Mitochondrial Medicine and the Neurodegenerative Mitochondriopathies

{kind=link}
{kind=link}
Abstract
1. Neurodegenerative Diseases Are Mitochondriopathies
1.1. Brief overview of the neurodegenerative diseases
1.2. Mitochondrial perturbation occurs in multiple neurodegenerative diseases
2. Mitochondrial Medicine
2.1. Definition and overview of mitochondrial medicine targets
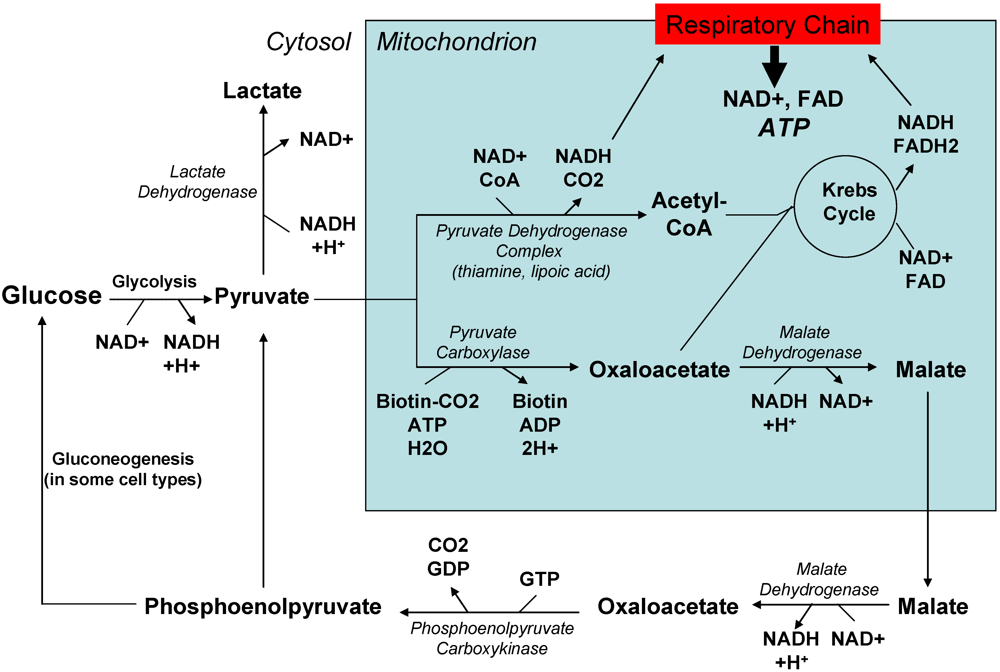
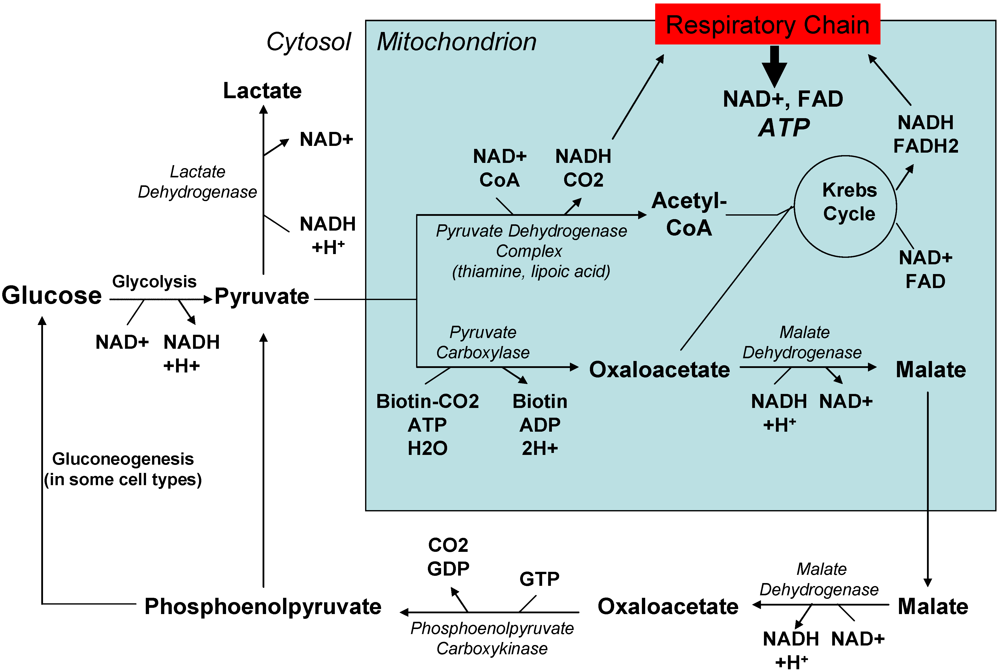
2.2. Past and current experience
3. Ongoing and Future Efforts To Develop Neurodegenerative Disease Mitochondrial Medicine
3.1. New drugs for existing targets
3.2. Enhancing aerobic metabolism
3.2.1. With mitochondrial biogenesis
3.2.2. Without mitochondrial biogenesis
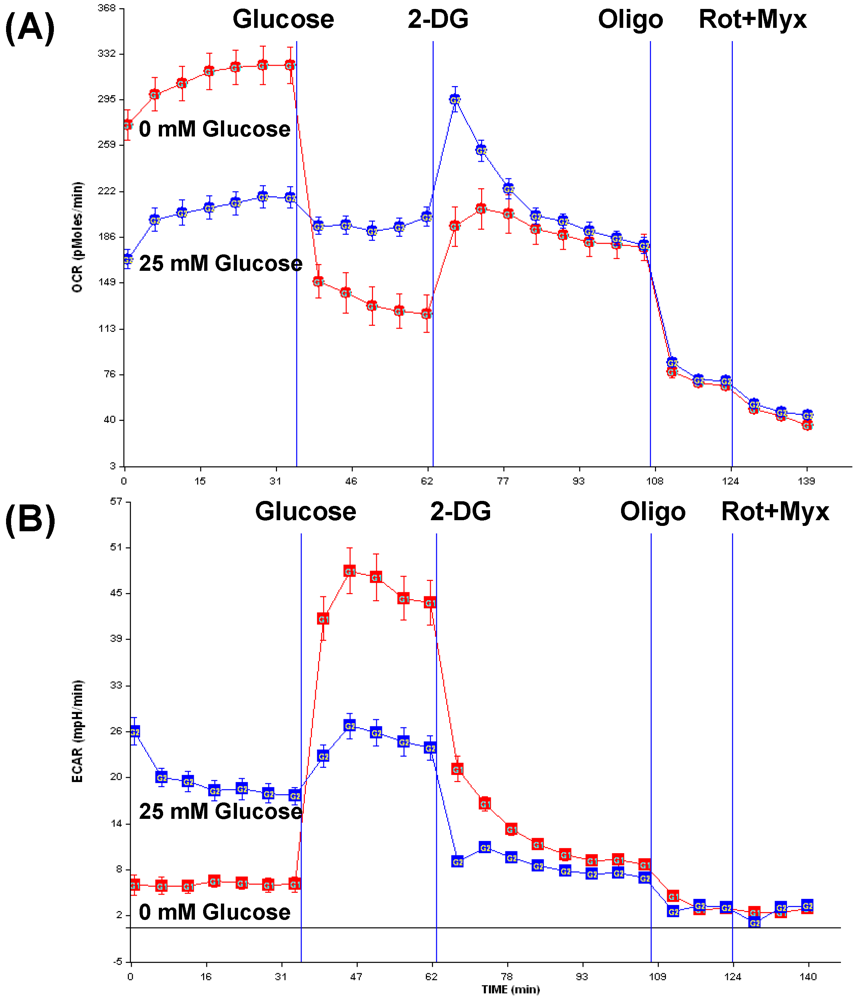
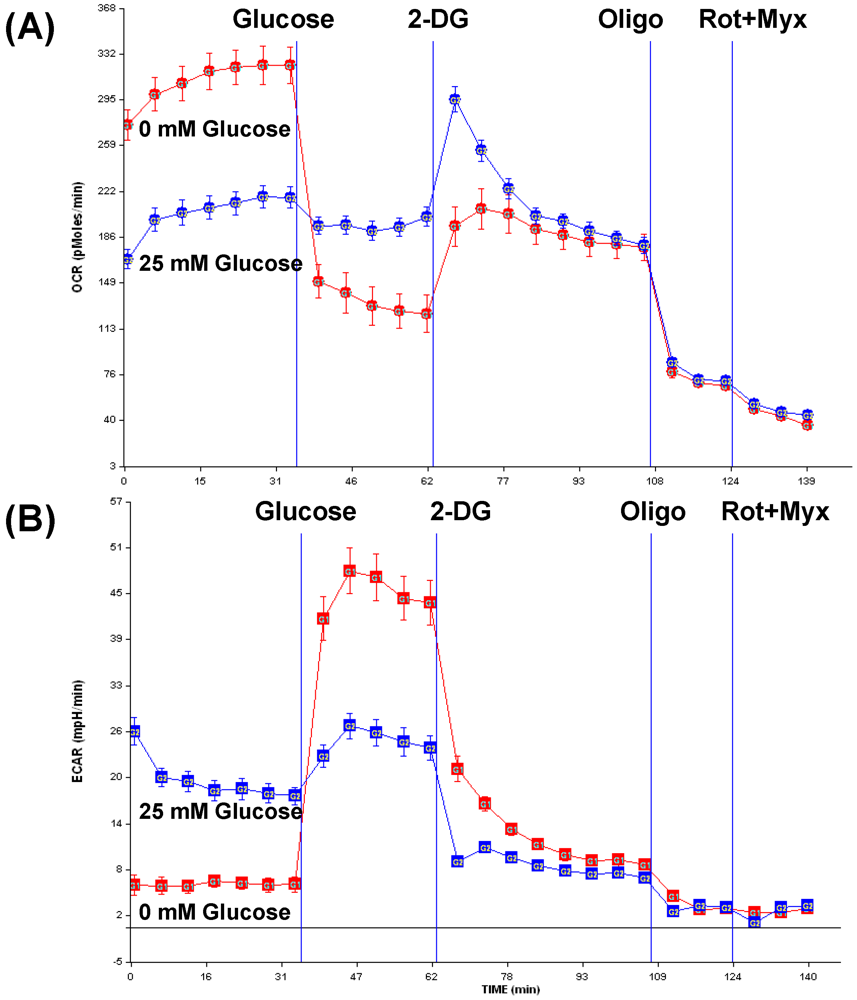
3.3. Opportunities for combination therapy
4. Conclusions
Acknowledgements
References and Notes
- Swerdlow, R.H. Is aging part of Alzheimer's disease, or is Alzheimer's disease part of aging? Neurobiol. Aging 2007, 28, 1465–1480. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Galasko, D.; Kosaka, K.; Perry, E.K.; Dickson, D.W.; Hansen, L.A.; Salmon, D.P.; Lowe, J.; Mirra, S.S.; Byrne, E.J.; Lennox, G.; Quinn, N.P.; Edwardson, J.A.; Ince, P.G.; Bergeron, C.; Burns, A.; Miller, B.L.; Lovestone, S.; Collerton, D.; Jansen, E.N.; Ballard, C.; de Vos, R.A.; Wilcock, G.K.; Jellinger, K.A.; Perry, R.H. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 1996, 47, 1113–1124. [Google Scholar]
- Swerdlow, R.H. Treating neurodegeneration by modifying mitochondria: Potential solutions to a "complex" problem. Antioxid. Redox Signal 2007, 9, 1591–1603. [Google Scholar]
- Swerdlow, R.H. The Neurodegenerative Mitochondriopathies. J. Alzheimers Dis. 2009. [Google Scholar]
- Swerdlow, R.H.; Kish, S.J. Mitochondria in Alzheimer's disease. Int. Rev. Neurobiol. 2002, 53, 341–385. [Google Scholar]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology 1990, 40, 1302–1303. [Google Scholar]
- Swerdlow, R.H. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J. Neurosci. Res. 2007, 85, 3416–3428. [Google Scholar]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Cybrids in Alzheimer's disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; Shimohama, S.; Cash, A.D.; Siedlak, S.L.; Harris, P.L.; Jones, P.K.; Petersen, R.B.; Perry, G.; Smith, M.A. Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; Quinn, J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; Gunn-Moore, F.J.; Vonsattel, J.P.; Arancio, O.; Chen, J.X.; Yan, S.D. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; Trinchese, F.; Liu, S.; Gunn-Moore, F.; Lue, L.F.; Walker, D.G.; Kuppusamy, P.; Zewier, Z.L.; Arancio, O.; Stern, D.; Yan, S.S.; Wu, H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 2004, 304, 448–452. [Google Scholar]
- Anandatheerthavarada, H.K.; Devi, L. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer's disease. Neuroscientist 2007, 13, 626–638. [Google Scholar]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar]
- Gabuzda, D.; Busciglio, J.; Chen, L.B.; Matsudaira, P.; Yankner, B.A. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 1994, 269, 13623–13628. [Google Scholar]
- Khan, S.M.; Cassarino, D.S.; Abramova, N.N.; Keeney, P.M.; Borland, M.K.; Trimmer, P.A.; Krebs, C.T.; Bennett, J.C.; Parks, J.K.; Swerdlow, R.H.; Parker, W.D., Jr.; Bennett, J.P., Jr. Alzheimer's disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann. Neurol. 2000, 48, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar]
- Swerdlow, R.H.; Khan, S.M. The Alzheimer's disease mitochondrial cascade hypothesis: An update. Exp. Neurol. 2009, 218, 308–315. [Google Scholar]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar]
- Arduino, D.M.; Esteves, A.R.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial Metabolism Modulation: A New Therapeutic Approach for Parkinson's Disease. CNS Neurol. Disord. Drug Targets 2009. [Google Scholar]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar]
- Swerdlow, R.H.; Parks, J.K.; Miller, S.W.; Tuttle, J.B.; Trimmer, P.A.; Sheehan, J.P.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Origin and functional consequences of the complex I defect in Parkinson's disease. Ann. Neurol. 1996, 40, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Domingues, A.F.; Ferreira, I.L.; Januario, C.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial function in Parkinson's disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008, 8, 219–228. [Google Scholar]
- Esteves, A.R.; Arduino, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Oxidative Stress involvement in alpha-synuclein oligomerization in Parkinsons disease cybrids. Antioxid. Redox Signal 2008. [Google Scholar]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; Taylor, R.W.; Turnbull, D.M. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar]
- Parker, W.D., Jr.; Parks, J.K. Mitochondrial ND5 mutations in idiopathic Parkinson's disease. Biochem. Biophys. Res. Commun. 2005, 326, 667–669. [Google Scholar]
- Smigrodzki, R.; Parks, J.; Parker, W.D. High frequency of mitochondrial complex I mutations in Parkinson's disease and aging. Neurobiol. Aging 2004, 25, 1273–1281. [Google Scholar]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson's disease. Exp. Neurol. 2009, 218, 235–246. [Google Scholar]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Trimmer, P.A.; Miller, S.W.; Maguire, D.J.; Sheehan, J.P.; Maguire, R.S.; Pattee, G.; Juel, V.C.; Phillips, L.H.; Tuttle, J.B.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Mitochondria in sporadic amyotrophic lateral sclerosis. Exp. Neurol. 1998, 153, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Parks, J.K.; Pattee, G.; Parker, W.D., Jr. Role of mitochondria in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 185–190. [Google Scholar]
- Albers, D.S.; Swerdlow, R.H.; Manfredi, G.; Gajewski, C.; Yang, L.; Parker, W.D., Jr.; Beal, M.F. Further evidence for mitochondrial dysfunction in progressive supranuclear palsy. Exp. Neurol. 2001, 168, 196–198. [Google Scholar]
- Swerdlow, R.H.; Golbe, L.I.; Parks, J.K.; Cassarino, D.S.; Binder, D.R.; Grawey, A.E.; Litvan, I.; Bennett, J.P., Jr.; Wooten, G.F.; Parker, W.D. Mitochondrial dysfunction in cybrid lines expressing mitochondrial genes from patients with progressive supranuclear palsy. J. Neurochem. 2000, 75, 1681–1684. [Google Scholar]
- Browne, S.E.; Beal, M.F. The energetics of Huntington's disease. Neurochem. Res. 2004, 29, 531–546. [Google Scholar]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar]
- Koutnikova, H.; Campuzano, V.; Foury, F.; Dolle, P.; Cazzalini, O.; Koenig, M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 1997, 16, 345–351. [Google Scholar]
- Pandolfo, M. Friedreich ataxia. Arch. Neurol. 2008, 65, 1296–1303. [Google Scholar]
- Luft, R. The development of mitochondrial medicine. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 8731–8738. [Google Scholar]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar]
- Aliev, G.; Liu, J.; Shenk, J.C.; Fischbach, K.; Pacheco, G.J.; Chen, S.G.; Obrenovich, M.E.; Ward, W.F.; Richardson, A.G.; Smith, M.A.; Gasimov, E.; Perry, G.; Ames, B.N. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J. Cell Mol. Med. 2009, 13, 320–333. [Google Scholar]
- Rosca, M.G.; Lemieux, H.; Hoppel, C.L. Mitochondria in the elderly: Is acetylcarnitine a rejuvenator? Adv. Drug Deliv. Rev. 2009. [Google Scholar]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F., Jr. Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51, 241–247. [Google Scholar] [PubMed]
- Adhihetty, P.J.; Beal, M.F. Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. Neuromolecular Med. 2008, 10, 275–290. [Google Scholar]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar]
- Crompton, M.; Virji, S.; Doyle, V.; Johnson, N.; Ward, J.M. The mitochondrial permeability transition pore. Biochem. Soc. Symp. 1999, 66, 167–179. [Google Scholar]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar]
- Haller, R.G.; Vissing, J. Drilling for energy in mitochondrial disease. Arch. Neurol. 2009, 66, 931–932. [Google Scholar]
- Mestre, T.; Ferreira, J.; Coelho, M.M.; Rosa, M.; Sampaio, C. Therapeutic interventions for disease progression in Huntington's disease. Cochrane Database Syst. Rev. 2009, CD006455. [Google Scholar]
- Swerdlow, R.; Marcus, D.M.; Landman, J.; Harooni, M.; Freedman, M.L. Brain glucose and ketone body metabolism in patients with Alzheimer's disease. Clin. Res. 1989, 37, 461A. [Google Scholar]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. (Lond) 2009, 6, 31. [Google Scholar]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; Kompoliti, K.; Perlmutter, J.S.; Reich, S.; Stern, M.; Watts, R.L.; Kurlan, R.; Molho, E.; Harrison, M.; Lew, M. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Pandolfo, M. Antioxidants and other pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2009, CD007791. [Google Scholar]
- Mariotti, C.; Solari, A.; Torta, D.; Marano, L.; Fiorentini, C.; Di Donato, S. Idebenone treatment in Friedreich patients: One-year-long randomized placebo-controlled trial. Neurology 2003, 60, 1676–1679. [Google Scholar]
- Stamelou, M.; Reuss, A.; Pilatus, U.; Magerkurth, J.; Niklowitz, P.; Eggert, K.M.; Krisp, A.; Menke, T.; Schade-Brittinger, C.; Oertel, W.H.; Hoglinger, G.U. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: A randomized, placebo-controlled trial. Mov. Disord. 2008, 23, 942–949. [Google Scholar]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 2002, 287, 3223–3229. [Google Scholar]
- Engelhart, M.J.; Ruitenberg, A.; Meijer, J.; Kiliaan, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Plasma levels of antioxidants are not associated with Alzheimer's disease or cognitive decline. Dement. Geriatr. Cogn. Disord. 2005, 19, 134–139. [Google Scholar]
- Boothby, L.A.; Doering, P.L. Vitamin C and vitamin E for Alzheimer's disease. Ann. Pharmacother. 2005, 39, 2073–2080. [Google Scholar]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; Schneider, L.S.; Thal, L.J. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [PubMed]
- Doody, R.S.; Stevens, J.C.; Beck, C.; Dubinsky, R.M.; Kaye, J.A.; Gwyther, L.; Mohs, R.C.; Thal, L.J.; Whitehouse, P.J.; DeKosky, S.T.; Cummings, J.L. Practice parameter: Management of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001, 56, 1154–1166. [Google Scholar] [PubMed]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8665–8670. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar]
- Zhu, S.; Stavrovskaya, I.G.; Drozda, M.; Kim, B.Y.; Ona, V.; Li, M.; Sarang, S.; Liu, A.S.; Hartley, D.M.; Wu, D.C.; Gullans, S.; Ferrante, R.J.; Przedborski, S.; Kristal, B.S.; Friedlander, R.M. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002, 417, 74–78. [Google Scholar]
- Gordon, P.H.; Moore, D.H.; Miller, R.G.; Florence, J.M.; Verheijde, J.L.; Doorish, C.; Hilton, J.F.; Spitalny, G.M.; MacArthur, R.B.; Mitsumoto, H.; Neville, H.E.; Boylan, K.; Mozaffar, T.; Belsh, J.M.; Ravits, J.; Bedlack, R.S.; Graves, M.C.; McCluskey, L.F.; Barohn, R.J.; Tandan, R. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomised trial. Lancet Neurol. 2007, 6, 1045–1053. [Google Scholar]
- Finkel, T. Reactive oxygen species and signal transduction. IUBMB Life 2001, 52, 3–6. [Google Scholar]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar]
- Armstrong, J.S. Mitochondrial medicine: Pharmacological targeting of mitochondria in disease. Br. J. Pharmacol. 2007, 151, 1154–1165. [Google Scholar]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer's disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer's disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [PubMed]
- Bachurin, S.O.; Shevtsova, E.P.; Kireeva, E.G.; Oxenkrug, G.F.; Sablin, S.O. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann. NY Acad. Sci. 2003, 993, 334–344, discussion 345-339. [Google Scholar]
- Navarro, A.; Boveris, A. The mitochondrial energy transduction system and the aging process. Am. J. Physiol. Cell Physiol. 2007, 292, C670–C686. [Google Scholar]
- Boveris, A.; Navarro, A. Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free Radic. Biol. Med. 2008, 44, 224–229. [Google Scholar]
- Civitarese, A.E.; Smith, S.R.; Ravussin, E. Diet, energy metabolism and mitochondrial biogenesis. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 679–687. [Google Scholar]
- Fernstrom, M.; Tonkonogi, M.; Sahlin, K. Effects of acute and chronic endurance exercise on mitochondrial uncoupling in human skeletal muscle. J. Physiol. 2004, 554, 755–763. [Google Scholar]
- Hepple, R.T. Why eating less keeps mitochondria working in aged skeletal muscle. Exerc. Sport Sci. Rev. 2009, 37, 23–28. [Google Scholar]
- Holloszy, J.O. Adaptation of skeletal muscle to endurance exercise. Med. Sci. Sports 1975, 7, 155–164. [Google Scholar]
- Hood, D.A. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl. Physiol. Nutr. Metab. 2009, 34, 465–472. [Google Scholar]
- Hoppeler, H.; Fluck, M. Plasticity of skeletal muscle mitochondria: Structure and function. Med. Sci. Sports Exerc. 2003, 35, 95–104. [Google Scholar]
- Lambert, A.J.; Wang, B.; Yardley, J.; Edwards, J.; Merry, B.J. The effect of aging and caloric restriction on mitochondrial protein density and oxygen consumption. Exp. Gerontol. 2004, 39, 289–295. [Google Scholar]
- Weindruch, R.; Sohal, R.S. Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. N. Engl. J. Med. 1997, 337, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009, 8, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins-emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Spindler, S.R. Biological effects of calorie restriction: From soup to nuts. Ageing Res. Rev. 2009. [Google Scholar]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar]
- Norrbom, J.; S, E.W.; Gustafsson, T.; Rundqvist, H.; Jansson, E.; Sundberg, C. Training response of mitochondrial transcription factors in human skeletal muscle. Acta Physiol. (Oxf) 2009.
- McGinley, C.; Shafat, A.; Donnelly, A.E. Does Antioxidant Vitamin Supplementation Protect against Muscle Damage? Sports Med. 2009, 39, 1011–1032. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Romagnoli, M.; Arduini, A.; Borras, C.; Pallardo, F.V.; Sastre, J.; Vina, J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 2008, 87, 142–149. [Google Scholar]
- Onyango, I.G.; Lu, J.; Rodova, M.; Lezi, E.; Crafter, A.B.; Swerdlow, R.H. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta 2009. [Google Scholar]
- Ji, L.L.; Gomez-Cabrera, M.C.; Vina, J. Exercise and hormesis: Activation of cellular antioxidant signaling pathway. Ann. N. Y. Acad. Sci. 2006, 1067, 425–435. [Google Scholar]
- Anekonda, T.S.; Reddy, P.H. Neuronal protection by sirtuins in Alzheimer's disease. J. Neurochem. 2006, 96, 305–313. [Google Scholar]
- Guarente, L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 483–488. [Google Scholar]
- Harikumar, K.B.; Aggarwal, B.B. Resveratrol: A multitargeted agent for age-associated chronic diseases. Cell Cycle 2008, 7, 1020–1035. [Google Scholar]
- Bogacka, I.; Xie, H.; Bray, G.A.; Smith, S.R. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 2005, 54, 1392–1399. [Google Scholar]
- Ghosh, S.; Patel, N.; Rahn, D.; McAllister, J.; Sadeghi, S.; Horwitz, G.; Berry, D.; Wang, K.X.; Swerdlow, R.H. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol. Pharmacol. 2007, 71, 1695–1702. [Google Scholar]
- Strum, J.C.; Shehee, R.; Virley, D.; Richardson, J.; Mattie, M.; Selley, P.; Ghosh, S.; Nock, C.; Saunders, A.; Roses, A. Rosiglitazone induces mitochondrial biogenesis in mouse brain. J. Alzheimers Dis. 2007, 11, 45–51. [Google Scholar]
- Risner, M.E.; Saunders, A.M.; Altman, J.F.; Ormandy, G.C.; Craft, S.; Foley, I.M.; Zvartau-Hind, M.E.; Hosford, D.A.; Roses, A.D. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006, 6, 246–254. [Google Scholar]
- Ferguson, E.C.; Rathmell, J.C. New roles for pyruvate kinase M2: Working out the Warburg effect. Trends Biochem. Sci. 2008, 33, 359–362. [Google Scholar]
- Feron, O. Pyruvate into lactate and back: From the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Swerdlow, R.H. Mitochondrial Medicine and the Neurodegenerative Mitochondriopathies. Pharmaceuticals 2009, 2, 150-167. https://doi.org/10.3390/ph2030150
Swerdlow RH. Mitochondrial Medicine and the Neurodegenerative Mitochondriopathies. Pharmaceuticals. 2009; 2(3):150-167. https://doi.org/10.3390/ph2030150
Chicago/Turabian StyleSwerdlow, Russell H. 2009. "Mitochondrial Medicine and the Neurodegenerative Mitochondriopathies" Pharmaceuticals 2, no. 3: 150-167. https://doi.org/10.3390/ph2030150
APA StyleSwerdlow, R. H. (2009). Mitochondrial Medicine and the Neurodegenerative Mitochondriopathies. Pharmaceuticals, 2(3), 150-167. https://doi.org/10.3390/ph2030150