.; Opala, G.; Tan, E.K.; Drozdzik, M. Analysis of LRRK 2 G 2019 S and I 2020 T mutations in Parkinson's disease. Neurosci. Lett. 2005, 390, 1–3. [Google Scholar]
- Hernandez, D.; Paisan, R.C.; Crawley, A.; Malkani, R.; Werner, J.; Gwinn, H.K.; Dickson, D.; Wavrant Devrieze, F.; Hardy, J.; Singleton, A. The dardarin G 2019 S mutation is a common cause of Parkinson's disease but not other neurodegenerative diseases. Neurosci. Lett. 2005, 389, 137–139. [Google Scholar]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun, F.H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; Morrison, A.D.; Wood, N.W.; Downward, J.; Martins, L.M. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009, 16, 449–464. [Google Scholar]
- Strauss, K.M.; Martins, L.M.; Plun, F.H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Bornemann, A.; Wolburg, H.; Downward, J.; Riess, O.; Schulz, J.B.; Krüger, R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar]
- Song, D.D.; Shults, C.W.; Sisk, A.; Rockenstein, E.; Masliah, E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp. Neurol. 2004, 186, 158–172. [Google Scholar]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A.; Rocha, M.; Jackson, L.V.; Hersch, S.; Sulzer, D.; Przedborski, S.; Burke, R.; Hen, R. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar]
- Darios, F.; Corti, O.; Lücking, C.B.; Hampe, C.; Muriel, M.P.; Abbas, N.; Gu, W.J.; Hirsch, E.C.; Rooney, T.; Ruberg, M.; Brice, A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet 2003, 12, 517–526. [Google Scholar]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar]
- Kuroda, Y.; Mitsui, T.; Kunishige, M.; Shono, M.; Akaike, M.; Azuma, H.; Matsumoto, T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum. Mol. Genet. 2006, 15, 883–895. [Google Scholar]
- Chung, K.K.; Dawson, V.L.; Dawson, T.M. New insights into Parkinson's disease. J. Neurol. 2003, 250, 15–24. [Google Scholar]
- Whitworth, A.J.; Theodore, D.A.; Greene, J.C.; Benes, H.; Wes, P.D.; Pallanck, L.J. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc. Natl. Acad. Sci. USA 2005, 102, 8024–8029. [Google Scholar]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; Dawson, V.L. Mitochondrial localization of the Parkinson's disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar]
- Takahashi, N.K.; Niki, T.; Taira, T.; Iguchi, A.S.M.; Ariga, H. Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson's disease patients. Biochem. Biophys. Res. Commun. 2004, 320, 389–397. [Google Scholar]
- Li, H.M.; Niki, T.; Taira, T.; Iguchi, A.S.M.; Ariga, H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic. Res. 2005, 39, 1091–1099. [Google Scholar]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You, T.A.J.; Kalia, S.K.; Horne, P.; Westaway, D.; Lozano, A.M.; Anisman, H.; Park, D.S.; Mak, T.W. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar]
- Inden, M.; Taira, T.; Kitamura, Y.; Yanagida, T.; Tsuchiya, D.; Takata, K.; Yanagisawa, D.; Nishimura, K.; Taniguchi, T.; Kiso, Y.; Yoshimoto, K.; Agatsuma, T.; Koide, Y.S.; Iguchi, A.S.M.; Shimohama, S.; Ariga, H. PARK7 DJ-1 protects against degeneration of nigral dopaminergic neurons in Parkinson's disease rat model. Neurobiol. Dis. 2006, 24, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Andres, M.E.; Perier, C.; Zhang, L.; Blanchard, F.B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; Dawson, T.M.; Dawson, V.L. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar]
- Nural, H.; He, P.; Beach, T.; Sue, L.; Xia, W.; Shen, Y. Dissembled DJ-1 high molecular weight complex in cortex mitochondria from Parkinson's disease patients. Mol. Neurodegener. 2009, 4, 23:1–23:9. [Google Scholar]
- Yang, Y.; Gehrke, S.; Haque, M.E.; Imai, Y.; Kosek, J.; Yang, L.; Beal, M.F.; Nishimura, I.; Wakamatsu, K.; Ito, S.; Takahashi, R.; Lu, B. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 13670–13675. [Google Scholar]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; Taylor, R.W.; Turnbull, D.M. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar]
- Simon, D.K.; Pulst, S.M.; Sutton, J.P.; Browne, S.E.; Beal, M.F.; Johns, D.R. Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology 1999, 53, 1787–1793. [Google Scholar]
- Luoma, P.T.; Eerola, J.; Ahola, S.; Hakonen, A.H.; Hellström, O.; Kivistö, K.T.; Tienari, P.J.; Suomalainen, A. Mitochondrial DNA polymerase gamma variants in idiopathic sporadic Parkinson disease. Neurology 2007, 69, 1152–1159. [Google Scholar]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; Hoffer, B.; Cullheim, S.; Mohammed, A.H.; Olson, L.; Larsson, N.G. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar]
- Liang, C.L.; Wang, T.T.; Luby, P.K.; German, D.C. Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson's disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar]
- Milakovic, T.; Johnson, G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar]
- Díaz, H.M.; Hernández, F.; Martín, A.E.; Gómez, R.P.; Morán, M.A.; Castaño, J.G.; Ferrer, I.; Avila, J.; Lucas, J.J. Neuronal induction of the immunoproteasome in Huntington's disease. J. Neurosci. 2003, 23, 11653–11661. [Google Scholar]
- Bowman, A.B.; Yoo, S.Y.; Dantuma, N.P.; Zoghbi, H.Y. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum. Mol. Genet. 2005, 14, 679–691. [Google Scholar]
- Maynard, C.J.; Böttcher, C.; Ortega, Z.; Smith, R.; Florea, B.I.; Díaz, H.M.; Brundin, P.; Overkleeft, H.S.; Li, J.Y.; Lucas, J.J.; Dantuma, N.P. Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc. Natl. Acad. Sci. USA 2009, 106, 13986–13991. [Google Scholar]
- Jenkins, B.G.; Koroshetz, W.J.; Beal, M.F.; Rosen, B.R. Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy. Neurology 1993, 43, 2689–2695. [Google Scholar]
- Kuhl, D.E.; Phelps, M.E.; Markham, C.H.; Metter, E.J.; Riege, W.H.; Winter, J. Cerebral metabolism and atrophy in Huntington's disease determined by 18FDG and computed tomographic scan. Ann. Neurol. 1982, 12, 425–434. [Google Scholar]
- Kuhl, D.E.; Metter, E.J.; Riege, W.H.; Markham, C.H. Patterns of cerebral glucose utilization in Parkinson's disease and Huntington's disease. Ann. Neurol. 1984, 15, 119–125. [Google Scholar] [PubMed]
- Parker, W.D., Jr.; Boyson, S.J.; Luder, A.S.; Parks, J.K. Evidence for a defect in NADH: Ubiquinone oxidoreductase (complex I) in Huntington's disease. Neurology 1990, 40, 1231–1234. [Google Scholar]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy, A.F.; Cooper, J.M; Schapira, A.H. Mitochondrial defect in Huntington's disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Bowling, A.C; MacGarvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington's disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar]
- Benchoua, A.; Trioulier, Y.; Zala, D.; Gaillard, M.C.; Lefort, N.; Dufour, N.; Saudou, F.; Elalouf, J.M.; Hirsch, E.; Hantraye, P.; Déglon, N.; Brouillet, E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol. Biol. Cell 2006, 17, 1652–1663. [Google Scholar]
- Guidetti, P.; Charles, V.; Chen, E.Y.; Reddy, P.H.; Kordower, J.H.; Whetsell, W.O., Jr.; Schwarcz, R.; Tagle, D.A. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp. Neurol. 2001, 169, 340–350. [Google Scholar]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar]
- Moreno, S.R. Regulation of oxidative phosphorylation in mitochondria by external free Ca2+ concentrations. J. Biol. Chem. 1985, 260, 4028–4034. [Google Scholar]
- Luthi, C.R.; Strand, A.D.; Hanson, S.A.; Kooperberg, C.; Schilling, G.; La Spada, A.R.; Merry, D.E.; Young, A.B.; Ross, C.A.; Borchelt, D.R.; Olson, J.M. Polyglutamine and transcription: Gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum. Mol. Genet. 2002, 11, 1927–1937. [Google Scholar]
- Sugars, K.L.; Rubinsztein, D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003, 19, 233–238. [Google Scholar]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; Snyder, S.H.; Sawa, A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron 2005, 47, 29–41. [Google Scholar]
- Kiaei, M. Peroxisome Proliferator-Activated Receptor-gamma in Amyotrophic Lateral Sclerosis and Huntington's Disease. PPAR Res. 2008, 2008, 1–8. [Google Scholar]
- Sasaki, S.; Iwata, M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology 1996, 47, 535–540. [Google Scholar]
- Higgins, C.M.; Jung, C.; Xu, Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4, 1–14. [Google Scholar]
- Jaarsma, D.; Rognoni, F.; Van, D.W.; Verspaget, H.W.; Haasdijk, E.D.; Holstege, J.C. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001, 102, 293–305. [Google Scholar]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande, V.C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; Brännström, T.; Gredal, O.; Wong, P.C.; Williams, D.S.; Cleveland, D.W. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar]
- Sasaki, S.; Warita, H.; Murakami, T.; Abe, K.; Iwata, M. Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2004, 107, 461–474. [Google Scholar]
- Mattiazzi, M.; D'Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint, B.M.; Manfredi, G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar]
- Nguyen, K.T.; García, C.L.E.; Barrett, J.N.; Barrett, E.F.; David, G. The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc. Natl. Acad. Sci. USA 2009, 106, 2007–2011. [Google Scholar]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; Shaw, C.E.; Leigh, P.N.; Miller, C.C.; Grierson, A.J. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar]
- Vande, V.C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar]
- Takeuchi, H.; Kobayashi, Y.; Ishigaki, S.; Doyu, M.; Sobue, G. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J Biol Chem 2002, 277, 50966–50972. [Google Scholar]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar]
- Devaskar, S.U.; Giddings, S.J.; Rajakumar, P.A.; Carnaghi, L.R.; Menon, R.K.; Zahm, D.S. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J. Biol. Chem. 1994, 269, 8445–8454. [Google Scholar] [PubMed]
- Schechter, R.; Holtzclaw, L.; Sadiq, F.; Kahn, A.; Devaskar, S. Insulin synthesis by isolated rabbit neurons. Endocrinology 1988, 123, 505–513. [Google Scholar]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in th central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar]
- Havrankova, J.; Roth, J.; Brownstein, M.J. Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J. Clin. Invest. 1979, 64, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar]
- Zhao, W.Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell Endocrinol. 2001, 177, 125–134. [Google Scholar]
- Cole, A.R.; Astell, A.; Green, C.; Sutherland, C. Molecular connexions between dementia and diabetes. Neurosci. Biobehav. Rev. 2007, 31, 1046–1063. [Google Scholar]
- Rodgers, E.E.; Theibert, A.B. Functions of PI 3-kinase in development of the nervous system. Int. J. Dev. Neurosci. 2002, 20, 187–197. [Google Scholar]
- Gerozissis, K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur. J. Pharmacol. 2008, 585, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Wine, R.N.; McPherson, C.A.; Harry, G.J. IGF-1 and pAKT Signaling Promote Hippocampal CA1 Neuronal Survival Following Injury to Dentate Granule Cells. Neurotox. Res. 2009, 16, 280–292. [Google Scholar]
- Bondy, C.A.; Cheng, C.M. Signaling by insulin-like growth factor 1 in brain. Eur. J. Pharmacol. 2004, 490, 25–31. [Google Scholar]
- Gasparini, L.; Netzer, W.J.; Greengard, P.; Xu, H. Does insulin dysfunction play a role in Alzheimer's disease? Trends Pharmacol. Sci. 2002, 23, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Lee, V.M. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J. Biol. Chem. 1997, 272, 19547–19553. [Google Scholar]
- Blüher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le, B.Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; Kadowaki, T.; Krone, W.; Schröder, H.; Brüning, J.C.; Schubert, M. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer's disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [PubMed]
- Selcher, J.C.; Atkins, C.M.; Trzaskos, J.M.; Paylor, R.; Sweatt, J.D. A necessity for MAP kinase activation in mammalian spatial learning. Learn. Mem. 1999, 6, 478–490. [Google Scholar]
- Atkins, C.M.; Selcher, J.C.; Petraitis, J.J.; Trzaskos, J.M.; Sweatt, J.D. The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1998, 1, 602–609. [Google Scholar]
- Toyoda, H.; Zhao, M.G.; Xu, H.; Wu, L.J.; Ren, M.; Zhuo, M. Requirement of extracellular signal-regulated kinase/mitogen-activated protein kinase for long-term potentiation in adult mouse anterior cingulate cortex. Mol. Pain 2007, 3, 1–15. [Google Scholar]
- Ito, I.A.; Kakegawa, W.; Yuzaki, M. ERK1/2 but not p38 MAP kinase is essential for the long-term depression in mouse cerebellar slices. Eur. J. Neurosci. 2006, 24, 1617–1622. [Google Scholar]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [PubMed]
- Park, C.R.; Seeley, R.J.; Craft, S.; Woods, S.C. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol. Behav. 2000, 68, 509–514. [Google Scholar]
- Babri, S.; Badie, H.G.; Khamenei, S.; Seyedlar, M.O. Intrahippocampal insulin improves memory in a passive-avoidance task in male wistar rats. Brain Cogn. 2007, 64, 86–91. [Google Scholar]
- Craft, S.; Asthana, S.; Newcomer, J.W.; Wilkinson, C.W.; Matos, I.T.; Baker, L.D.; Cherrier, M.; Lofgreen, C.; Latendresse, S.; Petrova, A.; Plymate, S.; Raskind, M.; Grimwood, K.; Veith, R.C. Enhancement of memory in Alzheimer disease with insulin and somatostatin, but not glucose. Arch. Gen. Psychiatry 1999, 56, 1135–1140. [Google Scholar]
- Fehm, H.L.; Perras, B.; Smolnik, R.; Kern, W.; Born, J. Manipulating neuropeptidergic pathways in humans: A novel approach to neuropharmacology? Eur. J. Pharmacol. 2000, 405, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar]
- Benedict, C.; Hallschmid, M.; Schultes, B.; Born, J.; Kern, W. Intranasal insulin to improve memory function in humans. Neuroendocrinology 2007, 86, 136–142. [Google Scholar]
- Nelson, T.J.; Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharmacol. 2008, 585, 76–87. [Google Scholar]
- Craft, S.; Watson, G.S. Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurol. 2004, 3, 169–178. [Google Scholar]
- Raizada, M.K.; Shemer, J.; Judkins, J.H.; Clarke, D.W.; Masters, B.A.; LeRoith, D. Insulin receptors in the brain: Structural and physiological characterization. Neurochem. Res. 1988, 13, 297–303. [Google Scholar]
- Wilcox, B.J.; Matsumoto, A.M.; Dorsa, D.M.; Baskin, D.G. Reduction of insulin binding in the arcuate nucleus of the rat hypothalamus after 6-hydroxydopamine treatment. Brain Res. 1989, 500, 149–155. [Google Scholar]
- Figlewicz, D.P.; Patterson, T.A.; Zavosh, A.; Brot, M.D.; Roitman, M.; Szot, P. Neurotransmitter transporters: Target for endocrine regulation. Horm. Metab. Res. 1999, 31, 335–339. [Google Scholar]
- Kovacs, P.; Hajnal, A. In vivo electrophysiological effects of insulin in the rat brain. Neuropeptides 2009, 43, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.; Antony, S.; Paulose, C.S. Increased glutamate receptor gene expression in the cerebral cortex of insulin induced hypoglycemic and streptozotocin-induced diabetic rats. Neuroscience 2008, 156, 298–304. [Google Scholar]
- Zheng, W.H.; Quirion, R. Glutamate acting on N-methyl-D-aspartate receptors attenuates insulin-like growth factor-1 receptor tyrosine phosphorylation and its survival signaling properties in rat hippocampal neurons. J. Biol. Chem. 2009, 284, 855–861. [Google Scholar]
- De la Monte, S.M. Insulin resistance and Alzheimer's disease. BMB Rep. 2009, 42, 475–481. [Google Scholar]
- Ristow, M. Neurodegenerative disorders associated with diabetes mellitus. J. Mol. Med. 2004, 82, 510–529. [Google Scholar]
- Cole, G.M.; Frautschy, S.A. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer's Disease. Exp. Gerontol. 2007, 42, 10–21. [Google Scholar]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.W.; Kovacina, K.S.; Craft, S. Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer's disease is associated with the apolipoprotein E-epsilon4 allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar]
- Leissring, M.A.; Farris, W.; Chang, A.Y.; Walsh, D.M.; Wu, X.; Sun, X.; Frosch, M.P.; Selkoe, D.J. Enhanced proteolysis of beta amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 2003, 40, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Ye, Z.; Kholodenko, D.; Seubert, P.; Selkoe, D.J. Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J. Biol. Chem. 1997, 272, 6641–6646. [Google Scholar]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; Hof, P.R.; Pasinetti, G.M. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J. 2004, 18, 902–904. [Google Scholar]
- Martín, D.; Salinas, M.; López, V.R.; Serrano, E.; Recuero, M.; Cuadrado, A. Effect of the Alzheimer amyloid fragment Abeta(25-35) on Akt/PKB kinase and survival of PC12 cells. J. Neurochem. 2001, 78, 1000–1008. [Google Scholar]
- Lee, H.K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol. Biol. Cell 2009, 20, 1533–1544. [Google Scholar]
- Leroy, K.; Boutajangout, A.; Authelet, M.; Woodgett, J.R.; Anderton, B.H.; Brion, J.P. The active form of glycogen synthase kinase-3beta is associated with granulovacuolar degeneration in neurons in Alzheimer's disease. Acta Neuropathol. 2002, 103, 91–99. [Google Scholar]
- Lucas, J.J.; Hernández, F.; Gómez, R.P.; Morán, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001, 20, 27–39. [Google Scholar]
- Salkovic, P.M.; Tribl, F.; Schmidt, M.; Hoyer, S.; Riederer, P. Alzheimer-like changes in protein kinase B and glycogen synthase kinase-3 in rat frontal cortex and hippocampus after damage to the insulin signalling pathway. J. Neurochem. 2006, 96, 1005–1015. [Google Scholar]
- Grünblatt, E.; Salkovic, P.M.; Osmanovic, J.; Riederer, P.; Hoyer, S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J. Neurochem. 2007, 101, 757–770. [Google Scholar]
- Hong, M.; Lee, V.M. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J. Biol. Chem. 1997, 272, 19547–19553. [Google Scholar]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Honolulu-Asia Aging Study. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer's amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; De la, M.S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [PubMed]
- Sandyk, R. The relationship between diabetes mellitus and Parkinson's disease. Int. J. Neurosci. 1993, 69, 125–130. [Google Scholar]
- Schwab, R.S. Progression and prognosis in Parkinson's disease. J. Nerv. Ment. Dis. 1960, 130, 556–566. [Google Scholar]
- Moroo, I.; Yamada, T.; Makino, H.; Tooyama, I.; McGeer, P.L.; McGeer, E.G.; Hirayama, K. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson's disease. Acta Neuropathol. 1994, 87, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Liang, Y.; Cincotta, A.H. Intracerebroventricular administration of bromocriptine ameliorates the insulin-resistant/glucose-intolerant state in hamsters. Neuroendocrinology 1999, 69, 160–166. [Google Scholar]
- Figlewicz, D.P.; Szot, P.; Chavez, M.; Woods, S.C.; Veith, R.C. Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res. 1994, 644, 331–334. [Google Scholar]
- Figlewicz, D.P.; Brot, M.D.; McCall, A.L.; Szot, P. Diabetes causes differential changes in CNS noradrenergic and dopaminergic neurons in the rat: A molecular study. Brain Res. 1996, 736, 54–60. [Google Scholar]
- Murzi, E.; Contreras, Q.; Teneud, L.; Valecillos, B.; Parada, M.A.; De Parada, M.P.; Hernandez, L. Diabetes decreases limbic extracellular dopamine in rats. Neurosci. Lett. 1996, 202, 141–144. [Google Scholar]
- Park, C.R. Cognitive effects of insulin in the central nervous system. Neurosci. Biobehav. Rev. 2001, 25, 311–323. [Google Scholar]
- Farrer, L.A. Diabetes mellitus in Huntington disease. Clin. Genet. 1985, 27, 62–67. [Google Scholar]
- Podolsky, S.; Leopold, N.A. Abnormal glucose tolerance and arginine tolerance tests in Huntington's disease. Gerontology 1977, 23, 55–63. [Google Scholar]
- Lalić, N.M.; Marić, J.; Svetel, M.; Jotić, A.; Stefanova, E.; Lalić, K.; Dragasević, N.; Milicić, T.; Lukić, L.; Kostić, V.S. Glucose homeostasis in Huntington disease: Abnormalities in insulin sensitivity and early-phase insulin secretion. Arch. Neurol. 2008, 65, 476–480. [Google Scholar]
- Humbert, S.; Bryson, E.A.; Cordelières, F.P.; Connors, N.C.; Datta, S.R.; Finkbeiner, S.; Greenberg, M.E.; Saudou, F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev. Cell. 2002, 2, 831–837. [Google Scholar]
- Yamamoto, A.; Cremona, M.L.; Rothman, J.E. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 2006, 172, 719–731. [Google Scholar]
- Colin, E.; Régulier, E.; Perrin, V.; Dürr, A.; Brice, A.; Aebischer, P.; Déglon, N.; Humbert, S.; Saudou, F. Akt is altered in an animal model of Huntington's disease and in patients. Eur. J. Neurosci. 2005, 21, 1478–1488. [Google Scholar]
- Pradat, P.F.; Bruneteau, G.; Gordon, P.H.; Dupuis, L.; Bonnefont, R.D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.M.; Jardel, C.; Coussieu, C.; Forestier, N.L.; Lacomblez, L.; Loeffler, J.P.; Meininger, V. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2009, 1–6. [Google Scholar]
- Vincent, A.M.; Mobley, B.C.; Hiller, A.; Feldman, E.L. IGF-I prevents glutamate-induced motor neuron programmed cell death. Neurobiol. Dis. 2004, 16, 407–416. [Google Scholar]
- Kaspar, B.K.; Lladó, J.; Sherkat, N.; Rothstein, J.D.; Gage, F.H. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003, 301, 839–842. [Google Scholar]
- Papaconstantinou, J. Insulin/IGF-1 and ROS signaling pathway cross-talk in aging and longevity determination. Mol. Cell Endocrinol. 2009, 299, 89–100. [Google Scholar]
- De la, M.S.M.; Wands, J.R. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: Relevance to Alzheimer's disease. J. Alzheimers Dis. 2005, 7, 45–61. [Google Scholar] [PubMed]
- Puche, J.E.; García, F.M.; Muntané, J.; Rioja, J.; González, B.S.; Castilla, C.I. Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology 2008, 149, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Sena, C.; Seiça, R.; Oliveira, C.R. Insulin protects against amyloid beta-peptide toxicity in brain mitochondria of diabetic rats. Neurobiol. Dis. 2005, 18, 628–637. [Google Scholar]
- Moreira, P.I.; Rolo, A.P.; Sena, C.; Seiça, R.; Oliveira, C.R.; Santos, M.S. Insulin attenuates diabetes-related mitochondrial alterations: A comparative study. Med. Chem. 2006, 2, 299–308. [Google Scholar]
- Sanderson, T.H.; Kumar, R.; Sullivan, J.M.; Krause, G.S. Insulin blocks cytochrome c release in the reperfused brain through PI3-K signaling and by promoting Bax/Bcl-XL binding. J. Neurochem. 2008, 106, 1248–1258. [Google Scholar]
- Bijur, G.N.; Jope, R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003, 87, 1427–1435. [Google Scholar]
- Feinstein, D.L. Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes Technol. Ther. 2003, 5, 67–73. [Google Scholar]
- Correia, S.; Carvalho, C.; Santos, M.S.; Seiça, R.; Oliveira, C.R.; Moreira, P.I. Mechanisms of action of metformin in type 2 diabetes and associated complications: An overview. Mini Rev. Med. Chem. 2008, 8, 1343–1354. [Google Scholar]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar]
- Baker, P.R.; Lin, Y.; Schopfer, F.J.; Woodcock, S.R.; Groeger, A.L.; Batthyany, C.; Sweeney, S.; Long, M.H.; Iles, K.E.; Baker, L.M.; Branchaud, B.P.; Chen, Y.E.; Freeman, B.A. Fatty acid transduction of nitric oxide signaling: Multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 2005, 280, 42464–42475. [Google Scholar]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer's disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar]
- Storer, P.D.; Xu, J.; Chavis, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: Implications for multiple sclerosis. J. Neuroimmunol. 2005, 161, 113–122. [Google Scholar]
- Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J. Neurosci. Res. 2005, 81, 403–411. [Google Scholar]
- Heneka, M.T.; Feinstein, D.L.; Galea, E.; Gleichmann, M.; Wüllner, U.; Klockgether, T. Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J. Neuroimmunol. 1999, 100, 156–168. [Google Scholar]
- Zhao, X.; Ou, Z.; Grotta, J.C.; Waxham, N.; Aronowski, J. Peroxisome-proliferator-activated receptor-gamma (PPARγ) activation protects neurons from NMDA excitotoxicity. Brain Research 2006, 1073/1074, 460–469. [Google Scholar] [CrossRef]
- Wang, Y.L.; Frauwirth, K.A.; Rangwala, S.M.; Lazar, M.A.; Thompson, C.B. Thiazolidinedione Activation of Peroxisome Proliferator-activated Receptor γ Can Enhance Mitochondrial Potential and Promote Cell Survival. J. Biol.Chem. 2002, 277, 31781–31788. [Google Scholar]
- Fuenzalida, K.; Quintanilla, R.; Ramos, P.; Piderit, D.; Fuentealba, R.A.; Martinez, G.; Inestrosa, N.C.; Bronfman, M. Peroxisome Proliferator-activated Receptor γ Up-regulates the Bcl-2 Anti-apoptotic Protein in Neurons and Induces Mitochondrial Stabilization and Protection against Oxidative Stress and Apoptosis. J. Biol.Chem. 2007, 282, 37006–37015. [Google Scholar] [PubMed]
- Wu, J.S.; Lin, T.N.; Wu, K.K. Rosiglitazone and PPAR-gamma overexpression protect mitochondrial membrane potential and prevent apoptosis by upregulating anti-apoptotic Bcl-2 family proteins. J. Cell Physiol. 2009, 220, 58–71. [Google Scholar]
- Ghosh, S.; Patel, N.; Rahn, D.; McAllister, J.; Sadeghi, S.; Horwitz, G.; Berry, D.; Wang, K.X.; Swerdlow, R.H. The Thiazolidinedione Pioglitazone Alters Mitochondrial Function in Human Neuron-Like Cells. Mol. Pharmacol. 2007, 71, 1695–1702. [Google Scholar]
- Heneka, M.T.; Sastre, M.; Dumitrescu, O.L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O'Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar]
- Bernardo, A.; Minghetti, L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar]
- Heneka, M.T.; Landreth, G.E. PPARs in the brain. Biochim. Biophys. Acta 2007, 1771, 1031–1045. [Google Scholar]
- Yan, Q.; Zhang, J.; Liu, H.; Babu, K.S.; Vassar, R.; Biere, A.L.; Citron, M.; Landreth, G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J. Neurosci. 2003, 23, 7504–7509. [Google Scholar]
- Sastre, M.; Dewachter, I.; Landreth, G.E.; Willson, T.M.; Klockgether, T.; van Leuven, F.; Heneka, M.T. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J. Neurosci. 2003, 23, 9796–9804. [Google Scholar]
- Inestrosa, N.C.; Godoy, J.A.; Quintanilla, R.A.; Koenig, C.S.; Bronfman, M. Peroxisome proliferator-activated receptor gamma is expressed in hippocampal neurons and its activation prevents beta-amyloid neurodegeneration: Role of Wnt signaling. Exp. Cell Res. 2005, 304, 91–104. [Google Scholar]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer's disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar]
- Kummer, M.P.; Heneka, M.T. PPARs in Alzheimer’s Disease. PPAR Res. 2008, 2008, 1–8. [Google Scholar]
- Strum, J.C.; Shehee, R.; Virley, D.; Richardson, J.; Mattie, M.; Selley, P.; Ghosh, S.; Nock, C.; Saunders, A.; Roses, A. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J. Alzheimers Dis. 2007, 11, 45–51. [Google Scholar]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; Kahn, S.E.; Keeling, M.L.; Craft, S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar]
- Risner, M.E.; Saunders, A.M.; Altman, J.F.; Ormandy, G.C.; Craft, S.; Foley, I.M.; Zvartau, H.M.E.; Hosford, D.A.; Roses, A.D. Rosiglitazone in Alzheimer's Disease Study Group. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006, 6, 246–254. [Google Scholar] [PubMed]
- Chalmanov, V.; Vurbanova, M. Diabetes mellitus in parkinsonism patients. Vutr. Boles. 1987, 26, 68–73. [Google Scholar]
- Takahashi, M.; Yamada, T.; Tooyama, I.; Moroo, I.; Kimura, H.; Yamamoto, T.; Okada, H. Insulin receptor mRNA in the substantia nigra in Parkinson's disease. Neurosci. Lett. 1996, 204, 201–204. [Google Scholar]
- Mattson, M.P.; Pedersen, W.A.; Duan, W.; Culmsee, C.; Camandola, S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann. N. Y. Acad. Sci. 1999, 893, 154–175. [Google Scholar]
- Hunter, R.L.; Bing, G. Agonism of Peroxisome Proliferator Receptor–Gamma may have Therapeutic Potential for Neuroinflammation and Parkinson’s Disease. Current Neuropharmacology 2007, 5, 35–46. [Google Scholar]
- Breidert, T.; Callebert, J.; Heneka, M.T.; Landreth, G.; Launay, J.M.; Hirsch, E.C. Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson's disease. J. Neurochem. 2002, 82, 615–624. [Google Scholar]
- Dehmer, T.; Heneka, M.T.; Sastre, M.; Dichgans, J.; Schulz, J.B. Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J. Neurochem. 2004, 88, 494–501. [Google Scholar]
- Quinn, L.P.; Crook, B.; Hows, M.E.; Vidgeon, H.M.; Chapman, H.; Upton, N.; Medhurst, A.D.; Virley, D.J. The PPARgamma agonist pioglitazone is effective in the MPTP mouse model of Parkinson's disease through inhibition of monoamine oxidase B. Br. J. Pharmacol. 2008, 154, 226–233. [Google Scholar]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar]
- Hunter, R.L.; Choi, D.Y.; Ross, S.A.; Bing, G. Protective properties afforded by pioglitazone against intrastriatal LPS in Sprague-Dawley rats. Neurosci. Lett. 2008, 432, 198–201. [Google Scholar]
- Xing, B.; Liu, M.; Bing, G. Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-kappaB and JNK activation and suppression of COX-2 activity. J. Neuroimmunol. 2007, 192, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Xin, T.; Hunter, R.L.; Bing, G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J. Neuroinflammation 2008, 5, 1–11. [Google Scholar]
- Jung, T.W.; Lee, J.Y.; Shim, W.S.; Kang, E.S.; Kim, S.K.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against acetaldehyde-induced cytotoxicity. Biochem. Biophys. Res. Commun. 2006, 340, 221–227. [Google Scholar]
- Jung, T.W.; Lee, J.Y.; Shim, W.S.; Kang, E.S.; Kim, S.K.; Ahn, C.W.; Lee, H.C.; Cha, B.S. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J. Neurol. Sci. 2007, 253, 53–60. [Google Scholar]
- Podolsky, S.; Leopold, N.A.; Sax, D.S. Increased frequency of diabetes mellitus in patients with Huntington's chorea. Lancet 1972, 1, 1356–1358. [Google Scholar]
- Hurlbert, M.S.; Zhou, W.; Wasmeier, C.; Kaddis, F.G.; Hutton, J.C.; Freed, C.R. Mice transgenic for an expanded CAG repeat in the Huntington’s disease gene develop diabetes. Diabetes 1999, 48, 649–651. [Google Scholar]
- Andreassen, O.A.; Dedeoglu, A.; Stanojevic, V.; Hughes, D.B.; Browne, S.E.; Leech, C.A.; Ferrante, R.J.; Habener, J.F.; Beal, M.F.; Thomas, M.K. Huntington's disease of the endocrine pancreas: Insulin deficiency and diabetes mellitus due to impaired insulin gene expression. Neurobiol. Dis. 2002, 11, 410–424. [Google Scholar]
- Quintanilla, R.A.; Jin, Y.N.; Fuenzalida, K.; Bronfman, M.; Johnson, G.V. Rosiglitazone treatment prevents mitochondrial dysfunction in mutant huntingtin-expressing cells: Possible role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in the pathogenesis of Huntington disease. J. Biol. Chem. 2008, 283, 25628–25637. [Google Scholar]
- McGill, J.K.; Beal, M.F. PGC, lalpha, a new therapeutic target in Huntington's disease? Cell 2006, 127, 465–468. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; Simon, D.K.; Bachoo, R.; Spiegelman, B.M. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; Cui, L.; Beyer, R.P.; Easley, C.N.; Smith, A.C.; Krainc, D.; Luquet, S.; Sweet, I.R.; Schwartz, M.W.; La Spada, A.R. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab 2006, 4, 349–362. [Google Scholar]
- Weydt, P.; Soyal, S.M.; Gellera, C.; Didonato, S.; Weidinger, C.; Oberkofler, H.; Landwehrmeyer, G.B.; Patsch, W. The gene coding for PGC-1alpha modifies age at onset in Huntington's Disease. Mol. Neurodegener. 2009, 4, 3:1–3:6. [Google Scholar]
- Taherzadeh, F.E.; Saft, C.; Andrich, J.; Wieczorek, S.; Arning, L. PGC-1alpha as modifier of onset age in Huntington disease. Mol. Neurodegener. 2009, 4, 1–4. [Google Scholar]
- Parker, J.A.; Arango, M.; Abderrahmane, S.; Lambert, E.; Tourette, C.; Catoire, H.; Ne´ri, C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet. 2005, 37, 349–350. [Google Scholar]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar]
- Heneka, M.T.; Landreth, G.E.; Hüll, M. Drug insight: Effects mediated by peroxisome proliferator-activated receptor-gamma in CNS disorders. Nat. Clin. Pract. Neurol. 2007, 3, 496–504. [Google Scholar] [PubMed]
- Schütz, B.; Reimann, J.; Dumitrescu, O.L.; Kappes, H.K.; Landreth, G.E.; Schürmann, B.; Zimmer, A.; Heneka, M.T. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. 2005, 25, 7805–7812. [Google Scholar]
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 191, 331–336. [Google Scholar]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cardoso, S.; Santos, R.; Correia, S.; Carvalho, C.; Zhu, X.; Lee, H.-G.; Casadesus, G.; Smith, M.A.; Perry, G.; Moreira, P.I. Insulin and Insulin-Sensitizing Drugs in Neurodegeneration: Mitochondria as Therapeutic Targets. Pharmaceuticals 2009, 2, 250-286. https://doi.org/10.3390/ph2030250
Cardoso S, Santos R, Correia S, Carvalho C, Zhu X, Lee H-G, Casadesus G, Smith MA, Perry G, Moreira PI. Insulin and Insulin-Sensitizing Drugs in Neurodegeneration: Mitochondria as Therapeutic Targets. Pharmaceuticals. 2009; 2(3):250-286. https://doi.org/10.3390/ph2030250
Chicago/Turabian StyleCardoso, Susana, Renato Santos, Sonia Correia, Cristina Carvalho, Xiongwei Zhu, Hyoung-Gon Lee, Gemma Casadesus, Mark A. Smith, George Perry, and Paula I. Moreira. 2009. "Insulin and Insulin-Sensitizing Drugs in Neurodegeneration: Mitochondria as Therapeutic Targets" Pharmaceuticals 2, no. 3: 250-286. https://doi.org/10.3390/ph2030250
APA StyleCardoso, S., Santos, R., Correia, S., Carvalho, C., Zhu, X., Lee, H.-G., Casadesus, G., Smith, M. A., Perry, G., & Moreira, P. I. (2009). Insulin and Insulin-Sensitizing Drugs in Neurodegeneration: Mitochondria as Therapeutic Targets. Pharmaceuticals, 2(3), 250-286. https://doi.org/10.3390/ph2030250